

Abstract
Biochemical studies of purified and dissected fungal polyketide synthase and nonribosomal peptide synthetase (PKS-NRPS) hybrid enzymes involved in biosynthesis of pseurotin and aspyridone indicate that one α-methylation step during polyketide synthesis is a prerequisite and a key checkpoint for chain transfer between PKS and NRPS modules. In the absence of the resulting γ-methyl feature, the completed polyketide intermediate is offloaded as an α-pyrone instead of being aminoacylated by the NRPS domain. These examples illustrate that precisely timed tailoring domain activities play critical roles in the overall programming of the iterative PKS (and NRPS) functions.
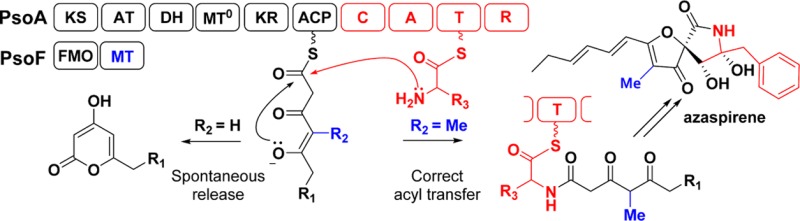
Fungal polyketide synthase-nonribosomal peptide synthetase (PKS-NRPS) are large (∼450 kDa) and multidomain (∼10 catalytic domains) enzymes that synthesize complex natural products with incompletely resolved programming rules.1 Products of fungal PKS-NRPS pathways are exemplified by pseurotin A (1),2 azaspirene (2),3 aspyridone,4 fusarin C,5 and tenellin6 (Figure 1a). The N-terminal PKS portion is a highly reducing PKS (HRPKS) in which a single set of catalytic domains operates iteratively to synthesize a reduced polyketide. The polyketide chain is transferred to the C-terminal NRPS module that condenses the acyl chain with an amino acid to yield an aminoacylated adduct that can be subjected to further downstream transformations.7 Since the lone acyl carrier protein (ACP) of PKS is loaded with different acyl groups during the iterative PKS functions, timely and correct recognition of the polyketide substrate by the condensation (C) domain of the NRPS is crucial to ensure product fidelity.8 Dissecting the mechanisms of acyl intermediate transfer between PKS and NRPS is important in advancing our understanding of this enigmatic family of megasynthetases and can enable reprogramming for biosynthetic applications.
Figure 1.

Methylation-dependent acyltransfer by fungal PKS-NRPS. (a) Selected γ-methylation containing polyketide–nonribosomal peptide natural products produced from fungi. The final position of the γ-methyl group in the polyketide portion is boxed. (b) Proposed biosynthetic pathway of azaspirene 2 and preaspyridone 3 illustrating the key γ-methyl containing intermediates 4 and 9, respectively. The KS domain of PsoA and ApdA use propionyl-CoA and malonyl-CoA as the starter unit to initiate polyketide synthesis, respectively. Domains labeled with 0 are nonfunctional and are complemented with in trans partners. The white arrow represents one round of Claisen-like chain elongation catalyzed by the KS domain.
Pseurotin A (1) is an immunosuppressive spirocyclic compound produced by Aspergillus fumigatus.2b The biosynthesis of 1 proceeds via the key intermediate azaspirene (2),2b,3b itself an angiogenesis inhibitor (Figure 1b). Compound 2 was proposed to be derived from the intermediate 5, which is formed when a γ-methyltriketodiene pentaketide chain 4 is condensed with l-phenylalanine (l-Phe) and reductively released from PsoA.2b The biosynthetic pathway of 1 was uncovered by genetic knockouts and intermediate analyses,2b,3b,9 which revealed the intriguing presence of a bifunctional flavin monooxygenase (FMO)-methyltransferase (MT) enzyme PsoF. Using a β-oxo-acyl-S-N-acetylcysteamine (S-NAC) mimic, PsoF was suggested to methylate the polyketide chain in trans to the PsoA, the PKS-NRPS of the pathway.3b The in trans modification, which becomes the γ-methyl substituent of the completed polyketide chain 4, appears to be important for the production of 1 and 2, as inactivation of PsoF led to 400-fold decrease in titer of the corresponding, desmethyl version of 1 in A. fumigatus.3b,9 Therefore, we hypothesized that the MT domain function of PsoF may be important for acyl transfer between the PKS and NRPS modules of PsoA and overall turnover of PsoA.
To investigate the programming rule of PsoA and associated enzymes, we reconstituted the biosynthesis of 2 in Saccharomyces cerevisiae strain BJ5464-NpgA10 through the coexpression of PsoA, PsoF, a hydrolase involved in product release PsoB, and a membrane-bound oxidase PsoG. These four enzymes were proposed to be required for the formation of 2.3b Extraction of a three-day yeast culture revealed the production of 2 (m/z 370 [M + H]+) when compared to the authentic standard (Figure 2a). Removal of PsoG, which is responsible for hydroxylation of the spirocycle to yield 2, led to the production of two compounds 7 and 8 with the same molecular formula (C21H23NO4, m/z 354 [M + H]+, Figures S2 and S3, Supporting Information), both of which have similar UV absorption spectra as 2. Although the structures of these compounds were not determined due to low abundance, we putatively assigned one of the two compounds to be the spirocyclic compound as shown in Figure 1b. This intermediate can be derived from the 5-exo epoxide opening of 6. Formation of the epoxide intermediate 6 from the tetramic acid intermediate 5 is proposed to be catalyzed by the FMO domain of PsoF. Removal of PsoF or excision of the MT domain of PsoF led to complete loss of product accumulation at selected ion monitoring detection limits (Figure S2, iv and v, Supporting Information). The yeast reconstitution experiment is therefore consistent with the genetic analyses in A. fumigatus and demonstrates that recombinant PsoA retained the expected programming rules.
Figure 2.

Analysis of PsoA functions in S. cerevisiae and in vitro. (a) ESI-MS analyses of reconstitution of azaspirene 2 biosynthesis in BJ5464-NpgA: (i) untransformed BJ5464-NpgA control; (ii) BJ5464-NpgA transformed with vectors that encode PsoA, PsoB, PsoF, and PsoG; (iii) authentic standard of 2. (b) LC–MS analyses of in vitro reconstitution of PsoA, PsoF, and PsoB with selected cofactors and building blocks. All assays are performed with PsoA, NADPH, and malonyl-CoA in the presence of (i) propionyl-CoA; (ii) no additional substrate; (iii) propionyl-CoA, l-Phe and ATP; (iv) propionyl-CoA, PsoF and SAM; (v) propionyl-CoA, PsoF, l-Phe and ATP; (vi) propionyl-CoA, PsoF, SAM, l-Phe and ATP; and (vii) propionyl-CoA, PsoF, SAM, l-Phe, ATP and PsoB. (c) Pyrone products 10–12 released from PsoA.
The yeast reconstitution results also confirmed a distinguishing biochemical feature of PsoA compared to other known PKS-NRPS, which is the incorporation of a propionyl starter unit that is reflected in 1 and 2.11 All characterized fungal HRPKSs are primed with acetyl unit which can either be derived from acetyl-CoA directly or through the decarboxylation of malonyl-CoA.10a The incorporation of a propionyl starter unit therefore points to unexpected level of programming rule by PsoA. In other PKS systems where different starter units are incorporated, additional enzymatic machineries, such as starter unit acyltransferase, are typically present to facilitate the priming step.12 To examine this unusual feature of PsoA, as well as the importance of γ-methyl in 4 on PsoA function, the intact PKS-NRPS (436 kDa) was purified to homogeneity at 3 mg/L (Figure S1, Supporting Information) and subjected to assays in the presence of different combinations of substrates, cofactors, and accessory enzymes.
We first examined the effect of various alkyl acyl-CoA on the priming and catalytic iterations of PsoA. In the absence of l-Phe that is required for acyl transfer to the NRPS module, we expected the 1,3,5-triketopentaketide 4 to be cyclized and released as a diene-containing α-pyrone. In an assay with 0.5 mM propionyl-CoA, 1 mM malonyl-CoA, and 2 mM NADPH, we observed efficient production of an unsaturated α-pyrone 10 (m/z 193 [M + H]+, Figure 2b, i). The λmax (344 nm) of 10 is identical to that of 11, which was previously characterized and structurally verified.4a,10a The incorporation of four malonyl-extender units in 10 was verified through the incorporation of [2-13C]-malonyl-CoA and the corresponding increase in mass of 10 by 4 amu (Figure S4, Supporting Information). This suggested that PsoA alone is able to incorporate the propionyl starter unit. The formation of 10 also shows that unmethylated tetraketide can be chain-extended by the PKS module to form γ-desmethyl version of 4. Hence this methylation step has no direct effect on the functions of the PKS module. When propionyl-CoA was removed, the production of 10 was replaced with 11, albeit with significantly lower turnover (Figure 2b, ii). Incubation of PsoA with acetyl-, isobutyryl-, hexanoyl-, and octanoyl-CoA similarly led to the production of differently primed pyrone products, albeit all at much lower yields compared to that of 10 (Figures S4 and S5, Supporting Information). A competition assay of PsoA in the presence of equimolar amounts of different acyl-CoAs led to the near-exclusive formation of 10 (Figure S4, Supporting Information), confirming that PsoA indeed preferentially uses the C3 starter unit to initial polyketide synthesis.
Intriguingly, additional l-Phe to the reaction with propionyl-CoA still resulted in the formation of 10 (Figure 2b, iii), and no phenylalanine-containing product was formed by PsoA. This points to the failure of the NRPS module to recognize the polyketide product that is unmethylated at the γ-position. Base hydrolysis or inclusion of PsoB, a hydrolase that was implicated to aid the release of aldehyde product, similarly did not lead to detection of any aminoacylated products. This is consistent with our initial hypothesis that the γ-methyl group present in the PKS product 4 may be essential in facilitating chain transfer between the PKS and NRPS modules.
We then examined the effect of including PsoF in the reaction. In the absence of l-Phe, addition of PsoF and SAM (1 mM) to the assay led to the production of a new pyrone 12 (Figure 2b, iv), which has the same λmax as 10, but with an increase in molecular weight of 14 mu that is consistent with the incorporation of an additional methyl group. MS/MS fragmentation analysis supports 12 as the 4-methylpyrone product (Figure S6, Supporting Information), which can be spontaneously cyclized from the PKS product 4. When l-Phe was also included, we observed the emergence of a pair of compounds that has the same retention times and masses as 7 and 8 observed from the in vivo studies extracts (Figure 2b, vi, and Figure S2, Supporting Information). Further inclusion of PsoB led to the production of 7 and 8 as the dominant products of the in vitro assay (Figure 2b, vii), suggesting that the PsoA enzymatic machinery is fully functional under the in vitro conditions. Due to difficulties associated with reconstituting the integral membrane protein PsoG in vitro, we were not able to achieve the complete biosynthesis of 2 in vitro.
The PsoA assays therefore strongly indicate methylation by PsoF on the polyketide precursor is critical in chain transfer to the NRPS module. To prove this, a direct assay with the NRPS module using the S-NAC version of 4 would be unambiguous.13 However, the 1,3,5-triketo moiety in 4 makes the acyl-S-NAC derivative intrinsically unstable and can rapidly undergo pyrone formation to yield 12. In the absence of proper substrates to interrogate NRPS gatekeeping functions, we turned to using the ApdA PKS-NRPS from the aspyridone pathway4 to examine whether γ-methyl dependent chain transfer is present in other fungal PKS-NRPSs.
ApdA synthesizes the tetramic acid preaspyridone 3 en route to aspyridone in A. nidulans (Figure 1b).4 The function of PKS-NRPS was previously reconstituted in vitro in which we showed the PKS module synthesizes a 4,6-dimethyl-3-oxooctanoyl 9 precursor that is aminoacylated with l-tyrosine by the NRPS module. We showed that similar to PsoA, exclusion of SAM (hence eliminating the MT function) did not lead to unmethylated version of 3.4a Given that the acyl-S-NAC compounds mimicking 9 are synthetically accessible, the ApdA NRPS serves as an excellent model system for confirming the γ-methyl dependent acyl transfer hypothesized for the PsoA and ApdA systems.
We tested the acyl transfer with a panel of linear (13a–c)14 and β-oxoacyl-γ-methyl-S-NAC substrates (14a–c) ranging in size between C7 and C9 (Figure 3) as variants of 9, the natural substrate of ApdA NRPS. Compounds 14a–c were synthesized from α-methyl carboxylic acids, via the corresponding α-methylacyl-Meldrum’s acids (Supporting Information, Figure S7–9).15 Each substrate was supplied to the recombinant ApdA NRPS module together with l-tyrosine and ATP. Each of 14a–c supported the synthesis of a new tetramic acid product (15a–c) that has the same UV absorption as 3 (Figure 3, i–iii; Figure S10, Supporting Information). The different retention times of the products are also consistent with the chain length of the supplied precursors. In contrast, no tetramic acid product was observed when the linear precursors 13a–c were used (Figure 3, iv–vi). These results therefore confirmed that the γ-methyl present in 9 is essential for substrate recognition by the ApdA NRPS module, a property that may be shared by the PsoA NRPS toward 4. The ApdA NRPS, however, is not sensitive to the ε-methyl substitution present in the natural substrate 9, thereby pinpointing the gatekeeping recognition to the γ position. Although the NRPS displays preferential recognition toward the substrate 14b that is the same chain length as 9, overall the NRPS displayed considerable chain length tolerances, converting both the shorter C7 substrate 14c and the longer C9 substrate 14a to the corresponding tetramic acid product. The conversion of shorter methylated triketide (C6) substrate was not observed. This is consistent with the intact ApdA assay, where no acyl transfer of the earlier intermediate to the NRPS module was observed.4a
Figure 3.

LC–MS analyses of the reactions of ApdA NRPS module supplied with various S-NAC substrates. (i–iii) Tetramic acid products were detected when β-ketoacyl-γ-methyl S-NAC (14a–c) were supplied as substrates; (iv–vi) no product was formed when β-ketoacyl S-NAC (13a–c) were used as substrates.
The in vitro reconstitution results with both PsoA and ApdA point to a strict gatekeeping mechanism by their NRPS modules. Specifically the condensation (C) domains maintain fidelity of these megasynthases to ensure desired methyl substituent is installed during PKS function. In the absence of γ-methyl moiety, the polyketide chain is stalled on the ACP domain and released as the pyrone spontaneously. The methylation-dependent programming rule is analogous to that found in LovB megasynthase, in which a downstream enoylreducase LovC requires methylation of the triketide to function properly.9 Failure of LovC to function on the unmethylated substrate leads to product offloading and premature release.9 It remains unclear, however, whether the methylation steps are programmed to serve as biochemical checkpoints or if the resulting methyl groups play essential roles in natural product functions.
Acknowledgments
This work was supported by the National Institutes of Health (1DP1GM106413 to Y.T.) and the Japan Society for the Promotion of Science (JSPS) (grant to K.W.). NMR instrumentation was supported by the NSF equipment grant CHE-1048804.
Supporting Information Available
Experimental details and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∥ Y.Z. and W.X. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Fisch K. M. RSC Adv. 2013, 3, 18228. [Google Scholar]; b Boettger D.; Hertweck C. ChemBioChem 2013, 14, 28. [DOI] [PubMed] [Google Scholar]; c Cox R. J. Org. Biomol. Chem. 2007, 5, 2010. [DOI] [PubMed] [Google Scholar]; d Chooi Y. H.; Tang Y. J. Org. Chem. 2012, 77, 9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bloch P.; Tamm C.; Bollinger P.; Petcher T. J.; Weber H. P. Helv. Chim. Acta 1976, 59, 133. [DOI] [PubMed] [Google Scholar]; b Maiya S.; Grundmann A.; Li X.; Li S. M.; Turner G. ChemBioChem 2007, 8, 1736. [DOI] [PubMed] [Google Scholar]
- a Asami Y.; Kakeya H.; Onose R.; Yoshida A.; Matsuzaki H.; Osada H. Org. Lett. 2002, 4, 2845. [DOI] [PubMed] [Google Scholar]; b Tsunematsu Y.; Fukutomi M.; Saruwatari T.; Noguchi H.; Hotta K.; Tang Y.; Watanabe K. Angew. Chem., Int. Ed. 2014, 53, 8475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Xu W.; Cai X.; Jung M. E.; Tang Y. J. Am. Chem. Soc. 2010, 132, 13604. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bergmann S.; Schumann J.; Scherlach K.; Lange C.; Brakhage A. A.; Hertweck C. Nat. Chem. Biol. 2007, 3, 213. [DOI] [PubMed] [Google Scholar]; c Wasil Z.; Pahirulzaman K. A. K.; Butts C.; Simpson T. J.; Lazarus C. M.; Cox R. J. Chem. Sci. 2013, 4, 3845. [Google Scholar]
- a Gelderblom W. C.; Jaskiewicz K.; Marasas W. F.; Thiel P. G.; Horak R. M.; Vleggaar R.; Kriek N. P. Appl. Environ. Microbiol. 1988, 54, 1806. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Song Z.; Cox R. J.; Lazarus C. M.; Simpson T. T. ChemBioChem 2004, 5, 1196. [DOI] [PubMed] [Google Scholar]
- a Eley K. L.; Halo L. M.; Song Z. S.; Powles H.; Cox R. J.; Bailey A. M.; Lazarus C. M.; Simpson T. J. ChemBioChem 2007, 8, 289. [DOI] [PubMed] [Google Scholar]; b Mcinnes A. G.; Smith D. G.; Wat C. K.; Vining L. C.; Wright J. L. C. J. Chem. Soc., Chem. Commun. 1974, 281. [Google Scholar]
- a Halo L. M.; Heneghan M. N.; Yakasai A. A.; Song Z.; Williams K.; Bailey A. M.; Cox R. J.; Lazarus C. M.; Simpson T. J. J. Am. Chem. Soc. 2008, 130, 17988. [DOI] [PubMed] [Google Scholar]; b Liu X. Y.; Walsh C. T. Biochemistry 2009, 48, 8746. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kakule T. B.; Sardar D.; Lin Z. J.; Schmidt E. W. ACS Chem. Biol. 2013, 8, 1549. [DOI] [PubMed] [Google Scholar]
- a Boettger D.; Bergmann H.; Kuehn B.; Shelest E.; Hertweck C. ChemBioChem 2012, 13, 2363. [DOI] [PubMed] [Google Scholar]; b Kakule T. B.; Lin Z.; Schmidt E. W. J. Am. Chem. Soc. 2014, 10.1021/ja511087p. [DOI] [PubMed] [Google Scholar]; c Fisch K. M.; Bakeer W.; Yakasai A. A.; Song Z.; Pedrick J.; Wasil Z.; Bailey A. M.; Lazarus C. M.; Simpson T. J.; Cox R. J. J. Am. Chem. Soc. 2011, 133, 16635. [DOI] [PubMed] [Google Scholar]
- Wiemann P.; Guo C. J.; Palmer J. M.; Sekonyela R.; Wang C. C. C.; Keller N. P. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 17065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ma S. M.; Li J. W.; Choi J. W.; Zhou H.; Lee K. K.; Moorthie V. A.; Xie X.; Kealey J. T.; Da Silva N. A.; Vederas J. C.; Tang Y. Science 2009, 326, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lee K. K.; Da Silva N. A.; Kealey J. T. Anal. Biochem. 2009, 394, 75. [DOI] [PubMed] [Google Scholar]
- Mohr P.; Tamm C. Tetrahedron 1981, 37Supplement 1201. [Google Scholar]
- a Crawford J. M.; Dancy B. C. R.; Hill E. A.; Udwary D. W.; Townsend C. A. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 16728. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Crawford J. M.; Vagstad A. L.; Ehrlich K. C.; Townsend C. A. Bioorg. Chem. 2008, 36, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Waldman A. J.; Balskus E. P. Org. Lett. 2014, 16, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Bao W. L.; Sheldon P. J.; Wendt-Pienkowsi E.; Hutchinson C. R. J. Bacteriol. 1999, 181, 4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- We acknowledge that the in trans assay using small molecules may not fully represent the in cis interactions present in intact systems. However, the direct assay with acyl-SNAC can still provide a measure of the relative substrate specificity of the C domain towards different acyl chains.
- Zhou H.; Gao Z.; Qiao K.; Wang J.; Vederas J. C.; Tang Y. Nat. Chem. Biol. 2012, 8, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Oikawa Y.; Sugano K.; Yonemitsu O. J. Org. Chem. 1978, 43, 2087. [Google Scholar]; b Arthur C.; Cox R. J.; Crosby J.; Rahman M. M.; Simpson T. J.; Soulas F.; Spogli R.; Szafranska A. E.; Westcott J.; Winfield C. J. ChemBioChem 2002, 3, 253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.