

Abstract
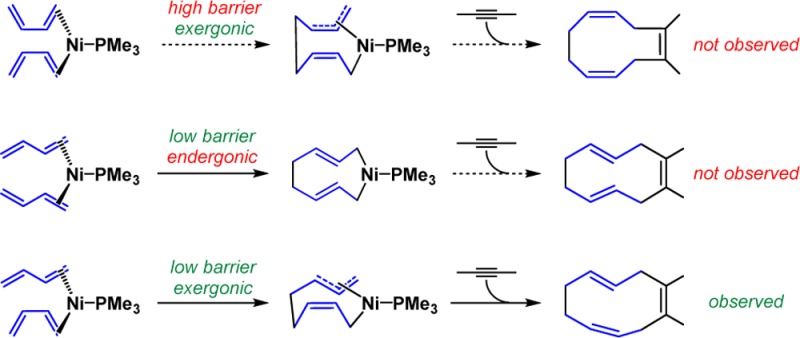
Density functional theory (DFT) calculations with B3LYP and M06 functionals elucidated the reactivities of alkynes and Z/E selectivity of cyclodecatriene products in the Ni-catalyzed [4 + 4 + 2] cycloadditions of dienes and alkynes. The Ni-mediated oxidative cyclization of butadienes determines the Z/E selectivity. Only the oxidative cyclization of one s-cis to one s-trans butadiene is facile and exergonic, leading to the observed 1Z,4Z,8E-cyclodecatriene product. The same step with two s-cis or s-trans butadienes is either kinetically or thermodynamically unfavorable, and the 1Z,4E,8E- and 1Z,4Z,8Z-cyclodecatriene isomers are not observed in experiments. In addition, the competition between the desired cooligomerization and [2 + 2 + 2] cycloadditions of alkynes depends on the coordination of alkynes. With either electron-deficient alkynes or alkynes with free hydroxyl groups, the coordination of alkynes is stronger than that of dienes, and alkyne trimerization prevails. With alkyl-substituted alkynes, the generation of alkyne-coordinated nickel complex is much less favorable, and the [4 + 4 + 2] cycloaddition occurs.
Introduction
As an ubiquitous and important family of molecules, terpenes, provide flavors, fragrances, medicines, and commercial products.1 Although terpenes possess a seemingly endless variety of architectural complexities, nature is able to utilize simple five-carbon moieties to build the tens of thousands of different members of the terpene family.2 The biosynthesis of terpenes often occurs in a unified fashion as a “two-phase” process:3 (1) in the “cyclase” phase, small linear hydrocarbon phosphate building blocks are coupled together, followed by both enzymatic and nonenzymatic cyclizations and rearrangements; (2) in the “oxidase” phase, the oxidations of alkenes and carbon–hydrogen bonds result in large structural diversity.
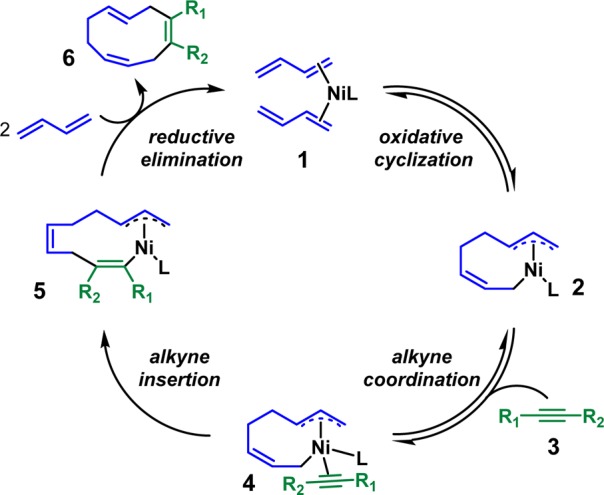
Inspired by the early work on Ni-catalyzed diene oligomerizations from Wilke,4 Heimbach,5 and others,6 the Baran group proposed a Ni-catalyzed diene/alkyne cooligomerization that could mimic Nature’s cyclase approach to terpenes (Scheme 1).7 From the nickel–butadiene complex 1, the oxidative cyclization of dienes gives the nine-membered ring intermediate 2. This intermediate, 2, undergoes alkyne insertion to produce the 11-membered ring intermediate 5. Subsequent reductive elimination generates the desired 10-membered ring product 6.
Scheme 1. Proposed Ni-Catalyzed Diene/Alkyne Cooligomerization.
Extensive experimental explorations of ligand, substrate, and other conditions led to successive butadiene/alkyne cooligomerizations with intriguing reactivities and selectivities:7 (i) only the 1Z,4Z,8E-cyclodecatriene is generated, while the 1Z,4E,8E- and 1Z,4Z,8Z-isomers are not observed (Scheme 2a); (ii) the [2 + 2 + 2] cycloadditions of alkynes are competitive with electron-deficient alkynes or alkynes with free hydroxyl group (Scheme 2b, 2c).8 In order to contribute to the future development of this methodology,9 we explored the mechanism, reactivities, and selectivities of the Ni-catalyzed diene/alkyne cooligomerization with density functional theory (DFT) calculations.10
Scheme 2. Selected Examples of Ni-Catalyzed Butadiene/Alkyne Cooligomerization and Competitive [2 + 2 + 2] Cycloadditions of Alkynes.

Computational Details
Geometry optimizations, vibrational frequencies, and thermal energy corrections were performed with the B3LYP functional, and a 6-31G(d) basis set for all main group elements and SDD basis set for nickel, as implemented in Gaussian 09.11 Energies were evaluated with the M06 method12 and a 6-311+G(2d,p) basis set for all main group elements and SDD basis set for nickel. All reported free energies involve zero-point vibrational energy corrections and thermal corrections to Gibbs free energy at 298 K.13 Extensive conformational searches have been conducted to make sure that the most stable conformers are located, and only the most stable conformers are discussed in this work.
Results and Discussion
1. Mechanism of Butadiene/Alkyne Cooligomerization
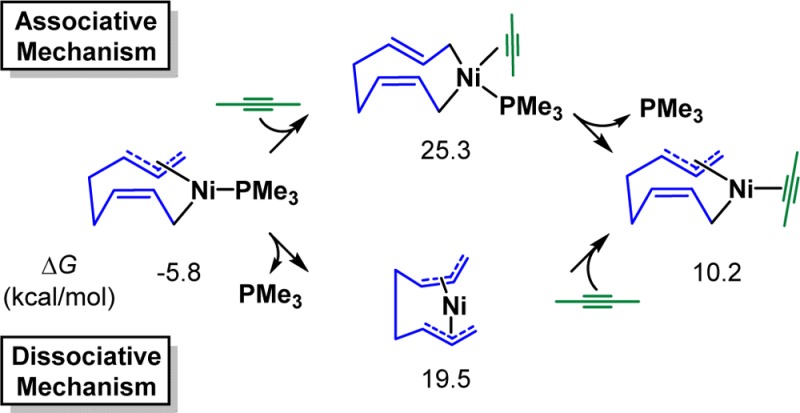
The mechanism of Ni-catalyzed diene/alkyne cooligomerization of butadiene and 2-butyne as the model substrates and PMe3 as the model ligand was first explored.14 The calculated free energy profile of the productive pathway is shown in Figure 1, and optimized structures of selected intermediates and transition states are shown in Figure 2. From the [(PMe3)Ni(butadiene)2] complex 15, the facile oxidative cyclization of one s-cis and one s-trans butadiene via TS16 generates the nine-membered ring intermediate 17 (see later text for discussions on Z/E selectivity).15 The coordination of 2-butyne produces the unstable tetracoordinate intermediate 18, and the subsequent alkyne insertion via TS19 is very unfavorable with a 45.4 kcal/mol overall barrier with respect to the intermediate 17. Alternatively, a ligand exchange between 2-butyne and PMe3 occurs to produce the intermediate 20.16 Despite the endergonic ligand exchange, the alkyne insertion via TS21 only requires a 25.5 kcal/mol barrier with respect to 17, generating the 11-membered ring intermediate 22. From 22, the reductive elimination through TS23 is unfavorable with a barrier of 31.2 kcal/mol. The alternative reductive elimination with PMe3 coordination via TS25 is much more favorable, producing the cyclodecatriene-coordinated nickel complex 26, and the product liberation regenerates the butadiene–nickel complex 15. Therefore, the rate-determining step of the catalytic cycle is the alkyne insertion and the overall reaction barrier is 25.5 kcal/mol from the resting state 17 to the alkyne insertion transition state TS21. In addition, the ligand dissociation and recoordination are essential in this reaction. The phosphine ligand dissociates first in order to undergo a facile alkyne insertion via TS21 and recoordinates to facilitate the reductive elimination through TS25.
Figure 1.

Gibbs free energy profile of [Ni(PMe3)]-catalyzed butadiene/2-butyne cooligomerization (energies computed at the level of M06/6-311+G(2d,p),SDD//B3LYP/6-31G(d),SDD).
Figure 2.

Optimized structures of selected intermediates and transition states of [Ni(PMe3)]-catalyzed butadiene/2-butyne cooligomerization.
2. Origins of Z/E Selectivity
Based on the productive pathway with one s-cis and one s-trans butadienes, we studied the Z/E selectivity of cyclodecatriene products when two s-cis or s-trans butadienes undergo the same cooligomerization with 2-butyne. The reaction with one s-cis and one s-trans butadiene produces the observed 1Z,4Z,8E-cyclodecatriene, and the other two pathways generate the Z/E isomers. The computed free energy profiles are shown in Figure 3. The first step of the catalytic cycle, Ni-mediated oxidative cyclization of butadienes, determines the Z/E selectivity. The oxidative cyclization with two s-cis butadienes (via TS16-cc) requires a 25.7 kcal/mol barrier, making this pathway unfavorable. In addition, the same cyclization with two s-trans butadienes is very endergonic, generating the unstable nine-membered ring intermediate 17-tt. This leads to the 34.6 kcal/mol overall barrier for the subsequent alkyne insertion, making the reaction with two s-trans butadienes unfavorable. Only the oxidative cyclization with one s-cis and one s-trans butadienes is facile and exergonic, leading to a productive pathway.
Figure 3.

Gibbs free energy profiles of [Ni(PMe3)]-catalyzed cooligomerization involving s-cis and s-trans butadienes and 2-butyne (Gibbs free energies in kcal/mol).
Figure 4 shows the optimized structures and relative free energies of the transition states and nine-membered ring intermediates of the Ni-mediated oxidative cyclization step.17 Comparing the transition states, TS16-cc is at least 10.1 kcal/mol less stable than the others. This results from the weak coordination of the dienes in TS16-cc. In order to form the terminal C–C bond in the oxidative cyclization, one of the coordinating double bonds of dienes in TS16-cc is distorted to be perpendicular to the Ni1–C2–P10 plane (highlighted in green in TS16-cc of Figure 4). This distortion significantly weakens the coordination of this double bond and destabilizes the transition state.18 In contrast, when the oxidative cyclization occurs with s-trans butadienes, TS16 and TS16-tt maintain the planar coordination of both dienes and are much more stable than TS16-cc, leading to the achievable barriers. Comparing the generated nine-membered ring intermediates, 17-tt is much less stable than 17 and 17-cc. Because of the two trans-double bonds in the nine-membered ring of 17-tt, this complex cannot maintain the η3 coordination as in the other two intermediates. This makes 17-tt much less stable than 17 and 17-cc. Therefore, only the oxidative cyclization with one s-cis and one s-trans butadienes is facile and exergonic, leading to the productive pathway.
Figure 4.

Optimized structures and relative free energies (transition states are compared to TS16, and intermediates are compared to 17) of transition states and generated intermediates of [Ni(PMe3)]-mediated oxidative cyclization of s-cis and s-trans butadienes.
3. Competition with Alkyne Trimerization
We also studied the competing [2 + 2 + 2] cycloadditions of alkynes, using 2-butyne as the model substrate.19,20 The free energies for the most favorable pathway are shown in Figure 5. From the nickel–diene complex 15, the endergonic ligand exchange of alkynes generates the intermediate 27. The oxidative cyclization of 2-butynes via TS28 involves a 27.7 kcal/mol overall barrier with respect to 15, producing the nickellacyclopentadiene intermediate 29. From 29, the coordination of 2-butyne and subsequent insertion through TS31 are facile, generating the intermediate 32. The reductive elimination via TS33 only requires a barrier of 9.1 kcal/mol and produces the aryl ring-coordinated complex 34. Therefore, the rate-limiting step of the [2 + 2 + 2] cycloaddition is the oxidative cyclization, and this trimerization of 2-butyne requires a 27.7 kcal/mol overall barrier, which is much less favorable than the [4 + 4 + 2] cycloaddition of butadienes and 2-butyne.
Figure 5.

Free energy changes of the most favorable pathway of [Ni(PMe3)]-catalyzed [2 + 2 + 2] cycloaddition of 2-butynes.
We studied the same trimerization reaction with electron-deficient alkyne (using dimethyl acetylene dicarboxylate as example) and alkyne with free hydroxyl group (using 2-methyl-3-butyn-2-ol as example), and the free energy changes are shown in Figure 6. The ligand exchange between alkynes and butadienes determines the competition between the [4 + 4 + 2] and [2 + 2 + 2] cycloadditions. With 2-butyne, the ligand exchange between alkynes and butadienes is endergonic by 8.4 kcal/mol, making the subsequent oxidative cyclization of alkynes much less favorable as compared to the cooligomerization pathway with butadiene (Figure 1). In contrast, the same step is exergonic by 18.4 kcal/mol with dimethyl acetylene dicarboxylate and exergonic by 9.3 kcal/mol with 2-methyl-3-butyn-2-ol. These exergonic coordinations of alkynes make the [2 + 2 + 2] cycloadditions competitive, leading to the benzene derivatives, as found in the experiments.
Figure 6.

Gibbs free energy profiles of [Ni(PMe3)]-catalyzed [2 + 2 + 2] cycloadditions with 2-butyne, dimethyl acetylene dicarboxylate, and 2-methyl-3-butyn-2-ol.
Figure 7 shows the optimized structures of the alkyne-coordinated complexes, 27, 27b, and 27c. Electron-deficient alkynes, such as dimethyl acetylene dicarboxylate in 27b, have much stronger coordination to electron-rich nickel(0) as compared to 2-butyne. This strong coordination of electron-deficient alkynes makes the substrate exchange from 16b to 27b exergonic, leading to the favorable alkyne trimerization. In addition, when the alkyne has free hydroxyl group, there is an intramolecular hydrogen bond that stabilizes the complex 27c. This makes the formation of 27c favorable, also leading to the productive [2 + 2 + 2] cycloadditions. Therefore, when electron-deficient alkynes, or alkynes with a free hydroxyl group are used, the alkyne coordination of nickel is stronger than the diene coordination. This favorable coordination alters the energetics such that the [2 + 2 + 2] trimerization of alkynes competes with the [4 + 4 + 2] reaction.
Figure 7.
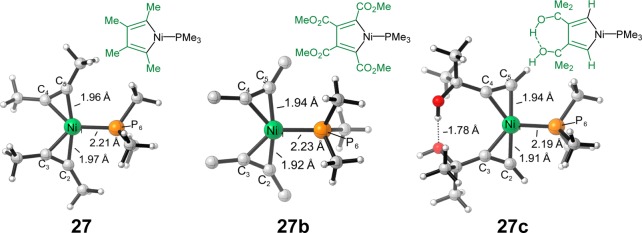
Optimized structures of [(PMe3)Ni(alkyne)2] complexes, 27 for 2-butyne, 27b for dimethyl acetylene dicarboxylate, and 27c for 2-methyl-3-butyn-2-ol (only the α-carbons of the ester groups in 27b are shown for simplicity).
Conclusions
We have studied the mechanism, reactivities, and selectivities of Ni-catalyzed [4 + 4 + 2] cycloadditions of dienes and alkynes through density functional theory (DFT) calculations. The reaction is found to proceed through the oxidative cyclization of dienes to form the nickel–phosphine complex. This nine-membered ring intermediate dissociates the phosphine ligand and then undergoes rate-determining alkyne insertion, affording the 11-membered ring intermediate. In order to undergo a facile reductive elimination, the phosphine ligand recoordinates to nickel and facilitates the formation of 1Z,4Z,8E-cyclodecatriene products.
We found that the Z/E selectivity of the cyclodecatriene products is determined by the diene oxidative cyclization step. Only the oxidative cyclization between one s-cis and one s-trans butadiene is facile and exergonic. The step with two s-cis butadienes requires much higher barrier due to the weak coordination of dienes, and the step with two s-trans butadienes generates high energy nine-membered ring intermediates. Therefore, the oxidative cyclization with two s-cis or s-trans butadienes are both much less favorable than the reaction with one s-cis and one s-trans butadienes either kinetically or thermodynamically, resulting in the 1Z,4Z,8E-cyclodecatriene products.
We also investigated the competing [2 + 2 + 2] cycloaddition of alkynes with [Ni(PMe3)] as the same active catalyst and found that the alkyne coordination determines the feasibility of the trimerization of alkynes. With electron-deficient alkynes or alkynes with free hydroxyl groups, the coordination of alkynes is much stronger as compared to the coordination of dienes, and the side reaction prevails. In contrast, the alkyl-substituted alkynes coordinate more weakly, and the generation of alkyne-coordinated nickel complex is less favorable. In these cases, the [4 + 4 + 2] cycloaddition of dienes and alkynes occurs.
Acknowledgments
We are grateful to the National Science Foundation (Grant No. CHE-1361104 (K.N.H.)) and the National Institutes of Health (Grant No. GM-097444 (P.S.B.)) for financial support of this research. We are grateful to the German Academic Exchange Service, DAAD (postdoctoral fellowship to D.C.G.G.), the NSF (predoctoral fellowship to D.H.), and Amgen (predoctoral fellowship to D.H.). Calculations were performed on the Hoffman2 Cluster at UCLA and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (Grant No. OCI-1053575).
Supporting Information Available
Free energy profiles of unfavorable [2 + 2 + 2] cycloadditions with butadienes, Cartesian coordinates, and energies of DFT-computed stationary points and saddle points. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Weitz H. M.; Loser E.. Isoprene. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, 2002; pp 1–20. [Google Scholar]; b Breitmaier E.Terpenes: Flavors, Fragrances, Pharmaca, Pheromones; Wiley-VCH: Weinheim, 2006; pp 1–9, 24–50. [Google Scholar]; c Nicolaou K. C.; Montagnon T.. Molecules That Changed the World; Wiley-VCH: Weinheim, 2006. [Google Scholar]
- a Sacchettini J. C.; Poulter C. D. Science 1997, 277, 1788. [DOI] [PubMed] [Google Scholar]; b Maimone T. J.; Baran P. S. Nat. Chem. Biol. 2007, 3, 396. [DOI] [PubMed] [Google Scholar]
- a Kellogg B. A.; Poulter C. D. Curr. Opin. Chem. Biol. 1997, 1, 570. [DOI] [PubMed] [Google Scholar]; b Ishihara Y.; Baran P. S. Synlett 2010, 12, 1733. [Google Scholar]; c Chen K.; Baran P. S. Nature 2009, 459, 824. [DOI] [PubMed] [Google Scholar]; d Chen K.; Ishihara Y.; Morón Galán M.; Baran P. S. Tetrahedron 2010, 66, 4738. [Google Scholar]; e Ishihara Y.; Mendoza A.; Baran P. S. Tetrahedron 2013, 69, 5685. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Jørgensen L.; McKerrall S. J.; Kuttruff C. A.; Ungeheuer F.; Felding J.; Baran P. S. Science 2013, 341, 878. [DOI] [PubMed] [Google Scholar]; g Wilde N. C.; Isomura M.; Mendoza A.; Baran P. S. J. Am. Chem. Soc. 2014, 136, 4909. [DOI] [PMC free article] [PubMed] [Google Scholar]; h McKerrall S. J.; Jorgensen L.; Kuttruff C. A.; Ungeheuer F.; Baran P. S. J. Am. Chem. Soc. 2014, 136, 5799. [DOI] [PubMed] [Google Scholar]
- a Bogdanovic B.; Kröner M.; Wilke G. Liebigs Ann. Chem. 1966, 699, 1. [DOI] [PubMed] [Google Scholar]; b Heimbach P.; Wilke G. Liebigs Ann. Chem. 1969, 727, 183. [Google Scholar]; c Brenner W.; Heimbach P.; Wilke G. Liebigs Ann. Chem. 1969, 727, 194. [Google Scholar]; d Bogdanovic B.; Heimbach P.; Kröner M.; Wilke G.; Brandt J. Liebigs Ann. Chem. 1969, 727, 143. [Google Scholar]; e Heimbach P.; Wilke G. Liebigs Ann. Chem. 1969, 727, 183. [Google Scholar]; f Barnett B.; Büssemeier B.; Heimbach P.; Jolly P. W.; Krüger C.; Tkatchenko I.; Wilke G. Tetrahedron Lett. 1972, 15, 1457. [Google Scholar]; g Benn R.; Büssemeier B.; Holle S.; Jolly P. W.; Mynott R.; Tkatchenko I.; Wilke G. J. Organomet. Chem. 1985, 279, 63. [Google Scholar]; h Wilke G. Angew. Chem., Int. Ed. 1988, 27, 185. [Google Scholar]; i Wilke G.; Eckerle A.. Cyclooligomerizations and Cyclo-cooligomerizations of 1,3-Dienes. In Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Two Volumes; Cornils B., Herrmann W., Eds.; Wiley-VCH: Weinheim, 1996; Vol. 1, pp 368–382. [Google Scholar]
- a Heimbach P.; Hey H.-J. Angew. Chem., Int. Ed. 1970, 9, 528. [Google Scholar]; b Heimbach P. Angew. Chem., Int. Ed. 1973, 12, 975–989. [Google Scholar]; c Heimbach P. J. Synthetic Org. Chem. 1973, 31, 300. [Google Scholar]; d Brenner W.; Heimbach P.; Ploner K.-J.; Thömel F. Liebigs Ann. Chem. 1973, 1882. [Google Scholar]
- a Kiji J.; Masuy K.; Furukawa J. Bull. Chem. Soc. Jpn. 1971, 44, 1956. [Google Scholar]; b Wender P. A.; Ihle N. C. J. Am. Chem. Soc. 1986, 108, 4678. [Google Scholar]; c Wender P. A.; Snapper M. L. Tetrahedron Lett. 1987, 28, 2221. [Google Scholar]; d Wender P. A.; Ihle N. C.; Correia C. R. D. J. Am. Chem. Soc. 1988, 110, 5904. [Google Scholar]; e Wender P. A.; Jenkins T. E. J. Am. Chem. Soc. 1989, 111, 6432. [Google Scholar]
- Holte D.; Gotz D. C.; Baran P. S. J. Org. Chem. 2012, 77, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For related Ni-catalyzed [2 + 2 + 2] cycloadditions of alkynes, see:; a Chopade P. R.; Louie J. Adv. Synth. Catal. 2006, 348, 2307. [Google Scholar]; b Ogoshi S.; Nishimura A.; Ohashi M. Org. Lett. 2010, 12, 3450. [DOI] [PubMed] [Google Scholar]; c Domínguez G.; Pérez-Castells J. Chem. Soc. Rev. 2011, 40, 3430. [DOI] [PubMed] [Google Scholar]; d Okamoto S.; Sugiyama Y. Synlett 2013, 24, 1044. [Google Scholar]
- For related experimental studies on the Ni-catalyzed cycloadditions of dienes and diynes, see:; a Ikeda S.; Watanabe H.; Sato Y. J. Org. Chem. 1998, 63, 7026. [DOI] [PubMed] [Google Scholar]; b Shanmugasundaram M. W.; Wu M.-S.; Jeganmohan M.; Huang C.-W.; Cheng C.-H. J. Org. Chem. 2002, 67, 7724. [DOI] [PubMed] [Google Scholar]; c Louie J.; Gibby J. E.; Farnworth M. V.; Tekavec T. N. J. Am. Chem. Soc. 2002, 124, 15188. [DOI] [PubMed] [Google Scholar]; d Duong H. A.; Cross M. J.; Louie J. J. Am. Chem. Soc. 2004, 126, 11438. [DOI] [PubMed] [Google Scholar]; e Tekevac T. N.; Arif A. M.; Louie J. Tetrahedron 2004, 60, 7431. [Google Scholar]; f Tekevac T. N.; Louie J. Org. Lett. 2005, 7, 4037. [DOI] [PubMed] [Google Scholar]; g Wender P. A.; Christy J. P. J. Am. Chem. Soc. 2007, 129, 13402. [DOI] [PubMed] [Google Scholar]; h Saito N.; Shiotani K.; Kinbara A.; Sato Y. Chem. Commun. 2009, 4284. [DOI] [PubMed] [Google Scholar]; i Stolley R.; Maczka M.; Louie J. Eur. J. Org. Chem. 2011, 3815. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Ohashi M.; Takeda I.; Ikawa M.; Ogoshi S. J. Am. Chem. Soc. 2011, 133, 18018. [DOI] [PubMed] [Google Scholar]; k Kumar P.; Prescher S.; Louie J. Angew. Chem., Int. Ed. 2011, 50, 10694. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Horie H.; Kurahashi T.; Matsubara S. Chem. Commun. 2012, 48, 3866. [DOI] [PubMed] [Google Scholar]; m Nishimura A.; Ohashi M.; Ogoshi S. J. Am. Chem. Soc. 2012, 134, 15692. [DOI] [PubMed] [Google Scholar]; n Kumar P.; Zhang K.; Louie J. Angew. Chem., Int. Ed. 2012, 51, 8602. [DOI] [PMC free article] [PubMed] [Google Scholar]; o Brusoe A. T.; Edwankar R. V.; Alexanian E. J. Org. Lett. 2012, 14, 6096. [DOI] [PubMed] [Google Scholar]; p Thakur A.; Facer M. E.; Louie J. Angew. Chem., Int. Ed. 2013, 52, 12161. [DOI] [PMC free article] [PubMed] [Google Scholar]; q Morimoto M.; Nishida Y.; Miura T.; Murakami M. Chem. Lett. 2013, 42, 550. [Google Scholar]; r Noucti N. N.; Alexanian E. J. Angew. Chem., Int. Ed. 2013, 52, 8424. [DOI] [PubMed] [Google Scholar]
- For related mechanistic studies on the Ni-catalyzed cycloadditions of alkenes and alkynes, see:; a Musaev D. G.; Froese R. D.; Svensson M.; Morokuma K. J. Am. Chem. Soc. 1997, 119, 367. [Google Scholar]; b Eisch J. J.; Ma X.; Han K. I.; Gitua J. N.; Krüger C. Eur. J. Inorg. Chem. 2001, 77. [Google Scholar]; c Li J.; Jia G.; Lin Z. Organometallics 2008, 27, 3892. [Google Scholar]; d Xu R.; Winget P.; Clark T. Eur. J. Inorg. Chem. 2008, 2874. [Google Scholar]; e Yu H.; Fu Y. Chem.—Eur. J. 2012, 18, 16765. [DOI] [PubMed] [Google Scholar]; f Komagawa S.; Wang C.; Morokuma K.; Saito S.; Uchiyama M. J. Am. Chem. Soc. 2013, 135, 14508. [DOI] [PubMed] [Google Scholar]; g Li Y.; Lin Z. Organometallics 2013, 32, 3003. [Google Scholar]; h Hong X.; Liu P.; Houk K. N. J. Am. Chem. Soc. 2013, 135, 1456. [DOI] [PubMed] [Google Scholar]; i Poater A.; Vummaleti S. V. C.; Cavallo L. Organometallics 2013, 32, 6330. [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Mont-gomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Keith T.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dap-prich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision D.01; Gaussian Inc.: Wallingford, CT, 2013. [Google Scholar]
- For the superior performance of M06 method on the organometallic systems, see:; a Zhao Y.; Truhlar D. G. Theor. Chem. Acc. 2008, 120, 215. [Google Scholar]; b Averkiev B. B.; Truhlar D. G. Catal. Sci. Technol. 2011, 1, 1526. [Google Scholar]; c Gusev D. G. Organometallics 2013, 32, 4239. [Google Scholar]
- Because the solvent is not necessarily required in the experiments,7 the solvation correction is not applied in this work.
- For a related computational study on nickel-catalyzed cyclodimerization of butadiene, see:; a Tobisch S.; Ziegler T. J. Am. Chem. Soc. 2002, 124, 13290. [DOI] [PubMed] [Google Scholar]; For another related computational study on phosphorus-based nickel catalysts, see:; b Heyndrickx W.; Occhipinti G.; Minenkov Y.; Jensen V. R. Chem.—Eur. J. 2011, 17, 14628. [DOI] [PubMed] [Google Scholar]
- The bis-ligated pathway is found much less favorable as compared to the monoligated pathway.
-
The ligand exchange
from 17 to 20 could occur by dissociative
or associative mechanisms, and the dissociative mechanism is more
favorable. In addition, solvent participation could facilitate the
ligand exchange, see:Xu L.; Hilton M. J.; Zhang X.; Norrby P. O.; Wu Y.-D.; Sigman M. S.; Wiest O.
J. Am. Chem. Soc.
2014, 136, 1960. [DOI] [PMC free article] [PubMed] [Google Scholar];
- The geometries of TS16, TS16-cc, and TS16-tt were also calculated with the B3LYP-D3 method, and the relative energies of the reoptimized geometries, TS16′, TS16-cc′, and TS16-tt′, are similar to those of the original geometries. (ΔΔG(TS16′) = 0.0 kcal/mol, ΔΔG(TS16-cc′) = 12.5 kcal/mol, and ΔΔG(TS16-tt′) = 1.9 kcal/mol).
- For related computational studies on the role of distortion in organometallic reactions, see:; a Garcia Y.; Schoenebeck F.; Legault C. Y.; Merlic C. A.; Houk K. N. J. Am. Chem. Soc. 2007, 129, 12664. [DOI] [PubMed] [Google Scholar]; b Legault C. Y.; Garcia Y.; Merlic C. A.; Houk K. N. J. Am. Chem. Soc. 2009, 131, 6632. [DOI] [PMC free article] [PubMed] [Google Scholar]; c van Zeist W.-J.; Bickelhaupt F. M. Org. Biomol. Chem. 2010, 8, 3118. [DOI] [PubMed] [Google Scholar]; d van Zeist W.-J.; Bickelhaupt F. M. Dalton Trans. 2011, 40, 3028. [DOI] [PubMed] [Google Scholar]; e Poater J.; Feixas F.; Bickelhaupt F. M.; Solà M. Phys. Chem. Chem. Phys. 2011, 13, 20673. [DOI] [PubMed] [Google Scholar]; f Yang Y.-F.; Cheng G.-J.; Liu P.; Leow D.; Sun T. Y.; Chen P.; Zhang X.; Yu J. Q.; Wu Y.-D.; Houk K. N. J. Am. Chem. Soc. 2014, 136, 344. [DOI] [PubMed] [Google Scholar]; g Hong X.; Liang Y.; Houk K. N. J. Am. Chem. Soc. 2014, 136, 2017. [DOI] [PubMed] [Google Scholar]; h Fernández I.; Bickelhaupt F. M. Chem. Soc. Rev. 2014, 43, 4953. [DOI] [PubMed] [Google Scholar]
- For a related computational study on Ni-catalyzed cycloaddition of alkynes, see:Straub B. F.; Gollub C. Chem.—Eur. J. 2004, 10, 3081. [DOI] [PubMed] [Google Scholar]
- The [2 + 2 + 2] cycloaddition of 2-butynes is more favorable compared with the [2 + 2 + 2] cycloaddition of two butadienes and one 2-butyne and the [2 + 2 + 2] cycloaddition of one butadiene and two 2-butynes. The free energy profiles of these unfavorable [2 + 2 + 2] cycloadditions are shown in Figures S1 and S2 of the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.