

Abstract
Glucocorticoid (GC) resistance is a severe problem associated with various inflammatory diseases. Previous studies have shown that repeated social stress induces GC resistance in innate immune cells, but the underlying molecular mechanisms have not been fully elucidated. Therefore, the purpose of this study was to examine potential underlying molecular mechanism(s) of repeated social defeat (RSD) stress on GC resistance in splenic macrophages. It was hypothesized that mRNA expression of receptors for GC and nuclear translocating-associated regulators in splenic macrophages would be affected by RSD, and that these changes would be associated with epigenetic modification. The data showed that the mRNA expression of GC and mineralocorticoid receptors were significantly decreased in splenic macrophages by RSD. RSD also induced a significantly decreased mRNA expression in FK506-binding protein 52 (FKBP52), consequently resulting in a significantly increased ratio of FKBP51 to FKBP52. Moreover, DNA methyltransferases 3a and 3b showed a significant decrease in their mRNA expression in the RSD group as did mRNA expression of histone deacetyltransferase 2. The RSD group also showed a significantly reduced quantity of methylated DNA in splenic macrophages. Based on microRNA (miRNA) profiling data, it was determined that RSD induced significantly increased expression of 9 different miRNAs that were predicted to interact with mRNAs of the GC receptor (6 miRNAs), mineralocorticoid receptor (3 miRNAs) and FKBP52 (2 miRNAs). Spearman correlation analysis revealed significantly strong correlations between the expression of 2 miRNAs and their target mRNA expression for GC receptors. Among these miRNAs, we verified direct effects of miRNA-29b and -340 overexpression on mRNA expression of GC receptors in L929 cells. The overexpression of miRNA-29b or -340 in L929 cells significantly reduced LPS-induced overexpression of GC receptors. In conclusion, this study provides evidence that epigenetic regulation, such as DNA methylation and miRNA expression, may play a role in the RSD-induced GC resistance that we have observed in splenic macrophages.
Keywords: glucocorticoid insensitivity, chronic social stress, macrophages, miRNA, innate immunity, epigenetics
Introduction
An accumulating body of evidence shows socially stressed individuals are less healthy psychologically and physically (Fagundes et al., 2013; House et al., 1988; Jaremka et al., 2013; Powell et al., 2013; Voorhees et al., 2013). In addition to the development of mood disorders, chronically stressed individuals respond less well to vaccination, are more susceptible of infectious challenges, and have slower wound healing responses (Gouin et al., 2010; Kiecolt-Glaser et al., 1995). These behavioral and physiological alterations are driven by activation of the hypothalamic-pituitary-adrenal (HPA) axis and sympathetic nervous system.
Immune regulation is an important aspect of development of the appropriate immune response to challenge. Cells of the innate and adaptive arms of the immune response express receptors for hormones produced by the HPA axis. Steroid hormone glucocorticoids (GCs) are crucial regulators of inflammation and immunity and exert their effects on target cells mostly through intracellular receptors. Steroids are recognized as effective treatment for many inflammatory and immune diseases (Stratakis et al., 1994; Yang et al., 2012). It is now known that chronic social stress can modulate innate immune cells, especially CD11b+ macrophages, to become desensitized to GC, and that immune modulation consequently induces psychological and behavioral alterations (Avitsur et al., 2001; Glaser et al., 2001; Sheridan et al., 2000; Stark et al., 2001).
Previous data indicate that RSD-induced GC resistance in splenocytes continued for at least 10 days past the cessation of the stressor (Avitsur et al., 2002). Moreover, the target cell population for RSDinduced GC resistance was determined to be CD11b+ splenocytes (also referred as splenic macrophages) (Stark et al., 2001), although the underlying molecular mechanisms for the RSD-induced GC resistance in splenic macrophages were not fully known. A study (Quan et al., 2003a) investigated potential underlying mechanisms of RSD-induced GC resistance and reported that nuclear translocation of GC-receptor complexes was impaired in splenic macrophages, providing a molecular mechanism for Stark and colleagues' findings (2001). Because nuclear translocation of the GC-receptor complex is a critical step in GC-induced gene regulation, it was important to elucidate how RSD impaired nuclear translocation.
Immunophilin FK506 binding proteins (FKBPs) regulate the nuclear translocation of GC-receptor complexes and nuclear translocation depends on binding of the GC-receptor complex to either FKBP51 or FKBP52 (Galigniana et al., 2012; Sivils et al., 2011; Stechschulte and Sanchez, 2011). Because FKBP51 blocks the translocation of the complex to the nucleus while FKBP52 facilitates the nuclear translocation, a lower ratio of FKBP51 to FKBP52 predicts the successful nuclear translocation of GC-receptor complex (Davies et al., 2002; Sivils et al., 2011). While previous studies provided evidence that RSD-induced GC resistance in splenic macrophages was associated with impaired nuclear translocation, no study has investigated the effects of RSD on the expression of FKBP51 and FKBP52 in splenic macrophages. In this study, we examined the underlying molecular mechanisms of RSD-induced GC resistance by focusing on the mRNA and protein expression of GC receptors in splenic macrophages in addition to investigating potential epigenetic mechanisms. First, to examine global modification mechanisms, we investigated the effects of RSD on the expression of chromatin epigenetic regulatory enzymes, such as DNA methyltransferases that play a role in DNA methylation and histone deacetylase 2 that plays a role in deacetylation on histones, particularly on GC binding sites in target genes (Yang et al., 2012). A previous study indicated that RSD significantly decreased GC receptor protein expression, but had no effect on pre-mRNA molecules, indicating potential molecular regulation between the stages of premRNA expression and mRNA translation (Quan et al., 2003a). Consequently, we investigated a role of translational epigenetic regulators by analyzing the expression of small non-coding RNAs “microRNAs” in splenic macrophages.
Methods
Animals
Male C57BL/6 (5 – 6 weeks old) and CD-1 (12 months old, retired breeders) mice were obtained from Charles River Laboratories (Wilmington, MA) and were housed in a facility accredited by the American Association for Accreditation of Laboratory Animal Care. All C57BL/6 mice were housed in cohorts of three with ad libitum access to water and food and maintained on12:12-h light-dark cycle (dark cycle started at 18:00). Mice were allowed to acclimate for at least 7 – 8 days before they were exposed to any experimental procedures. While a rare occurrence (less than 5%; Wohleb et al., 2011), any mouse showing severe wounding was excluded from the experiments. Cages of 3 mice were randomly assigned to RSD or control group. Mice were used for the analyses of: 1) mRNA expression of GC regulated targets, 2) microRNA profiling, and 3) correlation between the expression of mRNAs and selected miRNAs. All procedures were approved by the Ohio State University Institutional Laboratory Animal Care and Use Committee.
Repeated Social Defeat (RSD) Protocol
RSD was performed as described previously (Avitsur et al., 2001). In brief, an intruder (male CD-1 mouse) was introduced into a cage of established male cohorts of C57BL/6 mice for 2 hours between 17:00 and 19:00 for 6 consecutive days. The intruder normally started to attack the resident mice within 5 min and the residents exhibited submissive behavior, including upright posture, fleeing, and huddling (Avitsur et al., 2001; Stark et al., 2001). If the intruder did not attack within 5 min from the beginning of the session, or if the intruder was defeated by any of the cage residents, the intruder was replaced with a new intruder. Different intruders were used on consecutive nights. The health status of all mice in the RSD group was checked daily in the morning and just before the initiation of RSD. If mice were severely injured or moribund, they were excluded from the study. Home-cage control mice were maintained as cohorts of three mice per cage in a separate room without any disruption until sacrifice, and the health status of these mice was checked daily.
Isolation of CD11b+ splenocytes
On the morning following the last RSD cycle (approximately post 14 h post RSD), all mice from control and RSD groups were sacrificed by CO2 asphyxiation. Spleens were immediately harvested from mice into ice-cold Hanks' balanced salt solution (HBSS). Their body weight and spleen mass were recorded. Splenocytes were isolated from each spleen as described previously (Quan et al., 2003a). In brief, spleens were transferred into ice-cold HBSS-containing plastic bags, and a Stomacher machine was used for 60 sec at high speed to produce a single cell suspension. Cells were transferred into 50 mLconical tubes and were centrifuged at 1,800 rpm for 10 min at 4 °C. Cell pellets were then incubated at room temperature for 2 min with 1mL Red Blood Cell Lysis buffer (0.16M NH4Cl, 10mM KHCO3, 0.13 mM EDTA) to eliminate red blood cells. After the 2-min incubation, 19ml HBSS/10% heat-inactivated fetal bovine serum (FBS) was added to the tube and centrifuged at 1,800 rpm for 10 min at 4 °C. Each pellet was then resuspended in ice-cold HBSS and filtered through a sterile 70 μm nylon cell strainer. Samples were then centrifuged at 1,800 rpm for 10 min at 4°C, and the pellets were resuspended in icecold MACs buffer, containing phosphate buffered saline (PBS, pH 7.2), 0.5% bovine serum albumin (BSA) and 2mM ethylenediaminetetraacetic acid (EDTA). Using a Beckman Coulter Z2 particle counter, total cell number was determined and CD11b magnetic MicroBead positive selection was performed as described in the manufacturer's protocol (Miltenyi Biotec Inc, Auburn, CA) The unlabeled splenocytes were eluted first, and CD11b+ splenocytes were isolated according to the protocol. After counting the number of CD11b+ splenocytes, cells were centrifuged at 1,800 rpm for 10 min at 4 °C. The CD11b+ splenocyte pellets were resuspended into RNAlater medium (Life Technologies, Grand Island, NY) and stored at 4°C overnight. It is important to note that the isolated CD11b+ splenocyte subset includes all myelomonocytic cell types (e.g. macrophages, granulocytes, natural killer cells, dendritic cells).
Isolation of Total RNAs
mirVana™ miRNA isolation Kit (Ambion®, Life Technologies, CA) was used to isolate total RNAs from splenic macrophages (CD11b+ cells) according to the manufacturer's protocol. In brief, after the RNAlater was removed, splenic macrophages were quickly disrupted in Lysis/Binding solution. Cells lysates were then incubated with miRNA Homogenate Additive for 10 min on ice followed by adding equal volumes of Acid-Phenol: Chloroform, vortexing for 30 – 60 sec, and centrifuging at 10,000 × g for 5 min at 4 °C to separate the aqueous and organic phases. The aqueous phase was carefully transferred into a new tube, and 1.25 volumes of room temperature 100% ethanol was added to the aqueous phase. The mixture of the lysate and ethanol was passed through a filter cartridge by centrifugation. The filter cartridge was washed with 700 μL miRNA Wash Solution 1 and then washed twice with 500 μL Wash Solution 2/3. Preheated nuclease-free water (95 °C) was quickly passed through the cartridge to elute total RNAs. The quality of total RNAs was detected spectrophotometrically, and the inclusion criteria for this study were greater or equal to 1.9 (A260/A280) and 1.8 (A260/A230). Total RNA was stored at – 80 °C until the further analysis.
microRNA Expression Profiling
The NanoString nCounter Mouse miRNA Expression Assay Kit (NanoString® Technologies, Inc.) was used to profile more than 600 mouse and mouse-associated viral microRNAs (miRNAs), derived from miRBase, in splenic macrophages (CD11b+ cells). The NanoString nCounter Mouse miRNA assay is a direct global profiling analysis of individual miRNAs in a single reaction without amplification. All sample preparation was performed based on the manufacturer's instructions (NanoString® Technologies, Inc., Seattle, WA), and the total RNA samples (at least 250 ng) were sent to the Nucleic Acid Facility, at the James Cancer Hospital and Solove Research Institute, (Ohio State University, Columbus, OH) to perform the miRNA Expression Assay as previously described (Geiss et al., 2008).
In brief, total RNA contaminants were determined based on the ratios of A260/A280 and A260/A230. 100 ng total RNAs for each sample were used as input for nCounter miRNA sample preparation reactions. A specific DNA tag was ligated onto the 3′ end of each mature miRNA to prepare small RNA samples according to manufacturer's protocol (NanoString® Technologies, Inc.). The tags were used to normalize the melting temperatures of the miRNAs and provided identification for each of miRNA species in the sample. Excess tags were then removed and the remaining sample was hybridized with a panel of miRNA: tag-specific nCounter capture and barcoded reporter probes. Hybridization reactions were induced by incubation at 64 °C for 18 hours, and then hybridized probes were purified. The purified probes were immobilized on a streptavidin-coated cartridge using the nCounter Prep Station (NanoString® Technologies, Inc.). Individual fluorescent barcodes were counted and target RNA molecules present in each sample were quantified by using nCounter Digital Analyzer (NanoString® Technologies, Inc.). For each assay, 600 fields of view were scanned (a high-density scan) for each sample.
Reverse Transcription PCR and Quantitative Real Time PCR for mRNA Analysis
Total RNA was reverse-transcribed into cDNA according to the High-Capacity cDNA Reverse Transcription Kit Protocol (Applied Biosystems). RNA was reverse-transcribed as per manufacturer's protocol in a 20 μL reaction volume, containing 1.5 μg RNA in the 10uL 2X master mix. cDNA was stored at − 20 °C until subsequent amplification.
Quantitative real time PCR (qPCR) was then performed according to the TaqMan® Gene Expression Assays protocol (Life Technologies) to analyze gene expression of chemokine C-C motif ligand 2 (ccl2, Mm00441242_m1), glucocorticoid receptor (nr3c1, Mm00433832_m1), mineralocorticoid receptors (nr3c2, Mm01241596_m1), FKBP51 (fkbp5, Mm00487401_m1), FKBP52 (fkbp4, Mm00487391_m1), DNA methyltransferase 1 (dnmt1, Mm01151063_m1), DNMT 3a (dnmt3a, Mm00432881_m1), DNMT 3b (dnmt3b, Mm01240113_m1), histone deacetylase 2 (hdac2, Mm00515108_m1), and glyceraldehydes-3-phosphate dehydrogenase (gapdh, Mm99999915_g1). All samples were run in duplicates, and data were analyzed using Applied Biosystems software using cycle threshold (CT) values. Fold change (2- ΔΔCT) was calculated in addition to calculate relative fold change to the average control 2-ΔΔCT values.
Reverse Transcription PCR and Quantitative Real Time PCR for miRNA Analysis
Micro RNAs from total RNA samples were reverse-transcribed into cDNAs according to the TaqMan® MicroRNA Reverse Transcription Kit Protocol (Life technologies). Briefly, after all reagents were thawed on ice, 10ng RNA sample was combined with 5X RT primer in a 0.2-mL polypropylene reaction tube and incubated at 85 °C for 5 min then at 60 °C for 5 min followed by placing it on ice. The RT master mix, containing 100 mM dNTPs (with dTTP), 10X Reverse Transcription Buffer, RNase Inhibitor (20 μ/μL), MultiScribe™ Reverse Transcriptase (50 U/μL), and Nuclease-free water, was added into the tube containing denatured RNA and RT primer in the ratio of 7 μL RT master mix : 8 μL denatured RNA and RT primer. The mixture was incubated on ice for 5 min, and then reverse transcribed according to the manufacturer's protocol: incubation at 16 °C for 30 min, at 42°C for 30 min and at 85°C for 5 min followed by a hold at 4 °C. cDNA of each miRNA was stored at − 20 °C until the next PCR amplification.
The expression of 3 miRNAs (Table-1) in splenic macrophages was verified by stem loop quantitative PCR (qPCR) analysis according to the TaqMan® MicroRNA Assay Protocol (Life Technologies). In brief, after all reagents were first thawed on ice, PCR cocktail was prepared with 2X TaqMan® Universal PCR Master Mix II, 20X TaqMan® Small RNA Assay and Nuclease-free water. cDNA of each miRNA was mixed with the PCR cocktail in the ratio of 1:15 in the final PCR reaction, and then qPCR was run according to the manufacturer's protocol: holding at 50 °C for 2 min and at 95 °C for 10 min, followed by 40 cycles of the incubation for 15 sec at 95 °C and at 60 °C for 60 sec. SYBR® Green Assay (Fast SYBR ® Green Master Mix, Life technologies) was used as control. All samples were run in duplicates, and data were analyzed using Applied Biosystems software using cycle threshold (CT) values. Using CT values, the fold change (2-ΔΔCT) was calculated as in addition to calculate relative fold change to the average control 2- ΔΔCT values.
Table-1. miRNA and stem-loop sequence information for qRT-PCR miRNA validation.
| miRNA | Mature miRNA Sequence |
Stem-loop Sequence | Chromosome Location |
|---|---|---|---|
| 20a | 5′–UAAAGUGCUUA UAGUGCAGGUAG–3′ | 5′–GUGUGAUGUGACAGCUUCUGUAGCACUAAAGUGCUU AUAGUGCAGGUAGUGUGUAGCCAUCUACUGCAUUACGA GCACUUAAAGUACUGCCAGCUGUAGAACUCCAG–3′ | Chr. 14: 115044157 - 115044263 [+] |
| 29b | 5′–UAGCACCAUUUGAAAUCAGUGUU–3′ | 5′–AGGAAGCUGGUUUCAUAUGGUGGUUUAGAUUUAAAU AGUGAUUGUCUAGCACCAUUUGAAAUCAGUGUUCU–3′ | Chr. 6: 31063023 - 31063093 [-] |
| 5′–CUUCUGGAAGCUGGUUUCACAUGGUGGCUUAGAUUU UUCCAUCUUUGUAUCUAGCACCAUUUGAAAUCAGUGUU UUAGGAG–3′ | Chr. 1: 195037040 - 195037120 [+] | ||
| 340 | 5′–UUAUAAAGCAA UGAGACUGAUU–3′ | 5′–CAAUUGUACUUGGUGUGAUUAUAAAGCAAUGAGACU GAUUGUCAUAUGUCGUUUGUGGGAUCCGUCUCAGUUAC UUUAUAGCCAUACCUGGUAUCUUA–3′ | Chr. 11: 50069702 - 50069799 [+] |
DNA Extraction & Analysis of Methylated DNA
DNA was extracted from CD11b+ splenocytes by using FitAmp™ blood and cultured cell DNA extraction kit (Epigentek Group Inc., NY), and then the status of global DNA methylation at the 5 position of cytosine (5-mC) was detected by using MethylFlash™ methylated DNA quantification kit (Epigentek Group Inc., NY) according to the manufacturer's protocols. In brief, CD11b+ splenocytes were washed with PBS and treated with the DNA digestion buffer followed by an incubation at 65 °C for 15 min. After the 15-min incubation, cells were transferred to F-Spin column, washed DNA 3 times, and, then, eluted DNA that was stored at − 20 °C until the next analysis. One-hundred forty nanograms of DNA was incubated with binding solution at 37 °C for 90 min to bind to assay well, followed by washing 3 times with 1X wash buffer. Capture antibody was diluted with 1X wash buffer at 1:1,000 ratio, and 50 μL of diluted capture antibody was added and incubated at room temperature for 60 min, followed by washing 3 times with 1X wash buffer. Detection antibody was diluted with 1X wash buffer at 1:2,000 ratio, and 50 μL of the diluted detection antibody was then added to each well and incubated at room temperature for 30 min. After washing 4 times, enhancer solution was also diluted at 1:5,000 ratio, and 50 μL of the diluted solution was added to each well and incubated at room temperature for 30 min. After 5-time washing with 1X wash buffer, 100 μL of developer solution was added and incubated at room temperature for 5 min in a dark room followed by adding stop solution. The absorbance was read on a microplate reader at 450 nm at 9 min. All samples and controls (positive and negative) were run in duplicates.
Analysis of GC receptor protein expression in CD11b+ splenocytes
Single cell suspensions of splenocytes were used for flow cytometric analyses of CD11b (eBioscience, San Diego, CA) and GC receptor (Santa Cruz Biotechnology, Inc., Dallas, TX) expression (n = 6/group). In brief, washed splenocytes, blocked FcγII/III receptors (BD Biosciences, San Jose, CA) for 15 min at 4 °C, and washed the cells. Cells were incubated with CD11b antigen for 30 min at 4 °C and washed 2 times. Resuspended cells were fixed and permeabilized by BD Cytofix/Cytoperm™ Fixation/Permeabilization kit (BD Biosciences, San Jose, CA), followed by 2-time wash in 1X BD Perm/Wash™ buffer. Cells resuspended were incubated with GC receptor antigen for 30 min at 4 °C and washed 2 times with 1X BD Perm/Wash™ buffer. Because the GC receptor primary antibody is not conjugated, splenocytes were incubated in secondary antibody (PE; goat anti-mouse IgG1-PE, Santa Cruz Biotechnology, Inc., Dallas, TX). Non-specific, isotype-matched antibodies were used to assess nonspecific binding in combination with unstained cells and blocking control. A Becton-Dickinson FACSCaliber four color cytometer was used to determine antigen expression, and 10,000 events were recorded for each sample and control. FlowJo software (ver. 7.6, Ashland, OR) was used to analyze data, and gating for CD11b and GC receptor was determined based on non-specific binding of appropriate negative isotype stained controls.
miRNA overexpression in L929 cell line
To confirm whether a particular miRNA regulated its predicted target gene expression (nr3c for miR-29b and 340), miRNA-29b and miRNA-340 were overexpressed in L929 cell lines by using Ambion® Pre-miR™ miRNA Precursors (mmu-miR-29b-1-5p and hsa-miR-340-5p, respectively) and Lipofectamine® RNAiMAX Reagent (InvitrogenTM), according to the manufacture's protocol. In brief, L929 cells (2 × 105 cells/well) were seeded in a 24-well plate with Dulbecco's Modified Eagle Medium (DMEM, High Glucose, GlutaMAX™, HEPES, Gibco®) with 10% heat-inactivated FBS and 1% Penicillin-Streptomycin (Gibco®). On the next day, 4.5 μL Lipofectamine® RNAiMAX Reagent and 500 pmol miRNA precursors were individually diluted in 25 μL DMEM, High Glucose, GlutaMAX™, HEPES (Gibco®). The diluted miRNA precursor was mixed with the diluted Lipofectamine® RNAiMAX Reagent (1:1 ratio) and incubated for 5 min at room temperature. After the 5-min incubation, the miRNA precursor-Lipofectamine complex was added to cells (final concentrations of miRNA precursor and Lipofectamine were 15 pmol and 4.5 μL, respectively) and then incubated in the incubator (5% CO2 and 37 °C). After the post 1-day incubation, cells were aspirated, treated with the fresh medium contained LPS (1μg/mL), and incubated for 2 days. After 2 days, cells were washed with PBS, and total RNAs were isolated by mirVana™ miRNA isolation Kit (Ambion®) according to the manufacturer's protocol (see the protocol for isolation total RNAs).
Statistical Analysis
To analyze miRNA profiling data, the abundance of miRNA was quantified using the NanoString nCounter gene-expression system (Geiss et al., 2008). The data were normalized to the sum of positive control counts (technical normalization), to the top 300 most highly expressed miRNAs (global sum biological normalization) and to negative control counts (background normalization). Normalized data was then used for statistical analysis in the Student's t test (two-tailed) to detect statistical significance between RSD and control groups. JMP® statistical discovery software (SAS Institute Inc., Ver. 10.0.2) was used to create a heatmap for selected miRNAs with a Green-to-Black-to-Red color theme.
For data from qPCR, flow cytometric and methylated DNA analyses, Shapiro-Wilk test was run to determine the normality of each variable and F-test was used as equal variance test. If the equal variance test failed, then a two-tailed unequal variance t test (Welch's t test) was used to determine statistical significance. Otherwise, Student's t test (two-tailed) was used to determine statistical significance between RSD and control groups. Spearman correlation coefficient was calculated to determine the association between variables. For miRNA overexpression data from the cell study, oneway ANOVA with post-hoc test (Student-Newman-Keuls method) was used to determine statistical significance. For this study, data were represented as mean ± standard error of mean (SEM), and p< 0.05 was considered statistically significant.
Results
Body weights and splenic mass
Shapiro-Wilk normality tests for variables of body weight, splenic mass and the ratio of splenic mass to body weight passed, but the equal variance F tests for splenic mass and the ratio of splenic mass to body weight failed (p< 0.05). The Mann-Whitney Rank Test was performed to determine significance on dependent variables of splenic mass and the ratio. RSD induced a 2.3-fold increase in splenic mass and a 2.2-fold increase in the ratio of splenic mass to body weight (n = 9/group, p< 0.0001, Table 2).
Table-2. Effects of 6 RSD cycles on dependent variables of body weight, splenic mass and CCL2 mRNA expression in splenic macrophages (n = 8 – 9/group).
| Dependent variable | Control | RSD | two-tailed p value |
|---|---|---|---|
|
| |||
| Body weight (g) | 23.65 ± 0.38 | 25.28 ± 0.65 | 0.0457 |
| Splenic mass (mg) | 65.11 ± 1.78 (median = 66) | 149.89 ± 6.10 (median = 151) | < 0.0001* |
| Ratio of splenic mass to body weight | 0.00275 ± 0.00006 (median = 0.00276) | 0.00598 ± 0.00032 (median = 0.00612) | < 0.0001* |
| CCL2 mRNA expression (2-ΔΔCTvalues) | 1.111 ± 0.064 (median = 1.138) | 2.574 ± 0.206 (median = 2.471) | < 0.05* |
Two-tailed unequal variance t-test was used to calculate p values because equal variance F tests for splenic mass, the ratio and CCL2 mRNA expression failed
Data are represented as mean ± standard error of mean
Gene and protein expression
To examine whether RSD induced a similar increase in inflammatory cytokines in CD11b+ cells as we reported in a previous study (Wohleb et al., 2013), we first determined mRNA expression of inflammatory cytokine C-C motif ligand 2 (CCL2) in splenic macrophages (CD11b+ splenocytes). A significant increase in CCL2 mRNA expression in splenic macrophages was detected in the RSD group (mean ± SED: 2.574 ± 0.206) compared to the control group (1.111 ± 0.064) (n = 8/group, p< 0.05, Table 2). RSD induced a significant reduction in mRNA expression of both GC (nr3c1) and mineralocorticoid (nr3c2) receptors in splenic macrophages (Figure 1-A). mRNA expression of FKBP51 (fkbp5) in splenic macrophages did not result in a statistical difference between two groups, but RSD induced a significant reduction in the FKBP52 mRNA expression (fkbp4) in splenic macrophages (Figure 1-B). The pattern led to a significantly higher ratio of FKBP51 to FKBP52 in RSD splenic macrophages than that in the control group (Figure 1-C).
Figure 1.
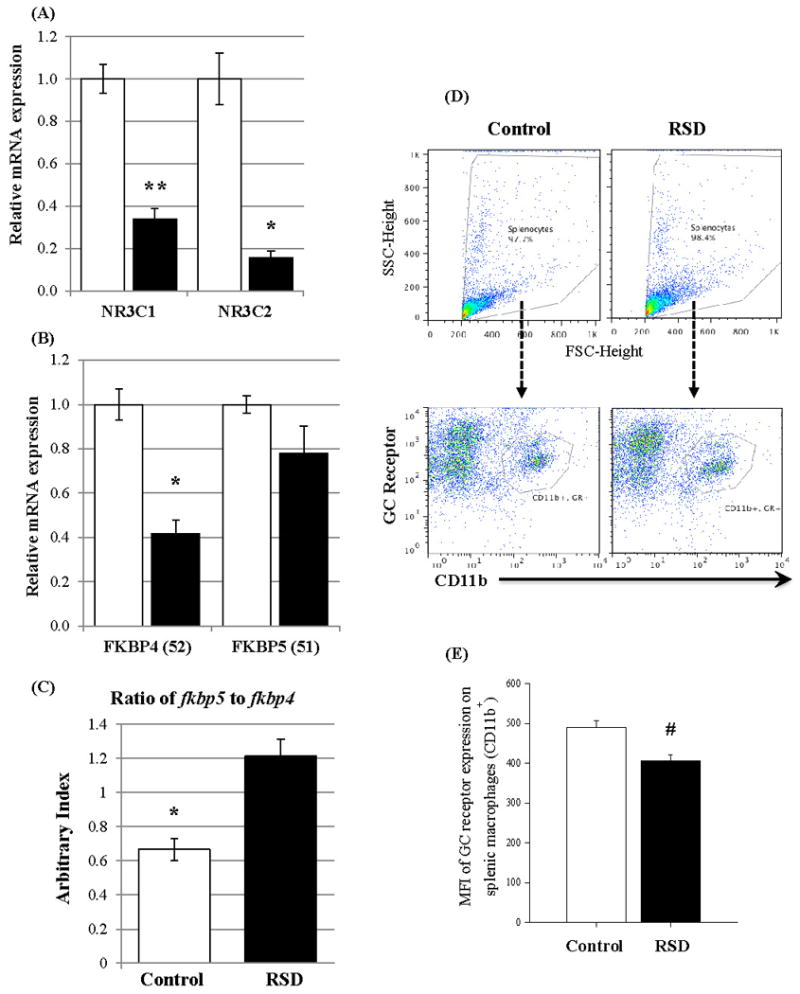
Effects of the 6-cycles of RSD on expression of glucocorticoid-associated proteins in splenic macrophages (CD11b+ cells) in the control (clear bar) and RSD (filled bar) groups.
(A) mRNA expression of glucocorticoid (nr3c1) and mineralocorticoid (nr3c2) receptors (n = 8/group). Equal Variance F test for the dependent variable of mineralocorticoid receptors failed (p <0.05). The median values for the control and RSD groups were 1.057 and 0.160 respectively, and the p value calculated by two-tailed unequal variance t test was 0.0015 (mean ± SEM).
(B) Results of FKBP51 (fkbp5) and FKBP52 (fkbp4) expression (n = 6/group). Equal Variance F test for the dependent variable of mineralocorticoid receptors failed (p <0.05). The median values for the control and RSD groups were 0.988 and 0.751 respectively, and the p value calculated by two-tailed unequal variance t test was 0.139 (mean ± SEM).
(C) Ratio of FKBP51 (fkbp5) and FKBP52 (fkbp4) mRNA expression (mean ± SEM).
(D) Gating examples of CD11b and GC receptor on splenocytes in control and RSD groups. Splenocytes were first gated based on plotting FSC vs SCC (top), and flow cytometric analyses of CD11b and GC receptor expression were performed (bottom).
(E) Protein expression of GC receptors (n = 6/group) on splenic macrophages. Protein expression of GC receptors in CD11b+ splenocytes was determined by using MFI of GC receptors, and the p value was calculated by two-tailed equal variance t test (mean ± SEM).
#: p = 0.003; *: p ≤ 0.001; **: p ≤ 0.0001
To confirm the decreased mRNA expression of GC receptor in CD11b+ splenocytes also results in a decrease in its protein expression, flow cytometric analyses of CD11b and GC receptor expression in splenocytes were performed and a mean fluorescence intensity (MFI) of GC receptor on CD11b+ splenocytes was calculated. There was no difference in the average number (1820 vs 1432, p= 0.181) or mean percentile (18.6% vs 14.6%, p=0.177) of CD11b+/GC receptor+ splenocytes between control and RSD groups, respectively (Figure 1-D). However, as expected, the MFI of GC receptors on CD11b+ splenocytes in the RSD group was significantly decreased (489.3 vs 406.8, p= 0.0032, Figure 1-E).
To elucidate potential mechanisms by which the RSD decreased the gene expression of GC receptor, mineralocorticoid receptor, and FKBP52, we investigated the possibility of DNA epigenetic repression by the gene expression of DNA methyltransferases that induce global DNA repressive modification. As shown in Figure 2-A, the mRNA expression of DNA methyltransferases DNMT3a and DNMT3b in splenic macrophages were significantly decreased by RSD, while RSD did not affect DNMT1 expression. To investigate a possibility of histone epigenetic repression, we analyzed the expression of histone deacetylase 2 (HDAC2) that deacetylates and induces histones to become inaccessible for transcription factors and other associated proteins. Exposure to RSD resulted in a significant decreased HDAC2 mRNA expression compared to the control group (Figure 2-B).
Figure 2.
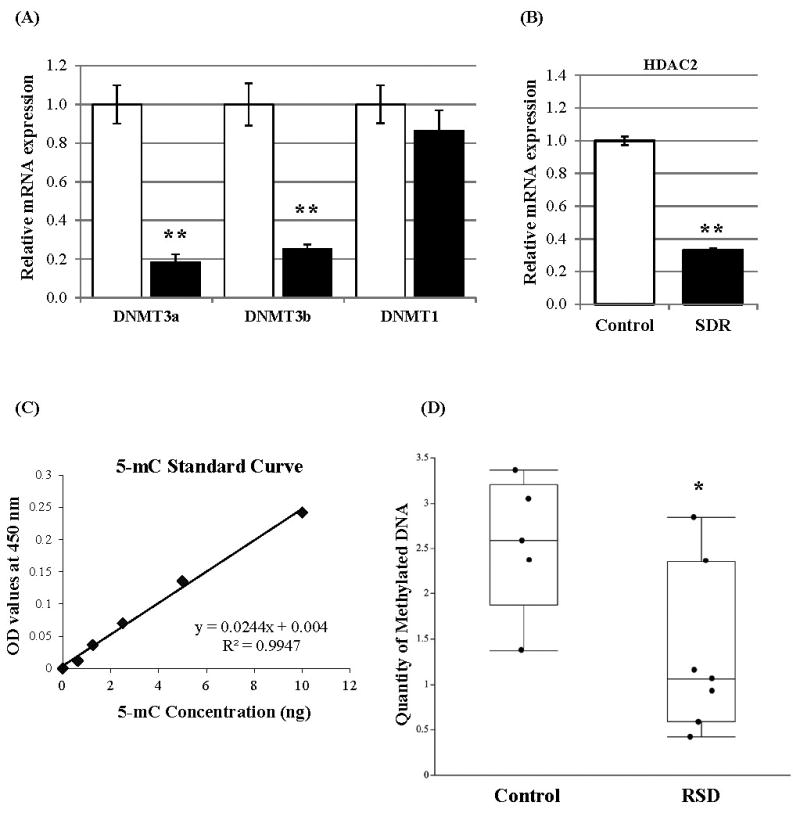
Effects of the 6-cycles of RSD on mRNA expression of chromatin modification-associated proteins and status of DNA methylation in splenic macrophages (CD11b+ cells).
(A) mRNA expression of DNA methyltransferases DNMT3a, DNMT3b, and DNMT1 in the control (clear bar) and RSD (filled bar) groups (mean ± SEM, n = 8/group)
(B) mRNA expression of histone deacetylase 2 (mean ± SEM)
(C) Standard curve plot for OD values observed at 450 nm vs. concentrations of control methylated DNA (5-mC)
The coefficient of determination (R2) for the linear regression is 0.9947.
(D) The absolute amount of methylated DNA in splenic macrophages (CD11b+ cells) in control and RSD groups (mean ± SEM)
*: p< 0.001; **: p< 0.0001
Using the 2-ΔΔCT values for each variable (FKBP51, FKBP52, FKBP51/FKBP52, GC receptor and mineralocorticoid receptor), the Spearman correlation coefficients were calculated (Table-3). The results showed a significant correlation for GC receptor with FKBP52 and the ratio of FKBP51 to FKBP52. We found a similar significant correlation for mineralocorticoid receptor with these variables. The results showed a significant negative correlation between two nuclear receptors and the ratio of FKBP51 to FKBP52. No other correlation between FKBP51 and any other variable was significant.
Table-3. Spearman Correlation coefficients between mRNA expression of glucocorticoid regulatory/associated proteins (n = 12 – 16/group).
| Spearman correlation coefficient (ρ) | FKBP52 |
FKBP51 FKBP52 |
GR | MR |
|---|---|---|---|---|
| FKBP51 | 0.438 | -0.105 | 0.259 | 0.193 |
| FKBP52 | -0.860** | 0.944** | 0.734* | |
| FKBP51/FKBP52 | -0.958** | -0.846** | ||
| GR | 0.876** | |||
| MR |
p < 0.01
p < 0.0001
Abbreviations: glucocorticoid receptor (GR) and mineralocorticoid receptor (MR)
Status of DNA methylation in splenic macrophages
To clarify whether RSD decreased the quantity of methylated DNA in splenic macrophages, the absolute amount of 5-mC in splenic macrophages from RSD and control groups was investigated. First, a linear regression (r2= 0.9947, Figure 2-C) from the standard curve plotted for the observed OD values and the 5-mC control concentrations was calculated. As expected, the absolute quantity of 5-mC was significantly decreased in RSD group (p= 0.0361, Figure 2-D). The control group contained 1.82% methylated DNA in the whole DNA, but the RSD group resulted in a 0.95% methylated DNA in CD11b+ splenocytes that results in a 47.5% reduction in the total amount of methylated DNA in splenic macrophages by RSD from the control group.
microRNA expression
Although we measured mRNA expression of certain epigenetic enzymes and global status of DNA methylation, we expected to generate data that supported suppression of gene expression and an increased amount of methylated DNA. However, the results did not support the finding of reduced expression of the GC receptor, the mineralocorticoid receptors and FKBP52. There is another epigenetic regulator known as microRNAs (miRNAs) that interacts to regulate mRNA and functions as a posttranscriptional regulator of gene expression. Therefore, we investigated whether RSD induced miRNAs that regulate gene expression of GC receptor (nr3c1), mineralocorticoid receptor (nr3c2) and FKBP52 (fkbp4). To determine if RSD induced the expression of miRNAs that were predicted to interact with these mRNAs, we searched an internet-based database (DIANA-microT-CDS database, Ver. 5.0, http://www.microrna.gr/microT-CDS) to seek out potential miRNAs. Then, we conducted a miRNA expression profiling analysis. Because the Diana lab suggested using a 0.6 of search threshold for sensitive analysis for Mus musculus, we set up the least search threshold as 0.60 to find miRNAs that were predicted to interact with GC receptor mRNA (search threshold: 0.70, result miRNA #: 132 miRNAs), mineralocorticoid receptor mRNA (search threshold: 0.70, result miRNA #: 125miRNAs), and FKBP52 mRNA (search threshold: 0.60, result miRNA #: 65 miRNAs). Subsequently, when we searched our miRNA expression profiling data for these miRNAs we found significantly increased expression of 8 different miRNAs (Figure 3-A) that were predicted to interact with mRNAs of GC receptor (6 miRNAs), mineralocorticoid receptor (3 miRNAs), and FKBP52 (2 miRNAs). miRNA-29b, 340-5p, 30b, 139-5p, 103, and 361 were predicted to interact with GC receptor mRNA. Among them, miRNA-30b and 361 were also predicted to interact with mineralocorticoid receptor mRNA. Prediction scores, provided from the DIANA-microT-CDS database (version 5.0), for the interactions of GC receptor mRNA with miRNA-29b and miRNA-340-5p were 0.70 and 0.87 respectively (70% and 87% possibility for each miRNA to interact to GC receptor mRNA; provided in the parentheses in Figure 3-A). As shown in Figure 3-A, RSD induced an increased expression of miRNA-29b by 44.2%, whereas RSD induced an increased expression of miRNA-340-5p by 83.9%.
Figure 3.
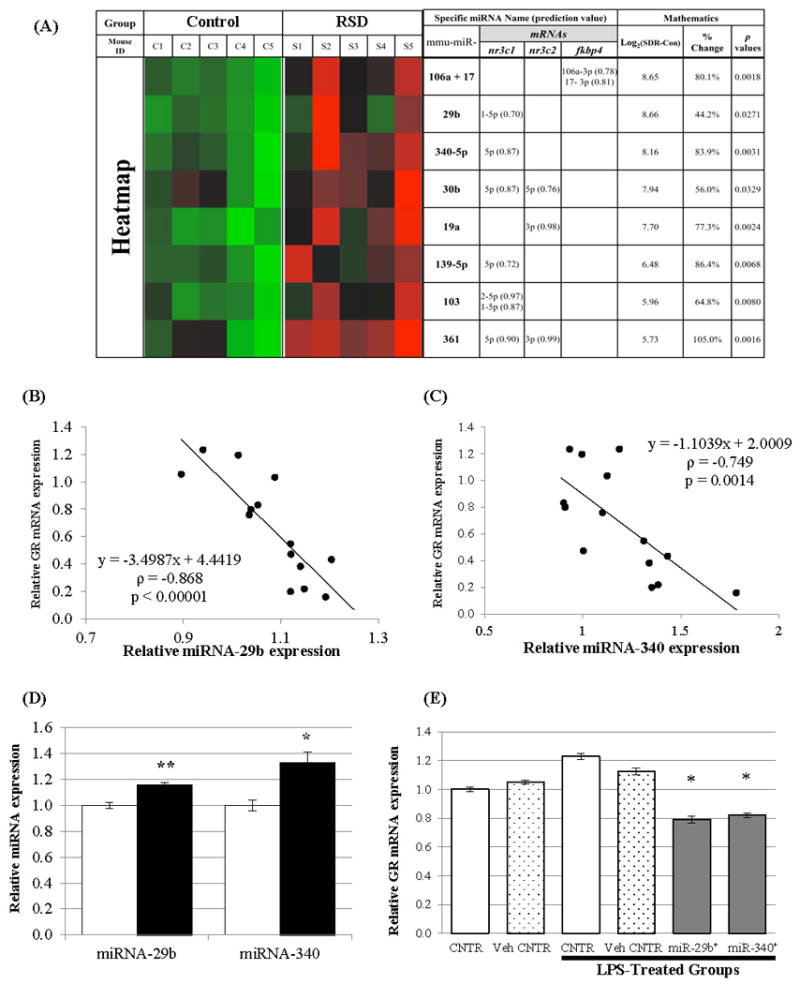
Effects of the 6-cycles of RSD on miRNA expression in splenic macrophages (CD11b+ cells) and effects of miRNA overexpression in glucocorticoid receptor expression in L929 cells.
(A) The heatmap for selected miRNAs that were recognized from the DIANA-microT-CDS database (Ver. 5.0, http://www.microrna.gr/microT-CDS) and also showed significant differences in their expression between control and RSD groups (n = 5/group, p< 0.05 by two-tailed t test). A miRNA with less than 50 counts from both control and RSD was excluded. The heatmap (cell plot) was generated by JMP® statistical discovery software (SAS Institute Inc., Ver. 10.0.2, 64 bit Edition) with a Green-to-Black-to-Red color theme. The miTG score (prediction score) for each miRNA was given in parentheses.
(B), (C) Spearman correlation coefficients of miRNA expression with their target mRNAs. The expression of miRNA-29b (B) and miRNA-340 (C) were all significantly correlated with the expression of their target mRNAs for glucocorticoid receptor (GR, nr3c1). Linear regression equations for each miRNA expression with glucocorticoid receptor (y) were included on the plot with Spearman correlation coefficients (ρ) and p values (n = 14).
(D) Verification of two selected miRNA expression in splenic macrophages in the control (clear bar) and RSD (filled bar) groups (mean ± SEM, n = 8/group). miRNAs were chosen if it was predicted to interact with nr3c1 mRNA with more than 100 counts in both groups. The expression level of each miRNA was normalized to that of the SYBR Green detection.
*: p= 0.00271; **: p< 0.0001
(E) Effects of miRNA-29b and 340 overexpression on glucocorticoid receptor (nr3c1) expression in L929 cells (mean ± SEM, n = 4/group). One way ANOVA test resulted in a significant group difference (p < 0.001), and post-hoc test (Student-Newman-Keuls Method) was performed (*: p< 0.01 vs. control, vehicle control, LPS-control, and LPS-vehicle control groups).
To verify the profiling data for these miRNAs, first, we analyzed the relationship between each miRNA expression and their target mRNA expression by calculating Spearman correlation coefficients using their 2-ΔΔCT values (Figure 3-B & C). We, next, selected 2 miRNAs that were predicted to interact with GC receptor mRNA and confirmed a significantly increased expression of miRNA-29b and 340 in splenic macrophages of the RSD group (Figure 3-D). Overall, we detected significant inverse correlations between the expression of each miRNA (miRNA-340-5p and miRNA-29b), which was increased by RSD, and their target GC receptor mRNA expression.
To confirm the biological effects of these miRNAs, miRNA-29b and 340 were separately overexpressed in L929 cells using Ambion® Pre-miR™ miRNA Precursors (hsa-miR-29b-1-5p and hsamiR-340-5p, respectively). To stimulate the expression of GC receptors that are expressed at a very low level under normal condition, LPS (1μg/mL) was added into cells transfected with miRNA precursors. As shown in Figure 3-E, LPS-stimulated GC receptor nr3c1 mRNA expression was significantly reduced when the miRNA-29b or -340 were overexpressed (p < 0.01).
Discussion
Glucocorticoids are effective anti-inflammatory agents that are used to treat various inflammatory diseases, but this treatment may not be as effective in individuals who have experienced repeated or chronic psychological stress (Avitsur et al., 2001; Bauer et al., 2009; Miller et al., 2002; Sheridan et al., 2000). Our previous studies have shown that repeated social defeat induces GC resistance (Stark et al., 2001) and a functional reduction in GC receptors (Quan et al., 2003a) in splenic macrophages. Immunologic changes induced by RSD included splenomegaly marked by an increase in CD11b+ splenocytes and increased expression of inflammatory chemokines and cytokines (Avitsur et al., 2005). Our results showed a significant increase in body weight. Interestingly, we cannot find any article that actually reported rodent' body weight data using a social stress paradigm, but recent publication from human data showed stress promotes obesity (Kiecolt-Glaser et al., 2014).
To elucidate the underlying mechanisms of RSD on GC resistance in splenic macrophages, we first examined the hypothesis that gene expression of receptors for GC would be negatively affected by RSD: GC receptor expression would be reduced in splenic macrophages, while the expression of mineralocorticoid receptor might be increased or unchanged. We found that exposure to 6 cycles of RSD induced significant reductions in the mRNA expression of both GC and mineralocorticoid receptors in splenic macrophages. We also found that the protein expression of GC receptor in CD11b+ splenocytes was reduced by 6 cycles of RSD, and this is consist with a previous finding (Quan et al., 2003a) showing the decreased protein expression of GC receptor in splenic macrophages (CD11b+ cells) by 6 cycles of RSD. Although the decreased expression of mineralocorticoid receptor in splenic macrophages was not expected, for which future studies must be conducted to elucidate underlying mechanisms, the RSDinduced reduction of the GC receptor expression was consistent with other studies reporting similar expression pattern of GC receptor in difference tissues (Chiba et al., 2012; Witzmann et al., 2012). Chiba et al. (2012), for example, reported that chronic stress in a rodent model induced a significant reduction in GC receptors in the prefrontal cortex. Although we did not determine the GC receptor's binding affinity/sensitivity for glucocorticoids, it was reported that GC resistance was associated with reduced receptor sensitivity (Miller et al., 2002; Quan et al., 2003a; Sheridan et al., 2000). The present study showed a significant reduction in the GC receptor expression in both mRNA and protein levels that may suggest a potential underlying mechanism for the stress-induced GC insensitivity in splenic macrophages. Therefore, this result, in concert with our previous findings (Avitsur et al., 2002), suggests that RSD may affect GC receptors quantitatively and qualitatively in splenic macrophages, consequently resulting in the occurrence of GC resistance.
Numerous published studies show that GC resistance is associated with a reduction in nuclear translocation of GC-receptor complexes, furthermore, it is established that FKBP51 and FKBP52 play a critical role in nuclear translocation of GC receptors (Davies and Sanchez, 2005; De Iudicibus et al., 2011; Jaaskelainen et al., 2011; Wochnik et al., 2005). We examined the hypothesis that RSD would induce an increase in the mRNA ratio of FKBP51 to FKBP52, resulting in a decline in nuclear translocation of the complexes. Because an increase in FKBP51 expression relative to FKBP52 expression is reported to be associated with GC resistance, we calculated a mRNA expression ratio of FKBP51 to FKBP52. Our results indicated that the RSD-induced increased ratio of FKBP51 to FKBP52 was due to a significant decrease in the FKBP52 expression in splenic macrophages. Because the nuclear translocation of GC-receptor complexes is signaled by replacing immunophilin FKBP51 with FKBP52 (Jaaskelainen et al., 2011), it is reasonable to expect that the nuclear translocation of receptor-GC complexes would be reduced because of the significant diminution of FKBP52 expression in response to RSD. This finding may explain our previous observation (Quan et al., 2003a) that GC resistance was associated with impaired nuclear translocation of GC-receptor complexes in splenic macrophages from mice exposed to the 6 cycles of RSD. Furthermore, it has been shown that the higher level of FKBP51 expression results in a lower probability of FKBP52 binding to the GC-receptor and reduced nuclear translocation of GC-receptor complex (Holownia et al., 2009; Paakinaho et al., 2010; Woodruff et al., 2007). One interesting finding from the present study was that FKBP51 expression did not increase in response to RSD, suggesting that GC resistance was unrelated to increased FKBP51 expression. On the other hand, RSD, which was previously shown to induce GC resistance in splenic macrophages (Quan et al., 2003b), prompted no change in FKBP51 expression but a significant decline in FKBP52 expression. However, because we only reported here mRNA expression of FKBP51 and FKBP52, this important difference needs to be further studied in future experiments. Thus, although only mRNA levels (not protein levels) of FKBP52 and FKBP51 were determined and causality was not determined in the current study, we reported that RSD-induced GC resistance was associated with an increased mRNA ratio of FKBP51 to FKBP52, possibly resulting in the impaired nuclear translocation of GC-receptor complexes.
In a follow-up experiment, we examined the mechanisms behind RSD-induced decreases in mRNA expression of GC receptor and FKBP52. We hypothesized that RSD-induced changes might be caused by increased chromatin methylation that inhibited or reduced gene expressions. To test this hypothesis, we first investigated the expression of DNA methyltransferase (DNMT) 3a and 3b, and DNMT1 that are known to be capable of methylating genetic sites and of maintaining methylated sites, respectively. Our qPCR data indicated no significant group difference in DNMT1 expression, suggesting no effect of RSD on the capacity to maintain methylated sites. However, RSD significantly decreased the expression of DNMT3a and DNMT3b, indicating that a reduced amount of methylated sites occurred. To confirm that decreased mRNA expression of DNMT3a and DNMT3b was associated with a reduced amount of methylated sites, the quantity of methylated DNA was analyzed, and we here report that the amount of methylated DNA in splenic macrophages was significantly reduced by RSD. These results suggest an increase in the overall rate of gene transcription, which does not explain the data that showed a significant reduction of gene transcription product “mRNAs.” The results also showed that more “new” methylated sites occurred in the control group compared to the RSD group which was unexpected. However, there is a study consistent with these results. Elliott et al. (2010) recently reported that DNMT3b expression in a brain region (paraventricular nucleus) was significantly decreased 1h after the last cycle of 10 days of social defeat, but they did not suggest a possible underlying mechanism for the change in DNMT3b expression. Because our results on DNA methyltransferases and methylated DNA quantity represent an overall picture in DNA transcription mechanisms, it is still possible to suggest that higher methylation occurs in specific genetic sites, especially promoter and gene sites for GC receptor (nr3c1) and FKBP52 (fkbp4), to inhibit their gene decoding transcriptional process. Although, in this study, we did not investigate chromatin methylation on the specific DNA/histone sites, the current data suggest in general that RSD induces more gene transcription with the exception of transcription for glucocorticoid receptors and FKBP52.
We next hypothesized that RSD might be driving repressive histone modification, resulting in decreased gene transcription. For this study, we analyzed mRNA expression of histone deacetylase 2 (HDAC2) that is known to be recruited in response to increased promoter methylation (Uchida et al., 2011) because of its capacity to deacetylate lysine residues on local histones, especially those related to simple negative glucocorticoid response element (nGRE) on target genes (Ito et al., 2006). When the GC receptor binds to specific DNA sequences, especially nGREs on target genes, it recruits HDACs to provoke deacetylation, and this deacetylation by HDACs is known to correlate with improved glucocorticoid sensitivity (Kassel and Herrlich, 2007). Among different subtypes of HDACs, we chose to analyze HDAC2 expression because HDAC2's association with GC resistance has been well characterized (Kadmiel and Cidlowski, 2013; Kassel and Herrlich, 2007). Our data resulted in a significant decrease in HDAC2 expression in splenic macrophages from the RSD group. The RSDinduced reduction in HDAC2 mRNA expression raised the possibility that more histones would become acetylated, which also suggested that more genetic sites wrapped around acetylated histones were available for transcription. Although the decreased HDAC2 mRNA expression in splenic macrophages by RSD was not expected, this result is consistent with Elliott et al., (2010) showing that a 10-day social defeat paradigm induced a significant decrease in HDAC2 expression in the paraventricular nucleus. The decrease in HDAC2 expression indicates in general that RSD causes splenic macrophages to have less capacity to repress histone activation and transcriptional activation, and, therefore, yield increased gene expression because histone acetyltransferase is inversely associated with HDAC (Guo et al., 2011). Moreover, the literature on GC sensitivity and HDAC2 showed the positive correlation between HDAC2 quantity and GC sensitivity in macrophages (Kadmiel and Cidlowski, 2013; Kassel and Herrlich, 2007). In this case, RSD-induced decrease in HDAC2 mRNA expression may be explainable for RSD-induced GC resistance. However, since we did not report its protein expression and expression of other subtypes; future studies are needed. Based on our results showing both the reduced amount of methylated DNA and decreased mRNA expression of DNMT3a, DNMT3b and HDAC2, it is likely that RSD induced chromatin activation, higher levels of transcriptional activation due to at least in part decreased DNA methylation status and, therefore, more gene expression. This suggestion is supported, especially, by recent findings (Chen et al., 2012; Ito et al., 2000; Malhotra et al., 2011) that HDAC2 plays a critical role in the suppressive action of GC-GC receptor complexes by increasing deacetylation on certain sites of previously activated genes, especially inflammatory genes.
We investigated the effects of RSD on miRNA expression in splenic macrophages. miRNAs are small non-coding RNAs that are known to play an important role in regulating target mRNAs by mRNA repression and degradation. We report that RSD induced significantly increased expression of 9 miRNAs that were predicted to interact with mRNAs of the GC receptor (6 miRNAs), mineralocorticoid receptor (3 miRNAs) and FKBP52 (2 miRNAs). By qPCR, we verified 2 different miRNAs (miR-29b and miR-340) that were predicted to interact with mRNA expression of GC receptor. The results showed a similar pattern of enhanced expression as shown in the miRNA profiling data. Their target mRNA expression of GC receptor was negatively correlated with the expression of each miRNA (miR-29b or miR-340). To verify a direct, biological effect of each miRNA on their target gene expression, we conducted an in vitro study, in which miR-29b or 340 was overexpressed in L929 cells to determine whether the mRNA expression of their target gene (GC receptor) was influenced by the miRNA. LPS-induced expression of GC receptor was significantly blocked by the overexpression of miR-29b and 340. This result confirmed direct effects of these miRNA on GC receptor gene expression. This result indicated that RSD-induced GC resistance in splenic macrophages was, at least partially, stimulated by the miRNAs. Although a recent study (Ma et al., 2011) showed that miR-29b suppresses innate and adaptive immune responses and suggested that miR-29b plays an important role in immune regulation, very few studies have been published to investigate roles of these GC receptor-targeted miRNAs in immune cells, including macrophages. Therefore, it is necessary to elucidate, in future investigations, roles of these miRNA in immunity in addition to investigating whether the direct influence of these miRNAs on their target mRNAs is due to their direct binding capability to 3′UTR of GC receptor mRNA.
In this study, we examined the underlying molecular mechanisms of RSD-induced GC resistance in splenic macrophages. This study provided evidence of a molecular mechanistic explanation for GC resistance in splenic macrophages. The data showed that RSD induced a significant decrease in mRNA expression of the GC receptor, mineralocorticoid receptor, FKBP52, DNMT3a, DNMT3b and HDAC2 and a significant increase in the mRNA ratio of FKBP51 to FKBP52. Moreover, RSD also significantly modified methylated DNA status and miRNA expression in splenic macrophages. For the miRNA modification, the data showed that the expression of specific miRNAs that were predicted to interact with mRNAs for the GC receptor, mineralocorticoid receptor and FKBP52 were significantly enhanced by RSD. The verifying analyses for these potential miRNAs also showed a significantly strong correlation with their predicted target mRNA expression of GC receptors, suggesting a potential role of miRNAs in RSD-induced GC resistance in splenic macrophages. As summarized in Figure 4, our data provide evidence that chronic social stress stimulates epigenetic regulation, such as DNA methylation status and miRNA modifications. In particular, we demonstrated that RSD stimulated translational repressive modification by increasing the expression of certain miRNAs that have the ability to decrease the expression of GC receptors and FKBP52, and as a consequence could result in the occurrence of GC resistance in splenic macrophages. However, further studies are needed to evaluate specific epigenetic modification induced by RSD, such as specific genes in which methylation levels are modified, and binding affinities of each miRNA to their predicted mRNAs to confirm the role of these miRNAs in GC intracellular regulation.
Figure 4.
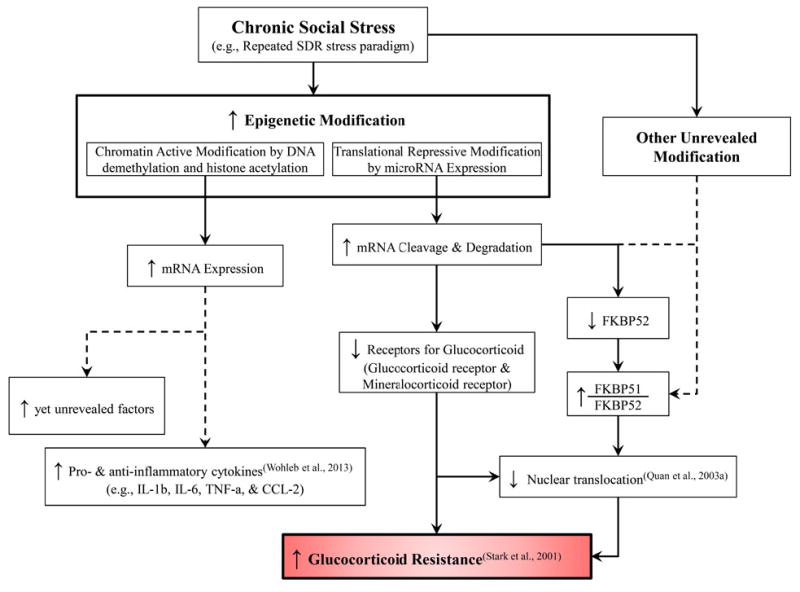
Diagram of the hypothesized underlying mechanisms for the RSD-induced glucocorticoid resistance in splenic macrophages (CD11b+ cells). For the glucocorticoid resistance induced by chronic social stress, chromatin modification includes DNA and histone methylation/demethylation and histone acetylation/deacetylation based on the present study.
Highlights.
Repeated social stress decreased nr3c1 and fkbp52 expression in splenic macrophage.
Repeated social stress induced epigenetic modification in splenic macrophages
Repeated social stress significantly decreased the amount of methylated DNA in splenic macrophages.
Repeated social stress induced significantly increased expression of six microRNAs.
microRNA-29b and 340 overexpression diminished GC receptor mRNA expression.
Acknowledgments
This study was supported by grants funded through NIH/NIMH: R01MH093473 and R01MH097243 (to Dr. John F. Sheridan). Dr. Seung Ho Jung was supported as a postdoctoral fellow on an NIDCR T32DE014320 training grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Avitsur R, Stark JL, Dhabhar FS, Padgett DA, Sheridan JF. Social disruption-induced glucocorticoid resistance: kinetics and site specificity. J Neuroimmunol. 2002;124:54–61. doi: 10.1016/s0165-5728(02)00010-3. [DOI] [PubMed] [Google Scholar]
- Avitsur R, Stark JL, Sheridan JF. Social stress induces glucocorticoid resistance in subordinate animals. Horm Behav. 2001;39:247–257. doi: 10.1006/hbeh.2001.1653. [DOI] [PubMed] [Google Scholar]
- Bauer ME, Jeckel CM, Luz C. The role of stress factors during aging of the immune system. Annals of the New York Academy of Sciences. 2009;1153:139–152. doi: 10.1111/j.1749-6632.2008.03966.x. [DOI] [PubMed] [Google Scholar]
- Chen Y, Watson AM, Williamson CD, Rahimi M, Liang C, Colberg-Poley AM, Rose MC. Glucocorticoid receptor and histone deacetylase-2 mediate dexamethasone-induced repression of MUC5AC gene expression. Am J Respir Cell Mol Biol. 2012;47:637–644. doi: 10.1165/rcmb.2012-0009OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba S, Numakawa T, Ninomiya M, Richards MC, Wakabayashi C, Kunugi H. Chronic restraint stress causes anxiety- and depression-like behaviors, downregulates glucocorticoid receptor expression, and attenuates glutamate release induced by brain-derived neurotrophic factor in the prefrontal cortex. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39:112–119. doi: 10.1016/j.pnpbp.2012.05.018. [DOI] [PubMed] [Google Scholar]
- Davies TH, Ning YM, Sanchez ER. A new first step in activation of steroid receptors: hormone-induced switching of FKBP51 and FKBP52 immunophilins. The Journal of biological chemistry. 2002;277:4597–4600. doi: 10.1074/jbc.C100531200. [DOI] [PubMed] [Google Scholar]
- Davies TH, Sanchez ER. Fkbp52. Int J Biochem Cell Biol. 2005;37:42–47. doi: 10.1016/j.biocel.2004.03.013. [DOI] [PubMed] [Google Scholar]
- De Iudicibus S, Franca R, Martelossi S, Ventura A, Decorti G. Molecular mechanism of glucocorticoid resistance in inflammatory bowel disease. World J Gastroenterol. 2011;17:1095–1108. doi: 10.3748/wjg.v17.i9.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott E, Ezra-Nevo G, Regev L, Neufeld-Cohen A, Chen A. Resilience to social stress coincides with functional DNA methylation of the Crf gene in adult mice. Nat Neurosci. 2010;13:1351–1353. doi: 10.1038/nn.2642. [DOI] [PubMed] [Google Scholar]
- Fagundes CP, Glaser R, Kiecolt-Glaser JK. Stressful early life experiences and immune dysregulation across the lifespan. Brain behavior and immunity. 2013;27:8–12. doi: 10.1016/j.bbi.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galigniana NM, Ballmer LT, Toneatto J, Erlejman AG, Lagadari M, Galigniana MD. Regulation of the glucocorticoid response to stress-related disorders by the Hsp90-binding immunophilin FKBP51. J Neurochem. 2012;122:4–18. doi: 10.1111/j.1471-4159.2012.07775.x. [DOI] [PubMed] [Google Scholar]
- Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, James JJ, Maysuria M, Mitton JD, Oliveri P, Osborn JL, Peng T, Ratcliffe AL, Webster PJ, Davidson EH, Hood L, Dimitrov K. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- Glaser R, MacCallum RC, Laskowski BF, Malarkey WB, Sheridan JF, Kiecolt-Glaser JK. Evidence for a shift in the Th-1 to Th-2 cytokine response associated with chronic stress and aging. J Gerontol A Biol Sci Med Sci. 2001;56:M477–482. doi: 10.1093/gerona/56.8.m477. [DOI] [PubMed] [Google Scholar]
- Gouin JP, Carter CS, Pournajafi-Nazarloo H, Glaser R, Malarkey WB, Loving TJ, Stowell J, Kiecolt-Glaser JK. Marital behavior, oxytocin, vasopressin, and wound healing. Psychoneuroendocrinology. 2010;35:1082–1090. doi: 10.1016/j.psyneuen.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Wu X, Ren L, Liu G, Li L. Epigenetic mechanisms of amyloid-beta production in anisomycin-treated SH-SY5Y cells. Neuroscience. 2011;194:272–281. doi: 10.1016/j.neuroscience.2011.07.012. [DOI] [PubMed] [Google Scholar]
- Holownia A, Mroz RM, Kolodziejczyk A, Chyczewska E, Braszko JJ. Increased FKBP51 in induced sputum cells of chronic obstructive pulmonary disease patients after therapy. Eur J Med Res. 2009;14(Suppl 4):108–111. doi: 10.1186/2047-783X-14-S4-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House JS, Landis KR, Umberson D. Social relationships and health. Science. 1988;241:540–545. doi: 10.1126/science.3399889. [DOI] [PubMed] [Google Scholar]
- Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol. 2000;20:6891–6903. doi: 10.1128/mcb.20.18.6891-6903.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Yamamura S, Essilfie-Quaye S, Cosio B, Ito M, Barnes PJ, Adcock IM. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J Exp Med. 2006;203:7–13. doi: 10.1084/jem.20050466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaaskelainen T, Makkonen H, Palvimo JJ. Steroid up-regulation of FKBP51 and its role in hormone signaling. Curr Opin Pharmacol. 2011;11:326–331. doi: 10.1016/j.coph.2011.04.006. [DOI] [PubMed] [Google Scholar]
- Jaremka LM, Fagundes CP, Glaser R, Bennett JM, Malarkey WB, Kiecolt-Glaser JK. Loneliness predicts pain, depression, and fatigue: understanding the role of immune dysregulation. Psychoneuroendocrinology. 2013;38:1310–1317. doi: 10.1016/j.psyneuen.2012.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. 2013;34:518–530. doi: 10.1016/j.tips.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassel O, Herrlich P. Crosstalk between the glucocorticoid receptor and other transcription factors: molecular aspects. Mol Cell Endocrinol. 2007;275:13–29. doi: 10.1016/j.mce.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Kiecolt-Glaser JK, Habash DL, Fagundes CP, Andridge R, Peng J, Malarkey WB, Belury MA. Daily Stressors, Past Depression, and Metabolic Responses to High-Fat Meals: A Novel Path to Obesity. Biol Psychiatry. 2014 doi: 10.1016/j.biopsych.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiecolt-Glaser JK, Marucha PT, Malarkey WB, Mercado AM, Glaser R. Slowing of wound healing by psychological stress. Lancet. 1995;346:1194–1196. doi: 10.1016/s0140-6736(95)92899-5. [DOI] [PubMed] [Google Scholar]
- Ma F, Xu S, Liu X, Zhang Q, Xu X, Liu M, Hua M, Li N, Yao H, Cao X. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nat Immunol. 2011;12:861–869. doi: 10.1038/ni.2073. [DOI] [PubMed] [Google Scholar]
- Malhotra D, Thimmulappa RK, Mercado N, Ito K, Kombairaju P, Kumar S, Ma J, Feller-Kopman D, Wise R, Barnes P, Biswal S. Denitrosylation of HDAC2 by targeting Nrf2 restores glucocorticosteroid sensitivity in macrophages from COPD patients. J Clin Invest. 2011;121:4289–4302. doi: 10.1172/JCI45144. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Miller GE, Cohen S, Ritchey AK. Chronic psychological stress and the regulation of proinflammatory cytokines: a glucocorticoid-resistance model. Health Psychol. 2002;21:531–541. doi: 10.1037//0278-6133.21.6.531. [DOI] [PubMed] [Google Scholar]
- Paakinaho V, Makkonen H, Jaaskelainen T, Palvimo JJ. Glucocorticoid receptor activates poised FKBP51 locus through long-distance interactions. Mol Endocrinol. 2010;24:511–525. doi: 10.1210/me.2009-0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell ND, Tarr AJ, Sheridan JF. Psychosocial stress and inflammation in cancer. Brain Behav Immun. 2013;30(Suppl):S41–47. doi: 10.1016/j.bbi.2012.06.015. [DOI] [PubMed] [Google Scholar]
- Quan N, Avitsur R, Stark JL, He L, Lai W, Dhabhar F, Sheridan JF. Molecular mechanisms of glucocorticoid resistance in splenocytes of socially stressed male mice. J Neuroimmunol. 2003a;137:51–58. doi: 10.1016/s0165-5728(03)00042-0. [DOI] [PubMed] [Google Scholar]
- Quan N, Avitsur R, Stark JL, He LL, Lai WM, Dhabhar F, Sheridan JF. Molecular mechanisms of glucocorticoid resistance in splenocytes of socially stressed male mice. Journal of Neuroimmunology. 2003b;137:51–58. doi: 10.1016/s0165-5728(03)00042-0. [DOI] [PubMed] [Google Scholar]
- Sheridan JF, Stark JL, Avitsur R, Padgett DA. Social disruption, immunity, and susceptibility to viral infection. Role of glucocorticoid insensitivity and NGF. Ann N Y Acad Sci. 2000;917:894–905. doi: 10.1111/j.1749-6632.2000.tb05455.x. [DOI] [PubMed] [Google Scholar]
- Sivils JC, Storer CL, Galigniana MD, Cox MB. Regulation of steroid hormone receptor function by the 52-kDa FK506-binding protein (FKBP52) Current opinion in pharmacology. 2011;11:314–319. doi: 10.1016/j.coph.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark JL, Avitsur R, Padgett DA, Campbell KA, Beck FM, Sheridan JF. Social stress induces glucocorticoid resistance in macrophages. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1799–1805. doi: 10.1152/ajpregu.2001.280.6.R1799. [DOI] [PubMed] [Google Scholar]
- Stechschulte LA, Sanchez ER. FKBP51-a selective modulator of glucocorticoid and androgen sensitivity. Current opinion in pharmacology. 2011;11:332–337. doi: 10.1016/j.coph.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratakis CA, Karl M, Schulte HM, Chrousos GP. Glucocorticosteroid resistance in humans. Elucidation of the molecular mechanisms and implications for pathophysiology. Annals of the New York Academy of Sciences. 1994;746:362–374. doi: 10.1111/j.1749-6632.1994.tb39257.x. discussion 374-366. [DOI] [PubMed] [Google Scholar]
- Uchida S, Hara K, Kobayashi A, Otsuki K, Yamagata H, Hobara T, Suzuki T, Miyata N, Watanabe Y. Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron. 2011;69:359–372. doi: 10.1016/j.neuron.2010.12.023. [DOI] [PubMed] [Google Scholar]
- Voorhees JL, Tarr AJ, Wohleb ES, Godbout JP, Mo X, Sheridan JF, Eubank TD, Marsh CB. Prolonged restraint stress increases IL-6, reduces IL-10, and causes persistent depressive-like behavior that is reversed by recombinant IL-10. PLoS One. 2013;8:e58488. doi: 10.1371/journal.pone.0058488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzmann SR, Turner JD, Meriaux SB, Meijer OC, Muller CP. Epigenetic regulation of the glucocorticoid receptor promoter 1(7) in adult rats. Epigenetics. 2012;7:1290–1301. doi: 10.4161/epi.22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wochnik GM, Ruegg J, Abel GA, Schmidt U, Holsboer F, Rein T. FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J Biol Chem. 2005;280:4609–4616. doi: 10.1074/jbc.M407498200. [DOI] [PubMed] [Google Scholar]
- Wohleb ES, Powell ND, Godbout JP, Sheridan JF. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:13820–13833. doi: 10.1523/JNEUROSCI.1671-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, Donnelly S, Ellwanger A, Sidhu SS, Dao-Pick TP, Pantoja C, Erle DJ, Yamamoto KR, Fahy JV. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U S A. 2007;104:15858–15863. doi: 10.1073/pnas.0707413104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Ray DW, Matthews LC. Current concepts in glucocorticoid resistance. Steroids. 2012;77:1041–1049. doi: 10.1016/j.steroids.2012.05.007. [DOI] [PubMed] [Google Scholar]