

Abstract
It is well known that lipids are heterogeneously distributed throughout the cell. Most lipid species are synthesized in the ER and then distributed to different cellular locations in order to create the distinct membrane compositions observed in eukaryotes. However, the mechanisms by which specific lipid species are trafficked to and maintained in specific areas of the cell are poorly understood and constitute an active area of research. Of particular interest is the distribution of phosphatidylserine (PS), an anionic lipid that is enriched in the cytosolic leaflet of the plasma membrane. PS transport occurs by both vesicular and non-vesicular routes, with members of the oxysterol binding protein family (Osh6 and Osh7) recently implicated in the latter route. In addition, the flippase activity of P4-ATPases helps build PS membrane asymmetry by preferentially translocating PS to the cytosolic leaflet. This asymmetric PS distribution can be used as a signaling device by the regulated activation of scramblases, which rapidly expose PS on the extracellular leaflet and play important roles in blood clotting and apoptosis. This review will discuss recent advances made in the study of phospholipid flippases, scramblases, and PS-specific lipid transfer proteins, as well as how these proteins contribute to subcellular PS distribution.
Keywords: P-type ATPase, flippase, scramblase, phosphatidylserine, lipid transfer protein, membrane asymmetry
Introduction
The plasma membrane and internal organelles of eukaryotic cells form an interconnected membrane system that maintains compositionally unique components (1). The trafficking of proteins from their site of synthesis to their final destinations has been extensively studied. However, less is known about the movement of lipids throughout the cell. Most lipid synthesis occurs within the cytosolic leaflet of the ER (2) and in order to achieve the unique organelle lipid compositions observed in eukaryotes, mechanisms must be in place to concentrate specific lipid species to different cellular membranes. For example, sterols and phosphatidylserine (PS) are highly concentrated at the plasma membrane (PM) and are maintained at low abundance in the ER despite being synthesized in this membrane (3,4). Even within an individual membrane bilayer, there are multiple levels of organization with lipid species asymmetrically distributed between the two leaflets (5,6) and heterogeneously distributed within a leaflet in the form of lipid rafts or nanodomains (7,8).
For most characterized eukaryotic cells, the extracellular leaflet of the PM is primarily composed of phosphatidylcholine (PC) and sphingolipids, whereas PS, phosphatidylethanolamine (PE), phosphatidylinositol (PI), and phosphoinositides are preferentially restricted to the cytosolic leaflet (9–12). This asymmetric structure, with a greater number of anionic lipids in the cytosolic leaflet and mostly neutral lipids in the extracellular leaflet, generates two surfaces with vastly different electrostatic potentials (13). The association of proteins with the membrane surface and the activity of integral membrane proteins can be strongly influenced by these membrane properties (14–17). The phospholipid gradient within the membrane is also used for signal transduction in a manner analogous to how ion gradients across the membrane are used. Regulated exposure of PS in the outer leaflet of the plasma membrane is an early event in apoptosis and serves as an “eat me” signal important for the orderly removal of cell corpses (18–20). PS exposure on red blood cells and platelets substantially accelerates clotting reactions and is therefore crucial for controlling thrombosis (21,22).
Three categories of proteins have been described that function in the formation and dissolution of membrane asymmetry: flippases, floppases, and scramblases (23,24). Flippases involved in creating membrane asymmetry are type-IV P-type ATPases (P4-ATPases) that catalyze the movement of specific phospholipid species from the extracellular leaflet to the cytosolic leaflet, whereas floppases are ABC-transporters that mediate the movement of phospholipids in the reverse direction (24,25). However, an exception to this categorization is the ABC-transporter ABCA4, which acts as a PE and retinylidene-PE flippase in outer segment membranes of rod and cone cells (26,27). Phospholipid transport mediated by P4-ATPases and ABC transporters is unidirectional and ATP-dependent. The P4-ATPases are clearly linked to the establishment of membrane asymmetry in multiple cell types and organisms, but the ABC transporters appear to play more specialized roles, such as the excretion of the lipid components of bile (24). In contrast, scramblases are ATP-independent and act to randomize lipid distribution by bidirectionally translocating lipids between leaflets (28). In addition to the lipid translocases, lipid transfer proteins (LTPs) are proposed to be involved in the localization of specific lipid species potentially by mediating non-vesicular transport of lipids at membrane contact sites (29,30). An LTP extracts a specific lipid species from a donor membrane and transports the lipid to an acceptor membrane, where it can be exchanged for a second lipid species (30,31).
This review will focus on exciting recent progress in the study of proteins that influence PS localization within the cell and control membrane asymmetry. Recent advances in this field have provided greater insight into the intracellular distribution of PS through the development and use of the GFP-Lact-C2 biosensor for PS (32). PS molecules traveling by vesicular transport through the secretory pathway appear to be initially enriched in the lumenal leaflet of the ER and cis-Golgi (32), but are then flipped to the cytosolic leaflet by P4-ATPases in the trans-Golgi network (TGN) and exocytic vesicles (Figure 1) (33,34). These vesicles would then have the appropriate membrane topology when they fuse to the plasma membrane. The P4-ATPases are now firmly established to catalyze phospholipid flippase activity and important new insights are emerging into how P4-ATPases have evolved the ability to recognize and flip a phospholipid substrate (35–40). The translocation of PS by a P4-ATPase was discovered to be crucial for vesicular transport pathways connecting the TGN and early endosomes (41). A pair of oxysterol binding protein homologs (Osh6 and Osh7) have recently been implicated in PS transfer, potentially providing a non-vesicular route for PS transport to the PM (42). A substantial body of evidence now indicates that TMEM16F is the phospholipid scramblase linked to coagulation reactions and the Xkr8/CED-8 family of proteins has recently been implicated in the exposure of PS in apoptotic cells (43,44). Most of these discoveries were made in just the last few years making this an exciting period for new insight into the mechanisms for establishing and disrupting membrane component distribution.
Figure 1.

Distribution of phosphatidylserine (PS) throughout the cell. (1) PS is synthesized on the cytosolic leaflet of the endoplasmic reticulum (ER). Rapid, energy-independent flip-flop occurs in the ER membrane, but PS is preferentially localized within the lumenal leaflet, either by retention through interaction with abundant lumenal components or because (2) PS in the cytosolic leaflet is transported by non-vesicular means from the ER to plasma membrane (PM) by lipid transport proteins (LTPs). (3) PS that travels through the secretory system remains within the lumenal leaflet until it is flipped by P4-ATPases in the trans-Golgi network (TGN) and early endosomes (EE) to the cytosolic leaflet. (4) P4-ATPases also flip PS at the PM, ensuring little PS is exposed in the extracellular leaflet. (5) When activated during the process of apoptosis or blood clotting, scramblases break down the lipid asymmetry of the PM, causing exposure of PS on the outer leaflet. (6) PS exposed on the extracellular leaflet of apoptotic cells acts as an “eat me” signal for engulfment by macrophages.
Intracellular Distribution of PS
Phospholipid synthesis occurs primarily in the cytosolic leaflet of the ER, and to preserve membrane stability of the ER, there must be a mechanism by which newly synthesized lipids are redistributed from the cytosolic leaflet to the lumenal leaflet. A number of labs have proposed that ER lipids are bidirectionally scrambled across the bilayer in an energy-independent fashion, presumably forming a symmetric membrane, although the mechanism remains unclear (2,45–48). In contrast, Higgins and Dawson reported that PS is enriched in the lumenal leaflet of the ER (49). A recent finding by Fairn, et al. supports the hypothesis of a non-symmetric ER (32). The Grinstein lab has developed a novel probe containing the C2 domain of lactadherin (Lact-C2) that specifically binds the biologically relevant PS isomer phosphatidyl-L-serine and no other lipids in a Ca2+-independent manner (50). Genetically encoded Lact-C2 labels the cytosolic surface of membranes with the highest concentration of the probe at the PM and with very little ER labeling. To provide access to the entire pool of PS, ultrathin sections of fast-frozen samples were overlaid with Lact-C2. A substantial increase in labeling of ER, Golgi, and mitochondria, was observed, indicating that PS is preferentially localized to the lumenal leaflet of the ER (32).
An unresolved question from the distribution of Lact-C2 is what happens to the PS in the cytosolic leaflet of the ER? One possibility is that PS flip-flops rapidly, but is selectively retained in the lumenal leaflet by Ca2+ and/or abundant ER resident proteins. Alternatively, PS in the cytosolic leaflet of the ER may be rapidly removed via non-vesicular lipid transport. Large amounts of lipids are exchanged between membranes during the processes of vesicle budding and fusion; however, lipid transport is still robust when vesicular trafficking is blocked by ATP depletion or by a reduction in temperature (51,52). The Voelker lab has identified several factors in yeast required for movement of PS from the ER to an endosomal compartment where it is decarboxylated to PE. These factors include a phosphatidylinositol 4-kinase (Stt4) and a Sec14 homolog (PstB2/Pdr17), which belongs to the LTP superfamily (53,54). LTPs are capable of catalyzing non-vesicular transport of lipids between membranes in vitro, but there has been significant controversy over if LTPs mediate bulk transfer of lipids and contribute to compositional differences between organelle membranes in vivo (55–59). PstB2 does not appear to bind or transfer PS directly and precisely how Stt4 and PstB2 contribute to PS transport is unknown.
The Gavin lab has recently characterized LTP binding to lipids in vivo to determine their specificities (42). In their analysis of 12 LTPs from budding yeast, they confirmed many known interactions such as the interaction between Osh4/Kes1 and sterol (42,60). Unexpectedly, they found that Osh6 and Osh7, which poorly bind to sterol in vitro, specifically bind PS and are able to transfer PS from donor to acceptor liposomes (42). Osh6/Osh7 localize to ER-PM contact sites, thus linking the main site of PS synthesis to the location where PS primarily accumulates. Deletion of both Osh6 and Osh7 reduces PM accumulation of PS by ~30% and causes PS to redistribute to the ER and vacuoles but has no effect on other lipid species examined. Remarkably, tethering Osh6 to the vacuolar membrane protein Vph1 by rapamycin-induced dimerization causes PS to redirect to the vacuole (42). Taken together, these results support the idea that Osh6/7 can transport PS in vivo, but it remains unclear how they mediate directional movement and concentration in the plasma membrane.
If PS is primarily enriched in the lumenal leaflet of the ER and cytosolic leaflet of the PM, PS must change sidedness somewhere along its journey. Colocalization experiments showed little overlap between Lact-C2 and a trans-Golgi cisternae marker, indicating that PS at the trans-Golgi is still largely constrained to the lumenal leaflet (32). However, significant colocalization occurred between Lact-C2 and a TGN marker, suggesting the TGN is the site of PS translocation, which is consistent with TGN localization of Drs2, the primary PS flippase in yeast (33,34,61,62) and indicates that mammals have one or more TGN-localized flippases (32). PS translocation at the TGN plays a crucial role in the budding of transport vesicles from this organelle (41), in addition to helping generate the asymmetric organization of the plasma membrane (34).
Mechanism of phospholipid translocation by P4-ATPases
P4-ATPases (flippases) are members of the evolutionarily conserved P-type ATPase family of membrane-bound pumps (63). Eukaryotes express multiple P4-ATPases including the well characterized homologs in budding yeast (Drs2, Dnf1, Dnf2, Dnf3, and Neo1), Caenorhabditis elegans (Tat-1 through Tat-6), mammals (ATP8A1 through ATP11C), and Arabidopsis (ALA1 through ALA12). All P-type ATPases have a similar structural organization, which consists of a membrane domain typically comprised of 10 transmembrane (TM) segments, an actuator domain, a phosphorylation domain, and a nucleotide-binding domain (64). Most of the P4-ATPases associate with a noncatalytic β subunit from the Cdc50 family, consisting of two transmembrane segments and a glycosylated ectodomain (65,66). Formation of the heterodimer is crucial for transport out of the ER and the β subunit also influences the catalytic activity of the pump (33,62) Unlike other ion-transporting P-type ATPases, P4-ATPases transport phospholipid from the exofacial leaflet to the cytosolic leaflet of the membrane – a flippase activity (67,39,40). By pumping PS and PE to the cytosolic leaflet, the P4-ATPases appear to be primarily responsible for establishing membrane asymmetry.
The mechanism of substrate recognition for cation pumps is well defined, but how the P4-ATPases evolved the ability to recognize and flip a phospholipid molecule across the membrane has been a quandary. Crystal structures of P2-ATPases revealed cation-binding sites in a small cavity formed by TM segment 4, 5, 6, and 8 in the Ca2+ ATPase (68) and by TM segment 4, 5, 6, 8, and 9 in the Na+/K+ ATPase (69). However, these canonical substrate binding pockets are likely too small to accommodate a bulky phospholipid substrate and the P4-ATPases lack the polar and charged residues found in the canonical site. This enigma of how P4-ATPases are able to flip lipids across the membrane has been termed the “giant substrate problem” (35,70). Until recently, the residues involved in recognizing and flipping phospholipid and even the path the phospholipid travels through the TM region to reach the opposite face of the membrane were a complete mystery.
Recent work from Baldridge and Graham addressed the P4-ATPase giant substrate problem by identifying residues important for phospholipid substrate specificity (35–37). This work was performed with two divergent P4-ATPases in Saccharomyces cerevisiae: Drs2 and Dnf1. Drs2 primarily flips PS and, to a lesser extent, PE (33,39), whereas Dnf1 was thought to primarily flip PC and PE (71,72). However, recent evidence suggests that Dnf1 preferentially flips lysophospholipids that lack the sn2 acyl chain (lyso-PC and lyso-PE) (37,73). Chimeras were created by transplanting TM segments of Drs2 into Dnf1, with the aim of altering the substrate preference of Dnf1 to that of Drs2 (35). They found that the Dnf1 chimera containing TM 3–4 of Drs2 had an ~8-fold increase in PS transport and decreased PC transport relative to Dnf1, suggesting that these segments are involved in substrate recognition. Further mapping revealed that the substitution of a single residue, Y618F in TM4, allows Dnf1 to flip 7-nitro-2–1,3-benzoxadiazol-4-yl phospholipid (NBD-PS) without altering NBD-PC/PE recognition. The reciprocal change, F511Y in TM4 of Drs2, abrogates PS flip and causes exposure of PS in the extracellular leaflet of the PM. In the cation pumps, TM4 forms part of the canonical substrate-binding pocket where a highly conserved, helix breaking proline (Pro) is followed by a glutamic acid (Glu) that coordinates ion substrates in both the Na+/K+-ATPase and Ca2+-ATPase (66). The TM4 Pro is also conserved in the P4-ATPases, but Dnf1/Drs2 has a PISLY/F motif rather than the PEGLX motif found in cation pumps. Thus, a Tyr or Phe four residues downstream of the Pro (P+4 position), representing the difference of a hydroxyl group, is an important determinant of PS recognition. Importantly, this residue is on the destination side of the membrane so it was clear from this initial study that more than one residue would be involved in PS recognition and that different Dnf1 residues were selecting PC and PE.
In subsequent work, Baldridge and Graham devised unbiased, genetic strategies for screening thousands of mutations targeted to the TM segments of Dnf1 for those that alter substrate specificity (36). More than 20 residues important for defining headgroup specificity were identified within TM segments 1, 2, 3 and 4, and the exofacial loop between TM3 and 4 (Figure 2). These residues cluster in two groups on opposing sides of the membrane, which were termed the entry and exit gates (36). The entry gate is composed of residues on the exofacial side where substrate would be selected for transport and loaded into the pump. The exit gate is composed of residues on the cytofacial side and represents a second site of selection prior to substrate release into the cytosolic leaflet. The gates act cooperatively, but imperfectly, such that neither gate was able to fully restrict PS flip when the opposite gate permitted it. Importantly, several residues at the exit gate are important for the recognition of lysophospholipid (a phospholipid lacking the sn2 acyl chain) versus diacylated phospholipid (37). For example, mutation of a conserved asparagine (N550S) in the exit gate allows Dnf1 to flip endogenous diacylated PS and restore PS asymmetry in drs2Δ cells. Most notably, the residues that are important for phospholipid selection do not correspond to the canonical substrate binding pocket seen in P2-type ATPases. For example, the first 3 TM segments of the cation pumps are not involved in substrate selection (68,69). Baldridge and Graham proposed a noncanonical pathway by which the phospholipid headgroup is translocated along a groove at the protein/membrane interface formed by TM segments 1, 3, and 4, while the acyl chains reorient within the surrounding lipid environment (36). This model provides a solution to the giant substrate problem because the entire phospholipid does not have to be accommodated within a binding pocket in the middle of the membrane domain.
Figure 2.
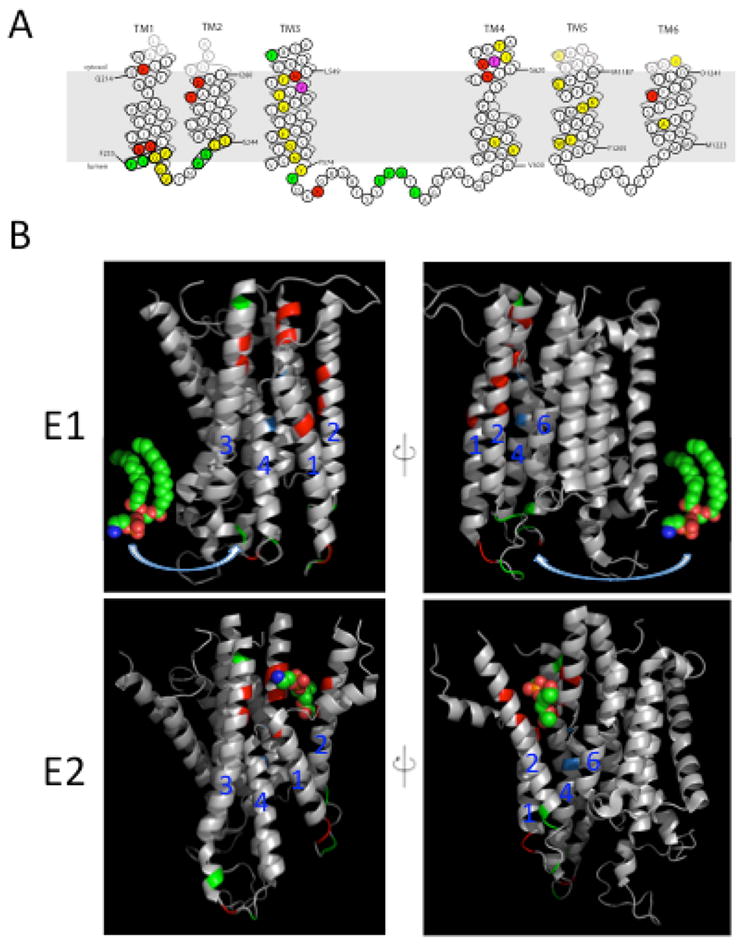
Amino acid residues in the membrane domain of Dnf1 that determine substrate specificity. (A) Topology of the first 6 transmembrane (TM) segments of Dnf1 highlighting residues important for substrate specificity and transport. Dnf1 normally recognizes and transports lyso-PC and lyso-PE, but specific point mutations changing the residues highlighted in red allow recognition of PS. Mutations in residues highlighted in green perturb recognition of PC without altering translocation of PE. Changes in only two residues, highlighted in purple, alter recognition of both PC and PS. Mutation of residues in yellow reduced activity without altering substrate specificity. (B) Two potential translocation pathways for flipping phospholipid substrate. The phospholipid headgroup may engage the entry gate residues in the E1 conformation, then slide along a grooved formed by TM segments 1, 3 and 4 (left) or TM 2, 4 and 6 (right) and dock in the exit gate in the E2 conformation. The crystal structures of the Ca2+-ATPase membrane domain in the E1 (PBD ISU4) and E2 (PDB 3W5C) conformational states are shown with the positions homologous to substrate-defining residues of the P4-ATPases highlighted. Red and green positions represent the Dnf1 residues shown in (A) involved in PS and PC recognition, respectively. Positions highlighted in blue represent the P+1 Ile and N359 in ATP8A2. The PE molecule in the E1 images derived from PDB 3B74 while the PE headgroup in the E2 images co-crystallized with the Ca2+-ATPase.
The P-type ATPases are characterized by the autocatalyzed formation of an aspartyl-phosphate intermediate within the P domain and subsequent hydrolysis of this bond during the ATPase cycle (74–76). These events are coupled to conformational changes (E1 → E1~P →E2~P → E2 → E1) that drive substrate transport against the prevailing concentration gradient. For the Na+/K+-ATPase, Na+ stimulates autophosphorylation and is pumped out of the cell during the E1 → E2~P transition. K+ then binds from the outside, stimulates dephosphorylation and is pumped into the cell during the E2~P → E1 transition. In contrast, the P4-ATPases autophosphorylate in the apparent absence of any substrate (E1 → E2~P), and phospholipid substrate stimulates dephosphorylation (E2~P → E1) (40,77,78). These observations suggest a model where phospholipid substrate binds the entry gate in either the E1 or E1~P conformation, but this interaction is not coupled to ATP binding or phosphorylation. TM1-2 are drawn up towards the cytosolic leaflet during the E1 to E2~P transition, presumably pulling the substrate headgroup with it, creating a path for migration of the headgroup into the exit gate. Exit gate binding is specific not only to the lipid headgroup, but also to the glycerol backbone (sphingolipids are not flipped) and to the acyl chain occupancy at the sn2 position (36,37). This binding event would stimulate dephosphorylation, release of substrate to the cytosolic leaflet, and resetting of the pump for another round of transport.
This model potentially provides insight into how the P4-ATPases evolved the ability to transport phospholipid substrate. The Ca2+-ATPase crystal structure in the E2 conformation (Protein Data Bank ID code 2AGV) has a bound PE in a position homologous to the exit gate in the P4-ATPases (79). This binding pocket closes in the E1 conformation, implying that the PE is a boundary lipid that enters and exits this site from the cytosolic leaflet with each round of ATP hydrolysis. The P4-ATPases may have evolved from a primordial cation pump through gene duplication and by acquiring the ability to load this phospholipid binding site from the opposite side of the membrane, thus flipping the phospholipid (63). Armed with a new substrate and a selective advantage, the P4-ATPases would have gradually lost the ability to pump cations.
However, an alternative model for phospholipid translocation has recently been proposed by the Molday and Andersen labs. Vestergaard, et al. investigated the consequences of mutating a highly conserved P+1 isoleucine (I364) in TM4 of the mammalian P4-ATPase ATP8A2 (38). This isoleucine residue corresponds to a glutamate in P2-ATPases that is crucial for cation transport (80,81). A missense mutation at the I364 position (I364M) was recently identified as the cause of cerebellar ataxia, mental retardation, and dysequilibrium syndrome (CAMRQ) in a Turkish family (82). Wabbler-lethal mice, which also have mutations in Atp8a2, have severe axonal degeneration (83,84), indicating that ATP8A2 is important in the development of the nervous system. Vestergaard, et al. found that mutations of I364 and N359, another TM4 residue, strongly perturbed flippase activity and the ability of PS to stimulate ATPase activity (38). In lieu of a P4-ATPase crystal structure, the researchers created two homology models of ATP8A2 based on the related P2-ATPase sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA) in two conformational states (E2P and E2). Molecular dynamic refinements of the models opened a groove bordered by TM1, TM2, TM4, and TM6 that allowed substantial water penetration. The groove was divided into two distinct pockets at the exoplasmic and cytoplasmic faces by a central cluster of hydrophobic residues, including I364. Vestergaard, et al. proposed that the hydrophobic cluster divides the hydrated groove into phospholipid entry and exit sites (38). Movement of the hydrophobic residues during the reaction cycle may cause the sequential formation and dissolution of the two sites to enable translocation of the phospholipid headgroup in an aqueous environment while the acyl chains follow passively in the lipid phase of the surrounding membrane (38).
The proposed route of translocation suggested by Vestergaard and colleagues differs from the one suggested by Baldridge and Graham, although it is consistent with the view that P4-ATPases use a noncanonical transport mechanism driven primarily by substrate interactions with the first four TM segments (36,38). The entry and exit gate residues that define substrate specificity could be accessible from the TM2-4-6 groove, depending on their actual side chain orientations and how deeply the phospholipid headgroup can enter the membrane domain. Limited sequence similarity between several membrane segments of P2 and P4-ATPases creates uncertainties in alignments and homology models. Ultimately, multiple P4-ATPase structures with substrate bound will be needed to assess the validity of the proposed models.
Distinct Mechanisms for Phospholipid Scrambling
Recent advances have also been made in our understanding of how membrane asymmetry is disrupted by scramblases. Activation of scramblase at the plasma membrane allows externalization of PS that is normally constrained within the cytosolic leaflet. PS exposure is a mediator of several biological processes, such as blood coagulation (21,22) and recognition of apoptotic cells by macrophages (18–20). The identity of the protein or proteins responsible for phospholipid scrambling activity has been hotly debated through the years. Patients with Scott syndrome (85,86) have a bleeding disorder caused by a lack of lipid scrambling in platelets due to mutations in the proposed scramblase TMEM16F (43). Previously it was thought that this scramblase activity was due to a protein named phospholipid scramblase (PLSCR) (87), This seems very unlikely, however, because PLSCR is not mutated in Scott syndrome patients (88) and Ca2+-induced PS exposure is unaffected in PLSCR1−/− mouse cells (89). Interestingly, patients with Scott syndrome have no defect in apoptotic PS exposure (90), suggesting that PS is exposed by two distinct mechanisms.
The Nagata lab identified TMEM16F as the best candidate for the elusive Ca2+-dependent scramblase by repeated fluorescence-activated cell sorting (FACS) of cell populations that overexpose PS in low Ca2+ conditions. A cDNA library was constructed from the PS-exposing cell line, and the mutation responsible was identified as TMEM16F D409G (43). This mutation causes hypersensitivity to normal intracellular levels of Ca2+, thereby triggering cells to constitutively expose PS and PE. Deletion of TMEM16F (TMEM16F−/−) in mice blocks PS exposure in response to ionophore stimulation, but does not prevent FasL-induced PS exposure (91). Consistent with this result, Scott syndrome patients with mutations in TMEM16F fail to expose PS in response to elevated Ca2+ concentration but expose PS normally in response to apoptotic signals (86). Interestingly, macrophages do not engulf viable PS-exposing TMEM16F D409G cells, rather only those that have undergone FasL-mediated apoptosis (92). Therefore, PS exposure is required, but not sufficient, for recognition and engulfment of cells by macrophages. These results indicate that there are two distinct pathways governing PS exposure in response to Ca2+ stimulation and caspase-dependent apoptosis.
However, it is still controversial if TMEM16 family members are in fact scramblases (43) or a channel that regulates the activity of an unidentified scramblase (93). The controversy is in part due to conflicting reports of TMEM16F heterologous expression inducing (43,91,94) or not inducing (93) Ca2+-dependent phospholipid scrambling in different cell lines. Recently, the Aspergillus fumigatus TMEM16F homologue afTMEM16 has been purified, reconstituted in proteoliposomes, and found to be a dual-function Ca2+-gated channel and Ca2+-dependent phospholipid scramblase (95). afTEM16F, like other TMEM16 family members (93,96), was found to be a nearly non-selective ion channel with a large pore. Scrambling activity was assayed by reconstituting afTMEM16 into liposomes containing trace amounts of NBD-phospholipids and measuring fluorescence loss due to quenching of the outer leaflet by dithionite (95,97). The total amount and rate of fluorescence quenching increased with increasing Ca2+ levels with a >20-fold increase in phospholipid scrambling in the presence of saturating Ca2+ (95). Mutations which charge neutralized a conserved di-acidic motif (D511A/E514A) in the Ca2+-binding site (93,98) decreased both ion and lipid transport, suggesting that a single Ca2+-binding site controls both functions (95). Interestingly, afTEM16F has independent pathways by which ions and lipids are transported. Inhibition of channel activity did not affect the rate of lipid transport and vice versa. However, not all TMEM16 homologues tested mediated phospholipid scrambling. Human TMEM16a and yeast Ist2 were able to transport ions, but not lipids, suggesting the TMEM16 family contains both channels and channels/scramblases (95). Human TMEM16F has not been purified due to instability issues so it remains to be seen if TMEM16F is definitively a dual-function channel/phospholipid scramblase.
Remarkably, the Nagata group was also able to identify a protein responsible for promoting PS exposure in response to apoptosis; this protein is XK family member Xkr8 (44). Xkr8, like TMEM16F, promotes scrambling of multiple lipid species with exposure of PS and PE. Xkr8 has a C. elegans homologue called CED-8 which has previously been implicated in the controlled timing of programmed cell death (99). Xkr8 repression prevents PS exposure and engulfment by macrophages during apoptosis. Cell lines that do not expose PS during apoptosis (PLB-985 and Raji lymphoma cells) have a severe deficiency in Xkr8 expression due to methylation in the Xkr8 promoter (44). Convincingly, transforming these cell lines with Xkr8 allowed cells to respond to apoptotic stimuli to expose PS and be engulfed by macrophages. Xkr8 was found to be activated by caspases with its caspase-3 cleavage site indispensable for activity. Xkr8 has no effect on Ca2+-induced exposure of PS, consistent with the proposed two mechanisms for PS exposure. While Xkr8 clearly plays an important role in apoptotic PS exposure, reconstitution of purified protein will be required to determine if Xkr8 directly scrambles lipids.
In addition to scramblase activity being turned on during apoptosis, flippase activity must also be turned off such that the disordering of lipid asymmetry is not corrected. Previous studies have reported apoptotic P4-ATPase inactivation, but the identity of the P4-ATPase and mechanism of inactivation remained unknown (19,100). Using a haploid human cell line mutagenized with a gene trap vector, the Nagata lab has recently identified cell populations defective in their ability to flip NBD-PS (101). Two genes identified in this screen were the P4-ATPase ATP11C and its β subunit CDC50A (101,102). ATP11C has been previously implicated in bile secretion (103), B cell development (104,105), and erythrocyte PS lipid asymmetry and lifespan (106). The ATP11C-CDC50A heterodimer may be the primary, although not the only, PS flippase present on the plasma membrane of several mammalian cell types. The researchers found that during FasL-induced apoptosis, ATP11C is cleaved by caspases (101). ATP11C has three caspase recognition sites in the nucleotide-binding domain. Mutation of all three sites prevented cleavage without altering the flippase activity of ATP11C. Unlike cells expressing wild-type ATP11C, cells expressing caspase resistant ATP11C (CasR) did not expose PS in response to FasL-induced apoptosis, and these cells were not engulfed by macrophages despite the apparent activation of Xkr8 scramblase activity and cell death (101). This result indicates that caspase-mediated inactivation of ATP11C is required for PS exposure, which is in turn required for engulfment. Thus, the inactivation of flippase activity is crucial for PS exposure during apoptosis and for elicitation of an immune response.
Concluding Remarks
Significant advancements have been made recently in understanding what proteins govern the distribution of phosphatidylserine within the cell. The development of a novel biosensor for PS has made it possible to map PS distribution and track its topology in route to the PM (32). Two models for how P4-ATPases may recognize and flip their phospholipid substrates have been proposed (35,36,38). Two distinct pathways have also been discovered for phospholipid scrambling in the cases of apoptosis (44) and blood coagulation (43). Mechanistic insight into how the flippases, scramblases and LTPs function is beginning to emerge, but many questions remain unanswered. For example, how is PS concentrated in the cytosolic leaflet of the plasma membrane despite intensive exchange of lipids with internal organelles by vesicular and non-vesicular routes? The phospholipid flippases and LTPs contribute to this localization although it is likely that other features of the plasma membrane, such as cytoskeletal contacts or abundant membrane proteins, also help “trap” PS at this site. If LTPs mediate non-vesicular ER to plasma membrane transfer of PS, how do they drive transport against the prevailing concentration gradient? How do flippases and scramblases move phospholipid from one leaflet to the other? How are these processes integrated and regulated? What are the cellular and physiological consequences of misregulation? Finally, how does one prevent or correct defects in these processes that lead to human disease? Future research in this field will hopefully answer these questions and continue to provide a better understanding of how the subcellular distribution of lipids is controlled.
Figure 3.
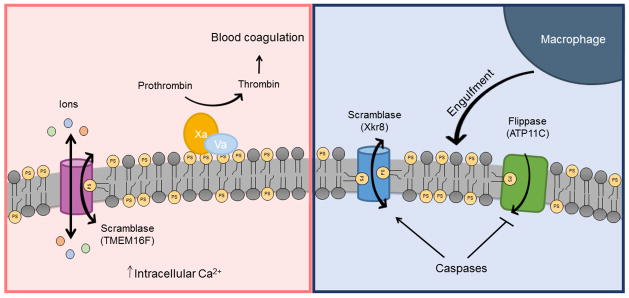
Two distinct pathways for phospholipid scrambling during blood coagulation and apoptosis. During the process of blood coagulation, an elevation in intracellular Ca2+ level stimulates scramblase activity on the PM of platelets (left). The scramblase functions as both a non-selective ion channel and phospholipid scramblase with two distinct pathways for ion and phospholipid transport. Phospholipid scrambling causes exposure of PS on the surface where it acts as a platform on which coagulation protein complexes assemble. During apoptosis, caspases both activate scramblase activity, proposed to be mediated by Xkr8, and inactivate flippase activity (Atp11C) by cleaving at their caspase-recognition sites (right). Phospholipid scrambling causes PS to be exposed on the surface of the apoptotic cell where it acts as an “eat me” signal for macrophages.
Synopsis.
The eukaryotic plasma membrane and internal organelles have unique lipid compositions. However, the mechanisms by which specific lipid species are trafficked to and maintained in specific areas of the cell are poorly understood and constitute an active area of research. Of particular interest is the distribution of the anionic lipid phosphatidylserine, which is enriched in the cytosolic leaflet of the plasma membrane. This review will discuss how phospholipid flippases, scramblases, and PS-specific lipid transfer proteins contribute to subcellular PS distribution.
Acknowledgments
We thank Nora Kayton of the Vanderbilt Editors’ Club for editing this review. Research in the Graham lab is supported by a grant from the National Institutes of Health (1R01GM107978).
References
- 1.Van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008 Feb;9(2):112–24. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bell RM, Ballas LM, Coleman RA. Lipid topogenesis. J Lipid Res. 1981 Mar 1;22(3):391–403. [PubMed] [Google Scholar]
- 3.Zinser E, Sperka-Gottlieb CD, Fasch EV, Kohlwein SD, Paltauf F, Daum G. Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J Bacteriol. 1991 Mar 1;173(6):2026–34. doi: 10.1128/jb.173.6.2026-2034.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zinser E, Daum G. Isolation and biochemical characterization of organelles from the yeast, Saccharomyces cerevisiae. Yeast. 1995 May 1;11(6):493–536. doi: 10.1002/yea.320110602. [DOI] [PubMed] [Google Scholar]
- 5.Bretscher MS. Membrane Structure: Some General Principles. Science. 1973 Aug 17;181(4100):622–9. doi: 10.1126/science.181.4100.622. [DOI] [PubMed] [Google Scholar]
- 6.Devaux PF. Protein Involvement in Transmembrane Lipid Asymmetry. Annu Rev Biophys Biomol Struct. 1992;21(1):417–39. doi: 10.1146/annurev.bb.21.060192.002221. [DOI] [PubMed] [Google Scholar]
- 7.Ariotti N, Fernández-Rojo MA, Zhou Y, Hill MM, Rodkey TL, Inder KL, et al. Caveolae regulate the nanoscale organization of the plasma membrane to remotely control Ras signaling. J Cell Biol. 2014 Mar 3;204(5):777–92. doi: 10.1083/jcb.201307055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malínská K, Malínský J, Opekarová M, Tanner W. Visualization of protein compartmentation within the plasma membrane of living yeast cells. Mol Biol Cell. 2003 Nov;14(11):4427–36. doi: 10.1091/mbc.E03-04-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bretscher MS. Phosphatidyl-ethanolamine: Differential labelling in intact cells and cell ghosts of human erythrocytes by a membrane-impermeable reagent. J Mol Biol. 1972 Nov 28;71(3):523–8. doi: 10.1016/s0022-2836(72)80020-2. [DOI] [PubMed] [Google Scholar]
- 10.Bergmann WL, Dressler V, Haest CWM, Deuticke B. Reorientation rates and asymmetry of distribution of lysophospholipids between the inner and outer leaflet of the erythrocyte membrane. Biochim Biophys Acta BBA - Biomembr. 1984 May 30;772(3):328–36. doi: 10.1016/0005-2736(84)90150-0. [DOI] [PubMed] [Google Scholar]
- 11.Gordesky SE, Marinetti GV, Love R. The reaction of chemical probes with the erythrocyte membrane. J Membr Biol. 1975;20(1–2):111–32. doi: 10.1007/BF01870631. [DOI] [PubMed] [Google Scholar]
- 12.Verkleij AJ, Zwaal RFA, Roelofsen B, Comfurius P, Kastelijn D, van Deenen LLM. The asymmetric distribution of phospholipids in the human red cell membrane. A combined study using phospholipases and freeze-etch electron microscopy. Biochim Biophys Acta BBA - Biomembr. 1973 Oct 11;323(2):178–93. doi: 10.1016/0005-2736(73)90143-0. [DOI] [PubMed] [Google Scholar]
- 13.Kay JG, Grinstein S. Phosphatidylserine-Mediated Cellular Signaling. In: Capelluto DGS, editor. Lipid-mediated Protein Signaling. Springer Netherlands; 2013. pp. 177–93. [DOI] [PubMed] [Google Scholar]
- 14.Andersen OS, Koeppe RE. Bilayer Thickness and Membrane Protein Function: An Energetic Perspective. Annu Rev Biophys Biomol Struct. 2007;36(1):107–30. doi: 10.1146/annurev.biophys.36.040306.132643. [DOI] [PubMed] [Google Scholar]
- 15.Phillips R, Ursell T, Wiggins P, Sens P. Emerging roles for lipids in shaping membrane-protein function. Nature. 2009 May 21;459(7245):379–85. doi: 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexander RT, Jaumouillé V, Yeung T, Furuya W, Peltekova I, Boucher A, et al. Membrane surface charge dictates the structure and function of the epithelial Na+/H+ exchanger. EMBO J. 2011 Feb 16;30(4):679–91. doi: 10.1038/emboj.2010.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iwamoto M, Oiki S. Amphipathic antenna of an inward rectifier K+ channel responds to changes in the inner membrane leaflet. Proc Natl Acad Sci U S A. 2013 Jan 8;110(2):749–54. doi: 10.1073/pnas.1217323110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992 Apr 1;148(7):2207–16. [PubMed] [Google Scholar]
- 19.Bratton DL, Fadok VA, Richter DA, Kailey JM, Guthrie LA, Henson PM. Appearance of Phosphatidylserine on Apoptotic Cells Requires Calcium-mediated Nonspecific Flip-Flop and Is Enhanced by Loss of the Aminophospholipid Translocase. J Biol Chem. 1997 Oct 17;272(42):26159–65. doi: 10.1074/jbc.272.42.26159. [DOI] [PubMed] [Google Scholar]
- 20.Schlegel RA, Williamson P. Phosphatidylserine, a death knell. Cell Death Differ. 2001 Jun;8(6):551–63. doi: 10.1038/sj.cdd.4400817. [DOI] [PubMed] [Google Scholar]
- 21.Bevers EM, Comfurius P, Zwaal RFA. Changes in membrane phospholipid distribution during platelet activation. Biochim Biophys Acta BBA - Biomembr. 1983 Dec 7;736(1):57–66. doi: 10.1016/0005-2736(83)90169-4. [DOI] [PubMed] [Google Scholar]
- 22.Lentz BR. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog Lipid Res. 2003 Sep;42(5):423–38. doi: 10.1016/s0163-7827(03)00025-0. [DOI] [PubMed] [Google Scholar]
- 23.Graham TR. Flippases and vesicle-mediated protein transport. Trends Cell Biol. 2004 Dec;14(12):670–7. doi: 10.1016/j.tcb.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 24.Daleke DL. Regulation of transbilayer plasma membrane phospholipid asymmetry. J Lipid Res. 2003 Feb 1;44(2):233–42. doi: 10.1194/jlr.R200019-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Daleke DL. Phospholipid Flippases. J Biol Chem. 2007 Jan 12;282(2):821–5. doi: 10.1074/jbc.R600035200. [DOI] [PubMed] [Google Scholar]
- 26.Quazi F, Lenevich S, Molday RS. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat Commun. 2012 Jun 26;3:925. doi: 10.1038/ncomms1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quazi F, Molday RS. Differential Phospholipid Substrates and Directional Transport by ATP-binding Cassette Proteins ABCA1, ABCA7, and ABCA4 and Disease-causing Mutants. J Biol Chem. 2013 Nov 29;288(48):34414–26. doi: 10.1074/jbc.M113.508812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sahu SK, Gummadi SN, Manoj N, Aradhyam GK. Phospholipid scramblases: An overview. Arch Biochem Biophys. 2007 Jun 1;462(1):103–14. doi: 10.1016/j.abb.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 29.Prinz WA. Bridging the gap: Membrane contact sites in signaling, metabolism, and organelle dynamics. J Cell Biol. 2014 Jun 23;205(6):759–69. doi: 10.1083/jcb.201401126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lev S. Non-vesicular lipid transport by lipid-transfer proteins and beyond. Nat Rev Mol Cell Biol. 2010 Oct;11(10):739–50. doi: 10.1038/nrm2971. [DOI] [PubMed] [Google Scholar]
- 31.Mesmin B, Bigay J, Moser von Filseck J, Lacas-Gervais S, Drin G, Antonny B. A Four-Step Cycle Driven by PI(4)P Hydrolysis Directs Sterol/PI(4)P Exchange by the ER-Golgi Tether OSBP. Cell. 2013 Nov 7;155(4):830–43. doi: 10.1016/j.cell.2013.09.056. [DOI] [PubMed] [Google Scholar]
- 32.Fairn GD, Schieber NL, Ariotti N, Murphy S, Kuerschner L, Webb RI, et al. High-resolution mapping reveals topologically distinct cellular pools of phosphatidylserine. J Cell Biol. 2011 Jul 25;194(2):257–75. doi: 10.1083/jcb.201012028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Natarajan P, Wang J, Hua Z, Graham TR. Drs2p-coupled aminophospholipid translocase activity in yeast Golgi membranes and relationship to in vivo function. Proc Natl Acad Sci U S A. 2004 Jul 20;101(29):10614–9. doi: 10.1073/pnas.0404146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alder-Baerens N, Lisman Q, Luong L, Pomorski T, Holthuis JCM. Loss of P4 ATPases Drs2p and Dnf3p Disrupts Aminophospholipid Transport and Asymmetry in Yeast Post-Golgi Secretory Vesicles. Mol Biol Cell. 2006 Apr 1;17(4):1632–42. doi: 10.1091/mbc.E05-10-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baldridge RD, Graham TR. Identification of residues defining phospholipid flippase substrate specificity of type IV P-type ATPases. Proc Natl Acad Sci U S A. 2012 Feb 7;109(6):E290–E298. doi: 10.1073/pnas.1115725109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baldridge RD, Graham TR. Two-gate mechanism for phospholipid selection and transport by type IV P-type ATPases. Proc Natl Acad Sci. 2013 Jan 29;110(5):E358–E367. doi: 10.1073/pnas.1216948110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baldridge RD, Xu P, Graham TR. Type IV P-type ATPases Distinguish Mono- versus Diacyl Phosphatidylserine Using a Cytofacial Exit Gate in the Membrane Domain. J Biol Chem. 2013 Jul 5;288(27):19516–27. doi: 10.1074/jbc.M113.476911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vestergaard AL, Coleman JA, Lemmin T, Mikkelsen SA, Molday LL, Vilsen B, et al. Critical roles of isoleucine-364 and adjacent residues in a hydrophobic gate control of phospholipid transport by the mammalian P4-ATPase ATP8A2. Proc Natl Acad Sci. 2014 Mar;24:201321165. doi: 10.1073/pnas.1321165111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou X, Graham TR. Reconstitution of phospholipid translocase activity with purified Drs2p, a type-IV P-type ATPase from budding yeast. Proc Natl Acad Sci. 2009 Sep 29;106(39):16586–91. doi: 10.1073/pnas.0904293106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coleman JA, Kwok MCM, Molday RS. Localization, Purification, and Functional Reconstitution of the P4-ATPase Atp8a2, a Phosphatidylserine Flippase in Photoreceptor Disc Membranes. J Biol Chem. 2009 Nov 20;284(47):32670–9. doi: 10.1074/jbc.M109.047415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu P, Baldridge RD, Chi RJ, Burd CG, Graham TR. Phosphatidylserine flipping enhances membrane curvature and negative charge required for vesicular transport. J Cell Biol. 2013 Sep 16;202(6):875–86. doi: 10.1083/jcb.201305094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maeda K, Anand K, Chiapparino A, Kumar A, Poletto M, Kaksonen M, et al. Interactome map uncovers phosphatidylserine transport by oxysterol-binding proteins. Nature. 2013 Sep 12;501(7466):257–61. doi: 10.1038/nature12430. [DOI] [PubMed] [Google Scholar]
- 43.Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature. 2010 Dec 9;468(7325):834–8. doi: 10.1038/nature09583. [DOI] [PubMed] [Google Scholar]
- 44.Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-Related Protein 8 and CED-8 Promote Phosphatidylserine Exposure in Apoptotic Cells. Science. 2013 Jul 26;341(6144):403–6. doi: 10.1126/science.1236758. [DOI] [PubMed] [Google Scholar]
- 45.Bishop WR, Bell RM. Assembly of the endoplasmic reticulum phospholipid bilayer: the phosphatidylcholine transporter. Cell. 1985 Aug;42(1):51–60. doi: 10.1016/s0092-8674(85)80100-8. [DOI] [PubMed] [Google Scholar]
- 46.Herrmann A, Zachowski A, Devaux PF. Protein-mediated phospholipid translocation in the endoplasmic reticulum with a low lipid specificity. Biochemistry (Mosc) 1990 Feb 1;29(8):2023–7. doi: 10.1021/bi00460a010. [DOI] [PubMed] [Google Scholar]
- 47.Buton X, Morrot G, Fellmann P, Seigneuret M. Ultrafast Glycerophospholipid-selective Transbilayer Motion Mediated by a Protein in the Endoplasmic Reticulum Membrane. J Biol Chem. 1996 Mar 22;271(12):6651–7. doi: 10.1074/jbc.271.12.6651. [DOI] [PubMed] [Google Scholar]
- 48.Sanyal S, Frank CG, Menon AK. Distinct flippases translocate glycerophospholipids and oligosaccharide diphosphate dolichols across the endoplasmic reticulum. Biochemistry (Mosc) 2008 Jul 29;47(30):7937–46. doi: 10.1021/bi800723n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Higgins JA, Dawson RMC. Asymmetry of the phospholipid bilayer of rat liver endoplasmic reticulum. Biochim Biophys Acta BBA - Biomembr. 1977 Nov 1;470(3):342–56. doi: 10.1016/0005-2736(77)90126-2. [DOI] [PubMed] [Google Scholar]
- 50.Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A, Grinstein S. Membrane Phosphatidylserine Regulates Surface Charge and Protein Localization. Science. 2008 Jan 11;319(5860):210–3. doi: 10.1126/science.1152066. [DOI] [PubMed] [Google Scholar]
- 51.Kaplan MR, Simoni RD. Intracellular transport of phosphatidylcholine to the plasma membrane. J Cell Biol. 1985 Aug;101(2):441–5. doi: 10.1083/jcb.101.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vance JE, Aasman EJ, Szarka R. Brefeldin A does not inhibit the movement of phosphatidylethanolamine from its sites for synthesis to the cell surface. J Biol Chem. 1991 May 5;266(13):8241–7. [PubMed] [Google Scholar]
- 53.Trotter PJ, Wu W-I, Pedretti J, Yates R, Voelker DR. A Genetic Screen for Aminophospholipid Transport Mutants Identifies the Phosphatidylinositol 4-Kinase, Stt4p, as an Essential Component in Phosphatidylserine Metabolism. J Biol Chem. 1998 May 22;273(21):13189–96. doi: 10.1074/jbc.273.21.13189. [DOI] [PubMed] [Google Scholar]
- 54.Wu W-I, Routt S, Bankaitis VA, Voelker DR. A New Gene Involved in the Transport-dependent Metabolism of Phosphatidylserine, PSTB2/PDR17, Shares Sequence Similarity with the Gene Encoding the Phosphatidylinositol/Phosphatidylcholine Transfer Protein, SEC14. J Biol Chem. 2000 May 12;275(19):14446–56. doi: 10.1074/jbc.275.19.14446. [DOI] [PubMed] [Google Scholar]
- 55.Zilversmit DB. Lipid transfer proteins: overview and applications. Methods Enzymol. 1983;98:565–73. doi: 10.1016/0076-6879(83)98183-1. [DOI] [PubMed] [Google Scholar]
- 56.Helmkamp GM., Jr Phospholipid transfer proteins: mechanism of action. J Bioenerg Biomembr. 1986 Apr;18(2):71–91. doi: 10.1007/BF00743477. [DOI] [PubMed] [Google Scholar]
- 57.Georgiev AG, Sullivan DP, Kersting MC, Dittman JS, Beh CT, Menon AK. Osh Proteins Regulate Membrane Sterol Organization but Are Not Required for Sterol Movement Between the ER and PM. Traffic. 2011;12(10):1341–55. doi: 10.1111/j.1600-0854.2011.01234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Graham TR, Burd CG. Coordination of Golgi functions by phosphatidylinositol 4-kinases. Trends Cell Biol. 2011 Feb;21(2):113–21. doi: 10.1016/j.tcb.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tripathi A, Nile AH, Bankaitis VA. Sec14-like phosphatidylinositol-transfer proteins and diversification of phosphoinositide signalling outcomes. Biochem Soc Trans. 2014 Oct 1;42(5):1383–8. doi: 10.1042/BST20140187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Raychaudhuri S, Im YJ, Hurley JH, Prinz WA. Nonvesicular sterol movement from plasma membrane to ER requires oxysterol-binding protein–related proteins and phosphoinositides. J Cell Biol. 2006 Apr 10;173(1):107–19. doi: 10.1083/jcb.200510084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hua Z, Fatheddin P, Graham TR. An Essential Subfamily of Drs2p-related P-Type ATPases Is Required for Protein Trafficking between Golgi Complex and Endosomal/Vacuolar System. Mol Biol Cell. 2002 Sep 1;13(9):3162–77. doi: 10.1091/mbc.E02-03-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen C-Y, Ingram MF, Rosal PH, Graham TR. Role for Drs2p, a P-Type Atpase and Potential Aminophospholipid Translocase, in Yeast Late Golgi Function. J Cell Biol. 1999 Dec 13;147(6):1223–36. doi: 10.1083/jcb.147.6.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Axelsen KB, Palmgren MG. Evolution of substrate specificities in the P-type ATPase superfamily. J Mol Evol. 1998 Jan;46(1):84–101. doi: 10.1007/pl00006286. [DOI] [PubMed] [Google Scholar]
- 64.Toyoshima C, Nakasako M, Nomura H, Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature. 2000 Jun 8;405(6787):647–55. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 65.Saito K, Fujimura-Kamada K, Furuta N, Kato U, Umeda M, Tanaka K. Cdc50p, a Protein Required for Polarized Growth, Associates with the Drs2p P-Type ATPase Implicated in Phospholipid Translocation in Saccharomyces cerevisiae. Mol Biol Cell. 2004 Jul 1;15(7):3418–32. doi: 10.1091/mbc.E03-11-0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sebastian TT, Baldridge RD, Xu P, Graham TR. Phospholipid flippases: building asymmetric membranes and transport vesicles. Biochim Biophys Acta. 2012 Aug;1821(8):1068–77. doi: 10.1016/j.bbalip.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tang X, Halleck MS, Schlegel RA, Williamson P. A subfamily of P-type ATPases with aminophospholipid transporting activity. Science. 1996 Jun 7;272(5267):1495–7. doi: 10.1126/science.272.5267.1495. [DOI] [PubMed] [Google Scholar]
- 68.Toyoshima C. Structural aspects of ion pumping by Ca2+-ATPase of sarcoplasmic reticulum. Arch Biochem Biophys. 2008 Aug 1;476(1):3–11. doi: 10.1016/j.abb.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 69.Shinoda T, Ogawa H, Cornelius F, Toyoshima C. Crystal structure of the sodium–potassium pump at 2.4_Å resolution. Nature. 2009 May 21;459(7245):446–50. doi: 10.1038/nature07939. [DOI] [PubMed] [Google Scholar]
- 70.Stone A, Williamson P. Outside of the box: recent news about phospholipid translocation by P4 ATPases. J Chem Biol. 2012 Jul 15;5(4):131–6. doi: 10.1007/s12154-012-0078-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pomorski T, Lombardi R, Riezman H, Devaux PF, van Meer G, Holthuis JCM. Drs2p-related P-type ATPases Dnf1p and Dnf2p Are Required for Phospholipid Translocation across the Yeast Plasma Membrane and Serve a Role in Endocytosis. Mol Biol Cell. 2003 Mar 1;14(3):1240–54. doi: 10.1091/mbc.E02-08-0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kato U, Emoto K, Fredriksson C, Nakamura H, Ohta A, Kobayashi T, et al. A Novel Membrane Protein, Ros3p, Is Required for Phospholipid Translocation across the Plasma Membrane in Saccharomyces cerevisiae. J Biol Chem. 2002 Oct 4;277(40):37855–62. doi: 10.1074/jbc.M205564200. [DOI] [PubMed] [Google Scholar]
- 73.Riekhof WR, Wu J, Gijón MA, Zarini S, Murphy RC, Voelker DR. Lysophosphatidylcholine Metabolism In Saccharomyces Cerevisiae The Role Of P-Type ATPases In Transport And A Broad Specificity Acyltransferase In Acylation. J Biol Chem. 2007 Dec 21;282(51):36853–61. doi: 10.1074/jbc.M706718200. [DOI] [PubMed] [Google Scholar]
- 74.Palmgren MG, Nissen P. P-type ATPases. Annu Rev Biophys. 2011;40:243–66. doi: 10.1146/annurev.biophys.093008.131331. [DOI] [PubMed] [Google Scholar]
- 75.Skou JC. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim Biophys Acta. 1957;23:394–401. doi: 10.1016/0006-3002(57)90343-8. [DOI] [PubMed] [Google Scholar]
- 76.Post RL, Kume S, Tobin T, Orcutt B, Sen AK. Flexibility of an Active Center in Sodium-Plus-Potassium Adenosine Triphosphatase. J Gen Physiol. 1969 Jul 1;54(1):306–26. doi: 10.1085/jgp.54.1.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ding J, Wu Z, Crider BP, Ma Y, Li X, Slaughter C, et al. Identification and functional expression of four isoforms of ATPase II, the putative aminophospholipid translocase. Effect of isoform variation on the ATPase activity and phospholipid specificity. J Biol Chem. 2000 Jul 28;275(30):23378–86. doi: 10.1074/jbc.M910319199. [DOI] [PubMed] [Google Scholar]
- 78.Jacquot A, Montigny C, Hennrich H, Barry R, le Maire M, Jaxel C, et al. Phosphatidylserine Stimulation of Drs2p·Cdc50p Lipid Translocase Dephosphorylation Is Controlled by Phosphatidylinositol-4-phosphate. J Biol Chem. 2012 Apr 13;287(16):13249–61. doi: 10.1074/jbc.M111.313916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Obara K, Miyashita N, Xu C, Toyoshima I, Sugita Y, Inesi G, et al. Structural role of countertransport revealed in Ca2+ pump crystal structure in the absence of Ca2+ Proc Natl Acad Sci U S A. 2005 Oct 11;102(41):14489–96. doi: 10.1073/pnas.0506222102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vilsen B, Andersen JP. Mutation to the Glutamate in the Fourth Membrane Segment of Na+,K+-ATPase and Ca2+-ATPase Affects Cation Binding from Both Sides of the Membrane and Destabilizes the Occluded Enzyme Forms. Biochemistry (Mosc) 1998 Aug 1;37(31):10961–71. doi: 10.1021/bi9802925. [DOI] [PubMed] [Google Scholar]
- 81.Olesen C, Picard M, Winther A-ML, Gyrup C, Morth JP, Oxvig C, et al. The structural basis of calcium transport by the calcium pump. Nature. 2007 Dec 13;450(7172):1036–42. doi: 10.1038/nature06418. [DOI] [PubMed] [Google Scholar]
- 82.Emre Onat O, Gulsuner S, Bilguvar K, Nazli Basak A, Topaloglu H, Tan M, et al. Missense mutation in the ATPase, aminophospholipid transporter protein ATP8A2 is associated with cerebellar atrophy and quadrupedal locomotion. Eur J Hum Genet. 2013 Mar;21(3):281–5. doi: 10.1038/ejhg.2012.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhu X, Libby RT, de Vries WN, Smith RS, Wright DL, Bronson RT, et al. Mutations in a P-Type ATPase Gene Cause Axonal Degeneration. PLoS Genet. 2012 Aug 9;8(8):e1002853. doi: 10.1371/journal.pgen.1002853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Van der Mark V, Elferink R, Paulusma C. P4 ATPases: Flippases in Health and Disease. Int J Mol Sci. 2013 Apr 11;14(4):7897–922. doi: 10.3390/ijms14047897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weiss HJ, Vicic WJ, Lages BA, Rogers J. Isolated deficiency of platelet procoagulant activity. Am J Med. 1979 Aug;67(2):206–13. doi: 10.1016/0002-9343(79)90392-9. [DOI] [PubMed] [Google Scholar]
- 86.Williamson P, Christie A, Kohlin T, Schlegel RA, Comfurius P, Harmsma M, et al. Phospholipid Scramblase Activation Pathways in Lymphocytes. Biochemistry (Mosc) 2001 Jul 1;40(27):8065–72. doi: 10.1021/bi001929z. [DOI] [PubMed] [Google Scholar]
- 87.Bassé F, Stout JG, Sims PJ, Wiedmer T. Isolation of an Erythrocyte Membrane Protein that Mediates Ca2+-dependent Transbilayer Movement of Phospholipid. J Biol Chem. 1996 Jul 19;271(29):17205–10. doi: 10.1074/jbc.271.29.17205. [DOI] [PubMed] [Google Scholar]
- 88.Zhou Q, Sims PJ, Wiedmer T. Expression of proteins controlling transbilayer movement of plasma membrane phospholipids in the B lymphocytes from a patient with Scott syndrome. Blood. 1998 Sep 1;92(5):1707–12. [PubMed] [Google Scholar]
- 89.Zhou Q, Zhao J, Wiedmer T, Sims PJ. Normal hemostasis but defective hematopoietic response to growth factors in mice deficient in phospholipid scramblase 1. Blood. 2002 Jun 1;99(11):4030–8. doi: 10.1182/blood-2001-12-0271. [DOI] [PubMed] [Google Scholar]
- 90.van Kruchten R, Mattheij NJA, Saunders C, Feijge MAH, Swieringa F, Wolfs JLN, et al. Both TMEM16F-dependent and TMEM16F-independent pathways contribute to phosphatidylserine exposure in platelet apoptosis and platelet activation. Blood. 2013 Mar 7;121(10):1850–7. doi: 10.1182/blood-2012-09-454314. [DOI] [PubMed] [Google Scholar]
- 91.Suzuki J, Fujii T, Imao T, Ishihara K, Kuba H, Nagata S. Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J Biol Chem. 2013 May 10;288(19):13305–16. doi: 10.1074/jbc.M113.457937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Segawa K, Suzuki J, Nagata S. Constitutive exposure of phosphatidylserine on viable cells. Proc Natl Acad Sci. 2011 Nov 29;108(48):19246–51. doi: 10.1073/pnas.1114799108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang H, Kim A, David T, Palmer D, Jin T, Tien J, et al. TMEM16F Forms a Ca2+-Activated Cation Channel Required for Lipid Scrambling in Platelets during Blood Coagulation. Cell. 2012 Sep 28;151(1):111–22. doi: 10.1016/j.cell.2012.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kmit A, van Kruchten R, Ousingsawat J, Mattheij NJA, Senden-Gijsbers B, Heemskerk JWM, et al. Calcium-activated and apoptotic phospholipid scrambling induced by Ano6 can occur independently of Ano6 ion currents. Cell Death Dis. 2013 Apr;4(4):e611. doi: 10.1038/cddis.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Malvezzi M, Chalat M, Janjusevic R, Picollo A, Terashima H, Menon AK, et al. Ca2+-dependent phospholipid scrambling by a reconstituted TMEM16 ion channel. Nat Commun. 2013;4:2367. doi: 10.1038/ncomms3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ferrera L, Scudieri P, Sondo E, Caputo A, Caci E, Zegarra-Moran O, et al. A minimal isoform of the TMEM16A protein associated with chloride channel activity. Biochim Biophys Acta. 2011 Sep;1808(9):2214–23. doi: 10.1016/j.bbamem.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McIntyre JC, Sleight RG. Fluorescence assay for phospholipid membrane asymmetry. Biochemistry (Mosc) 1991 Dec 1;30(51):11819–27. doi: 10.1021/bi00115a012. [DOI] [PubMed] [Google Scholar]
- 98.Yu K, Duran C, Qu Z, Cui Y-Y, Hartzell HC. Explaining Calcium-Dependent Gating of Anoctamin-1 Chloride Channels Requires a Revised Topology. Circ Res. 2012 Mar 30;110(7):990–9. doi: 10.1161/CIRCRESAHA.112.264440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stanfield GM, Horvitz HR. The ced-8 Gene Controls the Timing of Programmed Cell Deaths in C. elegans. Mol Cell. 2000 Mar;5(3):423–33. doi: 10.1016/s1097-2765(00)80437-2. [DOI] [PubMed] [Google Scholar]
- 100.Verhoven B, Schlegel RA, Williamson P. Mechanisms of phosphatidylserine exposure, a phagocyte recognition signal, on apoptotic T lymphocytes. J Exp Med. 1995 Nov 1;182(5):1597–601. doi: 10.1084/jem.182.5.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014 Jun 6;344(6188):1164–8. doi: 10.1126/science.1252809. [DOI] [PubMed] [Google Scholar]
- 102.Takatsu H, Baba K, Shima T, Umino H, Kato U, Umeda M, et al. ATP9B, a P4-ATPase (a Putative Aminophospholipid Translocase), Localizes to the trans-Golgi Network in a CDC50 Protein-independent Manner. J Biol Chem. 2011 Nov 4;286(44):38159–67. doi: 10.1074/jbc.M111.281006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Siggs OM, Schnabl B, Webb B, Beutler B. X-linked cholestasis in mouse due to mutations of the P4-ATPase ATP11C. Proc Natl Acad Sci. 2011 May 10;108(19):7890–5. doi: 10.1073/pnas.1104631108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Siggs OM, Arnold CN, Huber C, Pirie E, Xia Y, Lin P, et al. The P4-type ATPase ATP11C is essential for B lymphopoiesis in adult bone marrow. Nat Immunol. 2011 May;12(5):434–40. doi: 10.1038/ni.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yabas M, Teh CE, Frankenreiter S, Lal D, Roots CM, Whittle B, et al. ATP11C is critical for the internalization of phosphatidylserine and differentiation of B lymphocytes. Nat Immunol. 2011 May;12(5):441–9. doi: 10.1038/ni.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yabas M, Coupland LA, Cromer D, Winterberg M, Teoh NC, D’Rozario J, et al. Mice Deficient in the Putative Phospholipid Flippase ATP11C Exhibit Altered Erythrocyte Shape, Anemia and Reduced Erythrocyte Lifespan. J Biol Chem. 2014 Jun 4; doi: 10.1074/jbc.C114.570267. jbc.C114.570267. [DOI] [PMC free article] [PubMed] [Google Scholar]