

Abstract
Aporphines are attractive candidates for imaging D2 receptor function because, as agonists rather than antagonists, they are selective for the receptor in the high affinity state. In contrast, D2 antagonists do not distinguish between the high and low affinity states, and in vitro data suggests that this distinction may be important in studying diseases characterized by D2 dysregulation, such as schizophrenia and Parkinson’s disease. Accordingly, MCL-536 (R-(−)-N-n-propyl-2-(3-[18F]fluoropropanoxy-11-hydroxynoraporphine) was selected for labeling with 18F based on in vitro data obtained for the non-radioactive (19F) compound. Fluorine-18-labeled MCL-536 was synthesized in 70% radiochemical yield, >99% radiochemical purity and specific activity of 167 GBq/μmol (4.5 Ci/μmol) using p-toluenesulfonyl (tosyl) both as a novel protecting group for the phenol and a leaving group for the radiofluorination.
Keywords: aporphines, D2 agonist, neurological disorders, positron emission tomography, dopamine receptors, D2high, D2low, tosyl group, phenol protection, fluorine-18
Introduction
Dopamine is one of the most important neurotransmitters in the central nervous system, and irregularities of the dopaminergic system are implicated in many neuropsychiatric disorders such as Parkinson’s disease and schizophrenia. 1 Targets in the dopaminergic system suitable for imaging include the dopamine transporter (DAT) as well as the various dopamine (DA) receptors. Of particular interest in relation to dopaminergic dysfunction is the D2 receptor, which is mainly found postsynaptically. More specifically, in Parkinson’s disease and schizophrenia, D2 receptor status is thought to be altered with an increased fraction of the receptor in the high-affinity state.2,3,4,5,6
Positron Emission Tomography (PET) is a valuable tool in the evaluation of dopamine dysfunction because it allows for non-invasive in vivo assessment of receptor function. Thus, the importance of the D2 receptor in multiple neurological diseases has led to the development of several D2/3 receptor PET radioligands7 including [11C]methylspiperone,8 [11C]raclopride,9 and [18F]fallypride.10 However, these ligands are antagonists, and as such, they cannot distinguish between D2 receptors in high- and low-affinity states. The absence of the ability to make this distinction limits their use in the study of diseases such as schizophrenia, which is thought to involve differences in the state of the D2 receptor.
In order to circumvent this limitation and to more specifically address the role of the functional state of the D2 receptor in these neurological diseases, focus has recently turned to the development of agonist D2 PET ligands, which do have the potential to differentiate the high- and low-affinity states of the D2 receptor.11 An excellent review of D2/3 agonist PET radioligands was recently reported by Finnema et al.12 The majority of these ligands are 11C-labeled, including [11C]MNPA,13,14 [11C]NPA,15 and [11C]-(+)-PHNO.16 However, these compounds each have limitations that preclude their use as D2high selective radioligands, the most important being that none of these 11C-labeled radioligands have been shown to unambiguously identify the dopamine D2high receptor in vivo.17 The exact reasons are unknown, but may be related to these ligands’ relatively long dissociation half-lives from the receptor (from ~30 to ~600 s as measured in vitro), compared to the very short time it takes for the G protein to dissociate and flip the receptor from the high- to low-affinity state (<1 s).18 In addition, all of these radiotracers are labeled with 11C, and its short half-life (20 min) limits the tracer’s use to facilities with on-site cyclotrons, preventing their more widespread clinical use. The development of an effective 18F-labeled D2 agonist radioligand has, however, proven challenging. To date, there are no examples of an 18F-labeled D2high ligand.12,19 For example, N-alkyl radiofluorinated derivative of (+)-PHNO exhibited significantly decreased D2 binding compared to that of (+)-PHNO,16 possibly because of the location of the 18F label.19 Likewise, the radiofluorinated aporphine derivatives [18F]FNPA and [18F]FNEA, which are also fluorinated via N-alkylation, did not prove to be D2 agonists in vivo.20 There is a single report describing the aminotetralin D2 agonist [18F]5-OH-FPPAT, which does bind to the D2 receptor in vivo and shows a peak striatum:cerebellum binding ratio of 2.21 However, there have been no further reports on this compound. Thus, development of an 18F-labeled D2 agonist with high selectivity and specificity for the D2high receptor and high contrast between D2 rich and D2 poor areas remains an important, albeit elusive, goal.
Aporphines are an attractive class of compounds for this application as they exhibit high D2 receptor agonist activity and also include a potential 18F labeling site different from the N-alkyl site, where 18F-labeling has been shown to decrease D2high binding. Several 11C- and 18F-labeled aporphine analogs have been proposed as potential PET radioligands for the D2 receptors, and their distribution in brain and peripheral tissues of rats and non-human primates have been investigated.13d, 14,15,20, 21
The need for D2 ligands with even higher affinity and selectivity has led to the development of many novel aporphines. 22,23 We recently reported the synthesis and in vitro binding affinities for the D1, D2, and D3 high- and low affinity states of the twelve fluorinated (19F) aporphines and found that substituents in position 2 of the aporphine modulated the dopaminergic receptor potency and D2/D3 selectivity of such compounds.24 One of these analogs, MCL-536 (Figure 1), was found to possess high in vitro D2high affinity (Ki = 0.54 nM), excellent selectivity against other DA receptors, and ideal ClogP value (ideally less than 5). Further development as a 18F radioligand is reported here.
Figure 1.

MCL-536 and its affinities (Ki) for dopamine D2 receptors24
Experimental
General Synthetic Methods
1H (and 13C NMR) spectra were recorded at 300 MHz (75 MHz) on a Varian Mercury 300 spectrometer. Chemical shifts are given as δ value (ppm) downfield from tetramethylsilane as an internal reference. Elemental analyses, performed by Atlantic Microlabs (Atlanta, GA), were within 0.4% of theoretical values. Analytical thin-layer chromatography (TLC) was carried out on 0.2 μM Kieselgel 60F-254 silica gel aluminum sheets using methanol/dichloromethane (1/10) as the solvent (EM Science, Newark, NJ). Flash chromatography was used for the purification of products.
Chemicals
All chemicals were of ACS reagent grade and used without further purification unless otherwise specified. Iodopropane, 1,3-dihydroxypropane, triphenylphosphine, Kryptofix 222 (4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8] hexacosane; K222), hydrazine, and palladium on carbon (Pd/C) were purchased from Sigma-Aldrich (St. Louis, MO). p-Toluenesulfonyl chloride (TsCl) was purchased from Sigma-Aldrich and recrystallized from chloroform before use. Methanesulfonic acid, triethylamine, formic acid, 4-N-dimethylaminopyridine (DMAP), acetonitrile, anhydrous dimethylformamide (DMF), tert-butylchlorodimethylsilane (TBS), and acetone (extra-dry) were purchased from Acros Chemical (Thermo Fisher Scientific, Waltham, MA). Palladium acetate (Pd(OAc)2) was purchased from Strem Chemicals (Newburyport, MA). N-Phenyl-bis(trifluoromethyl)sulfonamide (PhNTf2) was purchased from Oakwood Chemical Company (West Columbia, SC). Anhydrous methanol (MeOH), anhydrous tetrahydrofuran (THF), and anhydrous dichloromethane (DCM) were purchased from EMD Chemical (EMD Millipore, Billerica, MA) in septum-sealed bottles. Iodoxybenzoic acid (IBX) was prepared as described in the literature.25 Sodium bicarbonate, potassium carbonate, anhydrous potassium fluoride, phosphorus pentoxide, chloroform, dichloromethane, methanol, and 5 N sodium hydroxide were purchased from Fisher Chemical (Hampton, NH). Morphine hydrochloride was supplied by Mallinckrodt (St. Louis, MO). MCL-536 (1) was prepared as described in the literature24 and used as reference materials for the indirect characterization of the 18F-labeled compounds.
Synthesis of Non-radioactive Compounds
3-Deoxy-N-n-propylnorthebainone (3)
was prepared as previously described.26 Briefly, to a flask containing N-n-propyl-3-deoxynormorphine (2) (859 mg, 2.89 mmol), anhydrous DMF (9 mL) was added and solution was stirred vigorously at RT. To the stirring solution, IBX (850 mg, 3.0 mmol) was added, and the reaction was monitored by TLC. After 2 h, a small additional portion of IBX (20 mg) was added.27 After another 2 h, only a trace of 2 was still observed, and the mixture was diluted with ethyl acetate (100 mL) and washed with saturated sodium bicarbonate solution (2 × 150 mL) and brine (150 mL). The organic layer was dried over sodium sulfate, filtered, and concentrated to produce an orange solid, which was purified by silica gel column chromatography using 1:20 MeOH:DCM to afford N-n-propyl-3-deoxynorthebainone (3) as a salmon-colored solid in 68% yield (580 mg, 1.96 mmol).
3-deoxy-N-n-propylnorthebainone (3): 1H NMR (300 MHz, CDCl3) δ 7.01 (d, J = 7.8 Hz, 1H), 6.74 – 6.54 (m, 3H), 6.06 (dd, J = 10.2, 2.9 Hz, 1H), 4.68 (s, 1H), 3.51 (dd, J = 5.2, 3.1 Hz, 1H), 3.21 (dd, J = 5.1, 2.7 Hz, 1H), 3.10 (d, J = 18.8 Hz, 1H), 2.98 – 2.82 (m, 1H), 2.70 (ddd, J = 12.2, 4.8, 1.7 Hz, 1H), 2.50 (td, J = 7.1, 3.3 Hz, 2H), 2.36 (dd, J = 18.8, 5.7 Hz, 1H), 2.25 (td, J = 12.1, 3.5 Hz, 1H), 2.06 (td, J = 12.2, 4.8 Hz, 1H), 1.84 – 1.72 (m, 1H), 1.54 (dq, J = 14.6, 7.3 Hz, 2H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 194.70, 157.08, 149.66, 134.29, 132.23, 128.60, 127.44, 119.00, 107.18, 87.63, 56.81, 56.73, 44.88, 42.87, 41.49, 33.93, 21.67, 20.73, 11.85.
(R)-11-hydroxy-2-(3-hydroxypropanoxy)-N-n-propylnoraporphine (4)
To a 20 mL vial were sequentially added: N-propyl-3-deoxynorthebainone 3 (400 mg, 1.36 mmol), 1,3-propanediol (1.2 mL), and methanesulfonic acid (10 mL). After briefly stirring at RT, the mixture was stirred at 95 ºC for 1 h. Upon cooling, the mixture was transferred to ice and water (400 mL) and stirred. The resulting solution was basified using 28% ammonium hydroxide to pH 9–10 and then thoroughly extracted with ethyl acetate (3 × 250 mL). The organic layer was washed with brine (200 mL), dried over sodium sulfate, filtered, and concentrated. The residue was purified by silica gel column chromatography using 1:10 MeOH:DCM to afford R-(−)-N-n-propyl-11-hydroxy-3-hydroxypropanoxyaporphine (4) as a pale solid in 64% isolated yield (256 mg, 0.87 mmol).
(R)-11-hydroxy-2-(3-hydroxypropanoxy)-N-n-propylnoraporphine (4): 1H NMR (300 MHz, CDCl3) δ 7.01 (d, J = 7.8 Hz, 1H), 6.74 – 6.54 (m, 3H), 6.06 (dd, J = 10.2, 2.9 Hz, 1H), 4.68 (s, 1H), 3.51 (dd, J = 5.2, 3.1 Hz, 1H), 3.21 (dd, J = 5.1, 2.7 Hz, 1H), 3.10 (d, J = 18.8 Hz, 1H), 2.98 – 2.82 (m, 1H), 2.70 (ddd, J = 12.2, 4.8, 1.7 Hz, 1H), 2.50 (td, J = 7.1, 3.3 Hz, 2H), 2.36 (dd, J = 18.8, 5.7 Hz, 1H), 2.25 (td, J = 12.1, 3.5 Hz, 1H), 2.06 (td, J = 12.2, 4.8 Hz, 1H), 1.84 – 1.72 (m, 1H), 1.54 (dq, J = 14.6, 7.3 Hz, 2H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 194.70, 157.08, 149.66, 134.29, 132.23, 128.60, 127.44, 119.00, 107.18, 87.63, 56.81, 56.73, 44.88, 42.87, 41.49, 33.93, 21.67, 20.73, 11.85.
(R)-11-hydroxy-N-n-propyl-2-(3-toluenesulfonyloxypropanoxy)noraporphine MCL-557 (5)
To a 20 mL vial under nitrogen atmosphere were added: R-(−)-N-n-propyl-11-hydroxy-3-hydroxypropanoxyaporphine (5) (100 mg, 0.283 mmol), anhydrous dichloromethane (3 mL), and triethylamine (90 μL,0.64 mmol). After briefly stirring the solution, a solution of tosyl chloride (61 mg, 0.320 mmol) in anhydrous dichloromethane (0.5 mL) was slowly added dropwise. After the reaction was observed to be complete by TLC, the solution was washed with saturated sodium bicarbonate (10 mL) and extracted with dichloromethane (1 × 5 mL). The combined organic phases were dried over sodium sulfate, filtered, and concentrated. The residue was purified by silica gel column chromatography using 1:20 MeOH:DCM to afford (R)-(−)-N-n-propyl-2-tosyloxypropanoxy-11-hydroxynoraporphine (5) as a brown oil in 49% isolated yield (70.4 mg, 0.139 mmol).
(R)-11-hydroxy-N-n-propyl-2-(3-toluenesulfonyloxypropanoxy)noraporphine MCL-557 (5): 1H NMR (300 MHz, CDCl3) δ 7.34 (d, J = 8.0 Hz, 1H), 7.29 – 7.18 (m, 3H), 7.15 (d, J = 7.1 Hz, 1H), 6.89 (d, J = 8.0 Hz, 2H), 6.54 (S, 1H), 4.19 – 3.97 (m, 2H), 3.88 (t, J = 5.9 Hz, 2H), 3.13 (dd, J = 11.0, 5.5 Hz, 1H), 3.03 – 2.88 (m, 2H), 2.77 (d, J = 9.9 Hz, 1H), 2.61 (d, J = 15.0 Hz, 2H), 2.28-2.24 (M, 2H), 2.25 )S, 3H), 2.06 (dd, J = 11.9, 5.9 Hz, 2H), 1.90 – 1.75 (m, 1H), 1.66 – 1.43 (m, 2H), 0.95 (t, J = 7.3 Hz, 3H). AWSV-032b: 13C NMR (75 MHz, CDCl3) δ 156.80, 145.85, 144.35, 138.94, 134.15, 131.29, 130.68, 128.71 (2C), 128.33, 128.18 (2C), 127.97, 127.85, 127.73, 127.04, 123.18, 114.42, 112.41, 65.84, 60.43, 58.89, 56.44, 48.98, 34.97, 32.00, 29.66, 29.25, 21.64, 19.51, 12.02.
(R)-N-n-propyl-11-toluenesulfonyloxy-2-(3-toluenesulfonyloxypropanoxy)noraporphine MCL-563 (6)
To a 20 mL vial under nitrogen atmosphere were sequentially added: (R)-(−)-N-n-propyl-2-hydroxypropanoxy-11-hydroxynoraporphine (4) (15.5 mg, 0.044 mmol), p-toluenesulfonyl chloride (26 mg, 0.137 mmol), DMAP (1 mg), and anhydrous dichloromethane (1 mL). After briefly stirring the solution, triethylamine (70 μL, 0.49 mmol) was added and the contents were stirred at RT for 4 h. When TLC analysis indicated the reaction was complete, the reaction mixture was applied directly to a silica gel column and purified using a 100% DCM to 1:40 MeOH:DCM gradient to afford (R)-N-n-propyl-11-toluenesulfonyloxy-2-(3-toluenesulfonyloxypropanoxy)noraporphine (6) as a greenish brown oil in 79% isolated yield (23 mg, 0.35 mmol).
(R)-N-n-propyl-11-toluenesulfonyloxy-2-(3-toluenesulfonyloxypropanoxy)noraporphine (MCL-563) (6): 1H NMR (300 MHz, CDCl3) δ 7.79 (d, J = 8.3 Hz, 2H), 7.26 (tt, J = 16.6, 8.9 Hz, 9H), 6.88 (d, J = 8.5 Hz, 2H), 6.45 (s, 1H), 5.30 (s, 1H), 4.40 – 4.21 (m, 2H), 3.98 (t, J = 5.9 Hz, 2H), 3.13 (dd, J = 11.2, 4.4 Hz, 1H), 3.08 – 2.91 (m, 2H), 2.81 (dd, J = 17.8, 11.1 Hz, 1H), 2.61 (d, J = 14.1 Hz, 2H), 2.51 – 2.39 (m, 2H), 2.35 (s, 3H), 2.27 (s, 3H), 2.32-2.22 (m, 3H), 2.21 – 2.05 (m, 2H), 1.73 – 1.43 (m, 3H), 0.96 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 156.57, 145.90, 144.74, 144.35, 138.98, 134.15, 132.80, 131.33, 130.67, 129.86, 128.73, 128.34, 128.14, 128.08, 127.90, 127.87, 127.06, 123.26, 113.78, 112.75, 67.16, 63.11, 58.91, 56.47, 48.99, 35.07, 29.38, 28.90, 21.65, 21.54, 19.64, 12.03.
(R)-2-(3-fluoropropanoxy)-N-n-propyl-11-toluenesulfonyloxy-noraporphine MCL-564 (7)
To a 1 mL Wheaton microreactor were added: MCL-536 (1) (3 mg, 8.4 μmol), p-toluenesulfonyl chloride (25 mg, 0.131 mmol), DMAP (1 mg, 0.082 mmol), and anhydrous dichloromethane (0.5 mL). Next, triethylamine (0.1 mL, 73 mg, 0.72 mmol) was added and contents were stirred overnight at RT. The next morning, the reaction was found to be complete by TLC. The reddish-brown reaction mixture was diluted with dichloromethane (6 mL) and washed sequentially with water and brine (10 mL each). The organic phase was filtered through sodium sulfate, concentrated, and purified by silica gel column chromatography using a gradient of 100% DCM to 1:50 to 1:40 MeOH:DCM to afford (R)-2-(3-fluoropropanoxy)-N-n-propyl-11-toluenesulfonyloxy-noraporphine MCL-564 (7) as a brownish residue in 50% isolated yield (2 mg, 3.9 μmol).
(R)-2-(3-fluoropropanoxy)-N-n-propyl-11-toluenesulfonyloxynoraporphine MCL-564 (7): HPLC > 95% pure. 1H NMR (300 MHz, CDCl3) δ 7.39 – 7.27 (m, 2H), 7.25 – 7.10 (m, 3H), 6.90 (d, J = 7.9 Hz, 2H), 6.54 (s, 1H), 4.76 (t, J = 5.9 Hz, 1H), 4.69 (dt, J = 47.1, 5.9 Hz, 1H), 4.61 (t, J = 5.8 Hz, 1H), 4.09 (t, J = 6.1 Hz, 2H), 3.11 (s, 1H), 2.97 (d, J = 10.9 Hz, 2H), 2.80 (s, 1H), 2.62 (d, J = 16.7 Hz, 2H), 2.33 (d, J = 19.9 Hz, 2H), 2.28 (s, 3H), 2.26 – 2.20 (m, 2H), 2.20 – 2.09 (m, 2H), 0.96 (t, J = 7.3 Hz, 3H).
Manual Synthesis of F-18-labeled R-(−)-N-n-propyl-2-(3-[18F]fluoropropanoxy-11-hydroxynoraporphine, [18F]MCL-536 (1)
Fluorine-18 in 0.3 ml [18O]H2O solution was purchased from the Brigham and Women’s Hospital BICOR. Anion exchange cartridges (QMA light) were purchased from Waters (Milford, MA) and rinsed with 1 mL saturated NaHCO3 solution, 3 mL H2O, and air-dried before use. The [18F]fluoride was isolated from H2[18O]O by passing the solution through the pre-treated QMA anion-exchange cartridge, drying the cartridge with air, and eluting with 1 mL K2CO3 (3.5 mg/ml) and K222 (20 mg/ml) in 10% water/acetonitrile (V/V) into a 5 ml V-vial (Pierce, Rockford, IL). The 18F solution was azeotropically dried by heating the solution to 90 °C in a heating block under a constant argon flow. After the first drying, 1 mL anhydrous CH3CN was added and the drying process was repeated twice.
MCL-563 (6) (0.5 mg) was added as a solid to the dried [18F]KF/K222 (370 MBq, 10 mCi) in a 5 mL V vial and 0.5 mL anhydrous CH3CN was added to dissolve the mixture. The vial was then capped and heated in a 100º C heating block for 15 min. The vial was cooled to room temperature, 0.2 ml 5 M NaOH solution was added, then the vial was recapped and heated at 100° C for 10 min. The reaction mixture was cooled and 0.2 ml 5 M HCl was added to neutralize the base. The entire reaction volume (0.9 mL) was injected onto a semi-prep HPLC column (10 × 150 mm, 5 μM, Grace Apollo C18) and eluted isocratically (30% acetonitrile containing 0.1% TFA and 70% water containing 0.1% TFA). The retention time of the starting material (MCL-563, 6) was 7.4 min, and the retention time of the product (MCL-536, 1) was 22.0 min. The total production time was approximately 2 h, the specific activity was 0.75 GBq/μmol, and the decay-corrected radiochemical yield was 70% (122 MBq, 3.3 mCi).
Automated Synthesis of fluorine-18 labeled R-(−)-N-n-propyl-2-(3-[18F]fluoropropanoxy-11-hydroxynoraporphine, [18F]MCL-536 (1)
[18F]Fluoride was purchased from Cardinal Health (Hartford, CT). Pre-conditioned anion exchange cartridges (Waters, QMA light) were purchased from ABX (Radeberg, Germany). The [18F]fluoride (approximately 18.5 GBq, 500 mCi) was isolated from H2[18O]O by passing the solution through the QMA cartridge, drying the cartridge with air, before eluting with a solution of potassium carbonate (1.2 mg) and Kryptofix 222 (10 mg) in 1 mL of acetonitrile/water (80/20, v/v) into the reaction vessel (RV1) of the TRACERlab® FXFN module. The solution was first evaporated by heating at 95 °C for 4 min under vacuum and helium flow. Acetonitrile (1 mL) was added and the evaporation was continued under the same conditions for 2 min. After a second addition of acetonitrile (1 mL), final evaporation was carried out at 95 °C for 2 min under vacuum and helium flow. After cooling RV1 to 60°C, a 2 mg solution of MCL-563 (6) in 1 mL of anhydrous acetonitrile was added to the RV1, which was sealed and heated at 95 °C for 10 min, then cooled to 70 °C. The deprotection solution (5 M NaOH/methanol, 50/50, v/v, 0.4 mL) was added and the reaction was stirred at 70 °C for 5 min. The reactor was cooled to 40° C and the contents of RV1 were transferred into the loop-loading vial (RV2) containing 1.0 mL of 1 M HCl. RV1 was rinsed with 1.5 mL of HPLC mobile phase and transferred into RV2. The contents of RV2 were transferred into the HPLC injector loop for purification.
Purification was performed by HPLC on a Phenomenex Luna C18(2) column (10 μm, 10 × 250 mm) eluted with a mixture of acetonitrile/water/trifluoroacetic acid, 30/70/0.1, v/v/v at a flow rate of 4 mL/min. The product fraction (Rt = 10 min) was collected in a round-bottom flask (Flask 1), containing 15 mL of diluted ascorbic acid solution. It should be noted that the tosylate precursor does not elute (Rt > 30 min) using this HPLC column and eluent. The resulting diluted product mixture was passed through a solid-phase extraction cartridge (SPE, Waters Sep-Pak® tC18 Light), and the cartridge was rinsed with 10 mL of diluted ascorbic acid solution. The radiolabeled product was eluted from the SPE cartridge with 1.0 mL of 200-proof USP grade ethanol into the formulation flask, containing the drug product matrix (15 mL of normal saline containing 0.7 mg/mL of ascorbic acid and 8 μg/mL of polysorbate-80). The cartridge was rinsed with a further portion of the formulation matrix (4 mL) and the rinse was added to the second flask. The resulting solution was passed through a sterilizing membrane filter (Millex LG 0.22 μm, Millipore) into a sterile, filter-vented vial (final product vial, FPV), obtaining 1.48 GBq (40 mCi, 11%) product in 52 min synthesis time.
The identity, chemical purity, and radiochemical purity of the final product were determined by HPLC using a Waters XBridge C18 column (5 μm, 4.6 × 250 mm) equipped with dual gamma and UV (245 nm) detectors and eluted with a mixture of methanol/5 mM ammonium acetate, 70/30 v/v at a flow rate of 1 mL/min and compared to a reference standard of MCL-536 (Rt = 9.3 min). The tosylate precursor does not elute (Rt > 20 min) using this HPLC column and eluent. The final product showed 100% radiochemical purity with a specific activity of 167 GBq/μmol (4.5 Ci/μmol) and total chemical impurity of 0.13 μg/mL.
Results and Discussion
Synthesis of [18F]MCL-536
We focused on preparing the phenol analog (MCL-536 (1)) as there are several advantages to the phenol derivatives over the catechol derivatives in this series. First, MCL-536 has very high D2high/D3 selectivity (185) and binding affinity to D2high (Ki = 0.54 nM).24 Second, the phenol derivatives are more stable than the catechols, which decompose quickly on synthesis and workup. Furthermore, we envisioned that the radiofluorination of the appropriate tosyl precursor of 1 might be accomplished in a single step, thus avoiding the need to use a protecting group on the phenol. The starting material for the synthesis was compound 4, the noraporphine containing both unprotected alcohol and phenol.
The monotosyl precursor (5) was prepared from 3-deoxy-N-propylnormorphine26 2 as outlined in Scheme 1. Oxidation with IBX in dimethylformamide (DMF)27 smoothly converted the normorphine derivative 2 into northebainone 3. Rearrangement of the latter by heating in methanesulfonic acid in the presence of 1,3-propanediol afforded R-(−)-11-hydroxy-2-(3-hydroxypropanoxy)-N-n-propylaporphine (4) in moderate yield (65%). Compound 4 was tosylated by employing a slight excess of p-toluenesulfonyl chloride in dichloromethane and triethylamine to afford 5 (MCL-557).
Scheme 1.
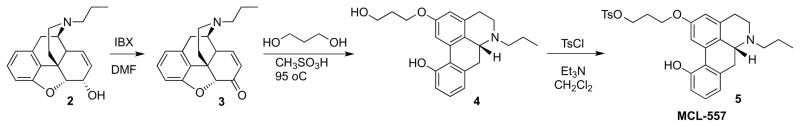
Synthesis of MCL-557
Unfortunately, all attempts to label 5 with 18F were unsuccessful. Despite multiple attempts under a wide variety of conditions, only unidentified polymerized products were produced, suggesting that the unprotected phenol was not stable enough to survive the radiofluorination conditions. We therefore turned our attention to identifying an appropriate protecting group for the phenol.
Several possible protecting groups were evaluated, including benzyl, tetrahydropyranyl ether, and acetyl. In all of these cases, the substrates decomposed during radiofluorination or during deprotection. The desired product was, however, ultimately obtained using tosyl as the protecting group (Scheme 2). While the alkyl tosylate readily participates in Sn2 fluorination, the aryl tosylate is inert to fluorination under these reaction conditions. An added advantage is that tosylation of both the alcohol and phenol takes place in the same reaction vessel. Thus, the diol 4 was converted to the ditosylate 6 (MCL-563) in 79% yield. Non-radioactive (19F) fluorination proceeded smoothly to afford MCL-564 (7), as confirmed by HPLC analysis using an independently synthesized sample of 7 as a reference. Hydrolysis in methanol and 5 M NaOH to remove the tosyl protecting group from the phenol moiety afforded the fluorinated phenol derivative, MCL-536 (1), in high and reproducible yields.
Scheme 2.
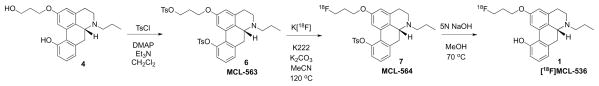
Synthesis of MCL-536 using a tosylate protecting group for the phenol
In contrast to previous efforts, compound 6 was readily labeled with 18F to produce [18F]MCL-536. Using a manual synthesis, the 18F-labeled compound was obtained in 70% decay-corrected yield in 2 h, but at relatively low specific activity (750 MBq/μmol, 20 mCi/μmol). Using an automated synthesis system and starting with a larger amount of 18F, we obtained [18F]MCL-536 in 11% yield with significantly higher specific activity of 167 GBq/μmol (4.5 Ci/μmol).
Conclusions
We developed a reliable, reproducible route for the synthesis of 11-hydroxy aporphines (MCL-536 (1)). We also validated the tosyl moiety as a protecting group for the phenol moiety that withstands radiofluorination conditions. To the best of our knowledge, this is the first example where the tosyl group is used simultaneously as a protecting group and a leaving group in a radiofluorination reaction. Studies are currently underway to evaluate the in vivo properties of this new 18F-labeled D2 agonist.
Acknowledgments
This work was supported by grant from the Branfman Family Foundation (JLN) and NIH NIDA Research Training Grant T-32-DA007252 (AWS). W. L. was supported as a Visiting Scholar by the China Scholarship Council.
References
- 1.(a) Bozzi Y, Borrelli YE. TRENDS in Neurosciences. 2006;29:167. doi: 10.1016/j.tins.2006.01.002. [DOI] [PubMed] [Google Scholar]; (b) Marsden CA. British J Pharm. 2006;147:S136. doi: 10.1038/sj.bjp.0706473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Seeman P. Clin Schizophrenia and related psychoses. 2008:351–355. [Google Scholar]; (b) Seeman P, Weinshenker D, Quirion R, Srivastava LK, Bhardwaj SK, Grandy DK, Premont RT, Sotnikova TD, Boksa P, El-Ghundi M, O’Dowd BF, George SR, Perreault ML, Männistö PT, Robinson S, Palmiter RD, Tallerico T. Proc Natl Acad Sci. 2005;102:3513. doi: 10.1073/pnas.0409766102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Seeman P. Synapse. 2009;63:186. doi: 10.1002/syn.20595. [DOI] [PubMed] [Google Scholar]; (d) Seeman P, Schwarz J, Chen J-F, Szechtman H, Perrault M, McKnight GS, Roder JC, Quirion R, Boksa P, Srivastava LK, Yanai L, Weinshenker D, Sumiyoshi T. 2006;60:319. doi: 10.1002/syn.20303. [DOI] [PubMed] [Google Scholar]; (d) Seeman P, McCormick PN, Kapur SS, editors. Synapse. 2007;61:263. doi: 10.1002/syn.20367. [DOI] [PubMed] [Google Scholar]; (f) Seeman P. CNS Neurosci & Ther. 2011;17:118. doi: 10.1111/j.1755-5949.2010.00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Seeman P, Kapur S. Schizophrenia: More dopamine, more D2 receptors. P Natl Acad Sci. 2000;97:7673. doi: 10.1073/pnas.97.14.7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corripio I, Escartí MJ, Portella MJ, Pérez V, Grasa E, Sauras RB, Alonso A, Safont G, Camacho MV, Dueñas R, Arranz B, San L, Catafau AM, Carrió I, Alvarez E. Eur Neuropsychopharmacol. 2011;12:861. doi: 10.1016/j.euroneuro.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Lieberman JA, Kane JM, Alvir JJ. Psychopharmacology. 1987;91:415. doi: 10.1007/BF00216006. [DOI] [PubMed] [Google Scholar]
- 5.Seeman P. Synapse. 2008;62:314. doi: 10.1002/syn.20499. [DOI] [PubMed] [Google Scholar]
- 6.(a) Seeman P. Synapse. 2007;61:1013. doi: 10.1002/syn.20453. [DOI] [PubMed] [Google Scholar]; (b) Seeman P, Ko F, Willeit M, McCormick P, Ginovart N. Synapse. 2005;58:122. doi: 10.1002/syn.20193. [DOI] [PubMed] [Google Scholar]; (c) Guttman M, Seeman P. J Neural Transmission. 1985;64:93. doi: 10.1007/BF01245971. [DOI] [PubMed] [Google Scholar]
- 7.Elsinga PH, Hatano K, Ishiwata K. Curr Med Chem. 2006;13:2139. doi: 10.2174/092986706777935258. [DOI] [PubMed] [Google Scholar]
- 8.Wagner HN, Jr, Burns HD, Dannals RF, Wong DF, Langstrom B, Duelfer T, Frost JJ, Ravert HT, Links JM, Rosenbloom SB, Lukas SE, Kramer AV, Kuhar MJ. Science. 1983;221:1264. doi: 10.1126/science.6604315. [DOI] [PubMed] [Google Scholar]
- 9.(a) Farde L, Ehrin E, Eriksson L, Greitz T, Hall H, Hedstrom CG, Litton JE, Sedvall G. Proc Natl Acad Sci USA. 1985;82:3863. doi: 10.1073/pnas.82.11.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Farde L, Hall H, Ehrin E, Sedvall G. Science. 1986;231:258. doi: 10.1126/science.2867601. [DOI] [PubMed] [Google Scholar]
- 10.Mukherjee J, Yang ZY, Brown T, Lew R, Wernick M, Ouyang X, Yasillo N, Chen CT, Mintzer R, Cooper M. Nucl Med Biol. 1999;26(5):519. doi: 10.1016/s0969-8051(99)00012-8. [DOI] [PubMed] [Google Scholar]
- 11.Skinbjerg M, Sibley DR, Javitch JA, Abi-Dargham A. Biochem Pharmacol. 2012;83:193. doi: 10.1016/j.bcp.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finnema SJ, Bang-Andersen B, Wikstrom HV, Halldin C. Curr Top Med Chem. 2010;10:1477. doi: 10.2174/156802610793176837. [DOI] [PubMed] [Google Scholar]
- 13.(a) Gao YG, Baldessarini RJ, Kula NS, Neumeyer JL. J Med Chem. 1990;33:1800. doi: 10.1021/jm00168a040. [DOI] [PubMed] [Google Scholar]; (b) Neumeyer JL, Gao YG, Kula NS, Baldessarini RJ. J Med Chem. 1990;33:3122. doi: 10.1021/jm00174a002. [DOI] [PubMed] [Google Scholar]; (c) Baldessarini RJ, Kula NS, Gao Y, Campbell A, Neumeyer JL. Neuropharmacol. 1991;30:97. doi: 10.1016/0028-3908(91)90049-h. [DOI] [PubMed] [Google Scholar]; (d) Skinbjerg M, Namkung Y, Halldin C, Innis RB, Sibley DR. Synapse. 2009;63:462. doi: 10.1002/syn.20626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Finnema SJ, Seneca N, Farde L, Shchukin E, Sovago J, Gulyas B, Wikstrom HV, Innis RB, Neumeyer JL, Halldin C. Nucl Med Biol. 2005;32:353. doi: 10.1016/j.nucmedbio.2005.01.007. [DOI] [PubMed] [Google Scholar]; (b) Seneca N, Zoghbi SS, Skinbjerg M, Liow JS, Hong J, Sibley DR, Pike VW, Halldin C, Innis RB. Occupancy of Dopamine D2/3 Receptors in Rat Brain by Endogenous Dopamine Measured With the Agonist Positron Emission Tomography Radioligand [11C]MNPA. Synapse. 2008;62:756. doi: 10.1002/syn.20549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hwang DR, Narendran R, Huang Y, Slifstein M, Talbot PS, Sudo Y, Van Berckel BN, Kegeles LS, Martinez D, Laruelle M. J Nucl Med. 2004;45:338. [PubMed] [Google Scholar]
- 16.(a) Willeit M, Ginovart N, Kapur S, Houle S, Hussey D, Seeman P, Wilson AA. Biol Psychiatry. 2006;59:389. doi: 10.1016/j.biopsych.2005.09.017. [DOI] [PubMed] [Google Scholar]; (b) Ginovart N, Galineau L, Willeit M, Mizrahi R, Bloomfield PM, Seeman P, Houle S, Kapur S, Wilson J. J Neurochem. 2006;97:1089. doi: 10.1111/j.1471-4159.2006.03840.x. [DOI] [PubMed] [Google Scholar]
- 17.(a) Peng T, Zysk J, Dorff P, Elmore CS, Strom P, Malmquist J, Ding M, Tuke D, Werkheiser J, Widzowski D, Mrzljak L, Maier D. Synapse. 2010;64:624. doi: 10.1002/syn.20771. [DOI] [PubMed] [Google Scholar]; (b) van Wieringen JP, Booij J, Shalgunov V, Elsinga P, Michel MC. Naunyn-Schmeideberg’s Arch Pharmacol. 2013;386:135. doi: 10.1007/s00210-012-0817-0. [DOI] [PubMed] [Google Scholar]
- 18.Seeman P. Synapse. 2012;66:88. doi: 10.1002/syn.20987. [DOI] [PubMed] [Google Scholar]
- 19.Vasdev N, Seeman P, Garcia A, Stableford WT, Nobrega JN, Houle S, Wilson AA. Nuc Med Biol. 2007;34:195. doi: 10.1016/j.nucmedbio.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Zijlstra S, Visser GM, Korf J, Vaalburg W. Appl Radiat Isot. 1993;44(4):651. doi: 10.1016/0969-8043(93)90127-v. [DOI] [PubMed] [Google Scholar]
- 21.Shi B, Narayanan TK, Christian BT, Chattopadhyay S, Mukherjee J. Nucl Med Biol. 2004;31:303. doi: 10.1016/j.nucmedbio.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 22.(a) Ramsby S, Neumeyer JL, Grigoriadis D, Seeman P. 1989;32:1198. doi: 10.1021/jm00126a009. [DOI] [PubMed] [Google Scholar]; (b) Neumeyer JL, Gao Y, Kula NS, Baldessarini RJ. J Med Chem. 1990;33:3122. doi: 10.1021/jm00174a002. [DOI] [PubMed] [Google Scholar]; (c) Søndergaard K, Kristensen JL, Gillings N, Begtrup M. Eur J Org Chem. 2005:4428. [Google Scholar]; (d) Zhang A, Csutoras C, Zong R, Neumeyer JL. Org Lett. 2005;7:3239. doi: 10.1021/ol051010d. [DOI] [PubMed] [Google Scholar]; (e) Si YG, Gardner MP, Tarazi FI, Baldessarini RJ, Neumeyer JL. Bioorg Med Chem Lett. 2008;18:3971. doi: 10.1016/j.bmcl.2008.06.016. [DOI] [PubMed] [Google Scholar]; (f) Si Y-G, Gardner MP, Tarazi FI, Baldessarini RJ, Neumeyer JL. J Med Chem. 2008;51:983. doi: 10.1021/jm701045j. [DOI] [PubMed] [Google Scholar]
- 23.Zhang A, Zhang Y, Branfman AR, Baldessarini RJ, Neumeyer JL. J Med Chem. 2007;50:171. doi: 10.1021/jm060959i. [DOI] [PubMed] [Google Scholar]
- 24.Sromek AW, Si YG, Zhang T, George SR, Seeman P, Neumeyer JL. ACS Med Chem Lett. 2011;2:189. doi: 10.1021/ml1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frigerio M, Santagostino M, Sputore S. J Org Chem. 1999;64:4537. [Google Scholar]
- 26.Csutoras C, Zhang A, Bidlack JM, Neumeyer JL. Bioorg Med Chem Lett. 2004;17:2687. doi: 10.1016/j.bmc.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 27.Søndergaard K, Laugard KJ, Palner M, Gillings N, Knudsen GM, Roth BL, Begtrup M. Org Biomol Chem. 2005;3:4077. doi: 10.1039/b507195j. [DOI] [PubMed] [Google Scholar]