

Abstract
Disease progression and recurrence are major barriers to surviving breast cancer. Understanding the etiology of recurrent or metastatic breast cancer and underlying mechanisms is critical for the development of new treatments and improved survival. Here, we report that two commonly over-expressed breast cancer oncogenes, Ron and DEK, cooperate to promote advanced disease through multi-pronged effects on β-catenin signaling. The Ron receptor is commonly activated in breast cancers, and Ron over-expression in human disease stimulates β-catenin nuclear translocation and is an independent predictor of metastatic dissemination. Dek is a chromatin-associated oncogene whose expression has been linked to cancer through multiple mechanisms, including β-catenin activity. We demonstrate here that Dek is a downstream target of Ron receptor activation in murine and human models. The absence of Dek in the MMTV-Ron mouse model led to a significant delay in tumor development, characterized by decreased cell proliferation, diminished metastasis, and fewer cells expressing cancer stem cell markers. Dek complementation of cell lines established from this model was sufficient to promote cellular growth and invasion in vitro and in vivo. Mechanistically, Dek expression stimulated the production and secretion of Wnt ligands to sustain an autocrine/paracrine canonical β-catenin signaling loop. Finally, we show that Dek over-expression promotes tumorigenic phenotypes in immortalized human mammary epithelial MCF10A cells and, in the context of Ron receptor activation, correlates with disease recurrence and metastasis in patients. Overall, our studies demonstrate that DEK over-expression, due in part to Ron receptor activation, drives breast cancer progression through the induction of Wnt/β-catenin signaling.
Keywords: DEK, RON, Wnt signaling, metastasis, breast cancer, tumor growth
Introduction
Despite high survival rates for early stage breast cancer, it is still the second-leading cause of cancer-related deaths in the United States. This is due to the poor survival associated with late stage, invasive disease and treatment resistant breast cancers. Most new diagnoses are invasive cancers, which have poor survival rates1 Therefore, it is important to identify proteins that predict disease relapse or progression and that also may be therapeutic targets.
Several reports identified the RON (Recepteur d’Origine Nantaise) receptor tyrosine kinase, a MET family member, as an important driver of breast tumorigenesis. RON is minimally expressed in normal breast epithelium but is highly expressed in approximately 50% of human breast cancers.2 RON activation from overexpression has been implicated in driving metastasis and correlates with poor prognosis.2, 3 Little is known about the molecular mechanisms through which RON promotes breast cancer proliferation and metastases. Recently, it was reported that RON activation leads to c-abl activation and PCNA localization to chromatin to drive proliferation.4 RON also activates numerous cytoplasmic signaling pathways, including Ras/MAPK, PI3K, and β-catenin through direct phosphorylation, and regulates many downstream targets.5–7 Here, we identify a novel nuclear effector of RON activation, the DEK oncogene.
DEK is a unique protein with no known paralogs or enzymatic function.8 It is predominantly chromatin-bound and has been implicated in numerous processes including DNA repair, replication, transcriptional regulation, and mRNA splicing.8–14 The role of DEK in these processes is unclear, but may be due to its histone chaperone and Su(var) activities that regulate chromatin organization.15, 16 Many reports have shown that DEK can function as a cofactor for transcriptional regulation; however, DEK can both repress and activate transcription of target genes. For example, several reports have shown that DEK inhibits NFκB/p65/RelA transcriptional activity17–19, decreases hTERT expression in leukemias,20 and can interact with PARP1 to restrict the accessibility of the transcription complexes to chromatin.21 Other reports indicate that DEK can also activate transcription by interacting with several transcription factors including, C/EBPα during myeloid differentiation,22 nuclear ecdysone receptor (EcR) in Drosophila,16 and AP-2α in rat liver and human cell lines.10 Interestingly, DEK can also be secreted and is an auto-antigen in patients with juvenile idiopathic arthritis and was detected in the urine of patients with bladder cancer.23–25 Macrophages can secrete DEK and neighboring cells can internalize it in a heparan sulfate dependent process where it then completes its normal nuclear functions.26
DEK is transcriptionally upregulated in several solid tumors and was initially discovered as a CAN (NUP214) fusion gene in acute myeloid leukemia.27–32 Transcriptional up-regulation is accomplished via the Rb/E2F pathway and steroid hormone signaling, but 6p22.3 amplifications have been noted.32–36 Cell culture, murine chemical carcinogenesis, and xenograft tumor models indicate that DEK over-expression promotes proliferation.29, 30, 32, 37 There are several possible molecular mechanisms for this DEK-mediated proliferation, including up-regulation of ΔNp63α and, in the case of the DEK-NUP214 fusion, mTORC1 activity.29, 30, 37, 38 Additional cell culture models implicate DEK in promoting cell invasion, in part due to β-catenin signaling.29, 32 However, the oncogenic functions of DEK have not been tested in vivo using genetically engineered mouse models. Furthermore, the molecular mechanisms through which full-length DEK drives proliferation and β-catenin signaling in breast cancer are unknown.
Here, we crossed Dek−/− mice with the MMTV-Ron murine breast cancer model to determine the requirement for Dek during breast cancer initiation and progression. We report that Dek is a downstream target of activated Ron signaling such that the MMTV-Ron model recapitulates DEK over-expression observed in some human breast cancers. The loss of Dek delayed tumor growth and decreased lung metastases. We generated Dek proficient and deficient breast cancer cell lines from the murine tumors and found that reconstitution of Dek in knockout cells rescued cellular growth and invasive phenotypes in vitro. Importantly, we present new evidence that Dek expressing cells can drive the proliferation of cells within the microenvironment through stimulated expression and secretion of Wnt4, Wnt7b, and Wnt10b. Finally, we translate the observed oncogenic DEK activities, and the combination of RON and DEK activation, to human breast cancer using 3D cell culture models, tissue microarrays, and patient survival data. We propose that DEK drives tumor growth and progression, particularly in the context of RON receptor activation, via Wnt ligand production and subsequent activation of the β-catenin pathway.
Results
The MMTV-Ron mouse model recapitulates DEK over-expression observed in breast cancer
The MMTV-Ron mouse is an established malignant breast cancer model wherein Ron expression is controlled by the MMTV mammary-specific promoter. Mice begin to develop tumors at 4–5 months of age with a high prevalence of lung metastases.3 Western blot analysis of primary tumors from MMTV-Ron mice compared to normal murine mammary gland tissue revealed Dek up-regulation in tumors (Figure 1A). Furthermore, western blot analysis of mammary epithelial cells (MECs) isolated from a non-transgenic (WT) and a non-tumor bearing MMTV-Ron transgenic mouse also revealed Dek upregulation in the transgenic mammary gland (Figure 1B). To determine whether Ron activation specifically induced Dek expression, R7 mammary tumor cells previously isolated from a MMTV-Ron tumor were treated with the Ron ligand, hepatocyte growth factor-like protein (HGFL; Figure S1).3, 39 Ron activation with HGFL resulted in increased Dek mRNA and protein (Figure 1C and 1D), indicating that Dek is a downstream target of Ron. These findings show that the MMTV-Ron (Rontg) model is appropriate for studying the role of Dek over-expression during tumorigenesis in vivo.
Figure 1. Dek expression is up-regulated in the MMTV-Ron murine breast cancer model.

(A) Dek is over-expressed in MMTV-Ron mammary gland tumors compared to normal mammary gland tissue as determined by western blot analysis. Actin is used as a loading control. (B) Dek is over-expressed in primary mammary epithelial cells (“MECs”) from pre-neoplastic MMTV-Ron mammary glands compared to non-transgenic mouse mammary glands, as determined by western blotting. Cytokeratin 14 (CK14) is used as a loading control demonstrating equal amounts of epithelial cells in the lysates. Relative expression as determined by densitometry is shown. (C) Dek is transcriptionally up-regulated in R7 cells, a cell line generated from a MMTV-Ron tumor, following Ron receptor activation with HGFL. Quantitative RT-PCR was performed at the given time points post-stimulation with HGFL. Dek expression is normalized to actin and relative to the 0 hour time point. (D) Dek protein levels are elevated following HGFL stimulation of the Ron receptor in R7 cells. Western blot analysis was performed on whole cell lysates and analyzed for Dek and actin expression.
The absence of Dek delays tumor development due to diminished proliferation
The Dek knockout allele was introduced into the MMTV-Ron mouse to generate Dek deficient (RontgDek−/−) mice. RontgDek−/− mice were significantly delayed in palpable tumor development compared to Dek proficient RontgDek+/+ controls (Figure 2A). There were no significant differences in tumor incidence or in the number of tumors that eventually developed (Figure 2A and data not shown). We previously published that DEK depletion by shRNA in MDA-MB-468 human breast cancer cells results in smaller xenograft tumors associated with decreased tumor proliferation.29 To determine if decreased proliferation accounted for the delayed tumor onset in RontgDek−/− mammary glands, pre-neoplastic glands were analyzed for BrdU incorporation as a proliferation marker. Glands from RontgDek−/− mice had substantially fewer BrdU-positive cells when compared with control mice (Figure 2B). To directly assess the necessity of Dek for tumor cell growth, cell lines were established from tumors isolated from independent mice. Dek expression was subsequently decreased by shRNA (Deksh) in RontgDek+/+ derived tumors, and murine Dek (mDek) was exogenously expressed in cell lines generated from RontgDek−/− tumors (see Western blots depicted as insets in Figure 2C). In all cell lines tested, depleting Dek in RontgDek+/+ cell lines decreased growth rates (Figure 2C, top row) whereas complementing Dek expression in RontgDek−/− cell lines increased growth rates (Figure 2C, bottom row). In addition, a trend was observed wherein cells derived from RontgDek−/− tumors grew more slowly than cells from RontgDek+/+ tumors, based on comparisons of vector transduced RontgDek+/+ “NTsh” and RontgDek−/− “R780” control cells (Figure 2C, black lines).
Figure 2. Dek promotes tumor growth and increases cell proliferation in vivo and in vitro.

(A) RontgDek−/− (MMTV-Ron and Dek−/−) mice exhibit delayed tumor onset compared to RontgDek+/+ mice. Time to tumor detection for RontgDek−/− mice was 232.1 days ± 10.27 (N=14) compared to 193.6 days ± 4.206 (N=23; mean ± SEM; ***p<0.0001, Log-rank test) (B) Pre-neoplastic mammary glands from RontgDek+/+ mice (24.16% ± 2.728, N=11) are more proliferative than glands from RontgDek−/− mice (9.401% ± 1.562 N=8) **p=0.0006, as determined by unpaired two-tailed t-test) Mice were IP injected with BrdU before sacrifice. Immunohistochemistry for BrdU incorporation was performed on pre-neoplastic glands. Representative images are shown. Results are quantified in the graph on the right as the percentage of BrdU positive cells in the tissue section. (C) Dek expression levels positively correlate with cellular growth rate in Rontg tumor-derived cell lines. Cell lines were generated from three RontgDek+/+ and three RontgDek−/− tumors from independent mice. A Dek shRNA (Deksh2) was introduced into RontgDek+/+ cells to decrease Dek expression or a murine Dek (mDek) over-expression construct was retrovirally transduced into RontgDek−/− to complement the loss of Dek.. Western blots in the inset show changes in Dek expression (D) with alpha-tubulin (T) as a loading control. Cells were counted over the time periods shown for each cell line. (D) Tumors from RontgDek−/− mice (N=4) have fewer cells with BCSC cell surface markers compared to RontgDek+/+ tumors (N=3). Flow cytometry was used to quantify the percentage of Lin−/CD49f+/CD24+/CD44low cells. (E) Dek expression promotes the growth of mammospheres. RontgDek+/+ cell line (R7) transduced with Deksh2 (“+Deksh2”) or NTsh control, and RontgDek−/− cell lines (RD147, RD219, RD238 and RD271) transduced with a mDek construct (“+mDek”) or control R780 vector were plated in non-adherent mammosphere cultures and measured after nine days to calculate volume. Data is presented as fold-change compared to control (NTsh or R780) spheres from triplicate experiments.
Dek expression positively correlates with breast cancer stem cell (BCSC) phenotypes
We have previously shown that DEK expression correlates with the size of the BCSC population in human MCF7 cells.29 To test this finding in murine samples, we analyzed first passage cancer cells isolated from the RontgDek+/+ and RontgDek−/− tumors for known cell surface murine BCSC markers by flow cytometry (Lin−/CD49f+/CD24+/CD44low).40–42 We detected significantly fewer Lin−/CD49f+/CD24+/CD44low cells in RontgDek−/− tumors compared to RontgDek+/+ samples (Figure 2D). To further our analysis, we plated RontgDek+/+ R7 NTsh/Deksh cells and four RontgDek−/− lines with empty vector or mDek for mammosphere formation. Although there were no differences in mammosphere numbers, a significant positive correlation between Dek expression and sphere volume was observed, which is in agreement with the notion that Dek drives proliferation (Figure 2E). There was also a positive correlation between Dek expression and the percentage of side population (SP) cells. Dek depletion in RontgDek+/+ R7 cells lowered the percentage of SP cells while reconstitution of Dek in a RontgDek−/− RD147 cells increased the percentage (Figure S2A). Interestingly, there also was a trend towards diminished xenograft tumor formation in cells derived from RontgDek−/− tumors (Figures S2B). Together, the data indicate that Dek contributes to tumor initiation and growth in the transgenic mouse model through enhanced proliferation and promotes BCSC phenotypes.
Dek expression promotes cancer metastasis in vivo and in vitro
Previous reports using in vitro transwell assays suggests that DEK conferred invasive potential to breast cancer cells via a β-catenin-dependent mechanism.29 However, an association between DEK expression and metastatic events in vivo had not yet been investigated. We examined metastases to the lungs and liver from RontgDek−/− and RontgDek+/+ mice. Of the mice examined, 100% of RontgDek+/+ and 83% of RontgDek−/− mice developed liver metastases. All mice Rontg mice examined had lung metastases, but RontgDek+/+ mice had more than double the number of lung metastases per animal when compared to RontgDek−/− mice (Figure 3A). Lung metastases were positive for cytokeratin 5 staining, indicating they were epithelial in origin (Figure S3A). One potential mechanism of metastasis is through the expression of matrix metalloproteases (MMPs), including the Wnt targets MMP2 and MMP9.43 Primary tumors and the cell lines showed no correlation between DEK expression and matrix metalloprotease (MMP)-2 or MMP-9 expression (Figure S3B and C). To test invasion in vitro, Dek proficient and deficient cell lines (described above) were analyzed using Matrigel transwell invasion assays. Dek expression enhanced the invasive potential in all cell lines tested (Figure 3B, see Figures 2C and 4A for Dek expression).
Figure 3. Dek expression supports breast cancer metastasis in vivo and in vitro.

(A) RontgDek+/+ mice have a greater metastatic lung tumor burden than RontgDek−/−. Lungs were harvested from six RontgDek−/− and five RontgDek+/+ mice with similar tumor volumes for the largest tumor and serial sections were microscopically analyzed for tumor metastases. Total tumor burden from all lung sections in wild type mice was 18.38 ± 1.754 compared to 8.183 ± 1.661 in knockout mice (p= 0.0023, unpaired two-tailed t-test). The average number of lung metastases per mouse is quantified and a representative H&E from each genotype is depicted below. (B) Dek expression correlates with cellular invasion in vitro. RontgDek+/+ cell lines (R7 and RD258) transduced with Deksh2 (“+Deksh2”) or NTsh control, and RontgDek−/− cell lines (RD147, RD219, RD238 and RD271) transduced with a mDek construct (“+mDek”) or control R780 vector were subjected to Matrigel transwell assays. Data is represented as fold change compared to the respective control for each cell line (NTsh or mDek) for triplicate experiments. Significance was calculated with a one-tailed unpaired t-test.
Figure 4. Dek expression stimulates Wnt ligand production to drive proliferation via a paracrine mechanism.

(A) Dek expression positively regulates Wnt4, Wnt7b, and Wnt10b transcription. Dek knockdown by Deksh2 in R7 cells downregulates Wnt expression whereas Dek complementation in RontgDek−/− cell lines up-regulates Wnt ligand expression. Dek and Wnt gene expression were analyzed by quantitative RT-PCR. The horizontal line represents the normalization relative to the respective control transduced cells. (B) Dek expression correlates with the secretion of Wnt10b. Conditioned media from RontgDek+/+ R7 NTsh and Deksh2 cells and RontgDek−/− RD147 R780 and mDek cells was concentrated and analyzed by western blot analysis for Wnt10b. (C) Complementation of Dek expression in RontgDek−/− RD147 increases β-catenin transcriptional activity. RD147 cells transduced with R780 and mDek (R780:mDek) were transfected with TOPflash and FOPflash luciferase reporter assay constructs. Luciferase expression represents β-catenin transcriptional activity. (D) Dek complementation in knockout cells results in increased expression of Wnt/β-catenin target genes, including cyclin D1. Western blot analysis of whole cell lysates from RontgDek−/− RD147 R780 and R780:mDek cells was performed using the antibodies shown. Relative expression as determined by densitometry is shown below each blot. (E) Dek-induced Wnt secretion increases the proliferation of neighboring Dek deficient cancer cells. GFP-negative parental RontgDek−/− RD147 cells were co-cultured with GFP-positive RD147 R780 or R780:mDek cells. BrdU incorporation was then analyzed by flow cytometry in GFP-negative cells as a measure of cell proliferation. Cells were also treated with IWP-2, a Wnt secretion inhibitor, prior to analysis. Inhibiting Wnt secretion partially rescues the pro-proliferative effect of neighboring Dek-expressing cells. The western blot inset depicts levels of secreted Wnt10b in conditioned media in untreated and IWP-2 treated RD147 R780 or R780:mDek GFP-positive cells. The images depicted are from different blots for Wnt10b.
Dek drives proliferation through Wnt signaling
We previously showed that DEK expression correlated with β-catenin activity in human breast cancer cell lines and that this activity drove cell invasion in Matrigel transwell assays.29 However, the mechanism for this regulation was unknown. Preliminary data from a quantitative RT-PCR array was utilized wherein we screened for expression differences of over 80 Wnt signaling pathway members in Dek+/+ versus Dek−/− mouse embryonic fibroblasts13 in order to select relevant Wnt/β-catenin pathway members for further analysis (Figure S4A–B). We identified Wnt4, Wnt7b, and Wnt10b to be differentially regulated by Dek expression (Figure 4A). Dek shRNA in R7 cells resulted in decreased Wnt expression whereas complementation of four RontgDek−/− cell lines with mDek resulted in increased Wnt expression. Wnt10b, a canonical Wnt ligand, was pursued due to its strong association with breast cancer development and proliferation.44, 45 The observed differences in Wnt10b mRNA correlated with differences in secreted protein levels detected in conditioned media (Figure 4B). The upregulation of Wnts also correlated with increased β-catenin transcriptional activity as determined by the TOP/FOP luciferase reporter assay (Figure 4C), a finding which agrees with our previous work with mouse embryonic fibroblasts.29 We also observed up-regulation of the pro-proliferative Wnt/β-catenin target gene cyclin D1 and increased protein levels of other Wnt pathway members (Figure 4D and S4C). To determine if Wnt signaling promoted Dek-mediated proliferation, GFP(−) parental RD147 cells (RontgDek−/−) were co-cultured in equal numbers with GFP(+) cells transduced with either R780 vector or R780:mDek. BrdU incorporation as a measure of proliferation was then assessed by flow cytometric anlaysis of the GFP(−) Dek deficient cells. Co-culturing RontgDek−/− RD147 parental cells with cells expressing murine Dek resulted in enhanced proliferation of the Dek deficient cells. Treatment with IWP-2, a Porcupine inhibitor which prevents Wnt secretion, significantly diminished the pro-proliferative effects of elevated Dek and Wnt expression, decreasing the proliferation of parental Dek-deficient RD147 cells close to control levels (Figure 4E).
Dek expression correlates with xenograft tumor growth, Wnt10b expression, and β-catenin activity in vivo
To determine if Dek regulates Wnt10b expression in vivo, RontgDek+/+ R7 cells transduced with NTsh or Deksh constructs were injected into the mammary gland to generate orthotopic tumors in female nude mice. Tumors from Deksh cells grew significantly slower than tumors from control (NTsh) cells (Figure 5A). Immunohistochemical staining demonstrated significantly diminished Wnt10b and activated β-catenin levels in Deksh tumors, suggesting attenuated β-catenin activity (Figure 5B, quantified in 5C). Together, this suggests that Dek-mediated tumor growth is associated with Wnt expression and subsequent β-catenin activation.
Figure 5. Dek depletion delays xenograft tumor growth and results in decreased Wnt10b and activated β-catenin expression in vivo.

(A) Dek depletion (Deksh2) in RontgDek+/+ R7 cells delays xenograft tumor growth. RontgDek+/+ R7 NTsh and Deksh2 cells were injected into inguinal mammary fat pads in nude mice and monitored for tumor growth. (B) Dek depletion inhibits Wnt/β-catenin activity in xenograft tumors. Immunohistochemical staining for Dek, Wnt10b, and activated β-catenin was performed on xenograft tumors from R7 NTsh and Deksh2 cells. (C) Quantification of data presented in (B). The relative staining intensity of activated β-catenin (N=5 tumors each) and Wnt10b (N=4 tumors each) was quantified as the product of staining intensity and percentage of positively-staining cells. Data is represented as fold-change. Activated β-catenin scores were (22.13±5.80) for R7 NTsh and (14.26±3.68) for R7 Deksh tumors and were determined based on positive staining nuclei while Wnt10b scores were (16.84±3.33) for R7 NTsh and (7.79±1.87) for R7 Deksh. Significance was calculated by comparing matched tumors harvested from the same mouse with a two-tailed paired t-test.
RON receptor activation increases DEK expression and DEK over-expression confers tumorigenic phenotypes to immortalized human mammary epithelial cells
We treated T47D cells with HGFL to determine if RON activation also up-regulates DEK in human breast cancer cell lines. DEK mRNA was up-regulated within hours of HGFL stimulation, resulting in elevated protein levels (Figure 6A–B). This further supports the hypothesis that DEK over-expression in breast cancer cell lines is induced by RON receptor activation.
Figure 6. Ron and DEK cooperate in human breast cancer to promote disease progression.

(A) DEK is a down-stream target gene of activated Ron signaling in T47D human breast cancer cells. T47D cells were treated with HGFL to activate Ron signaling and DEK expression was analyzed by quantitative RT-PCR. Expression was compared to GAPDH and normalized to expression in untreated cells. For the 24 hour time point, p=0.07 and significance was calculated with a one-tailed unpaired t-test. (B) DEK protein levels are elevated following HGFL-mediated activation of Ron signaling. T47D cells were treated with HGFL for the time periods shown and whole cell lysates were analyzed by western blotting. (C) DEK over-expression in MCF10A cells induces phenotypes of advanced breast cancer in 3D culture, including increased acinar size, cells present within the lumen, and cellular invasion. MCF10A cells transduced with R780 or R780:DEK (human DEK) were grown in Matrigel 3D culture (top and middle panels) to test for morphology or Matrigel-collagen 3D cultures to test for invasion (bottom panel). Immunofluorescence was performed on day 20 of culture and visualized with a Zeiss LSM510 scanning confocal microscope; the white size bar represents 10μm. Phase contrast images are also shown in the middle and bottom panels, and black arrows indicate invading cells in the bottom panel. The Golgi marker GM130 is used to mark cell apical-basal polarity, and white arrows highlight cells with de-regulated polarity. The percentage of cells with mislocalized GM130 per acinar structure is depicted below the images, which was calculated from triplicate experiments. (D) DEK and RON expression predict β-catenin expression levels in primary human breast cancer. Serial sections from two tissue microarrays of patient-derived breast infiltrating ductal carcinomas were stained by immunohistochemistry for DEK, RON, and β-catenin expression. Correlation was analyzed by χ2 testing. (E and F) Combined DEK and RON expression predict disease relapse in a cohort of 1000 un-medicated patients (p=0.00018) (E) and progression to distant metastatic disease in 1609 patients treated with systemic therapies (p=0.00098) (F). A meta-analysis of patient and gene expression data archived in Kaplan-Meier Plotter (www.kmplot.com) was performed to generate Kaplan-Meier curves. The numbers under each graph represent the number of patients at each time point.
To determine if DEK over-expression promoted tumorigenic phenotypes in human mammary cells similar to that observed in the mouse, we used 3D cultures of MCF10A immortalized human mammary epithelial cells transduced with control R780 vector or R780:DEK.29 This 3D human cell culture model closely mimics the structure of the mammary gland, with a spherical layer of polarized epithelium and a hollow lumen.46 DEK over-expression resulted in a hyperplastic phenotype, as evident by the enlarged acinar structures and filled lumens (Figure 6C, top and center panels). Wnt signaling is known to regulate cell and tissue polarity.47–49 Therefore, we assessed polarity in this three-dimensional context by staining for the apical surface marker, GM130. DEK over-expressing acini contained more cells with de- regulated polarity, as indicated by aberrant localization of GM130 (Figure 6C, top panel, white arrows and graph below). When the acini were cultured in a mix of Matrigel and collagen to promote invasive phenotypes, DEK over-expression led to the production of invasive laminin-V negative multi-cellular structures, which were not observed in control cells (Figure 6C, bottom panel and data not shown).
RON and DEK expression cooperate to promote β-catenin activity and disease progression in human primary breast cancers
Immunohistochemical staining of serial sections from two breast cancer microarrays was performed. Analyses were limited to infiltrating ductal carcinomas, and we determined that 54% (15 of 28) of samples expressed high levels of DEK while 46% (13 of 28) highly expressed RON. By themselves, the expression of each protein was suggestive of high β-catenin levels, indicating pathway activation (Figure S5A). When combined, 74% (31 of 42) of breast cancers expressed high levels of RON and/or DEK, which also was highly predictive of strong β-catenin staining (Figure 6D).
Finally, an in silico meta-analysis of patient and gene expression data archived in Kaplan-Meier Plotter (www.kmplot.com) was performed to assess the impact of DEK and RON expression on patient survival. In each case, high DEK expression was very predictive of poor patient outcome, the strength of the association was enhanced by adding RON expression (Figure S5B–D). Together, high DEK and RON expression were predictive of early relapse in breast cancer patients that did not receive systemic endocrine therapy or chemotherapy (Figure 6E). The combined markers also correlated with a higher risk for developing distant metastases in breast cancer patients treated with systemic therapies (Figure 6F) and with poor post-progression survival in all patients (Figure S5D).
Our results lead to a proposed model whereby elevated DEK expression results in the enhanced transcription and secretion of Wnt ligands that can promote cellular proliferation through a paracrine mechanism that is driven by β-catenin signaling (Figure 7). Furthermore, RON receptor tyrosine kinase activation is a novel mechanism for DEK up-regulation and, independently, can promote β-catenin stabilization and subsequent transcriptional activity (Figure 7).6 This combination of events converging upon the β-catenin pathway can then contribute to tumor growth and metastasis.
Figure 7. Model.
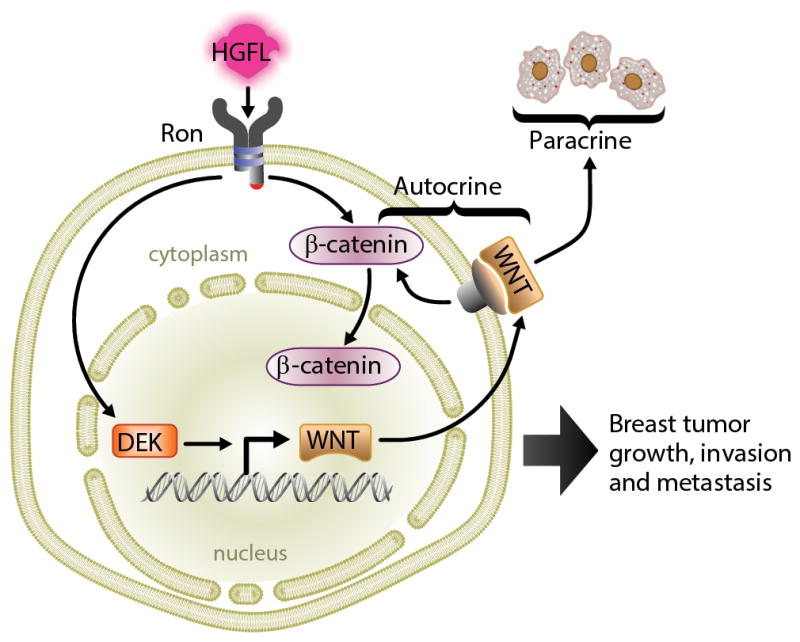
Ron stimulates Dek expression for sustained β-catenin signaling. Ron can directly activate intracellular β-catenin while Dek induces expression of Wnt ligands which are secreted into the tumor microenvironment to amplify β-catenin activity by autocrine and paracrine mechanisms.
Discussion
We previously published that Dek−/− mice developed fewer papillomas and were delayed in papilloma formation in a chemically-induced model of skin cancer.30 We provide the first report of the role of the DEK oncogene in a genetic murine model of malignant breast cancer. In this report, the absence of Dek did not prevent tumor formation and the mice ultimately developed similar numbers of tumors. However, Dek−/− mice show delayed tumor initiation and have significantly fewer proliferating cells in pre-neoplastic mammary glands. This is supported by the finding that proliferation could be rescued by exogenous Dek expression in independently generated cell lines from these Dek−/− tumors. Importantly, we also have observed the relationship between Dek expression and cell growth in an esophageal cancer cell line derived from a murine 4NQO chemical carcinogenesis model, indicating that this correlation is not limited to the MMTV-Ron model (Figure S6A).
In human cancers, RNAi-mediated DEK loss has been reported to cause apoptosis or senescence, as well as decreased proliferation in the surviving cells.29, 32, 34, 35, 50 Factors that determine the cellular response to DEK depletion are poorly understood, although p53, MCL-1, and p65 functionality have been implicated.18, 27, 50 The fact that Dek knockout mice are viable seems to be at odds with the findings in human cells. However, differentiated human keratinocytes are largely resistant to apoptosis and senescence after DEK depletion by shRNA expression. This suggests that the differentiated, largely quiescent tissues in the mouse may not be dramatically affected by the loss of Dek under controlled stress and pathogen-free conditions. It is of interest that mouse embryonic fibroblasts (MEFs) from Dek−/− mice are slightly less proliferative with elevated numbers of senescent cells under normal culture conditions compared to Dek+/+ MEFs, possibly due to increased baseline levels of DNA damage. However, knockout MEFs are significantly more senescent and apoptotic when stressed, such as treatment with etoposide.13 Therefore, we hypothesize that Dek/DEK expression promotes proliferation through Wnt signaling and ΔNp63 levels; however, the loss of Dek causes decreased mitogenic signaling as well as DNA damage which, when exacerbated by environmental stress and/or genetic background, leads to a dramatic apoptotic or senescent response.13, 29, 37
Molecular mechanism(s) through which DEK promotes proliferation are not well understood. Cell-free assays and immunoprecipitation experiments suggest that DEK may facilitate DNA replication.9, 14 DEK also promotes proliferation through ΔNp63 protein activities, but the effectors of this pathway remains unexplored.29, 51, 52 Our previous studies have shown that DEK expression correlates with proliferation by BrdU incorporation in xenograft tumors and organotypic raft cultures of skin, but the molecular drivers behind this finding were not identified.29, 52 We also published previously that DEK expression correlates with β-catenin activity in human breast cancer29 and we have observed this correlation in primary cells from human head and neck squamous cell carcinomas (HNSCCs; Figure S6B–C), thus supporting broad importance for the observed DEK-β-catenin signaling axis in solid tumors. Here, we connect our previous findings and identify a molecular mechanism through which DEK can promote proliferation and β-catenin activity – through the transcriptional regulation of Wnt4, Wnt7b, and Wnt10b. These three Wnts have been implicated in promoting oncogenic phenotypes and proliferation. In particular, Wnt10b is a known canonical Wnt ligand that prompts the stabilization and activation of β-catenin and can promote proliferation in triple- negative breast cancer.53 Wnt10b is an early marker of the mammary lineage, a crucial driver of mammary gland development, and supports mammary stem cell proliferation as suggested by regulation of mammary stem cell marker Sca-1.44, 54, 55 Wnt10b also is over-expressed in breast cancer cell lines and can induce transformation; furthermore MMTV-Wnt10b mice develop hyperplasia and excessive ductal branching, phenotypes which precede the development of adenocarcinomas.44, 45, 56 Wnt4 expression also supports basal mammary epithelial stem cell proliferation, promotes mammary gland development and branching during pregnancy, and is over-expressed in breast cancer cell lines, although it is not transforming by itself.56–58 Wnt7b is a non-canonical ligand whose up-regulation has been observed in other murine breast cancer models and was shown to promote epithelial proliferation in the lung.59–63 All three of these Wnts are known to be expressed in human breast tissue and were reported to be over- expressed in breast cancer.64 Additional work will be required to determine whether Dek- mediated transcription of Wnt ligands is due to the role of DEK in chromatin re-organization or as a transcriptional co-factor. 15, 16 Furthermore, we report DEK-mediated positive regulation of β-catenin in both breast cancer and HNSCCs, which suggests that this oncogenic mechanism may be relevant for several types of solid tumors.
In co-culturing experiments we found that Dek-expressing cells could promote the proliferation of neighboring Dek-deficient cells through a paracrine signaling mechanism. Importantly, inhibiting the secretion of Wnt ligands with the Porcupine inhibitor, IWP-2, dramatically reduced this pro-proliferative effect. This suggests that Dek expressing cells promote the proliferation of cells within the tumor microenvironment. It is also noteworthy that IWP-2 did not entirely eliminate the pro-proliferative effect observed in co-culture experiments, indicating that Dek expressing cells may be secreting other growth factors that drive the proliferation of neighboring cells or cell intrinsic factors may continue to promote proliferation in the presence of the inhibitor. Interestingly, this may explain why DEK is not expressed in every cell in primary invasive breast adenocarcinomas; only a fraction of the cells may need to express DEK in order to drive proliferation of the entire tumor.29 In addition, Wnt10b and Wnt4 drive mammary stem cell proliferation and Wnt/β-catenin activity is a well-known regulator of the normal and cancer stem cell populations. Future studies will determine if the positive effect of Dek expression on the size of the mouse and human breast cancer stem cell population is due to Wnt expression and β-catenin activation driving cancer stem cell proliferation.
This report highlights for the first time a role of Dek expression in an in vivo model of cancer metastasis. Although Dek loss did not prevent metastases, it significantly decreased metastatic tumor burden in the lungs and resulted in reduced liver metastases. We saw no correlation between Dek expression and the expression of Wnt target genes MMP2 and MMP9; however, this does not exclude other potential mechanisms. A possible alternative is that Dek may also regulate the expression of non-canonical Wnt ligands, like Wnt5a, which can activate the Rho/Rac planar cell polarity pathway to direct cell motility.65, 66 Interestingly, DEK loss was recently shown to negatively regulate Rho signaling in non-small cell lung cancer.67 In addition to lung metastases, we observed that Dek expression significantly promoted invasion in transwell assays in multiple murine breast cancer cell lines and in primary HNSCC cells (Figure S6D), which agrees with previous reports in human breast and lung cancer and indicates that DEK-mediated invasion is relevant to several cancer types.29, 32 Of interest, breast patient data associated high DEK expression with an increased risk for distant metastasis in previously node-negative patients. The overall finding is that while Dek is not required for metastasis, it promotes cellular invasion and the growth of metastatic tumors. Interestingly, Wnt signaling has also been implicated in cellular motility and cancer metastasis. In canine breast adenocarcinomas, Wnt7b was found to be up-regulated in metastatic cell lines isolated from the lungs compared to isogenic primary tumor cell lines.68 Wnt10b and Wnt4 are potent regulators of mammary gland branching, which leads us to hypothesize that they may be involved in DEK-mediated promotion of cancer cell motility when over-expressed in tumors.44, 58 We have already reported that DEK knockdown decreased invasion in vitro in human breast cancer cells, and invasion was rescued by the expression of a constitutively active β-catenin construct.29 This report, combined with our previous work, suggests that DEK-mediated activation of Wnt/β-catenin signaling, at least in part, promotes proliferation, invasion, and altered cell polarity.
Finally, we show that elevated DEK expression, partially due to RON activation, is relevant to human breast cancer. Exogenous expression of DEK in immortalized MCF10A cells grown in 3D culture induces tumorigenic phenotypes reminiscent of invasive adenocarcinomas. Furthermore, DEK expression can be induced by RON receptor activation with HGFL. This combination of RON and DEK expression are highly predictive of strong β-catenin staining by immunohistochemistry, disease relapse in patients not treated with systemic therapies, and distant metastases in patients that have received systemic therapies. This indicates that RON and DEK cooperate to promote breast cancer in mice and humans. Although the signaling events regulated by RON and DEK are not fully understood, both are known to activate Wnt/β-catenin signaling. Both also have been implicated in promoting the activity of other oncogenic signaling cascades linked to cancer progression, including the roles of DEK in regulating the function of p53 family members and DNA repair and of RON in promoting the activation of PI3K-Akt and Ras/MAPK pathways.13, 29, 52, 69 Combined, we propose that RON and DEK cooperate to drive breast cancer progression and relapse. Developing novel therapeutics that target these proteins could significantly slow tumor growth and limit the risk of metastasis or disease relapse, and may be particularly effective when combined with traditional therapies.
Materials and Methods
Cell Culture
T47D and MCF10A cells were obtained from the American Type Culture Collection and grown under recommended conditions. R7 cells are a murine mammary tumor line from a previously described MMTV-Ron mouse.3, 39 We established the remaining cell lines from mammary tumors that developed in the MMTV-Ron (Rontg) mice used in this study. Cell lines R7, RD258, and RD272 were generated from RontgDek+/+ tumors and cell lines RD147, RD219, RD238, and RD271 were from RontgDek−/− tumors. Briefly, tumor fragments were digested in 2 mg/ml collagenase in DMEM:F12 media for two hours at 37°C then washed six times in PBS with differential centrifugation to enrich for epithelial cells. Resulting organoids were grown in DMEM:F12 media supplemented with 10% FBS, 1% penicillin-streptomycin, 1% Fungizone, 2 mM L-glutamine, 5 μg/ml gentamicin, 10 ng/ml EGF, 10 μg/ml human recombinant insulin, 5 μg/ml transferrin, and 50 μM sodium selenite then passaged at least 20 times.
Cells were treated with 100 ng/ml recombinant HGFL protein (MSP, Cys 672 Ala; R&D Systems, Minneapolis, MN) for the indicated time points. To inhibit Wnt secretion, cells were treated with 10 μM IWP-2 (Sigma-Aldrich, St. Louis, MO) for 24 hours prior to analysis. For growth curves, cells were plated at equal density of 50,000 cells per well in 6-well plates, trypsinized, and counted in triplicate using a hemocytometer.
Three dimensional (3D) MCF10A cultures were generated and stained by immunofluorescence as previously described.46 MCF10A cells retrovirally transduced with R780 and R780:DEK also were previously described.29 Acinar structures were photographed at day 4 for invasive cultures (Matrigel and collagen) at 100x total magnification. Immunofluorescence was performed on day 20 of culture and visualized with a Zeiss LSM510 scanning confocal microscope; the white size bar represents 10μm.
Cells were transduced with retroviral constructs, R780 and R780:mDek containing a murine Dek construct, as described previously and sorted based on GFP expression30 or transduced with the lentiviral pLKO.1 constructs (Deksh represents pLKO.1_DEK832 in the Mission library; Sigma Aldrich and was previously described as “DEKsh2”29) and selected in 4 μg/ml puromycin. NTsh is a non-targeting control. All cell populations were analyzed within six passages of selection.
PCR
Total mRNA was isolated using Trizol extraction, then reverse transcribed using the Quantitect kit (Qiagen, Valencia, CA). cDNA was amplified with SYBR Green PCR master mix using an ABI-7500 quantitative PCR machine (Applied Biosystems, Carlsbad, CA) and analyzed using the ΔΔCt method. The following forward (F) and reverse (R) primer pairs were used at a concentration of 0.4 ng/μl each. Primers labeled with “m” were used for murine cells and those with an “h” denote human sequence.
mDek F: 5′-AACGTGGGTCAGTTCAGTGGC-3′
mDek R: 5′-TTCGCTGTTCACGCCTGACCT-3′
mActin F: 5′-GATATCGCTGCGCTGGTCGTC-3′
mActin R: 5′-ACCATCACACCCTGGTGCCTAG-3′
mWnt4 F: 5′-AGCAGGTGTGGCCTTTGCAGTGAC-3′
mWnt4 R: 5′-CGTTGTTGTGAAGATTCATGAGTGCC-3′
mWnt7b F: 5′-AGTGGATCTTTTACGTGTTTCTCTG-3′
mWnt7b R: 5′-CTGGTTGTAGTAGCCTTGCTTCTC-3′
mWnt10b F: 5′-AGAAGTTCTCTCGGGATTTCTTG-3′
mWnt10b R: 5′-CAAAGTAAACCAGCTCTCCAG-3′
hGAPDH F-5′-GGTCTCCTCTGACTTCAACA-3′
hGAPDH R-5′-ATACCAGGAAATGAGCTTGA-3′
hDEK F-5′-TGTTAAGAAAGCAGATAGCAGCACC-3′
hDEK R-5′-ATTAAAGGTTCATCATCTGAACTATCCTC-3′
Western blotting
Total protein was extracted with RIPA buffer and subjected to SDS-PAGE analysis prior to transfer to a PVDF membrane. Blots were probed with specific antibodies to DEK (BD Bioscience, San Jose, CA); Ron (C-20), Wnt10b [H70], and cyclin D1 [72-13G] (Santa Cruz, Dallas, TX); alpha-tubulin (Sigma), Actin C4 (gift of James Lessard, Cincinnati Children’s Hospital); Axin 2, MMP-2, and MMP-9 (Abcam, Cambridge, MA), cytokeratin 14 (Covance, Princeton, NJ), and GSK3β, phospho-ERK1/2, total ERK1/2, and TCF1 (Cell Signaling, Danvers, MA). Protein from primary mammary epithelial cells was collected from 18 week old nulliparous, non-tumor bearing Dek+/+ females by isolating the cells through collagenase digestion and differential centrifugation as described above immediately prior to the addition of lysis buffer. For secreted proteins, 3×106 cells were plated on 10cm dishes in complete media for 2 hours cells then washed in PBS and overlayed with media containing 1% FBS for 24 hours. Six milliliters of conditioned media was collected from each cell line and concentrated to 100 μl using Amicon Ultra-4 centrifugal filters with Ultracel-3 membrane (Millipore, Billerica, MA) then 2 μl was diluted into 18 μl of serum free media prior to western blotting. Western blots were analyzed by densitometry using Image J.70
Mice
Dek−/− mice (gift from Gerard Grosveld, St. Jude Children’s Research Hospital, Memphis, TN) were previously described and were backcrossed into an FVB/N background, which is the same background as the MMTV-Ron model.3, 30 MMTV-Ron male mice were bred to Dek+/− females then MMTV-Ron, Dek+/− male progeny were bred to Dek+/− females to generate RontgDek+/+ and RontgDek−/− females.3, 30 Females were continuously bred starting at six weeks old then monitored weekly for tumor development. Tumors were measured with digital calipers and volume was calculated as [(π/6)xLxW2].71, 72 Two hours before sacrifice, the mice were intraperitoneally injected with 600 μg of BrdU. At necropsy, the mice were weighed and the tumors were excised, measured, weighed, fixed in 4% paraformaldehyde, and embedded in paraffin. Portions of one tumor per mouse were used to generate novel RontgDek+/+ and RontgDek−/− cell lines (see above). Lungs and livers were examined for metastases as described previously.73
For xenograft studies, 5×105 cells suspended in PBS were injected into the inguinal mammary fat pads of nulliparous, 12-week old female athymic nude mice. Tumors were measured and collected as described above. Usage and handling of mice was performed with the approval of the Cincinnati Children’s Institutional Animal Care and Use Committee. All mice were housed in specific pathogen free housing with ad libitum access to food and water.
Immunohistochemistry
Paraformaldehyde fixed and paraffin embedded tissues were cut into 5 μm sections, subjected to sodium citrate antigen retrieval, and stained using the M.O.M. Peroxidase kit (Vector Labs, Burlingame, CA) and 3,3′-Diaminobenzidine (DAB). The following antibodies were used: DEK (1:60, BD Biosciences), BrdU (1:100, Molecular Probes, Invitrogen, Grand Island, NY), Wnt10b [H70] (1:50, Santa Cruz), MMP-2 and MMP-9 (1:100 and 1:200, respectively, Santa Cruz), cytokeratin-5 (1:200, Abcam), or activated β-catenin [8E7] (1:100, Millipore). Samples were counterstained with 0.1% Nuclear Fast Red (Poly Scientific, Bay Shore, NY) and preserved with Permount (Fisher Scientific, Pittsburgh, PA). Tissue microarrays were purchased from Imgenex (CBA2 and CBB2; Imgenex, San Diego, CA) for correlations of Ron, DEK, and β-catenin expression in human tissues and scored as previously described.6 For R7 xenograft tumors, staining intensity was quantified by the Threshold tool on Image J. The threshold for each section was set and normalized to each section’s respective total area of tissue as seen at 200x total magnification. Scores were calculated by multiplying the intensity score by the percentage of positive staining cells. The total number of cells counted per section ranged from 122 to 357. For activated β-catenin staining, cells were considered positive if the nucleus showed DAB staining.
Flow Cytometry
All flow cytometry was performed on a FACSCanto cytometer (BD Biosciences). Mouse cell surface marker staining was determined with the Lineage Cocktail kit and streptavidin-APC-Cy7 in combination with CD44-PE, CD49f-FITC, and CD24-APC antibodies (BD Biosciences). For co-culturing experiments, 2×105 cells of each genotype were plated in 60 mm dishes, grown overnight, then treated with 100 μM BrdU for 35 minutes before staining with the APC BrdU kit (BD Biosciences). BrdU incorporation was measured in GFP− and GFP+ cells.
Mammosphere Culture
Mammosphere cultures were performed and photographed as previously described.29 Image J was used to measure the radius and calculated sphere volume as (4/3)π*r3 for each mammosphere in triplicate experiments.
Invasion Assay
Matrigel transwell assays were used to determine invasive potential in vitro (BD Biosciences). Cells were seeded at low density in supplement-free media and complete media was placed into the bottom chamber as a chemoattractant. After 20 hours, invading cells were methanol-fixed and stained with Giemsa then manually counted. The data is represented as fold change relative to vector controls.
TOP/FOP Assay
Cells were transfected with 10 ng pRL-TK and 500ng of pSUPER(8x)TOPFlash or pSUPER(8x)FOPFlash using TransIT-LT1 reagent (Mirus Bio, Madison, WI) then assayed with the Dual-Luciferase Reporter Assay System (Promega, Madison, WI). Plasmids were a gift from Dr. Aaron Zorn (Cincinnati Children’s Hospital). Data is presented as the ratio of relative light units (RLUs) of TOPFlash to FOPFlash from triplicate experiments.
Kaplan-Meier Curves
Kaplan Meier plots were generated with KM plotter for Breast Cancer, 2014 version (www.kmplot.com), which is a publicly available online database of archived gene expression and patient survival data.74 We used the multigene classifier and mean expression of probes, 200934_at (DEK) and 205455_at (RON), and did not restrict analysis by subtype. We analyzed relapse free survival (RFS) in systemically untreated patients and distant metastasis free survival (DMFS) in patients treated with endocrine therapies or chemotherapies.
Statistics
Mouse tumor-free survival was analyzed using the Log-rank test. The χ2 test was used to identify correlations with protein expression using tissue microarrays. For all other experiments, an unpaired two-tailed Student’s t-test was used, unless otherwise noted. Error bars depict standard error of data collected from at least three experiments. Significance was set at p<0.05. One asterisk (*) indicates p<0.05, two asterisks (**) indicates p<0.01, and three asterisks (***) indicates p<0.001.
Supplementary Material
Acknowledgments
We acknowledge James Lessard and Gerard Grosveld for reagents, Kathryn Wikenheiser-Brokamp for technical assistance, and the Research Flow Cytometry Core at Cincinnati Children’s, supported by NIH AR-47363. L.M.P.V. is supported by the Ride Cincinnati Foundation for Breast Cancer Research in honor of Marlene Harris and Public Health Service grants F32CA139931, T32HL091805, and K12HD051953. Funding also was provided by Public Health Services grants R01CA116316 (S.I.W.), R01HL875109 (J.A.C), T32ES007250 (A.K.A.), T32CA117846-07 (P.K.W), and T32CA11784 (S.E.W and N.M.B.), Department of Defense grant W81XWH-12-1-0194 (S.I.W. and S.E.W), and the Veteran’s Administration grant BX000803 (S.E.W). J.S.-L. is supported by the Government of the Spanish Junta de Andalucia.
Footnotes
Conflicts of Interest The authors declare no conflicts of interest.
References
- 1.ACS. Breast Cancer Facts & Figures 2011–2012. Atlanta: American Cancer Society, Inc; 2011. [Google Scholar]
- 2.Maggiora P, Marchio S, Stella MC, Giai M, Belfiore A, De Bortoli M, et al. Overexpression of the RON gene in human breast carcinoma. Oncogene. 1998;16:2927–33. doi: 10.1038/sj.onc.1201812. [DOI] [PubMed] [Google Scholar]
- 3.Zinser GM, Leonis MA, Toney K, Pathrose P, Thobe M, Kader SA, et al. Mammary-specific Ron receptor overexpression induces highly metastatic mammary tumors associated with beta-catenin activation. Cancer Res. 2006;66:11967–74. doi: 10.1158/0008-5472.CAN-06-2473. [DOI] [PubMed] [Google Scholar]
- 4.Zhao H, Chen MS, Lo YH, Waltz SE, Wang J, Ho PC, et al. The Ron receptor tyrosine kinase activates c-Abl to promote cell proliferation through tyrosine phosphorylation of PCNA in breast cancer. Oncogene. 2013 doi: 10.1038/onc.2013.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Danilkovitch-Miagkova A. Oncogenic signaling pathways activated by RON receptor tyrosine kinase. Curr Cancer Drug Targets. 2003;3:31–40. doi: 10.2174/1568009033333745. [DOI] [PubMed] [Google Scholar]
- 6.Wagh PK, Gray JK, Zinser GM, Vasiliauskas J, James L, Monga SP, et al. beta-Catenin is required for Ron receptor-induced mammary tumorigenesis. Oncogene. 2011;30:3694–704. doi: 10.1038/onc.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leonis MA, Thobe MN, Waltz SE. Ron-receptor tyrosine kinase in tumorigenesis and metastasis. Future Oncol. 2007;3:441–8. doi: 10.2217/14796694.3.4.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Privette Vinnedge LM, Kappes F, Nassar N, Wells SI. Stacking the DEK: from chromatin topology to cancer stem cells. Cell Cycle. 2013;12:51–66. doi: 10.4161/cc.23121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alexiadis V, Waldmann T, Andersen J, Mann M, Knippers R, Gruss C. The protein encoded by the proto-oncogene DEK changes the topology of chromatin and reduces the efficiency of DNA replication in a chromatin-specific manner. Genes Dev. 2000;14:1308–12. [PMC free article] [PubMed] [Google Scholar]
- 10.Campillos M, Garcia MA, Valdivieso F, Vazquez J. Transcriptional activation by AP-2alpha is modulated by the oncogene DEK. Nucleic Acids Res. 2003;31:1571–5. doi: 10.1093/nar/gkg247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGarvey T, Rosonina E, McCracken S, Li Q, Arnaout R, Mientjes E, et al. The acute myeloid leukemia-associated protein, DEK, forms a splicing-dependent interaction with exon-product complexes. J Cell Biol. 2000;150:309–20. doi: 10.1083/jcb.150.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soares LM, Zanier K, Mackereth C, Sattler M, Valcarcel J. Intron removal requires proofreading of U2AF/3′ splice site recognition by DEK. Science. 2006;312:1961–5. doi: 10.1126/science.1128659. [DOI] [PubMed] [Google Scholar]
- 13.Kavanaugh GM, Wise-Draper TM, Morreale RJ, Morrison MA, Gole B, Schwemberger S, et al. The human DEK oncogene regulates DNA damage response signaling and repair. Nucleic Acids Res. 2011;39:7465–76. doi: 10.1093/nar/gkr454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Decaillet C, et al. FANCD2 Binds MCM Proteins and Controls Replisome Function upon Activation of S Phase Checkpoint Signaling. Mol Cell. 2013;51:678–90. doi: 10.1016/j.molcel.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 15.Kappes F, Waldmann T, Mathew V, Yu J, Zhang L, Khodadoust MS, et al. The DEK oncoprotein is a Su(var) that is essential to heterochromatin integrity. Genes Dev. 2011;25:673–8. doi: 10.1101/gad.2036411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawatsubashi S, Murata T, Lim J, Fujiki R, Ito S, Suzuki E, et al. A histone chaperone, DEK, transcriptionally coactivates a nuclear receptor. Genes Dev. 2010;24:159–70. doi: 10.1101/gad.1857410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim DW, Kim JY, Choi S, Rhee S, Hahn Y, Seo SB. Transcriptional regulation of 1-cys peroxiredoxin by the proto-oncogene protein DEK. Mol Med Report. 2010;3:877–81. doi: 10.3892/mmr.2010.346. [DOI] [PubMed] [Google Scholar]
- 18.Liu K, Feng T, Liu J, Zhong M, Zhang S. Silencing of the DEK gene induces apoptosis and senescence in CaSki cervical carcinoma cells via the up-regulation of NF-kappaB p65. Biosci Rep. 2012;32:323–32. doi: 10.1042/BSR20100141. [DOI] [PubMed] [Google Scholar]
- 19.Sammons M, Wan SS, Vogel NL, Mientjes EJ, Grosveld G, Ashburner BP. Negative regulation of the RelA/p65 transactivation function by the product of the DEK proto-oncogene. J Biol Chem. 2006;281:26802–12. doi: 10.1074/jbc.M600915200. [DOI] [PubMed] [Google Scholar]
- 20.Karam M, Thenoz M, Capraro V, Robin JP, Pinatel C, Lancon A, et al. Chromatin Redistribution of the DEK Oncoprotein Represses hTERT Transcription in Leukemias. Neoplasia. 2014;16:21–30. doi: 10.1593/neo.131658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gamble MJ, Fisher RP. SET and PARP1 remove DEK from chromatin to permit access by the transcription machinery. Nat Struct Mol Biol. 2007;14:548–55. doi: 10.1038/nsmb1248. [DOI] [PubMed] [Google Scholar]
- 22.Koleva RI, Ficarro SB, Radomska HS, Carrasco-Alfonso MJ, Alberta JA, Webber JT, et al. C/EBPalpha and DEK coordinately regulate myeloid differentiation. Blood. 2012;119:4878–88. doi: 10.1182/blood-2011-10-383083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mor-Vaknin N, Kappes F, Dick AE, Legendre M, Damoc C, Teitz-Tennenbaum S, et al. DEK in the synovium of patients with juvenile idiopathic arthritis: characterization of DEK antibodies and posttranslational modification of the DEK autoantigen. Arthritis Rheum. 2011;63:556–67. doi: 10.1002/art.30138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mor-Vaknin N, Punturieri A, Sitwala K, Faulkner N, Legendre M, Khodadoust MS, et al. The DEK nuclear autoantigen is a secreted chemotactic factor. Mol Cell Biol. 2006;26:9484–96. doi: 10.1128/MCB.01030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Datta A, Adelson ME, Mogilevkin Y, Mordechai E, Sidi AA, Trama JP. Oncoprotein DEK as a tissue and urinary biomarker for bladder cancer. BMC Cancer. 2011;11:234. doi: 10.1186/1471-2407-11-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saha AK, Kappes F, Mundade A, Deutzmann A, Rosmarin DM, Legendre M, et al. Intercellular trafficking of the nuclear oncoprotein DEK. Proc Natl Acad Sci U S A. 2013;110:6847–52. doi: 10.1073/pnas.1220751110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khodadoust MS, Verhaegen M, Kappes F, Riveiro-Falkenbach E, Cigudosa JC, Kim DS, et al. Melanoma proliferation and chemoresistance controlled by the DEK oncogene. Cancer Res. 2009;69:6405–13. doi: 10.1158/0008-5472.CAN-09-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kondoh N, Wakatsuki T, Ryo A, Hada A, Aihara T, Horiuchi S, et al. Identification and characterization of genes associated with human hepatocellular carcinogenesis. Cancer Res. 1999;59:4990–6. [PubMed] [Google Scholar]
- 29.Privette Vinnedge LM, McClaine R, Wagh PK, Wikenheiser-Brokamp KA, Waltz SE, Wells SI. The human DEK oncogene stimulates beta-catenin signaling, invasion and mammosphere formation in breast cancer. Oncogene. 2011;30:2741–52. doi: 10.1038/onc.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wise-Draper TM, Mintz-Cole RA, Morris TA, Simpson DS, Wikenheiser-Brokamp KA, Currier MA, et al. Overexpression of the cellular DEK protein promotes epithelial transformation in vitro and in vivo. Cancer Res. 2009;69:1792–9. doi: 10.1158/0008-5472.CAN-08-2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.von Lindern M, Fornerod M, van Baal S, Jaegle M, de Wit T, Buijs A, et al. The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia-specific dek-can mRNA. Mol Cell Biol. 1992;12:1687–97. doi: 10.1128/mcb.12.4.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shibata T, Kokubu A, Miyamoto M, Hosoda F, Gotoh M, Tsuta K, et al. DEK oncoprotein regulates transcriptional modifiers and sustains tumor initiation activity in high-grade neuroendocrine carcinoma of the lung. Oncogene. 2010;29:4671–4681. doi: 10.1038/onc.2010.217. [DOI] [PubMed] [Google Scholar]
- 33.Carro MS, Spiga FM, Quarto M, Di Ninni V, Volorio S, Alcalay M, et al. DEK Expression is controlled by E2F and deregulated in diverse tumor types. Cell Cycle. 2006;5:1202–7. doi: 10.4161/cc.5.11.2801. [DOI] [PubMed] [Google Scholar]
- 34.Privette Vinnedge LM, Ho SM, Wikenheiser-Brokamp KA, Wells SI. The DEK Oncogene Is a Target of Steroid Hormone Receptor Signaling in Breast Cancer. PLoS One. 2012;7:e46985. doi: 10.1371/journal.pone.0046985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wise-Draper TM, Allen HV, Thobe MN, Jones EE, Habash KB, Munger K, et al. The human DEK proto-oncogene is a senescence inhibitor and an upregulated target of high-risk human papillomavirus E7. J Virol. 2005;79:14309–17. doi: 10.1128/JVI.79.22.14309-14317.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evans AJ, Gallie BL, Jewett MA, Pond GR, Vandezande K, Underwood J, et al. Defining a 0. 5-mb region of genomic gain on chromosome 6p22 in bladder cancer by quantitative-multiplex polymerase chain reaction. Am J Pathol. 2004;164:285–93. doi: 10.1016/S0002-9440(10)63118-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adams AK, Hallenbeck GE, Casper KA, Patil YJ, Wilson KM, Kimple RJ, et al. DEK promotes HPV-positive and -negative head and neck cancer cell proliferation. Oncogene. 2014;0:1. doi: 10.1038/onc.2014.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanden C, Ageberg M, Petersson J, Lennartsson A, Gullberg U. Forced expression of the DEK-NUP214 fusion protein promotes proliferation dependent on upregulation of mTOR. BMC Cancer. 2013;13:440. doi: 10.1186/1471-2407-13-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McClaine RJ, Marshall AM, Wagh PK, Waltz SE. Ron receptor tyrosine kinase activation confers resistance to tamoxifen in breast cancer cell lines. Neoplasia. 2010;12:650–8. doi: 10.1593/neo.10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, et al. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993–7. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- 41.Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, et al. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–8. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- 42.Ma J, Lanza DG, Guest I, Uk-Lim C, Glinskii A, Glinsky G, et al. Characterization of mammary cancer stem cells in the MMTV-PyMT mouse model. Tumour Biol. 2012;33:1983–96. doi: 10.1007/s13277-012-0458-4. [DOI] [PubMed] [Google Scholar]
- 43.Wu B, Crampton SP, Hughes CC. Wnt signaling induces matrix metalloproteinase expression and regulates T cell transmigration. Immunity. 2007;26:227–39. doi: 10.1016/j.immuni.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lane TF, Leder P. Wnt-10b directs hypermorphic development and transformation in mammary glands of male and female mice. Oncogene. 1997;15:2133–44. doi: 10.1038/sj.onc.1201593. [DOI] [PubMed] [Google Scholar]
- 45.Miyoshi K, Rosner A, Nozawa M, Byrd C, Morgan F, Landesman-Bollag E, et al. Activation of different Wnt/beta-catenin signaling components in mammary epithelium induces transdifferentiation and the formation of pilar tumors. Oncogene. 2002;21:5548–56. doi: 10.1038/sj.onc.1205686. [DOI] [PubMed] [Google Scholar]
- 46.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–68. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 47.Habib SJ, Chen BC, Tsai FC, Anastassiadis K, Meyer T, Betzig E, et al. A localized Wnt signal orients asymmetric stem cell division in vitro. Science. 2013;339:1445–8. doi: 10.1126/science.1231077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karner C, Wharton KA, Carroll TJ. Apical-basal polarity, Wnt signaling and vertebrate organogenesis. Semin Cell Dev Biol. 2006;17:214–22. doi: 10.1016/j.semcdb.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 49.Gao B. Wnt regulation of planar cell polarity (PCP) Curr Top Dev Biol. 2012;101:263–95. doi: 10.1016/B978-0-12-394592-1.00008-9. [DOI] [PubMed] [Google Scholar]
- 50.Wise-Draper TM, Allen HV, Jones EE, Habash KB, Matsuo H, Wells SI. Apoptosis inhibition by the human DEK oncoprotein involves interference with p53 functions. Mol Cell Biol. 2006;26:7506–19. doi: 10.1128/MCB.00430-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res. 2004;2:371–86. [PubMed] [Google Scholar]
- 52.Wise-Draper TM, Morreale RJ, Morris TA, Mintz-Cole RA, Hoskins EE, Balsitis SJ, et al. DEK proto-oncogene expression interferes with the normal epithelial differentiation program. Am J Pathol. 2009;174:71–81. doi: 10.2353/ajpath.2009.080330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wend P, Runke S, Wend K, Anchondo B, Yesayan M, Jardon M, et al. WNT10B/beta-catenin signalling induces HMGA2 and proliferation in metastatic triple-negative breast cancer. EMBO Mol Med. 2013;5:264–79. doi: 10.1002/emmm.201201320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Veltmaat JM, Van Veelen W, Thiery JP, Bellusci S. Identification of the mammary line in mouse by Wnt10b expression. Dev Dyn. 2004;229:349–56. doi: 10.1002/dvdy.10441. [DOI] [PubMed] [Google Scholar]
- 55.Miranda-Carboni GA, Krum SA, Yee K, Nava M, Deng QE, Pervin S, et al. A functional link between Wnt signaling and SKP2-independent p27 turnover in mammary tumors. Genes Dev. 2008;22:3121–34. doi: 10.1101/gad.1692808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Benhaj K, Akcali KC, Ozturk M. Redundant expression of canonical Wnt ligands in human breast cancer cell lines. Oncol Rep. 2006;15:701–7. [PubMed] [Google Scholar]
- 57.Meier-Abt F, Milani E, Roloff T, Brinkhaus H, Duss S, Meyer DS, et al. Parity induces differentiation and reduces Wnt/Notch signaling ratio and proliferation potential of basal stem/progenitor cells isolated from mouse mammary epithelium. Breast Cancer Res. 2013;15:R36. doi: 10.1186/bcr3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robinson GW, Hennighausen L, Johnson PF. Side-branching in the mammary gland: the progesterone-Wnt connection. Genes Dev. 2000;14:889–94. [PubMed] [Google Scholar]
- 59.Shimizu H, Julius MA, Giarre M, Zheng Z, Brown AM, Kitajewski J. Transformation by Wnt family proteins correlates with regulation of beta-catenin. Cell Growth Differ. 1997;8:1349–58. [PubMed] [Google Scholar]
- 60.Huguet EL, McMahon JA, McMahon AP, Bicknell R, Harris AL. Differential expression of human Wnt genes 2, 3, 4, and 7B in human breast cell lines and normal and disease states of human breast tissue. Cancer Res. 1994;54:2615–21. [PubMed] [Google Scholar]
- 61.Kuorelahti A, Rulli S, Huhtaniemi I, Poutanen M. Human chorionic gonadotropin (hCG) up-regulates wnt5b and wnt7b in the mammary gland, and hCGbeta transgenic female mice present with mammary Gland tumors exhibiting characteristics of the Wnt/beta-catenin pathway activation. Endocrinology. 2007;148:3694–703. doi: 10.1210/en.2007-0249. [DOI] [PubMed] [Google Scholar]
- 62.Yin YJ, Katz V, Salah Z, Maoz M, Cohen I, Uziely B, et al. Mammary gland tissue targeted overexpression of human protease-activated receptor 1 reveals a novel link to beta-catenin stabilization. Cancer Res. 2006;66:5224–33. doi: 10.1158/0008-5472.CAN-05-4234. [DOI] [PubMed] [Google Scholar]
- 63.Rajagopal J, Carroll TJ, Guseh JS, Bores SA, Blank LJ, Anderson WJ, et al. Wnt7b stimulates embryonic lung growth by coordinately increasing the replication of epithelium and mesenchyme. Development. 2008;135:1625–34. doi: 10.1242/dev.015495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Howe LR, Brown AM. Wnt signaling and breast cancer. Cancer Biol Ther. 2004;3:36–41. doi: 10.4161/cbt.3.1.561. [DOI] [PubMed] [Google Scholar]
- 65.Florian MC, Nattamai KJ, Dorr K, Marka G, Uberle B, Vas V, et al. A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell ageing. Nature. 2013;503:392–6. doi: 10.1038/nature12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schlessinger K, Hall A, Tolwinski N. Wnt signaling pathways meet Rho GTPases. Genes Dev. 2009;23:265–77. doi: 10.1101/gad.1760809. [DOI] [PubMed] [Google Scholar]
- 67.Wang J, Sun L, Yang M, Luo W, Gao Y, Liu Z, et al. DEK depletion negatively regulates Rho/ROCK/MLC pathway in non-small cell lung cancer. J Histochem Cytochem. 2013;61:510–21. doi: 10.1369/0022155413488120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krol M, Polanska J, Pawlowski KM, Turowski P, Skierski J, Majewska A, et al. Transcriptomic signature of cell lines isolated from canine mammary adenocarcinoma metastases to lungs. J Appl Genet. 2010;51:37–50. doi: 10.1007/BF03195709. [DOI] [PubMed] [Google Scholar]
- 69.Wang MH, Zhang R, Zhou YQ, Yao HP. Pathogenesis of RON receptor tyrosine kinase in cancer cells: activation mechanism, functional crosstalk, and signaling addiction. J Biomed Res. 2013;27:345–356. doi: 10.7555/JBR.27.20130038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Euhus DM, Hudd C, LaRegina MC, Johnson FE. Tumor measurement in the nude mouse. J Surg Oncol. 1986;31:229–34. doi: 10.1002/jso.2930310402. [DOI] [PubMed] [Google Scholar]
- 72.Tomayko MM, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989;24:148–54. doi: 10.1007/BF00300234. [DOI] [PubMed] [Google Scholar]
- 73.Peace BE, Toney-Earley K, Collins MH, Waltz SE. Ron receptor signaling augments mammary tumor formation and metastasis in a murine model of breast cancer. Cancer Res. 2005;65:1285–93. doi: 10.1158/0008-5472.CAN-03-3580. [DOI] [PubMed] [Google Scholar]
- 74.Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123:725–31. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.