

Abstract
Successful embryonic implantation is the result of a receptive endometrium, a functional embryo at the blastocyst stage and a synchronized dialog between maternal and embryonic tissues. Successful implantation requires the endometrium to undergo steroid-dependent change during each menstrual cycle, exhibiting a short period of embryonic receptivity known as the window of implantation. The term “endometrial receptivity” was introduced to define the state of the endometrium during the window of implantation. It refers to the ability of the endometrium to undergo changes that will allow the blastocyst to attach, penetrate, and induce localized changes in the endometrial stroma. These changes are metabolically demanding, and glucose metabolism has been proven to be important for the preparation of the endometrium for embryo implantation. Obesity and polycystic ovary syndrome (PCOS) represent 2 common metabolic disorders that are associated with subfertility. The aim of this review is to summarize the effect of obesity and PCOS on endometrial receptivity at the time of implantation. Focus will be on metabolic alterations that regulate decidualization, including glucose metabolism, hyperinsulinemia, and hyperandrogenism.
Keywords: endometrial receptivity, implantation, PCOS, obesity, metabolism
Introduction
Successful embryonic implantation is the result of a receptive endometrium, a functional embryo at the blastocyst stage and a synchronized dialog between maternal and embryonic tissues.1,2 This phenomenon requires the endometrium to undergo steroid-dependent change during each menstrual cycle, exhibiting a short period of embryonic receptivity known as the “window of implantation”. During this progesterone-dominated phase, endometrial stromal cell differentiation occurs, where endometrial stromal cells undergo morphological and functional changes to differentiate into decidual cells that are receptive to embryonic implantation. This endometrial decidualization is characterized by changes in the endometrial cytoskeleton, glucose metabolism, and mediation of immune cells, cytokines, growth factors, chemokines, and adhesion molecules.3,4
The term “endometrial receptivity” was introduced to define the state of the endometrium during the window of implantation. It refers to the ability of the endometrium to undergo changes that will allow the blastocyst to attach, penetrate, and induce localized changes in the stroma.3 Identifying the specific molecular milieu that the blastocyst encounters when entering the uterine cavity provides clues as to the individual components released by the endometrium that may be critical for the maturation of the blastocyst and implantation.5 Thus, endometrial receptivity has been the focus of reproductive researchers in hope to identify defects that could be contributing to implantation failure. Researchers have explored various signaling proteins, interleukins, receptors, ligands, cytokines, and growth factors, unveiling multiple “markers of endometrial receptivity” such as prolactin, interleukin 15, and somatostatin that help identify differences among fertile and subfertile populations.5–7
The aforementioned dynamic changes that occur in the endometrium during the menstrual cycle are metabolically demanding,8 and glucose metabolism has been proven to be important for the preparation of the endometrium for embryo implantation.8–10 Specifically, it has been shown that endometrial stromal cell decidualization is dependent on increasing expression of glucose and facilitative glucose transporter (GLUT) 1.11 In addition, hyperinsulinemia and insulin resistance have been shown to inhibit markers of endometrial receptivity such as insulin-like growth factor (IGF) type 1 receptors, and cause hyperandrogenism.12–14 As decidualization is hormonally regulated, hyperandrogenism can effect proper decidualization, influencing differentiation pathways and affecting steroid-dependent metabolic enzymes.15
Obesity and polycystic ovary syndrome (PCOS) represent 2 common metabolic disorders that are associated with subfertility. According to the World Health Organization, in 2010, they estimated that 76.7% of females in the United States older than 15 years were considered overweight or obese (body mass index [BMI] > 25). This epidemic has reproductive implications, as obesity is associated with subfertility, increased miscarriage rates and, despite assisted reproductive technology, decreased implantation, pregnancy, and live birth rates.16–20 Specifically, obese women (BMI > 30) are 3 times more likely to suffer infertility than women with a normal BMI. In addition, obese women experience impaired fecundity, both in natural and assisted conception cycles.21 Studies have suggested that despite embryo quality, a significant reduction in implantation, pregnancy, and live birth rates have been identified as BMI increased, suggesting that the endometrium may be the main contributor in the impaired reproductive outcome of these patients.19 A recent clinical study from Spain demonstrates in a cohort of over 9,500 cycles that recipient obesity impairs the reproductive outcomes of oocyte donation from normal weight donors, suggesting a uterine receptivity explanation for the implantation failure and poor reproductive outcomes in obese women. Although the early clinical work in humans receiving donor oocytes reported conflicting results in regard to implantation, miscarriage, and pregnancy outcomes22–24 sparking an intense discussion,25 recent studies have confirmed a strong correlation between poor outcomes and recipient obesity.26 Furthermore, significant changes in uterine receptivity and markers of decidualization and implantation have been discovered in obese women, suggesting molecular mechanisms of endometrial dysfunction.27–30 Despite the fact that this has been disputed,22 the idea that obese patients have defective endometrial receptivity leading to altered implantation and live birth rates is probable.
PCOS is the most common endocrine disorder in women, with prevalence as high as 15% when diagnosed by Rotterdam criteria.31 Obesity complicates the diagnosis of PCOS with a reported 61% to 76% of patients with PCOS considered obese (BMI > 30) in the United States and Australia.31 Women with PCOS are subfertile, and this may be heightened by the effect of obesity, metabolic, inflammatory, and endocrine abnormalities on ovulatory function, oocyte quality, and endometrial receptivity.31 Miscarriage rates in patients with PCOS are generally considered to be comparable with those in other subfertile populations,31 with a miscarriage rate as high as 30% to 50% of all conceptions. Even when ovulation is pharmacologically restored, anovulatory patients have decreased cumulative pregnancy rates when compared with other subfertile populations32 and exhibit a higher rate of implantation failure and spontaneous miscarriage.30 Additionally, it is well established that the endometrium in patients with PCOS is dysfunctional, as women with PCOS are anovulatory or oligo-ovulatory and have suboptimal regulation by estrogen and suboptimal or absent progesterone, which puts them at increased risk for the development of endometrial hyperplasia and cancer.14,33–35 Gene expression studies to study this “progesterone resistance” in patients with PCOS show reduced expression of multiple endometrial receptivity-associated genes which may contribute to the observed subfertility. Additionally, genes associated with cellular proliferation and tumor suppressor genes were altered in PCOS endometrium, corresponding to phenotypes of endometrial hyperplasia and cancer.36,37 Investigation of endometrial receptivity in patients with PCOS at the time of implantation has led to the discovery of altered cellular endometrial milieu that is likely associated with subfertility and increased miscarriage rates. For example, patients with PCOS have dysregulated expression of markers of uterine receptivity, such as decreased expression of αvβ3 integrin, HOXA-10, and IGF binding protein 1 (IGFBP-1).1,38–40 In addition, the endometrium in women with PCOS have been found to overexpress androgen receptors and fail to downregulate estrogen receptor-α in the window of implantation.1,39,41
Taken together, obesity and PCOS alter endometrial receptivity. In addition, Bellver et al investigated the endometrial transcriptome during the window of implantation using gene expression microarray analysis and found that obese women have altered gene expression that is worsened by the diagnosis of PCOS.30 The aim of this review is to summarize the effect of obesity and PCOS on endometrial receptivity at the time of implantation. Focus will be on metabolic alterations that effect on the endometrium, including glucose metabolism, hyperinsulinemia, and hyperandrogenism.
Glucose Metabolism
Multiple studies have confirmed that glucose metabolism is important for endometrial preparation for embryonic implantation. Glucose metabolism is especially important in support of endometrial decidualization—the differentiation of the functionalis layer into the decidua rendering the endometrium capable of supporting a conceptus.8,9,42,43 The initial step in glucose utilization is cellular uptake, which is mediated in human and murine uteri by facilitative glucose transporters, commonly referred to as GLUTs, however, now known as the SLC2A family,9 specifically SLC2A 1,3,4, and 8. All SLC2As have shared characteristics but differ in tissue localization kinetics and substrate specificity.44
In murine endometrial stroma, expression of SLC2A1 has been found to increase throughout the process of decidualization in vitro and in vivo.11 Similarly, SLC2A3 is expressed in mouse and human uterine stroma and decidua and is a high-affinity GLUT, providing early nutritional support to the implanting blastocyst.9
SLC2A4 is an insulin-dependent GLUT.9 Insulin acts through the insulin and IGF-1 receptor to signal for SLC2A4 translocation to the cell surface in insulin-sensitive tissues such as muscle and fat.42 In rat skeletal muscle, defects in insulin-stimulated SLC2A4 translocation is responsible for the insulin resistance seen in obesity and type-2 diabetes, and expression of SLC2A4 is altered in adipose tissue in obesity and insulin-resistant states.9,45 The presence of SLC2A4 in the uterus has been contradictory. Initially, in 1999, expression of SLC2A4 was observed in rat uteri, and concentrations were far below those seen in rat skeletal muscle.46 In 2004, SLC2A4 was proven to be present in human endometrium based on messenger RNA (mRNA) expression, and immunohistochemical evidence confirmed it to be exclusively located in endometrial epithelial cells.47,48 Recent studies have quantified the presence of SLC2A4 in murine and human endometrial stromal cells and found it to be 100–1000-fold lower than other abundant SLC2As in rodents and to be below detection levels in human endometrial stroma.42 Importantly, some of the prior studies do not differentiate between the presence of SLC2A4 at the endometrium epithelium and the presence of that at the stroma. Thus, as our group has shown that stromal expression of SLC2A4 is low and, sometimes, below detection thresholds in human and rodents, and in 2004, immunohistochemical evidence was present only in the epithelial cells, the variation in reporting cell type and low detectable levels may explain the contradictory reports.
Most recently, in 2012, expression of SLC2A4 in human obese patients with PCOS was investigated. As SLC2A4 is insulin dependent and obesity and PCOS are insulin-resistant states, Zhai et al hypothesized that reduction of endometrial SLC2A4 leads to endometrial insulin resistance and may impair endometrial metabolism.49 Further, they investigated if metformin, an insulin sensitizer, would improve endometrial insulin resistance as assessed by endometrial mRNA expression and immunohistochemistry. They utilized endometrial biopsies and confirmed the presence of SLC2A4 protein and mRNA expression in human endometrium, finding increased expression in endometrial epithelial cells and little expression in stromal cells—shedding light onto the aforementioned controversy. They noted a statistically significant decrease in the expression of SLC2A4 mRNA and protein in obese patients with PCOS compared with that in obese control patients, which was significantly improved after 3 months of treatment with Metformin. In addition, expression of SLC2A4 was negatively correlated with homeostasis model assessment insulin resistance. Thus, they infer that the decrease in the expression of SLC2A4 in obese patients with PCOS is likely due to endometrial insulin resistance with the reduction of expression of SLC2A4, decreasing glucose transport, leading to abnormal cellular glucose utilization and, ultimately, abnormal endometrial differentiation and embryo implantation.49 The authors were unable to differentiate the effects of obesity from PCOS from normal weight women, as all participants enrolled (including controls) were obese. Their findings agree with Mioni et al, who, in 2004, concluded that SLC2A4 endometrial epithelial expression seems to be regulated by body weight and insulin.47 In the Mioni et al study, they found that SLC2A4 was significantly decreased in hyperinsulinemic patients with PCOS when compared to noromoinsulinemic patients with PCOS or controls. They also found that in normoinsulinemic patients with PCOS, expression of SLC2A4 was significantly decreased only in obese patients when compared with that in controls, and no difference was seen in lean normoinsulenimic patients with PCOS compared with that in controls.47 These findings agree with Mozzanga et al, who, in 2004, found that expression of SLC2A4 was significantly lower in obese patients with PCOS when compared with that in lean patients with PCOS and controls, and lean patients with PCOS and controls showed similar values.48 Thus, obesity and insulin seem to independently induce effects on endometrial epithelial cell expression of SLC2A4, more so than the hyperandrogenic state of PCOS. As we know, proper implantation requires a decidualized endometrium, and the full components of a receptive endometrium include the luminal epithelium, which undergoes apical surface specialization and expresses cell adhesion molecules that permit adherence of the blastocyst, glandular epithelial cells that secrete substances that support blastocyst development, decidual cells, large granular lymphocytes that modulate trophoblastic function, and stromal extracellular matrix that facilitates trophoblastic invasion.50 Further research can focus on using whole endometrial biopsies and investigating the effects of obesity and expression of SLCA4, as well as implantation.
Expression of SLC2A8 has been found in the murine and human uterus to dramatically increase during endometrial stromal decidualization, with the mRNA copy number increasing with in vitro decidualization.8,42 In contrast to SLC2A4, which is shuttled between intercellular storage compartments and the cell surface in response to a stimulus such as insulin or high glucose, SLC2A8 is located entirely intracellularly, except in blastocysts, where it translocates to the plasma membrane in response to insulin stimulation.9,44 It has been hypothesized that due to its intracellular location, SLC2A8 may be localized to the endoplasmic reticulum and play a role in providing the glucose necessary for glycosylation of proteins during endometrial decidualization.9 Recently, novel work by the Moley laboratory with creation of a SLC2A8-null female mouse has shown significant decrease in mRNA expression of markers of endometrial decidualization, cyclooxygenase-2 and prolactin-related protein, and incomplete decidualization of the uterus upon in vivo artificial stimulation.44 They went on to perform ovarian cross-transplantation studies, demonstrating that the decidualization phenotype is not a result of secondary effects from the ovary but lie in the embryo and endometrial aspects of implantation.44 This evidence suggests that metabolic disruption of the expression of SLC2A8 may explain poor implantation rates in metabolic derangements such as obesity and PCOS.
Hyperinsulinemia
Insulin resistance is a common metabolic feature of both obesity and PCOS. It is a condition in which endogenous or exogenously administered insulin has less than normal effect on fat, muscle, and liver. Decreased glucose utilization and increased hepatic gluconeogenesis result in increased blood glucose concentrations and compensatory hyperinsulinemia.51 It has been well documented that women with PCOS, independent of obesity, are insulin resistant and have compensatory hyperinsulinemia.52–54
The action of insulin is mediated via its receptor and 2 intracellular pathways—the phosphatidyl-inositol 3 kinase (PI3K) pathway, which mediates the metabolic effects of insulin, and the mitogen-activated protein kinase (MAPK) pathway, which mediates the proliferative actions of insulin. When insulin binds to its receptor, a conformational change is induced, resulting in tyrosine phosphorylation of the receptor and protein substrates which bind and activate PI3K and Akt, an effector molecule which induces signal transduction for glucose regulation and metabolism.55,56
In vitro, insulin has been found to inhibit production of the endometrial stromal product IGFBP-1, a recognized biomarker of decidualization.14 This recognition was one of the first suggestions that physiological levels of insulin play homeostatic roles for energy metabolism in the endometrium. This led to the belief that hyperinsulinemic states perturb the normal metabolic state of the endometrium and could contribute to the poor implantation and increased miscarriage rates.
Fornes et al went on to examine the expression of the molecules involved in the insulin pathway in endometria from women with PCOS with or without hyperinsulinemia.57 They found that certain insulin receptor substrates, including pAS160T642 and SLC2A4, were decreased in hyperinsulinemic women with PCOS compared with those in controls and nonhyperinsulinemic patients with PCOS as assessed by Western blot. As all of their patients with PCOS were overweight and most of the patients with PCOS with hyperinsulinemia were obese, they did not stratify their results to compare obesity and PCOS. Their work reiterates the decrease in the GLUT SLC2A4 and further suggests that this may account for impairment in glucose metabolism and homeostasis at the endometrial level. Taken together, the role of insulin resistance in the endometrium and derangement in glucose uptake likely is contributing to defects in endometrial receptivity and implantation defects as depicted in Figure 1.57
Figure 1.
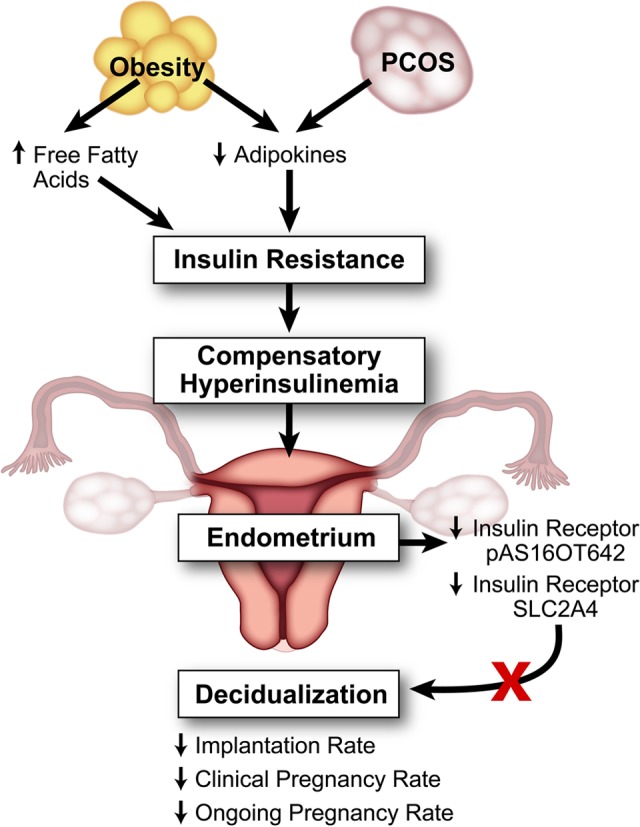
The effect of hyperinsulinemia on the endometrium in polycystic ovary syndrome (PCOS) and obesity. Obesity and PCOS lead to a compensatory hyperinsulinemic state, which affects endometrial homeostasis, leading to decreased insulin receptors and decreased production of decidualization biomarkers.
Recently, researchers found that insulin resistance lowers implantation rate in in vitro maturation–in vitro fertilization embryo transfer cycle (IVM-IVF-ET).46 Chang et al recruited patients with PCOS with insulin resistance, as determined by glucose tolerance test and homeostasis model assessment index, and non-hyperinsulinemic patients with PCOS undergoing IVM-IVF-ET and followed the outcomes. They found that insulin-resistant patients with PCOS had statistically significant decreases in implantation, clinical pregnancy, and ongoing pregnancy rates after controlling for age, BMI, and lipid profiles. Interestingly, embryo development was not affected in patients with insulin resistance, indicating that, in fact, insulin-resistant patients with PCOS have impaired endometrial receptivity.58
The pathophysiology behind insulin resistance is complex. Most authors believe that, in obese patients, insulin resistance is determined by liver accumulation of free fatty acids (FFAs) released by visceral fat. Adipose tissue stores and releases fatty acids in addition to synthesizing and releasing other active compounds such as interleukin 6, interleukin 1β, resistin, and angiotensin 2. Thus, an expanding fat mass (ie, obesity) releases increasing amounts of compounds, including FFAs, and when large amounts of FFA are released, insulin resistance results.59 Elevated plasma FFAs then inhibit insulin’s antilipolytic action, which further increases the rate of FFA release into the circulation.59 FFAs may also interfere with insulin stimulation of glucose transport by modulating GLUT transcription and mRNA stability as aforementioned.59 As adipose tissue mass expands and FFA release is increased, other mechanisms, in particular, dysfunctional regulation of adipokines, likely contribute to the pathologic state.60 Adipokines are hormones, cytokines, and other bioactive substances produced by adipose tissue which play a main role in the development of metabolic syndrome.61 Adiponectin is a protein secreted by the adipocytes in inverse proportion to adipose mass. Decreased adiponectin may induce insulin resistance and alter the lipid profile.37,38 Adiponectin is mostly produced in the subcutaneous fat and is low in patients with visceral obesity.62,63 It has been suggested that increased production of cytokines inside the visceral fat is the mechanism that inhibits subcutaneous production of adiponectin, possibly leading to insulin resistance.
Adiponectin plays an important role in regulating energy homeostasis, specifically lipid and glucose metabolism.64–66 Takemura et al found that adiponectin receptors are highly expressed in the human endometrium during the timeframe of the window of implantation.67 Kim et al further went on to examine the expression and hormonal regulation of adiponection and its receptors (AdipoR1 and AdipoR2) in mouse endometrial decidual cells.64 They observed that mouse endometrial stromal cells secrete adiponection when decidualized, adiponectin signaling was higher during implantation, and AdipoR1/R2 were localized in the decidual cells and embryo after implantation.52 This suggests that adiponectin is an integral part of proper endometrial decidualization and embryo implantation.
Systemic adiponection levels are reduced in obesity, and weight reduction leads to an increase in circulating adiponectin levels.60,68 Plasma adiponectin is also reduced in women with PCOS.66,69 To better understand hypoadiponectinemia and PCOS, Toulis et al published a systematic review comparing circulating adiponectin levels in women with PCOS matched for similar BMI. This analysis revealed that after controlling for BMI, adiponectin levels are lower in women with PCOS than in non-PCOS controls and that hypoadiponectinemia is probably related to insulin resistance in women with PCOS.70 Furthermore, studies suggest that women with PCOS treated with either metformin or pioglitazone show increased circulating adiponectin levels that correlate with reduction in insulin secretion and improvement of insulin action on glucose metabolism and lipid oxidation.71–73
As control of adiponectin levels may have therapeutic benefits, a recent systemic review by Palomba et al was published examining this exact topic. The review examined 10 randomized control trials, totaling 845 women with PCOS in the analysis, to assess the effects of metformin administration in infertile patients with PCOS who receive gonadotrophins for in vitro fertilization (IVF) and intracytoplasmic sperm injection (ICSI) cycles. They found that metformin administration in IVF/ICSI cycles had no effect on the rates of pregnancy and live birth; however, metformin administration reduced the risk of miscarriage and increased that of implantation.62 These results could be due to the observation that women with PCOS treated with metformin show increased circulating adiponectin levels and improved insulin action and possibly could be at the level of the endometrium, improving overall implantation rates.74 This hypothesis is supported by preliminary research by Jakubowicz, who performed a pilot study to investigate whether treatment with an insulin-sensitizing drug may prevent early pregnancy loss in PCOS. Preliminary results suggest a dramatic reduction in miscarriage rates in women with PCOS treated throughout pregnancy with metformin; however, no prospective controlled trials have been performed.75 This contrasts to the Legro et al study which compared clomiphene, metformin, or both for infertility in patients with PCOS. They found that the rates of first trimester pregnancy loss did not differ significantly among the groups, and that clomiphene was superior to metformin or combined treatment in terms of live birth rate.76 Further research in this area is warranted.
Hyperandrogenism
Hyperandrogenism is the key feature in PCOS and results primarily from excess androgen production in the ovaries and adrenals. The primary mechanisms driving increased ovarian androgen production are increased luteinizing hormone (LH) and hyperinsulinemia due to insulin resistance.77–79 Insulin excess binds to its receptor on the ovarian theca cell, enhancing LH- and IGF-I-stimulated androgen production.80 Insulin also acts indirectly to increase serum-free testosterone by inhibiting the hepatic production of sex hormone-binding globulin80,81 and enhances serum IGF-I bioactivity through suppressed IGF-binding protein production, perpetuating ovarian hyperandrogenism.80 Obesity increases hyperandrogenism, both independently and by exacerbating PCOS.82 The window of implantation occurs in the midsecretory phase of the menstrual cycle, which is a state characterized by low androgen levels.83 Hyperandrogenic states with persistently elevated androgens, such as PCOS and obesity, likely influence these processes.
Homeobox or HOX are developmental control genes essential for endometrial differentiation and receptivity. Both HOXA10 and HOXA11 mRNA are expressed in human endometrial epithelial and stromal cells with expression significantly higher during the window of implantation.1,84 Female mice with disruption of HOXA10 have infertility caused by uterine defects. They are ovulatory but are unable to support implantation.85 Cermik et al evaluated the effect of testosterone on expression of HOX in Ishikawa cells, a well-differentiated human endometrial adenocarcinoma cell line.40 They found that in vitro expression of HOXA10 was decreased by testosterone but not affected by dehydroepiandrosterone (DHEA) or dehydroepiandrosterone sulfate or insulin. The authors further went on to investigate expression of HOXA10 in the endometrium of hyperandrogenic patients with PCOS and found that these patients have significantly decreased expression of HOXA10 compared with that in controls.40 These results suggest that testosterone is a novel regulator of HOXA10 and has a negative impact on the expression of a gene essential for endometrial receptivity.
The Wilms tumor suppressor (WT1) gene is expressed in the window of implantation and has been shown to modulate androgen receptor expression.86,87 Gonzalez et al. studied endometrial biopsies from ovulatory patients with PCOS for endometrial expression of WT1 and downstream targets.83 They found increased androgen receptor expression in the endometria from ovulatory women with PCOS, expression of WT1 was downregulated during the window of implantation compared with that in controls, and endometrial stromal cells exposed to androgens in vitro showed downregulation of expression of WT1.83 These results further suggest a role of hyperandrogenism, antagonizing proper endometrial decidualization and receptivity as depicted in Figure 2.
Figure 2.
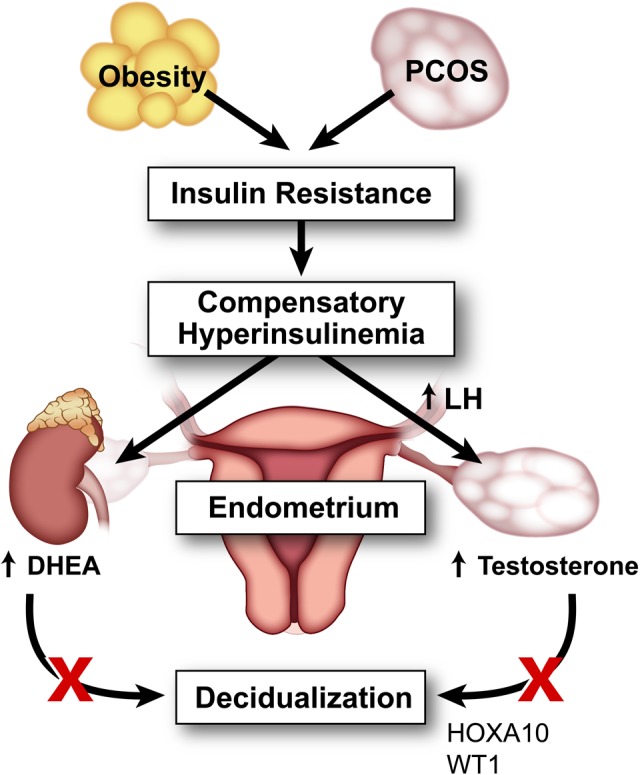
Effect of hyperandrogenism on endometrium in polycystic ovary syndrome (PCOS) and obesity. Polycystic ovary syndrome and obesity are hyperinsulinemic states which contribute to hyperandrogenism by enhancing luteinizing hormone (LH)-stimulated androgen production in the ovary and adrenal expression of dehydroepiandrosterone. This hyperandrogenism affects the window of implantation by decreasing gene expression of HOXA10 and WT1 and by affecting endometrial decidualization.
Finally, Frolova et al demonstrated that adequate glucose flux through the pentose phosphate pathway is essential for proper endometrial decidualization and that DHEA, a potent inhibitor of the pentose phosphate pathway, decreases endometrial decidualization both in vitro and in vivo.10 This finding provides mechanistic evidence that hyperandrogenism affects endometrial decidualization. As obesity and PCOS are both hyerandrogenic states, this mechanism could contribute to the increased rates of miscarriage and poor implantation rates seen in women with PCOS.
Conclusions and Future Prospects
The metabolic alterations that occur in obesity and PCOS alter endometrial receptivity and thus likely affect proper embryonic implantation, resulting in increased miscarriage rates and overall subfertility. Specifically, this review addresses the effect of glucose metabolism, compensatory hyperinsulinemia from insulin resistant state, and hyperandrogenism on the endometrium during the window of implantation. With mounting evidence that metabolic derangements alter endometrial receptivity, the need for mechanistic studies into energy utilization and decidualization is critical to the development of targeted therapies to improve subfertility and uterine-based reproductive disorders in this growing patient population.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: R01HD065435 to KHM;T32HD49305 to JT and KHM; and an ACOG Research Grant to MMBS.
References
- 1. Cakmak H, Taylor HS. Implantation failure: molecular mechanisms and clinical treatment. Hum Reprod Update. 2011;17 (2):242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Paulson RJ. Hormonal induction of endometrial receptivity. Fertil Steril. 2011;96 (3):530–535. [DOI] [PubMed] [Google Scholar]
- 3. Rashid NA, Lalitkumar S, Lalitkumar PG, Gemzell-Danielsson K. Endometrial receptivity and human embryo implantation. Am J Reprod Immunol. 2011;66 (suppl 1):23–30. [DOI] [PubMed] [Google Scholar]
- 4. Lessey BA. Assessment of endometrial receptivity. Fertil Steril. 2011;96 (3):522–529. [DOI] [PubMed] [Google Scholar]
- 5. Berlanga O, Bradshaw HB, Vilella-Mitjana F, Garrido-Gómez T, Simón C. How endometrial secretomics can help in predicting implantation. Placenta. 2011;32 (suppl 3):S271–S275. [DOI] [PubMed] [Google Scholar]
- 6. Donaghay M, Lessey BA. Uterine receptivity: alterations associated with benign gynecological disease. Semin Reprod Med. 2007;25 (6):461–475. [DOI] [PubMed] [Google Scholar]
- 7. Singh M, Chaudhry P, Asselin E. Bridging endometrial receptivity and implantation: network of hormones, cytokines, and growth factors. J Endocrinol. 2011;210 (1):5–14. [DOI] [PubMed] [Google Scholar]
- 8. Kim ST, Moley KH. Regulation of facilitative glucose transporters and AKT/MAPK/PRKAA signaling via estradiol and progesterone in the mouse uterine epithelium. Biol Reprod. 2009;81 (1):188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frolova AI, Moley KH. Glucose transporters in the uterus: an analysis of tissue distribution and proposed physiological roles. Reproduction. 2011;142 (2):211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Frolova AI, O'Neill K, Moley KH. Dehydroepiandrosterone inhibits glucose flux through the pentose phosphate pathway in human and mouse endometrial stromal cells, preventing decidualization and implantation. Mol Endocrinol. 2011;25 (8):1444–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frolova A, Flessner L, Chi M, Kim ST, Foyouzi-Yousefi N, Moley KH. Facilitative glucose transporter type 1 is differentially regulated by progesterone and estrogen in murine and human endometrial stromal cells. Endocrinology. 2009;150 (3):1512–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lathi RB, Hess AP, Tulac S, Nayak NR, Conti M, Giudice LC. Dose-dependent insulin regulation of insulin-like growth factor binding protein-1 in human endometrial stromal cells is mediated by distinct signaling pathways. J Clin Endocrinol Metab. 2005;90 (3):1599–1606. [DOI] [PubMed] [Google Scholar]
- 13. Nelson-Degrave VL, Wickenheisser JK, Hendricks KL, et al. Alterations in mitogen-activated protein kinase kinase and extracellular regulated kinase signaling in theca cells contribute to excessive androgen production in polycystic ovary syndrome. Mol Endocrinol. 2005;19 (2):379–390. [DOI] [PubMed] [Google Scholar]
- 14. Giudice LC, Dsupin BA, Irwin JC. Steroid and peptide regulation of insulin-like growth factor-binding proteins secreted by human endometrial stromal cells is dependent on stromal differentiation. J Clin Endocrinol Metab. 1992;75 (5):1235–1241. [DOI] [PubMed] [Google Scholar]
- 15. Leon L, Bacallao K, Gabler F, Romero C, Valladares L, Vega M. Activities of steroid metabolic enzymes in secretory endometria from untreated women with Polycystic Ovary Syndrome. Steroids. 2008;73 (1):88–95. [DOI] [PubMed] [Google Scholar]
- 16. Wise LA, Rothman KJ, Mikkelsen EM, Sørensen HT, Riis A, Hatch EE. An internet-based prospective study of body size and time-to-pregnancy. Hum Reprod. 2010;25 (1):253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van der Steeg JW, Steures P, Eijkemans MJ, et al. Obesity affects spontaneous pregnancy chances in subfertile, ovulatory women. Hum Reprod. 2008;23 (2):324–328. [DOI] [PubMed] [Google Scholar]
- 18. Boots C, Stephenson MD. Does obesity increase the risk of miscarriage in spontaneous conception: a systematic review. Semin Reprod Med. 2011;29 (6):507–513. [DOI] [PubMed] [Google Scholar]
- 19. Bellver J, Ayllón Y, Ferrando M, et al. Female obesity impairs in vitro fertilization outcome without affecting embryo quality. Fertil Steril. 2010;93 (2):447–454. [DOI] [PubMed] [Google Scholar]
- 20. Luke B, Brown MB, Stern JE, et al. Female obesity adversely affects assisted reproductive technology (ART) pregnancy and live birth rates. Hum Reprod. 2011;26 (1):245–252. [DOI] [PubMed] [Google Scholar]
- 21. Brewer CJ, Balen AH. The adverse effects of obesity on conception and implantation. Reproduction. 2010;140 (3):347–364. [DOI] [PubMed] [Google Scholar]
- 22. Jungheim ES, Schon SB, Schulte MB, DeUgarte DA, Fowler SA, Tuuli MG. IVF outcomes in obese donor oocyte recipients: a systematic review and meta-analysis. Hum Reprod. 2013;28(10):2720–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wattanakumtornkul S, Damario MA, Stevens Hall SA, Thornhill AR, Tummon IS. Body mass index and uterine receptivity in the oocyte donation model. Fertil Steril. 2003;80 (2):336–340. [DOI] [PubMed] [Google Scholar]
- 24. Bellver J, Rossal LP, Bosch E, et al. , Obesity and the risk of spontaneous abortion after oocyte donation. Fertil Steril. 2003;79 (5):1136–1140. [DOI] [PubMed] [Google Scholar]
- 25. Bellver J, Bosch E, Remohí J, Pellicer A. Evidence-based medicine is gaining momentum. Fertil Steril. 2005;84(5):1555–1556; author reply 1556-1557. [DOI] [PubMed] [Google Scholar]
- 26. Bellver J, Pellicer A, García-Velasco JA, Ballesteros A, Remohí J, Meseguer M. Obesity reduces uterine receptivity: clinical experience from 9,587 first cycles of ovum donation with normal weight donors. Fertil Steril. 2013;100 (4):1050–1058. [DOI] [PubMed] [Google Scholar]
- 27. Bellver J, Melo MA, Bosch E, Serra V, Remohí J, Pellicer A. Obesity and poor reproductive outcome: the potential role of the endometrium. Fertil Steril. 2007;88 (2):446–451. [DOI] [PubMed] [Google Scholar]
- 28. Dessolle L, Daraï E, Cornet D, et al. Determinants of pregnancy rate in the donor oocyte model: a multivariate analysis of 450 frozen-thawed embryo transfers. Hum Reprod. 2009;24 (12):3082–3089. [DOI] [PubMed] [Google Scholar]
- 29. DeUgarte DA, DeUgarte CM, Sahakian V. Surrogate obesity negatively impacts pregnancy rates in third-party reproduction. Fertil Steril. 2010;93 (3):1008–1010. [DOI] [PubMed] [Google Scholar]
- 30. Bellver J, Martínez-Conejero JA, Labarta E, et al. Endometrial gene expression in the window of implantation is altered in obese women especially in association with polycystic ovary syndrome. Fertil Steril. 2011;95(7):2335–2341, 2341 e1–e8. [DOI] [PubMed] [Google Scholar]
- 31. Fauser BC, Tarlatzis BC, Rebar RW, et al. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS): the Amsterdam ESHRE/ASRM-Sponsored 3 rd PCOS Consensus Workshop Group. Fertil Steril. 2012;97(1):28–38. e25. [DOI] [PubMed] [Google Scholar]
- 32. Dor J, Itzkowic DJ, Mashiach S, Lunenfeld B, Serr DM. Cumulative conception rates following gonadotropin therapy. Am J Obstet Gynecol. 1980;136 (1):102–105. [DOI] [PubMed] [Google Scholar]
- 33. Macklon NS, van der Gaast MH, Hamilton A, Fauser BC, Giudice LC. The impact of ovarian stimulation with recombinant FSH in combination with GnRH antagonist on the endometrial transcriptome in the window of implantation. Reprod Sci. 2008;15 (4):357–365. [DOI] [PubMed] [Google Scholar]
- 34. Hardiman P, Pillay OC, Atiomo W. Polycystic ovary syndrome and endometrial carcinoma. Lancet. 2003;361 (9371):1810–1812. [DOI] [PubMed] [Google Scholar]
- 35. Haoula Z, Salman M, Atiomo W. Evaluating the association between endometrial cancer and polycystic ovary syndrome. Hum Reprod. 2012;27 (5):1327–1331. [DOI] [PubMed] [Google Scholar]
- 36. Savaris RF, Groll JM, Young SL, et al. Progesterone resistance in PCOS endometrium: a microarray analysis in clomiphene citrate-treated and artificial menstrual cycles. J Clin Endocrinol Metab. 2011;96 (6):1737–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aghajanova L, Velarde MC, Giudice LC. Altered gene expression profiling in endometrium: evidence for progesterone resistance. Semin Reprod Med. 2010;28 (1):51–58. [DOI] [PubMed] [Google Scholar]
- 38. Suikkari AM, Ruutiainen K, Erkkola R, Seppälä M. Low levels of low molecular weight insulin-like growth factor-binding protein in patients with polycystic ovarian disease. Hum Reprod. 1989;4 (2):136–139. [DOI] [PubMed] [Google Scholar]
- 39. Apparao KB, Lovely LP, Gui Y, Lininger RA, Lessey BA. Elevated endometrial androgen receptor expression in women with polycystic ovarian syndrome. Biol Reprod. 2002;66 (2):297–304. [DOI] [PubMed] [Google Scholar]
- 40. Cermik D, Selam B, Taylor HS. Regulation of HOXA-10 expression by testosterone in vitro and in the endometrium of patients with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88 (1):238–243. [DOI] [PubMed] [Google Scholar]
- 41. Gregory CW, Wilson EM, Apparao KB, et al. Steroid receptor coactivator expression throughout the menstrual cycle in normal and abnormal endometrium. J Clin Endocrinol Metab. 2002;87 (6):2960–2966. [DOI] [PubMed] [Google Scholar]
- 42. Frolova AI, Moley KH. Quantitative analysis of glucose transporter mRNAs in endometrial stromal cells reveals critical role of GLUT1 in uterine receptivity. Endocrinology. 2011;152 (5):2123–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. von Wolff M, Ursel S, Hahn U, Steldinger R, Strowitzki T. Glucose transporter proteins (GLUT) in human endometrium: expression, regulation, and function throughout the menstrual cycle and in early pregnancy. J Clin Endocrinol Metab. 2003;88 (8):3885–3892. [DOI] [PubMed] [Google Scholar]
- 44. Adastra KL, Frolova AI, Chi MM, et al. Slc2a8 deficiency in mice results in reproductive and growth impairments. Biol Reprod. 2012;87 (2):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. James DE, Brown R, Navarro J, Pilch PF. Insulin-regulatable tissues express a unique insulin-sensitive glucose transport protein. Nature. 1988;333 (6169):183–185. [DOI] [PubMed] [Google Scholar]
- 46. Welch RD, Gorski J. Regulation of glucose transporters by estradiol in the immature rat uterus. Endocrinology. 1999;140 (8):3602–3608. [DOI] [PubMed] [Google Scholar]
- 47. Mioni R, Chiarelli S, Xamin N, et al. Evidence for the presence of glucose transporter 4 in the endometrium and its regulation in polycystic ovary syndrome patients. J Clin Endocrinol Metab. 2004;89 (8):4089–4096. [DOI] [PubMed] [Google Scholar]
- 48. Mozzanega B, Mioni R, Granzotto M, et al. Obesity reduces the expression of GLUT4 in the endometrium of normoinsulinemic women affected by the polycystic ovary syndrome. Ann N Y Acad Sci. 2004;1034:364–374. [DOI] [PubMed] [Google Scholar]
- 49. Zhai J, Liu CX, Tian ZR, Jiang QH, Sun YP. Effects of metformin on the expression of GLUT4 in endometrium of obese women with polycystic ovary syndrome. Biol Reprod. 2012;87 (2):29. [DOI] [PubMed] [Google Scholar]
- 50. Loke YW, King A, Burrows TD. Decidua in human implantation. Hum Reprod. 1995;10 (suppl 2):14–21. [DOI] [PubMed] [Google Scholar]
- 51. Moller DE, Flier JS. Insulin resistance--mechanisms, syndromes, and implications. N Engl J Med. 1991;325 (13):938–948. [DOI] [PubMed] [Google Scholar]
- 52. Dunaif A, Graf M. Insulin administration alters gonadal steroid metabolism independent of changes in gonadotropin secretion in insulin-resistant women with the polycystic ovary syndrome. J Clin Invest. 1989;83 (1):23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Steingold KA, Lobo RA, Judd HL, Lu JK, Chang RJ. The effect of bromocriptine on gonadotropin and steroid secretion in polycystic ovarian disease. J Clin Endocrinol Metab. 1986;62 (5):1048–1051. [DOI] [PubMed] [Google Scholar]
- 54. Burghen GA, Givens JR, Kitabchi AE. Correlation of hyperandrogenism with hyperinsulinism in polycystic ovarian disease. J Clin Endocrinol Metab. 1980;50 (1):113–116. [DOI] [PubMed] [Google Scholar]
- 55. Baptiste CG, Battista MC, Trottier A, Baillargeon JP. Insulin and hyperandrogenism in women with polycystic ovary syndrome. J Steroid Biochem Mol Biol. 2010;122 (1-3):42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Czech MP, Corvera S. Signaling mechanisms that regulate glucose transport. J Biol Chem. 1999;274 (4):1865–1868. [DOI] [PubMed] [Google Scholar]
- 57. Fornes R, Ormazabal P, Rosas C, et al. Changes in the expression of insulin signaling pathway molecules in endometria from polycystic ovary syndrome women with or without hyperinsulinemia. Mol Med. 2010;16 (3-4):129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chang EM, Han JE, Seok HH, Lee DR, Yoon TK, Lee WS. Insulin resistance does not affect early embryo development but lowers implantation rate in in vitro maturation-in vitro fertilization-embryo transfer cycle. Clin Endocrinol (Oxf). 2013;79 (1):93–99. [DOI] [PubMed] [Google Scholar]
- 59. Boden G. Obesity and free fatty acids. Endocrinol Metab Clin North Am. 2008;37(3):635–646, viii–ix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Palin MF, Bordignon VV, Murphy BD. Adiponectin and the control of female reproductive functions. Vitam Horm. 2012;90:239–287. [DOI] [PubMed] [Google Scholar]
- 61. Halberg NI, Wernstedt-Asterholm L, Scherer PE. The adipocyte as an endocrine cell. Endocrinol Metab Clin North Am. 2008;37(3):753–768, x–xi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Guerre-Millo M. Adiponectin: an update. Diabetes Metab. 2008;34 (1):12–18. [DOI] [PubMed] [Google Scholar]
- 63. Carmina E, Chu MC, Moran C, et al. Subcutaneous and omental fat expression of adiponectin and leptin in women with polycystic ovary syndrome. Fertil Steril. 2008;89 (3):642–648. [DOI] [PubMed] [Google Scholar]
- 64. Kim ST, Marquard K, Stephens S, Louden E, Allsworth J, Moley KH. Adiponectin and adiponectin receptors in the mouse preimplantation embryo and uterus. Hum Reprod. 2011;26 (1):82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med. 2001;7 (8):947–953. [DOI] [PubMed] [Google Scholar]
- 66. Dos Santos E, Serazin V, Morvan C, et al. Adiponectin and leptin systems in human endometrium during window of implantation. Fertil Steril. 2012;97(3):771–778. e1. [DOI] [PubMed] [Google Scholar]
- 67. Takemura Y, Osuga Y, Yamauchi T, et al. Expression of adiponectin receptors and its possible implication in the human endometrium. Endocrinology. 2006;147 (7):3203–3210. [DOI] [PubMed] [Google Scholar]
- 68. Weyer C, Tataranni PA, Bogardus C, Pratley RE. Insulin resistance and insulin secretory dysfunction are independent predictors of worsening of glucose tolerance during each stage of type 2 diabetes development. Diabetes Care. 2001;24 (1):89–94. [DOI] [PubMed] [Google Scholar]
- 69. Sir-Petermann T, Echiburú B, Maliqueo MM, et al. Serum adiponectin and lipid concentrations in pregnant women with polycystic ovary syndrome. Hum Reprod. 2007;22 (7):1830–1836. [DOI] [PubMed] [Google Scholar]
- 70. Toulis KA, Goulis DG, Farmakiotis D, et al. Adiponectin levels in women with polycystic ovary syndrome: a systematic review and a meta-analysis. Hum Reprod Update. 2009;15 (3):297–307. [DOI] [PubMed] [Google Scholar]
- 71. Agarwal N, Rice SP, Bolusani H, et al. Metformin reduces arterial stiffness and improves endothelial function in young women with polycystic ovary syndrome: a randomized, placebo-controlled, crossover trial. J Clin Endocrinol Metab. 2010;95 (2):722–730. [DOI] [PubMed] [Google Scholar]
- 72. Elkind-Hirsch K, Marrioneaux O, Bhushan M, Vernor D, Bhushan R. Comparison of single and combined treatment with exenatide and metformin on menstrual cyclicity in overweight women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93 (7):2670–2678. [DOI] [PubMed] [Google Scholar]
- 73. Glintborg D, Frystyk J, Højlund K, et al. Total and high molecular weight (HMW) adiponectin levels and measures of glucose and lipid metabolism following pioglitazone treatment in a randomized placebo-controlled study in polycystic ovary syndrome. Clin Endocrinol (Oxf). 2008;68 (2):165–174. [DOI] [PubMed] [Google Scholar]
- 74. Palomba S, Falbo A, La Sala GB. Effects of metformin in women with polycystic ovary syndrome treated with gonadotrophins for in vitro fertilisation and intracytoplasmic sperm injection cycles: a systematic review and meta-analysis of randomised controlled trials. BJOG. 2013;120 (3):267–276. [DOI] [PubMed] [Google Scholar]
- 75. Nestler JE, Stovall D, Akhter N, Iuorno MJ, Jakubowicz DJ. Strategies for the use of insulin-sensitizing drugs to treat infertility in women with polycystic ovary syndrome. Fertil Steril. 2002;77 (2):209–215. [DOI] [PubMed] [Google Scholar]
- 76. Johnson NP, Stewart AW, Falkiner J, et al. Clomiphene, metformin, or both for infertility in the polycystic ovary syndrome. N Engl J Med. 2007;356 (6):551–566. [DOI] [PubMed] [Google Scholar]
- 77. Nelson VL, Qin KN, Rosenfield RL, et al. The biochemical basis for increased testosterone production in theca cells propagated from patients with polycystic ovary syndrome. J Clin Endocrinol Metab. 2001;86 (12):5925–5933. [DOI] [PubMed] [Google Scholar]
- 78. Kumar A, Woods KS, Bartolucci AA, Azziz R. Prevalence of adrenal androgen excess in patients with the polycystic ovary syndrome (PCOS). Clin Endocrinol (Oxf). 2005;62 (6):644–649. [DOI] [PubMed] [Google Scholar]
- 79. Jakimiuk AJ, Weitsman SR, Navab A, Magoffin DA. Luteinizing hormone receptor, steroidogenesis acute regulatory protein, and steroidogenic enzyme messenger ribonucleic acids are overexpressed in thecal and granulosa cells from polycystic ovaries. J Clin Endocrinol Metab. 2001;86 (3):1318–1323. [DOI] [PubMed] [Google Scholar]
- 80. Dumesic DA, Richards JS. Ontogeny of the ovary in polycystic ovary syndrome. Fertil Steril. 2013;100 (1):23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Giallauria F, Palomba S, Vigorito C, et al. Androgens in polycystic ovary syndrome: the role of exercise and diet. Semin Reprod Med. 2009;27 (4):306–315. [DOI] [PubMed] [Google Scholar]
- 82. Fauser BC, Laven JS, Tarlatzis BC, et al. Sex steroid hormones and reproductive disorders: impact on women’s health. Reprod Sci. 2011;18 (8):702–712. [DOI] [PubMed] [Google Scholar]
- 83. Gonzalez D, Thackeray H, Lewis PD, et al. Loss of WT1 expression in the endometrium of infertile PCOS patients: a hyperandrogenic effect? J Clin Endocrinol Metab. 2012;97 (3):957–966. [DOI] [PubMed] [Google Scholar]
- 84. Du H, Sarno J, Taylor HS. HOXA10 inhibits Kruppel-like factor 9 expression in the human endometrial epithelium. Biol Reprod. 2010;83 (2):205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Satokata I, Benson G, Maas R. Sexually dimorphic sterility phenotypes in Hoxa10-deficient mice. Nature. 1995;374 (6521):460–463. [DOI] [PubMed] [Google Scholar]
- 86. Makrigiannakis A, Coukos G, Mantani A, et al. , Expression of Wilms' tumor suppressor gene (WT1) in human endometrium: regulation through decidual differentiation. J Clin Endocrinol Metab. 2001;86 (12):5964–5972. [DOI] [PubMed] [Google Scholar]
- 87. Anthony FW, Mukhtar DD, Pickett MA, Cameron IT. Progesterone up-regulates WT1 mRNA and protein, and alters the relative expression of WT1 transcripts in cultured endometrial stromal cells. J Soc Gynecol Investig. 2003;10 (8):509–516. [DOI] [PubMed] [Google Scholar]