

Abstract
Pancreatic cancer is one of the most fatal cancers and is associated with limited diagnostic and therapeutic modalities. Currently, gemcitabine is the only effective drug and represents the preferred first-line treatment for chemotherapy. However, a high level of intrinsic or acquired resistance of pancreatic cancer to gemcitabine can contribute to the failure of gemcitabine treatment. To investigate the underlying molecular mechanisms for gemcitabine resistance in pancreatic cancer, we performed label-free quantification of protein expression in intrinsic gemcitabine-resistant and - sensitive human pancreatic adenocarcinoma cell lines using our improved proteomic strategy, combined with filter-aided sample preparation, single-shot liquid chromatography-mass spectrometry, enhanced spectral counting, and a statistical method based on a power law global error model. We identified 1931 proteins and quantified 787 differentially expressed proteins in the BxPC3, PANC-1, and HPDE cell lines. Bioinformatics analysis identified 15 epithelial to mesenchymal transition (EMT) markers and 13 EMT-related proteins that were closely associated with drug resistance were differentially expressed. Interestingly, 8 of these proteins were involved in glutathione and cysteine/methionine metabolism. These results suggest that proteins related to the EMT and glutathione metabolism play important roles in the development of intrinsic gemcitabine resistance by pancreatic cancer cell lines.
Keywords: chemoresistance, gemcitabine, LC-MS/MS, pancreatic cancer, quantitative Proteomics
INTRODUCTION
Pancreatic cancer (PC) has a very poor prognosis, making it one of the five most common causes of cancer mortality world-wide (Li et al., 2004; Tuveson and Neoptolemos, 2012). Because the diagnosis of this disease often occurs at a late stage and shows poor responsiveness to chemotherapy and radiation therapy, it is associated with a dismal 5-year survival rate of less than 3% (Li et al., 2004). Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer, accounting for up to 90% of cases, and is thought to develop by a multistep process that involves intermediate precursor lesions known as pancreatic intraepithelial neoplasias (PanINs) (Hruban et al., 2000). PDAC arises as a consequence of mutations and/or the silencing of several oncogenes and tumor suppressor genes, including KRAS, TP53, CDKN2A, EGFR, and SMAD4, which lead to pancreatic tumor development and resistance to chemotherapeutic agents (Jones et al., 2008).
Gemcitabine (2′-deoxy-2′-difluorodeoxycytidine), the nucleoside analog, is the leading therapeutic for pancreatic cancer that is used to improve quality of life and overall survival (Burris et al., 1997). However, the median survival time of patients treated with gemcitabine is only 6.3 months (Carmichael et al., 1996). Intrinsic or acquired resistance to this drug is a major factor in the failure of this agent to control pancreatic cancer, and this has renewed the interest in developing novel therapeutic agents and combinations (Cao et al., 2013). Although several combination remedies with gemcitabine, as well as novel agents, have prolonged survival rates compared with gemcitabine alone (Cao et al., 2013), drug resistance and toxicity still decrease the overall clinical usefulness of these therapeutic agents. Therefore, a better understanding of the cellular and molecular mechanisms involved in gemcitabine resistance is imperative for improving drug efficiency and patient outcomes.
Tumor heterogeneity is a prominent feature of pancreatic malignancies, which represents a major challenge to studying PDAC (Kalluri and Weinberg, 2009; Li et al., 2004; Samuel and Hudson, 2012). Therefore, cell line models have become invaluable tools for PDAC research (Samuel and Hudson, 2012; Thu et al., 2014). Large-scale proteomic analyses using in vitro cell line models are needed to understand the molecular mechanisms underlying drug resistance and PDAC biology. However, few comparative analyses of pancreatic cell proteomes that underlie drug resistance and PDAC biology have been reported (Chen et al., 2011; Kuramitsu et al., 2010; Mori-Iwamoto et al., 2007; 2008; Poland et al., 2004; Zhou and Du, 2012).
The BxPC3 and PANC-1 cell lines are well established and widely used in research on the development of PDAC and drug resistance (Fryer et al., 2011; Huanwen et al., 2009; Rathos et al., 2012; Thu et al., 2014). Although there are many apparent contradictory reports regarding both the phenotype and genotype of these two cell lines, they have been designated KRAS-independent cell lines (Zimmermann et al., 2013). Interestingly, recent studies showed that BxPC3 cells with wild-type KRAS are the most sensitive to gemcitabine (Huanwen et al., 2009; Rathos et al., 2012). By contrast, PANC-1 cells, which express mutant KRAS, are the most resistant to gemcitabine. Furthermore, these two pancreatic cancer cell lines have different genotypes and phenotypes associated with the stage of tumor development (Table 1) (Deer et al., 2010; Thu et al., 2014). Hence, a comprehensive proteomics analysis of these cell lines would provide a valuable resource for studying the possible mechanisms associated with intrinsic gemcitabine resistance and PDAC development.
Table 1.
Clinical and molecular characteristics of 2 PDAC cell lines
| BxPC3 | PANC-1 | |
|---|---|---|
| Derivation | Primary tumor | Primary tumor |
| Differentiation | Moderate to poor | Poor |
| Metastasis | No | Yes |
| Tissue | Pancreatic adenocarcinoma | Pancreatic ductal epithelioid carcinoma |
| Karyotype | NA | Hypertriploid |
| Mutation | Wild type KRAS Mut CDKN2A, MAP2K4, SMAD4, and TP53 | Wild type SMAD4 Mut KRAS, CDKN2A, MAP2K4, and TP53 |
| KRAS dependence | KRAS independence | KRAS independence |
| Gemcitabine resistance | Sensitive | Resistance |
NA, information unavailable on ATCC website; PDAC, pancreatic ductal adenocarcinoma
In this study, we performed a comparative proteomic analysis between the PDAC cell lines (BxPC3 and PANC-1) and a normal cell line (HPDE) using an improved proteomic strategy. The approach included filter-aided sample preparation (FASP), single-shot LC-MS/MS, spectral counting using the distributed normalized spectral abundance factors (dNSAF), and a statistical approach using a power law global error model (PLGEM). We identified a total of 1931 proteins (FDR < 1%), of which 837 were differentially expressed in the PDAC cell lines. Finally, we validated the reliability of our proteomic data using western blotting for seven differentially expressed proteins (VIM, CDH1, CTNNB1, FGFBP1, IQGAP1, FLNB, and STAT3).
MATERIALS AND METHODS
Materials
Dulbecco’s Modified Eagle Medium (DMEM) and Roswell Park Memorial Institute (RPMI-1640) media were acquired from Welgene (Korea). Keratinocyte-SFM medium, fetal bovine serum (FBS), and a BCA Protein Assay Kit (reducing reagent compatible) were purchased from Pierce (USA). Amicon Ultra 0.5 ml-30 K was purchased from Millipore (UK). We acquired the tC18 Sep-Pak cartridge from Waters (USA). Sequence-grade modified trypsin and MTS assay kits were purchased from Promega (USA). Anti-vimentin (sc-7557), anti-STAT3 (sc-482), anti-cadherin 1 (sc-7870), anti-beta catenin (sc-7963), anti-IQGAP1 (sc-10792), and anti-filamin beta (sc-376241) were purchased from Santa Cruz Biotechnology (USA). Anti-FGFBP1 (ab67931) was purchased from Abcam (UK).
Cell culture
Human pancreatic cancer cell lines (BxPC3; CRL-1687, PANC-1; CRL-1469) were purchased from the American Tissue Culture Collection (ATCC, USA). The immortalized epithelial cell line (HPDE) that derived from normal human pancreatic duct was provided by Dr. Ming-Sound Tsao (Ontario Cancer Institute, Canada) (Furukawa et al., 1996; Makawita et al., 2011; Pramanik et al., 2011; Radulovich et al., 2008). PANC-1 cells were maintained in DMEM with 10% FBS, 100 U/ml penicillin, and 100 mg/mL streptomycin. BxPC3 cells were maintained in RPMI-1640 with 10% FBS, 100 U/ml penicillin, and 100 mg/mL streptomycin. HPDE cells were maintained in Keratinocyte-SFM media with bovine pituitary extract (BPE) and human recombinant EGF. All cells were cultured in a 37°C incubator with 5% CO2 and a humidified atmosphere.
IC50 assay
Each cell line was seeded in a 96-well cell culture plate at 2,500 cells per well. After 24 h, cells were treated with gemcitabine (0–20 nM and 0–1 μM for BxPC3 and PANC-1 cells, respectively). After treatment with gemcitabine, cells were incubated for 72 or 96 h, and then cell viability was analyzed using an MTS assay kit to assess the gemcitabine resistance of each cell line. All samples were analyzed in duplicate.
Sample preparation
Peptide samples were prepared using FASP. To digest proteins, the FASP procedure was performed for 200 μg proteins using a 30K Amicon filter (Millipore, UK) as described previously (Han et al., 2012; Min et al., 2014; Wisniewski et al., 2009). Trypsin, a universal digestion enzyme for shotgun proteomics (Choudhary and Mann, 2010; Fonslow et al., 2013; Gstaiger and Aebersold, 2009; Stergachis et al., 2011; Swaney et al., 2010), was used to digest proteins in the FASP. Digested samples were desalted using a tC18 Sep-Pak cartridge (Waters, USA). The desalted peptide samples were dried by Speed Vac and stored at −80°C until use in LC-MS/MS analysis.
LC-MS/MS analysis
Peptide samples were analyzed using an LTQ-velos mass spectrometer (Thermo Scientific, Germany) coupled to an EASY nano-LC system (Proxeon Biosystems, Denmark) as described previously (Han et al., 2012). For each sample, 10 μl sample dissolved in 50 μl solution A (2% acetonitrile and 0.1% formic acid) was loaded onto a nano-LC trap column (Zorbax 300SB-C18, 5 μm, 5 × 0.3 mm) and separated over a C18 analytical column (75 μm i.d. × 15 cm, 5 μm particle size). Peptides were separated using a 160 min gradient at 300 nl/min consisting of 5–30% solution B (100% acetonitrile and 0.1% formic acid) for 120 min, 30–40% solution B for 20 min, 40–90% solution B for 5 min, 90% solution B for 10 min, and 90-5% solution B for 5 min. The LTQ-velos mass spectrometer was set to scan masses ranging from 300 to 2000 m/z using a data-dependent scan mode for the 10 most abundant ions over a minimum threshold of 1000. The dynamic exclusion was set to the following parameters: repeat count, 1; repeat duration, 30 s; and exclusion duration, 60 s. The normalized collision energy was adjusted to 35%. All samples were analyzed in technical triplicates.
Database searches
The raw mass spectrometry files were searched using the SEQUEST search algorithm (SORCERER-SEQUEST, v.27, rev. 11) as previously described with some modifications (Han et al., 2012). SEQUEST search parameters were set to the following: fully tryptic peptides; missed cleavages, 2; parent ion tolerance, 1.5 Da; fragment ion mass tolerance, ± 0.5 Da; fixed modifications, carbamidomethyl cysteine; and variable modifications, methionine oxidation. SEQUEST search results were processed by Scaffold 3 software (Proteome Software, USA) to obtain peptide and protein identification information. A total of 27 LC-MS/MS runs were merged using Scaffold 3. Protein probability with at least two-peptides was set to 99%, and peptide probability was set to 95%. To adjust the false discovery rates (FDRs) to be <1.0% at peptide and protein level, XCorr scores were set at 1.8, 2.0, and 3.0 for singly, doubly, and triply charged peptides, respectively. The deltaCn scores were set to > 0.10.
Label-free quantitation and statistical analysis
For label-free quantitation, unweighted spectrum counts of all proteins identified were exported to an Excel file using Scaffold 3. Representative protein abundances were calculated by the dNSAF method as described previously (Zhang et al., 2010). We calculated the dNSAF using the following equation.
| I. |
| II. |
* uSpCi, unique spectrum counts of i peptide
* uSpCp, unique spectrum counts of proteins sharing i peptide
* sSpC, sum of double and triple charged spectrum counts
* L, number of amino acid for each protein
After conversion, dNSAF datasets for the three dataset comparisons (HPDE vs. BxPC3, HPDE vs. PANC-1, and BxPC3 vs. PANC-1) were imported into R 3.0.1 software for statistical analysis and converted into “exprSet” objects to allow for recognition by PLGEM software package (Bioconductor, v2.15.1) (Pavelka et al., 2008). Missing values were dealt with using “TrimAllZeroRows” and “ZeroMeanorSD” scripts. Data fitting was performed to estimate the “goodness of fit” of our datasets. P-values were obtained using the “plgem.pvalue” script, and then adjusted p-values were calculated using the “BH (Benjamini and Hochberg)” method based on the p-values that were obtained. After adjusted p-values were calculated, differentially expressed proteins were selected based on an adjusted p-value < 0.05.
Bioinformatics analysis
Analyses of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were performed using DAVID (Database for Annotation, Visualization, and Integrated Discovery) bioinformatics tools (Huang da et al., 2009). IPI accessions of target proteins were imported to the list category, and then we selected IPI_ID identifiers and Gene list types. GO analysis was used to assign the biological process, molecular function, and cellular localization. Significant pathways were selected based on p-values < 0.05. K-means clustering was performed using the GENE-E program (Broad Institute, USA) for significantly differentially expressed proteins based on PLGEM to discover protein functions based on protein expression patterns (Cheung et al., 2011). Among all 787 differentially expressed proteins, only 711 proteins were differentially expressed in two dataset comparisons (HPDE vs. BxPC3 and HPDE vs. PANC-1). We performed K-means clustering for these 711 proteins, which were grouped into six clusters that were calculated based on 2000 iterations.
Validation by Western blotting
To validate the mass spectrometric results, seven proteins were quantified by Western blotting. Western blotting was performed as described previously (Han et al., 2011). Briefly, each cell line was lysed using RIPA buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.4, 0.1% SDC, 1 mM EDTA, 1% NP-40, protease inhibitor cocktail, and 0.1 mM PMSF). We separated 30 g of proteins on 8–12% SDS-PAGE gels. Proteins were transferred to nitrocellulose membranes for 70 min at 100 V using wet-transfer techniques. Membranes were blocked with TBST containing 5% BSA for 2 h at room temperature and were then incubated with each primary antibody at 4°C overnight, based on optimized protein-antibody ratios; anti-vimentin, anti-STAT3, anti-cadherin 1, anti-beta catenin, anti-IQGAP1, and anti-filamin beta were diluted at a 1:1000 ratio, and anti-FGFBP1 was diluted at a 1:500 ratio. Membranes were washed with TBST five times and were subsequently incubated with HRP-conjugated secondary antibody (1:5000) at room temperature for 2 h. After washing with TBST five times, the membranes were developed using an ECL chemiluminescence system (GenDEPOT, USA), and visualized using a LAS-3000 system (Fujifilm, Japan).
Gene expression profiling and proteomic and genomic dataset comparisons
Gene expression profiling was performed as described in a previous study (Thu et al., 2014). In previous study, expression profiling for PDAC and HPDE cell lines was performed using Agilent 4 × 44K expression arrays (Agilent, Canada). Normalization of expression values for each array was calculated using the following formula: (median intensity - background intensity) / (median array intensity value). To reduce overestimation resulting from poor probe performance, the lowest 2% of genes, calculated based on expression values in the HPDE cell line, were eliminated.
To compare publically available genomic data with our proteomic data, our proteomic data were converted from IPI numbers (1931) to gene symbols (1910). Among 1910 gene symbols, 1695 gene symbols overlapped between the genomic (30,484 total gene symbols) and proteomic (1910 total gene symbols) data. Of these, 697 gene symbols for the differentially expressed in our proteomic dataset were subjected to Pearson correlation analysis. The fold-changes between dataset comparisons were transformed to log2 scales.
Evaluation of the quantitative proteomics platform using protein standards
To examine quantitative linearity, serially diluted ovalbumin samples were spiked in the matrix at fixed quantities (into 200 μg of PANC-1 cell lysate). Ovalbumin was serially diluted to 5.0, 2.5, 1.25, 0.625, 0.3125, and 0.15625 μg. All experimental steps were processed using conditions similar to those described above. All samples were analyzed in technical triplicates. The average dNSAF value for ovalbumin was used for each sample in technical triplicates to plot linear response curves.
RESULTS AND DISCUSSION
The overall scheme for the proteomic analysis of pancreatic cancer cell lines
Treatment of PC remains a therapeutic challenge because of the high degree of intrinsic and acquired resistance to chemotherapy and radiation therapy (Cao et al., 2013; Thu et al., 2014). Despite the improved understanding of the molecular pathways involved in pancreatic oncogenesis and chemoresistance, the clinical impact of targeted drugs, including gemcitabine, remains quite limited by the high degree of intrinsic and acquired drug resistance and the heterogeneous genetic makeup of PC (Thu et al., 2014). Therefore, understanding the underlying molecular mechanisms of intrinsic drug resistance in a well-established pancreatic cancer model system would provide valuable information to expand the treatment options for this deadly disease (Cao et al., 2013; Huanwen et al., 2009; Rathos et al., 2012).
Accordingly, we performed comparative proteomic analysis between normal pancreatic duct cells, HPDE cells, and two well-established pancreatic cancer cell lines, BxPC3 and PANC-1 cells. Interestingly, despite the presence of KRAS mutations (Zimmermann et al., 2013), these two KRAS-independent cancer cell lines are known to have different gemcitabine resistance profiles (Huanwen et al., 2009) and phenotypes (Deer et al., 2010; Thu et al., 2014) (Table 1).
To increase the numbers of proteins identified in the pancreatic cancer cell proteome, we processed proteins extracted from the cell lines using FASP, which effects high and stable recovery of peptides from protein samples (Wisniewski et al., 2009; 2011). After LC/MS analysis of the resultant peptides, dNSAF was used for relative label-free quantitation to obtain more accurate and reproducible quantitation results (Zhang et al., 2010). For the linear response curve, the R2 value was 0.9891 for six amounts of ovalbumin using unweighted spectrum counts, but it improved to 0.9903 using dNSAF values. Additionally, the average CV for ovalbumin for the six different “spiked” amounts improved after being transformed to dNSAF from 17.5% to 7.2%. These results indicate that the adoption of dNSAF improves the linearity and reliability of quantification (Supplementary Fig. S1). To estimate protein expression changes, we adapted a powerful statistical tool, PLGEM (Pavelka et al., 2008).
Quantified proteins were functionally annotated using bioinformatics tools, such as the DAVID bioinformatics resource. Additionally, we performed Western blot validation and comparison analysis with previously reported transcriptome data (Fig. 1A).
Fig. 1.
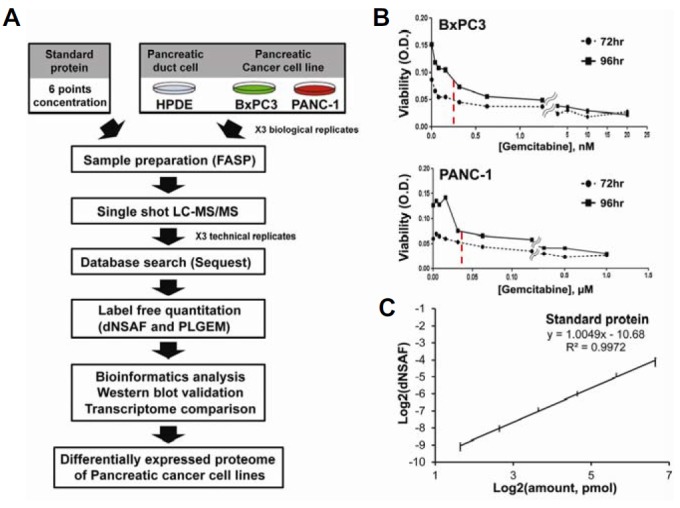
An overall flow-chart of the proteomics approach. (A) An overall schematic of this proteomics study. (B) IC50 values against gemcitabine. To test for chemoresistance of BxPC3 and PANC-1 cells against gemcitabine, IC50 assays were performed after the treatment of cells with gemcitabine for 72 and 96 h. The x- and y-axes represent the concentration of gemcitabine and cell viability (O.D.), respectively. The red dotted line indicates the average IC50 at two time points for each cell line. (C) Evaluation of linearity. To check the linearity of our quantitative platform, a linear response curve was drawn for six different amounts of ovalbumin (3.125, 6.25, 12.5, 25, 50, and 100 pmol) spiked into the same quantity of cell ly-sates. All samples were analyzed in technical triplicates and error bars represent the linear response curve for each point. The x- and y-axes represent the log2 value for the spiked amounts of ovalbumin and dNSAF, respectively. The R2 value was 0.9972.
To study the mechanism of gemcitabine resistance, we first generated BxPC3 and PANC-1 gemcitabine-resistant cell lines by exposing cells to varying concentrations of gemcitabine at two time points (72 and 96 h). The average IC50 value in BxPC3 cells was 0.1565 nM, while the average IC50 value in PANC-1 cells was 39.17 nM (Fig. 1B).
Next, we assessed the reproducibility of our label-free quantitative proteomic experiment by measuring concentrations of ovalbumin that were spiked into the peptide mixtures isolated from each cell line. The linearity curves for ovalbumin showed that our quantitative measurements were highly reproducible (R2 = 0.9903) (Fig. 1C) and the average coefficient of variation (CV) for the six amounts of spiked ovalbumin ranged from 1% to 13% (Supplementary Table S1). Additionally, the CVs of actin and GAPDH, selected as internal standards, were 5.2% and 8.7% for all 18 replicates, respectively. Furthermore, CV comparisons between traditional spectral counting and dNSAF indicated that the label-free quantitation method using dNSAF was more reproducible than the traditional method (Supplementary Fig. S1).
Generic characterization of the pancreatic cancer cell proteome
The 3 cell lines (HPDE, BxPC3, and PANC-1) were analyzed in triplicate. Using SEQUEST search engines and stringent filtering, a total of 1931 protein groups were identified at an FDR < 1%, of which 1307 (68%) were protein groups common to all three cell lines (Fig. 2A). Furthermore, 1581, 1672, and 1689 unique protein groups were identified in the HPDE, BxPC3, and PANC-1 cell lines, respectively.
Fig. 2.
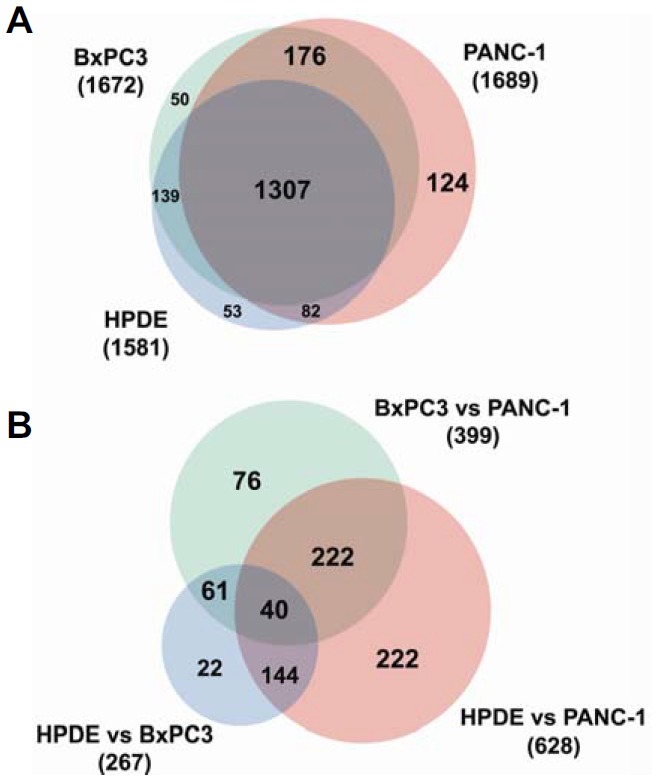
Venn diagrams of total identified proteins and differentially expressed proteins. (A) A Venn diagram of all proteins identified in our study. (B) A Venn diagram of the differentially expressed proteins identified in our study.
Among 1931 proteins, 1209 (63%) proteins were identified by more than 3 peptides, while 868 (45%) proteins were identified by more than 4 peptides, and 629 (33%) were identified by more than 5 peptides (Supplementary Fig. S2). More detailed proteomic information is provided in Supplementary Tables S2 and S3.
Label-free quantitation
For label-free quantification, the technical and biological variability was evaluated by calculating the correlation value (R2) with the representative dNSAF values of each protein after converting the unweighted spectrum counts that were obtained from Scaffold 3 to dNSAF values (Supplementary Table S3). Correlations for the technical and biological replicates in each cell line ranged from 0.8132 to 0.975, indicating that our proteomic strategy is highly reproducible. Additionally, the correlations for dataset comparisons were as follows: HPDE vs. BxPC3 ranged from 0.6777 to 0.7961, HPDE vs. PANC-1 ranged from 0.3325 to 0.4359, and BxPC3 vs. PANC-1 ranged from 0.4639 to 0.5998 (Supplementary Fig. S3A).
Next, to calculate the CV of the proteins that we identified, the spectral counts for groups of high-, medium-, and low-abundance proteins were ≥ 15, ≥ 5 and <15, > 0 and < 5, respectively. The average CV in the high- and medium-abundance protein groups was 11.7% and 20.0%, respectively. These CV values suggested that the measurements of these two groups were reliable, whereas the CV of the low-abundance group was relatively high (∼64.5%). The average CV for all proteins identified was ∼53.8% (Supplementary Fig. S3B). The CV for ovalbumin as the external standard and the CVs for actin and GAPDH, as the internal standards, were 25.4%, 17.3%, and 19.1% against the 27 datasets, respectively. The CVs for the external and internal standards suggest that this quantitative experiment has no bias or overestimation in quantitating proteins.
Finally, principal component analysis (PCA) was performed on a matrix consisting of the objects defined by 3 biological and 3 technical replicates of the 3 cell lines, and the variables were defined as the dNSAF values of each protein identified in each cell line. The PCA results revealed the correct segregation of the cell lines and each replicate into their corresponding groups, which had been generated based on component 1 (Supplementary Fig. S4), the component that accounts for the largest amount of variability in the system (59.8% in this case). Consequently, the correlations, CVs, and PCA results in the technical and biological replicates all indicated that our proteomic strategy is robust and highly reproducible.
To quantify differentially expressed proteins, pairwise PLGEM analysis was performed. The dNSAF values of 1931 proteins that were identified in 27 replicates were imported into PLGEM to select differentially expressed proteins. PLGEM has been regarded as a reliable model capable of handling large proteomic datasets. PLGEM has been proposed to improve the confidence of proteomic datasets. Pearson’s correlation was calculated between ln (row, Mean) values and the corresponding ln (row, S.D.) values for quality control, and an adjusted R2 value was obtained between the modeling points and the fitted PLGEM. Overall, the PLGEM fitted well on all dataset comparisons with correlation coefficients of 0.948 and 0.938, adjusted R2 values of 0.995 and 0.994, and fitted slope values 0.626 and 0.63 in HPDE-control and BxPC3-control datasets, respectively (Supplementary Figs. S5A, S5E, and S5I). If the fitted slope value was less than 0.5, or the Pearson correlation coefficient was less than 0.85, or the adjusted R2 value was less than 0.95, the dataset quality was considered to be poor. Additionally, all proteins identified in this study showed a normal distribution and broad dynamic range (Supplementary Figs. S5B, S5C, S5F, S5G, S5J, and S5K). In addition, the relationship between the standard normal distribution and the distribution about observed residuals showed a good correlation (Supplementary Figs. S5D, S5H, and S5L).
Finally, 837 of 1931 proteins were found to be significantly differentially expressed proteins with an adjusted p-value < 0.05 for at least one pair-wise comparison. After filtering based on 2-fold changes of dNSAF values, a total of 787 proteins (94%) ultimately met this criterion for at least one pair-wise comparison. Overall, 267, 628, and 399 proteins were detected in each pair-wise comparison (Fig. 2B). Detailed information about the differentially expressed proteins is provided in Supplementary Table S4.
Clustering of the differentially expressed protein signatures
To extract protein signatures that significantly segregated the three cell lines, we performed K-means clustering of the 711 differentially expressed proteins, which yielded 6 clusters. Each protein cluster was enriched in a specific pathway based on KEGG pathway analysis (Supplementary Figs. S6 and Supplementary Table S5). In clusters 0 and 5, proteins were down-regulated and up-regulated, respectively, in BxPC3 cells. The proteins that were included in cluster 0 were not significantly annotated to specific pathways; p < 0.05. Proteins that were included in cluster 5 were significantly enriched for seven pathways: ARVC (arrhythmogenic right ventricular cardiomyopathy), fatty acid metabolism, antigen processing and presentation, viral myocarditis, tight junction, adherens junction, and cardiac muscle contraction. In clusters 1 and 4, proteins were increased or decreased in PANC-1 cells compared with other cell lines, respectively. The proteins in cluster 1 were enriched for eight pathways: ribosome, aminoacyl-tRNA biosynthesis, pyrimidine metabolism, pathogenic Escherichia coli infection, glycoslysis/gluconeogenesis, drug metabolism, purine metabolism, and proteasome pathways. The proteins in cluster 4 were significantly associated with two pathways: the lysosome and Vibrio cholerae infection pathways. In cluster 2, proteins were down-regulated in both cancer cell lines, whereas proteins in cluster 3 were up-regulated. Proteins in cluster 2 were significantly associated with 18 pathways, including the arginine and proline metabolism, ECM-receptor interaction, TCA cycle, limonene and pinene degradation, and glycolysis/gluconeogenesis pathways. The proteins involved in cluster 3 were associated with two pathways: the ribosome and glycolysis/gluconeogenesis pathways.
Clustering results were consistent with the properties of each cell line. For example, cluster 1 mostly includes up-regulated proteins in PANC-1 cells that were enriched for ribosome, biosynthesis, and metabolism activities. By contrast, cluster 5 mostly includes proteins that are up-regulated in BxPC3 cells that were enriched for tight junction and adherens junction functions, which are closely related to epithelial cell properties. Both BxPC3 and PANC-1 cells are epithelial primary cancer cell lines, but BxPC3 cells exhibit epithelial properties, whereas PANC-1 cells exhibit more mesenchymal properties.
Functional analyses of the pathways associated with gemcitabine resistance
Previous studies have shown that gemcitabine resistance is closely associated with the acquisition of an epithelial-mesenchymal transition (EMT)-like phenotype by cancer cells (Kalluri and Weinberg, 2009). The EMT is characterized by the loss of cell-cell adhesion and the acquisition of cell motility, which leads to increased invasion ability (Kalluri and Weinberg, 2009). The progression of the EMT involves the loss of proteins involved in cell junctions, such as E-cadherin and claudins, and the expression of mesenchymal molecular markers, such as fibronectin, vimentin, and N-cadherin (Kalluri and Weinberg, 2009).
A total 15 of well-known EMT markers were identified in this study: ANPEP, ALCAM, DSG3, DSG2, KRT 14, KRT 19, KRT 8, CLDN1, VIM, CDH1, SDC1, CD44, ITGB1, NT5E, and DSP (Kalluri and Weinberg, 2009). Further, 13 proteins that are closely associated with the EMT phenotype were detected: CAV1, IQGAP1, ITGB4, ITGA6, CTNNB1, ACTN4, FLNA, FLNB, KRT18, MYH14, MYH9, MYL6, and PXN (Table 2).
Table 2.
List of EMT-related proteins and proteins involved in Glutathione pathway which annotated in our proteome study
| Pathway | IPI accession | Protein name | Gene symbol | Fold change | Note | ||
|---|---|---|---|---|---|---|---|
|
| |||||||
| HPDE Versus BxPC3 | HPDE Versus PANC-1 | BxPC3 Versus PANC-1 | |||||
| EMT pathway | IPI00221224 | Aminopeptidase N | ANPEP | 0 | 1000 | 1000 | Mesenchymal marker |
| IPI00015102 | Isoform 1 of CD166 antigen | ALCAM | 2.012 | 0.140 | 0.070 | Epithelial marker | |
| IPI00031547 | Desmoglein-3 | DSG3 | −1000 | −1000 | 0 | Epithelial marker | |
| IPI00028931 | Desmoglein-2 | DSG2 | 51.663 | −1000 | −1000 | Epithelial marker | |
| IPI00384444 | Keratin, type I cytoskeletal 14 | KRT14 | 0.003 | 0.002 | 0.947 | Epithelial marker | |
| IPI00479145 | Keratin, type I cytoskeletal 19 | KRT19 | 0.904 | 0.077 | 0.085 | Epithelial marker | |
| IPI00554648 | Keratin, type II cytoskeletal 8 | KRT8 | 0.959 | 0.625 | 0.652 | Epithelial marker | |
| IPI00000691 | Claudin-1 | CLDN1 | 1000 | 0 | −1000 | Epithelial marker | |
| IPI00009236 | Isoform Alpha of Caveolin-1 | CAV1 | 2.691 | 1.280 | 0.476 | ||
| IPI00009342 | Ras GTPase-activating-like protein IQGAP1 | IQGAP1 | 4.962 | 8.359 | 1.684 | ||
| IPI00216221 | Isoform Alpha-6X1A of Integrin alpha-6 | ITGA6 | 0.331 | −1000 | −1000 | ||
| IPI00013808 | Alpha-actinin-4 | ACTN4 | 8.172 | 12.875 | 1.575 | ||
| IPI00302592 | Isoform 2 of Filamin-A | FLNA | 2.756 | 1.943 | 0.705 | ||
| IPI00289334 | Isoform 1 of Filamin-B | FLNB | 5.026 | 1.918 | 0.382 | ||
| IPI00554788 | Keratin, type I cytoskeletal 18 | KRT18 | 1.289 | 0.401 | 0.311 | ||
| IPI00337335 | Isoform 1 of Myosin-14 | MYH14 | 6.322 | 1.030 | 0.163 | ||
| IPI00019502 | Isoform 1 of Myosin-9 | MYH9 | 3.257 | 1.178 | 0.362 | ||
| IPI00335168 | Isoform Non-muscle of Myosin light polypeptide 6 | MYL6 | 5.961 | 2.396 | 0.402 | ||
| IPI00220030 | Isoform Alpha of Paxillin | PXN | 0 | 1000 | 1000 | ||
| IPI00220845 | Isoform Beta-4A of Integrin beta-4 | ITGB4 | 0.251 | −1000 | −1000 | ||
| IPI00418471 | Vimentin | VIM | 0.654 | 47.428 | 72.513 | Mesenchymal marker | |
| IPI00017292 | Isoform 1 of Catenin beta-1 | CTNNB1 | 3.928 | 0.044 | 0.011 | ||
| IPI00025861 | Cadherin-1 | CDH1 | 1.392 | −1000 | −1000 | Epithelial marker | |
| IPI00002441 | Syndecan-1 | SDC1 | −1000 | −1000 | 0 | Mesenchymal marker | |
| IPI00418465 | Isoform 4 of CD44 antigen | CD44 | 1.011 | 5.672 | 5.609 | Mesenchymal marker | |
| IPI00217563 | Isoform Beta-1A of Integrin beta-1 | ITGB1 | 3.409 | 3.667 | 1.076 | Mesenchymal marker | |
| IPI00009456 | 5′-nucleotidase | NT5E | 1000 | 1000 | 2.628 | Mesenchymal marker | |
| IPI00013933 | Isoform DPI of Desmoplakin | DSP | 4.601 | 0.009 | 0.002 | Epithelial marker | |
| Glutathione pathway | IPI00216008 | Isoform Long of Glucose-6-phosphate 1-dehydrogenase | G6PD | 1.992 | 4.189 | 2.103 | |
| IPI00024266 | Microsomal glutathione S-transferase 3 | MGST3 | 0.045 | 0.473 | 10.406 | ||
| IPI00011118 | ribonucleoside-diphosphate reductase subunit M2 isoform 1 | hCG_23833 | 1000 | 1000 | 14.503 | ||
| IPI00005102 | Isoform 1 of Spermine synthase | SMS | −1000 | 3.156 | 1000 | Novel in this study | |
| IPI00221224 | Aminopeptidase N | ANPEP | 0 | 1000 | 1000 | Novel in this study | |
| IPI00010157 | S-adenosylmethionine synthase isoform type-2 | MAT2A | 0.202 | 2.788 | 13.829 | Novel in this study | |
| IPI00217966 | Isoform 1 of L-lactate dehydrogenase A chain | LDHA | 0.838 | 2.949 | 3.522 | Novel in this study | |
| IPI00219029 | Aspartate aminotransferase, cytoplasmic | GOT1 | 1000 | 1000 | 17.912 | Novel in this study | |
Note: remarkable substance for each protein
Twenty eight proteins were discovered as EMT-related proteins at previous papers and 8 proteins were annotated with Glutathione pathway which involved in drug resistance. The average values of fold changes for each compare set are shown at this table. The fold changes of extremely changed proteins are represented at −1000 and 1000, respectively. The proteins which were not identified for each compare set are represented at 0.
CDH1, an epithelial cell marker, was significantly downergulated, and VIM, a mesenchymal cell marker, was significantly upregulated in PANC-1 cells compared with BxPC3 cells. The suppression of CDH1 is a critical step in enhancing the invasiveness and EMT in carcinomas (Thiery et al., 2009). This result suggests that the EMT progressed further in PANC-1 versus BxPC3 cells.
Mechanistically, CAV1 mediates focal adhesion and cytoskeletal organization and promotes the EMT through the focal adhesion pathway (Bailey and Liu, 2008). In this study, the abundance of CAV1 increased ∼3-fold in PANC-1 cells. IQGAP1 regulates the actin cytoskeleton and resistance to gemcitabine (Jameson et al., 2013). Also, IQGAP1 mediates the disruption of CDH1 and promotes tumor progression (Cavallaro and Christofori, 2004). IQGAP1 was upregulated ∼5-fold in BxPC3 cells and ∼8-fold in PANC-1 cells.
SDC1 governs ECM-receptor interactions, cell-cell interactions, cell proliferation, and cell migration (Juuti et al., 2005), and its levels are depressed in a pancreatic cancer cell line (Gronborg et al., 2006). SDC1 had a similar expression pattern in this proteomic dataset, being markedly decreased in BxPC3 and PANC-1 cells.
ITGB1 has been linked to the invasiveness of pancreatic carcinoma, and its activity correlates with the invasiveness of pancreatic carcinoma cell lines, based on in vitro chemoinvasion assays (Arao et al., 2000). ITGB1 is increased ∼3-fold in 2 pancreatic cancer cell lines compared with HPDE cells. ITGB4-ITGA6 complexes are involved in epithelial cell migration (Mercurio et al., 2001). In this study, ITGB4 and ITGA6 were downregulated in both tumor cell lines.
CTNNB1 binds to the cytoplasmic tails of CDH1 (Reynolds et al., 1994), and declined ∼22-fold in PANC-1 cells. ACTN4 regulates the focal adhesion pathway (Wei et al., 2011) and was consistently upregulated in pancreatic cancer cell lines versus HPDE cells (∼8-fold in BxPC3 cells and ∼13-fold in PANC-1 cells). Filamin subunits participate in focal adhesion and cytoskeletal organization. Upregulation of FLNA and FLNB has been reported in pancreatic cancers and precancerous lesions (Buchholz et al., 2005; Chen et al., 2005; Sato et al., 2004; Yu et al., 2009). FLNA and FLNB were increased ∼3-fold and ∼5-fold, respectively, in BxPC3 cells.
The keratin family is associated with the differentiation of epithelial cancer cells (Seike et al., 2004). KRT18 expression is downregulated in pancreatic cancer cells (Yu et al., 2009) and declined by more than 2-fold in PANC-1 cells in this study. The myosin family is involved in cytoskeletal composition and cellular movement (Wei et al., 2011). We detected MYH6, MYH9, and MYL14 expression in this study, which were upregulated in pancreatic cancer. PXN is a marker of focal adhesion (Wei et al., 2011), which is a crucial pathway for the EMT (Bailey and Liu, 2008). PXN expression increased significantly in PANC-1 cells.
In summary, several EMT markers were identified as being differentially expressed between BxPC3 and PANC-1 cells, indicating that PANC-1 cells show decreased expression of epithelial markers and increased expression of mesenchymal markers. Additionally, analysis of the expression patterns of several proteins that were either directly or indirectly associated with the EMT revealed that PANC-1 cells acquired a phenotype that resembled the EMT more closely than did BxPC3 cells. This finding suggests that the expression of EMT-related proteins correlates with gemcitabine resistance.
With regard to therapeutics, the 28 differentially expressed proteins that we identified are potential targets for gemcitabine resistance in pancreatic cancer, because the EMT pathway is related to drug resistance (Arumugam et al., 2009; Singh and Settleman, 2010; Voulgari and Pintzas, 2009). In addition, these proteins have not been studied in earlier pancreatic cancer proteomic studies of drug resistance by mass spectrometry (Chen et al., 2011; Kuramitsu et al., 2010; 2012; Mori-Iwamoto et al., 2007; 2008; Poland et al., 2004; Zhou and Du, 2012), suggesting that these proteins, related to the EMT phenotype, are novel targets for the treatment of drug-resistant pancreatic cancer.
Furthermore, to investigate pathways that underlie the molecular mechanism of gemcitabine resistance, we analyzed differentially expressed proteins in BxPC3 and PANC-1 cells using the KEGG pathways database (Fig. 3). Most of the 178 up-regulated proteins in BxPC3 cells were linked to the fatty acid degradation, phenylalanine metabolism, tight junction, and adherens junction pathways (Fig. 3A). Notably, ten proteins linked to the tight junction and adherens junction pathways were also closely associated with the progression of the EMT, suggesting that increased expression of cell-cell adhesion proteins may be associated with increased gemcitabine sensitivity in pancreatic cancer cells (Supplementary Table S6).
Fig. 3.
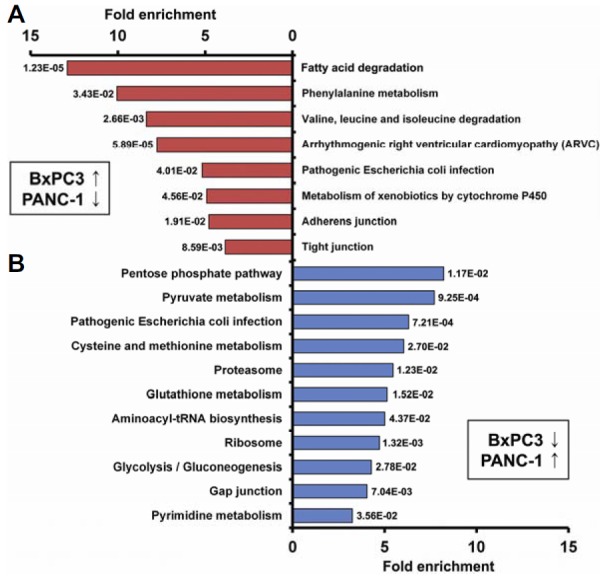
KEGG pathway analysis. KEGG pathway analysis results for comparison 1 (up-regulated proteins in BxPC3 cells compared with PANC-1 cells) (A), and for comparison 2 (up-regulated proteins in PANC-1 cells compared with BxPC3 cells) (B) are shown. The x- and y-axes represent the fold enrichment (indicating the magnitude of enrichment in our dataset against the population background based on analysis using DAVID bioinformatics tools) and the categories of pathways, respectively.
The majority of up-regulated proteins in PANC-1 cells are likely to be involved in metabolic pathways, such as pyruvate metabolism, the pentose phosphate pathway, glycolysis/gluconeogenesis, pyrimidine metabolism, and aminoacyl-tRNA biosynthesis (Fig. 3B and Supplementary Table S6), which is consistent with the relationship between gemcitabine resistance and metabolic pathways (Chen et al., 2011; Fryer et al., 2011; Kuramitsu et al., 2010).
Glutathione is a tripeptide thiol, composed of cysteine, glutamate, and glycine, which plays a critical role in DNA synthesis, multidrug and/or radiation resistance, and tumor surveillance (Griffith, 1999). Glutathione biosynthesis is critically dependent upon the metabolism of cysteine and methionine (Griffith, 1999). In several cancers, glutathione and cysteine/methionine metabolism have been previously associated with drug resistance (Lo et al., 2008). However, the characterization of these metabolism-related proteins in the development of gemcitabine resistance in pancreatic cancer cells has been limited (Lo et al., 2008; Locasale, 2013; Sato et al., 2011).
Interestingly, we observed that eight proteins linked to the glutathione and cysteine/methionine metabolism pathways were up-regulated in PANC-1 cells (Supplementary Table S7). In addition to the previously reported proteins (G6PD, MGST3, and hCG_23833) (Chen et al., 2011; Kuramitsu et al., 2010; Mori-Iwamoto et al., 2007; 2008; Poland et al., 2004; Zhou and Du, 2012), we were the first to identify the expression of five such metabolism-related proteins, SMS, ANPEP, MAT2A, LDHA, and GOT1, in this study. Thus, these 5 proteins could represent novel targets for gemcitabine resistance in pancreatic cancer. Detailed information about these proteins is presented in Supplementary Table S7.
Validation using western blotting and comparisons with transcriptome data
To validate our mass spectrometry quantification results, we measured changes in the abundance of seven proteins (VIM, CDH1, CTNNB1, FGFBP1, IQGAP1, FLNB, and STAT3) by Western blotting (Fig. 4A). The analyses of the three different cell lines showed similar changes in protein abundance by both mass spectrometry quantification and western blotting, supporting the reliability of our label-free quantitation results.
Fig. 4.
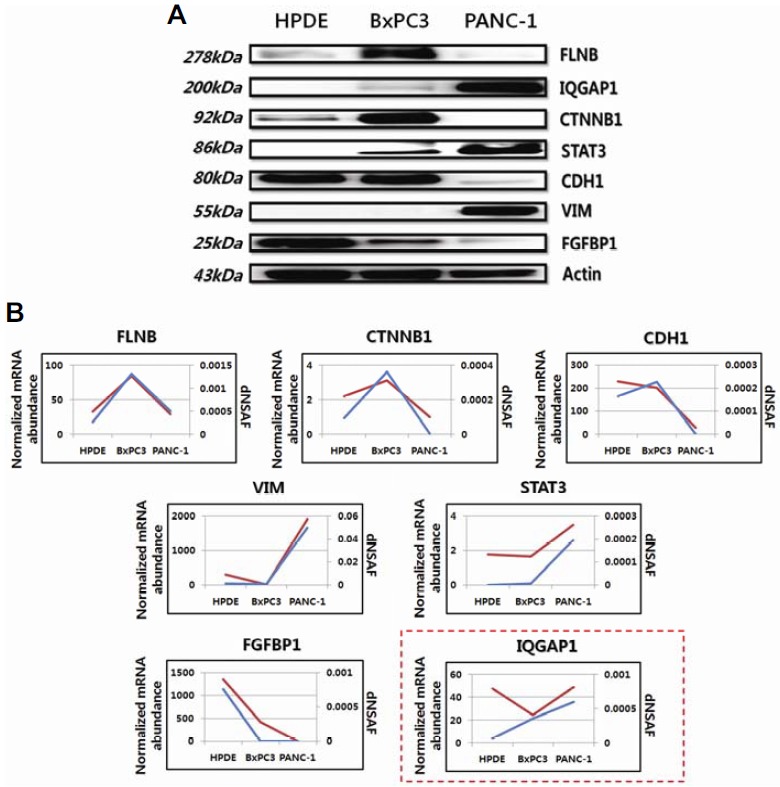
Western blotting and comparisons between genomic and proteomic data. (A) Western blotting to validate the label-free quantitation results. Actin was used as a loading control. Expression patterns of seven proteins (FLNB, CTNNB1, VIM, CDH1, IQGAP1, FGFBP1, and STAT3) for each cell line in Western blot assays were consistent with the label-free quantitation results. (B) A comparison between proteomic and genomic expression patterns of proteins measured in (A). In each plot, the blue and red lines indicate proteomic and genomic data, respectively. The expression patterns of 6 proteins (FLNB, CTNNB1, VIM, CDH1, FGFBP1, and STAT3) for each cell line were correlated with the genomic data, except for IQGAP1 (red-box). The y-axis represents normalized mRNA abundance for genomic data and dNSAF values for proteomic data.
We next assessed the correlation between our proteomic datasets and public gene expression datasets (Thu et al., 2014) for the three pancreatic cell lines. For three dataset comparisons (HPDE vs. BxPC3, HPDE vs. PANC-1, and BxPC3 vs. PANC-1), Pearson correlation analysis was performed. Overall correlations between the proteomic and transcriptomic data were calculated as 0.145, 0.435, and 0.528 for the three dataset comparisons (HPDE vs. BxPC3, HPDE vs. PANC-1, and BxPC3 vs. PANC-1), respectively (Supplementary Table S8). Notably, the mRNA expression patterns of genes encoding the seven proteins assessed by western blotting were significantly correlated with the label-free quantitation results (Fig. 4B).
In summary, a comprehensive proteomic analysis of model pancreatic cancer cell lines was conducted to investigate the molecular mechanisms of drug resistance and PC biology. We performed a comprehensive proteome analysis of two PDAC cell lines (BxPC3 and PANC-1) and a normal pancreatic ductal cell line (HPDE) using combined proteomic methods, including FASP, dNSAF, and PLGEM. We confidently identified 1931 proteins and a subset of proteins that may be associated with intrinsic gemcitabine resistance. We found that the majority of differentially expressed proteins are involved in tight junctions, adherens junctions, and glutathione and cysteine/methionine metabolism pathways, which are related to drug resistance in pancreatic cancer. We identified 15 known EMT markers and 13 EMT-related proteins in the comparison between BxPC3 and PANC-1 cells, suggesting that the EMT-related proteins may confer gemcitabine chemoresistance. Using well-validated pancreatic cell lines, we generated comprehensive proteome profiling data that can serve as baseline information associated with gemcitabine resistance in the major pancreatic cell lines. Further studies are needed to validate the functional roles of the other proteins that we identified in conferring gemcitabine resistance.
Acknowledgments
This work was supported by a National Research Foundation of Korea grant (No. 2011-0030740) and the Proteogenomic Research Program, funded by the Korea government [MISP]. This work was also supported by the Industrial Strategic Technology Development Program (#10045352), funded by the Ministry of Knowledge Economy (MKE, Korea). This research was also supported by a grant of the Korea Health Technology R&D Project through the KHIDI, funded by the Ministry of Health & Welfare, Republic of Korea (No. HI14C1277).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Arao S., Masumoto A., Otsuki M. Beta1 integrins play an essential role in adhesion and invasion of pancreatic carcinoma cells. Pancreas. 2000;20:129–137. doi: 10.1097/00006676-200003000-00004. [DOI] [PubMed] [Google Scholar]
- Arumugam T., Ramachandran V., Fournier K.F., Wang H., Marquis L., Abbruzzese J.L., Gallick G.E., Logsdon C.D., McConkey D.J., Choi W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820–5828. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey K.M., Liu J. Caveolin-1 up-regulation during epithelial to mesenchymal transition is mediated by focal adhesion kinase. J. Biol. Chem. 2008;283:13714–13724. doi: 10.1074/jbc.M709329200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz M., Braun M., Heidenblut A., Kestler H.A., Kloppel G., Schmiegel W., Hahn S.A., Luttges J., Gress T.M. Transcriptome analysis of microdissected pancreatic intraepithelial neoplastic lesions. Oncogene. 2005;24:6626–6636. doi: 10.1038/sj.onc.1208804. [DOI] [PubMed] [Google Scholar]
- Burris H.A., 3rd, Moore M.J., Andersen J., Green M.R., Rothenberg M.L., Modiano M.R., Cripps M.C., Portenoy R.K., Storniolo A.M., Tarassoff P., et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J. Clin. Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- Cao H., Le D., Yang L.X. Current status in chemotherapy for advanced pancreatic adenocarcinoma. Anticancer Res. 2013;33:1785–1791. [PubMed] [Google Scholar]
- Carmichael J., Fink U., Russell R.C., Spittle M.F., Harris A.L., Spiessi G., Blatter J. Phase II study of gemcitabine in patients with advanced pancreatic cancer. Br. J. Cancer. 1996;73:101–105. doi: 10.1038/bjc.1996.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallaro U., Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer. 2004;4:118–132. doi: 10.1038/nrc1276. [DOI] [PubMed] [Google Scholar]
- Chen R., Yi E.C., Donohoe S., Pan S., Eng J., Cooke K., Crispin D.A., Lane Z., Goodlett D.R., Bronner M.P., et al. Pancreatic cancer proteome: the proteins that underlie invasion, metastasis, and immunologic escape. Gastroenterology. 2005;129:1187–1197. doi: 10.1053/j.gastro.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Chen Y.W., Liu J.Y., Lin S.T., Li J.M., Huang S.H., Chen J.Y., Wu J.Y., Kuo C.C., Wu C.L., Lu Y.C., et al. Proteomic analysis of gemcitabine-induced drug resistance in pancreatic cancer cells. Mol. BioSyst. 2011;7:3065–3074. doi: 10.1039/c1mb05125c. [DOI] [PubMed] [Google Scholar]
- Cheung H.W., Cowley G.S., Weir B.A., Boehm J.S., Rusin S., Scott J.A., East A., Ali L.D., Lizotte P.H., Wong T.C., et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc. Natl. Acad. Sci. USA. 2011;108:12372–12377. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C., Mann M. Decoding signalling networks by mass spectrometry-based proteomics. Nat. Rev. 2010;11:427–439. doi: 10.1038/nrm2900. [DOI] [PubMed] [Google Scholar]
- Deer E.L., Gonzalez-Hernandez J., Coursen J.D., Shea J.E., Ngatia J., Scaife C.L., Firpo M.A., Mulvihill S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010;39:425–435. doi: 10.1097/MPA.0b013e3181c15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonslow B.R., Stein B.D., Webb K.J., Xu T., Choi J., Park S.K., Yates J.R., 3rd Digestion and depletion of abundant proteins improves proteomic coverage. Nat Methods. 2013;10:54–56. doi: 10.1038/nmeth.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer R.A., Barlett B., Galustian C., Dalgleish A.G. Mechanisms underlying gemcitabine resistance in pancreatic cancer and sensitisation by the iMiD lenalidomide. Anticancer Res. 2011;31:3747–3756. [PubMed] [Google Scholar]
- Furukawa T., Duguid W.P., Rosenberg L., Viallet J., Galloway D.A., Tsao M.S. Long-term culture and immortalization of epithelial cells from normal adult human pancreatic ducts transfected by the E6E7 gene of human papilloma virus 16. Am. J. Pathol. 1996;148:1763–1770. [PMC free article] [PubMed] [Google Scholar]
- Griffith O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999;27:922–935. doi: 10.1016/s0891-5849(99)00176-8. [DOI] [PubMed] [Google Scholar]
- Gronborg M., Kristiansen T.Z., Iwahori A., Chang R., Reddy R., Sato N., Molina H., Jensen O.N., Hruban R.H., Goggins M.G., et al. Biomarker discovery from pancreatic cancer secretome using a differential proteomic approach. Mol. Cell. Proteomics. 2006;5:157–171. doi: 10.1074/mcp.M500178-MCP200. [DOI] [PubMed] [Google Scholar]
- Gstaiger M., Aebersold R. Applying mass spectrometry-based proteomics to genetics, genomics and network biology. Nat. Revi. Genet. 2009;10:617–627. doi: 10.1038/nrg2633. [DOI] [PubMed] [Google Scholar]
- Han D., Moon S., Kim H., Choi S.E., Lee S.J., Park K.S., Jun H., Kang Y., Kim Y. Detection of differential proteomes associated with the development of type 2 diabetes in the Zucker rat model using the iTRAQ technique. J. Proteome Res. 2011;10:564–577. doi: 10.1021/pr100759a. [DOI] [PubMed] [Google Scholar]
- Han D., Moon S., Kim Y., Ho W.K., Kim K., Kang Y., Jun H., Kim Y. Comprehensive phosphoproteome analysis of INS-1 pancreatic beta-cells using various digestion strategies coupled with liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2012;11:2206–2223. doi: 10.1021/pr200990b. [DOI] [PubMed] [Google Scholar]
- Hruban R.H., Goggins M., Parsons J., Kern S.E. Progression model for pancreatic cancer. Clin. Cancer Res. 2000;6:2969–2972. [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huanwen W., Zhiyong L., Xiaohua S., Xinyu R., Kai W., Tonghua L. Intrinsic chemoresistance to gemcitabine is associated with constitutive and laminin-induced phosphorylation of FAK in pancreatic cancer cell lines. Mol. Cancer. 2009;8:125. doi: 10.1186/1476-4598-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson K.L., Mazur P.K., Zehnder A.M., Zhang J., Zarnegar B., Sage J., Khavari P.A. IQGAP1 scaffold-kinase interaction blockade selectively targets RAS-MAP kinase-driven tumors. Nat. Med. 2013;19:626–630. doi: 10.1038/nm.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S., Zhang X., Parsons D.W., Lin J.C., Leary R.J., Angenendt P., Mankoo P., Carter H., Kamiyama H., Jimeno A., et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juuti A., Nordling S., Lundin J., Louhimo J., Haglund C. Syndecan-1 expression--a novel prognostic marker in pancreatic cancer. Oncology. 2005;68:97–106. doi: 10.1159/000085702. [DOI] [PubMed] [Google Scholar]
- Kalluri R., Weinberg R.A. The basics of epithelialmesenchymal transition. J. Clin. Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramitsu Y., Taba K., Ryozawa S., Yoshida K., Zhang X., Tanaka T., Maehara S., Maehara Y., Sakaida I., Nakamura K. Identification of up- and down-regulated proteins in gemcitabine-resistant pancreatic cancer cells using two-dimensional gel electrophoresis and mass spectrometry. Anticancer Res. 2010;30:3367–3372. [PubMed] [Google Scholar]
- Kuramitsu Y., Wang Y., Taba K., Suenaga S., Ryozawa S., Kaino S., Sakaida I., Nakamura K. Heat-shock protein 27 plays the key role in gemcitabine-resistance of pancreatic cancer cells. Anticancer Res. 2012;32:2295–2299. [PubMed] [Google Scholar]
- Li D., Xie K., Wolff R., Abbruzzese J.L. Pancreatic cancer. Lancet. 2004;363:1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- Lo M., Ling V., Wang Y.Z., Gout P.W. The xccystine/glutamate antiporter: a mediator of pancreatic cancer growth with a role in drug resistance. Br. J. Cancer. 2008;99:464–472. doi: 10.1038/sj.bjc.6604485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale J.W. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer. 2013;13:572–583. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makawita S., Smith C., Batruch I., Zheng Y., Ruckert F., Grutzmann R., Pilarsky C., Gallinger S., Diamandis E.P. Integrated proteomic profiling of cell line conditioned media and pancreatic juice for the identification of pancreatic cancer biomarkers. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M111.008599. M111 008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercurio A.M., Rabinovitz I., Shaw L.M. The alpha 6 beta 4 integrin and epithelial cell migration. Curr. Opin. Cell Biol. 2001;13:541–545. doi: 10.1016/s0955-0674(00)00249-0. [DOI] [PubMed] [Google Scholar]
- Min H., Han D., Kim Y., Cho J.Y., Jin J., Kim Y. Label-free quantitative proteomics and N-terminal analysis of human metastatic lung cancer cells. Mol. Cells. 2014;37:457–466. doi: 10.14348/molcells.2014.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori-Iwamoto S., Kuramitsu Y., Ryozawa S., Mikuria K., Fujimoto M., Maehara S., Maehara Y., Okita K., Nakamura K., Sakaida I. Proteomics finding heat shock protein 27 as a biomarker for resistance of pancreatic cancer cells to gemcitabine. Int. J. Oncol. 2007;31:1345–1350. [PubMed] [Google Scholar]
- Mori-Iwamoto S., Kuramitsu Y., Ryozawa S., Taba K., Fujimoto M., Okita K., Nakamura K., Sakaida I. A proteomic profiling of gemcitabine resistance in pancreatic cancer cell lines. Mol. Med. Rep. 2008;1:429–434. [PubMed] [Google Scholar]
- Pavelka N., Fournier M.L., Swanson S.K., Pelizzola M., Ricciardi-Castagnoli P., Florens L., Washburn M.P. Statistical similarities between transcriptomics and quantitative shotgun proteomics data. Mol. Cell. Proteomics. 2008;7:631–644. doi: 10.1074/mcp.M700240-MCP200. [DOI] [PubMed] [Google Scholar]
- Poland J., Urbani A., Lage H., Schnolzer M., Sinha P. Study of the development of thermoresistance in human pancreatic carcinoma cell lines using proteome analysis. Electrophoresis. 2004;25:173–183. doi: 10.1002/elps.200305698. [DOI] [PubMed] [Google Scholar]
- Pramanik K.C., Boreddy S.R., Srivastava S.K. Role of mitochondrial electron transport chain complexes in capsaicin mediated oxidative stress leading to apoptosis in pancreatic cancer cells. PLoS One. 2011;6:e20151. doi: 10.1371/journal.pone.0020151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovich N., Qian J.Y., Tsao M.S. Human pancreatic duct epithelial cell model for KRAS transformation. Methods Enzymol. 2008;439:1–13. doi: 10.1016/S0076-6879(07)00401-6. [DOI] [PubMed] [Google Scholar]
- Rathos M.J., Joshi K., Khanwalkar H., Manohar S.M., Joshi K.S. Molecular evidence for increased antitumor activity of gemcitabine in combination with a cyclin-dependent kinase inhibitor, P276-00 in pancreatic cancers. J. Transl. Med. 2012;10:161. doi: 10.1186/1479-5876-10-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds A.B., Daniel J., McCrea P.D., Wheelock M.J., Wu J., Zhang Z. Identification of a new catenin: the tyrosine kinase substrate p120cas associates with E-cadherin complexes. Mo. Cell. Biol. 1994;14:8333–8342. doi: 10.1128/mcb.14.12.8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel N., Hudson T.J. The molecular and cellular heterogeneity of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2012;9:77–87. doi: 10.1038/nrgastro.2011.215. [DOI] [PubMed] [Google Scholar]
- Sato N., Fukushima N., Maitra A., Iacobuzio-Donahue C.A., van Heek N.T., Cameron J.L., Yeo C.J., Hruban R.H., Goggins M. Gene expression profiling identifies genes associated with invasive intraductal papillary mucinous neoplasms of the pancreas. Am. J. Pathol. 2004;164:903–914. doi: 10.1016/S0002-9440(10)63178-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato J., Kimura T., Saito T., Anazawa T., Kenjo A., Sato Y., Tsuchiya T., Gotoh M. Gene expression analysis for predicting gemcitabine resistance in human cholangiocarcinoma. J. Hepatobiliary Pancreat. Sci. 2011;18:700–711. doi: 10.1007/s00534-011-0376-7. [DOI] [PubMed] [Google Scholar]
- Seike M., Kondo T., Fujii K., Yamada T., Gemma A., Kudoh S., Hirohashi S. Proteomic signature of human cancer cells. Proteomics. 2004;4:2776–2788. doi: 10.1002/pmic.200300795. [DOI] [PubMed] [Google Scholar]
- Singh A., Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergachis A.B., MacLean B., Lee K., Stamatoyannopoulos J.A., MacCoss M.J. Rapid empirical discovery of optimal peptides for targeted proteomics. Nat. Methods. 2011;8:1041–1043. doi: 10.1038/nmeth.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney D.L., Wenger C.D., Coon J.J. Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J. Proteome Res. 2010;9:1323–1329. doi: 10.1021/pr900863u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery J.P., Acloque H., Huang R.Y., Nieto M.A. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Thu K.L., Radulovich N., Becker-Santos D.D., Pikor L.A., Pusic A., Lockwood W.W., Lam W.L., Tsao M.S. SOX15 is a candidate tumor suppressor in pancreatic cancer with a potential role in Wnt/beta-catenin signaling. Oncogene. 2014;33:279–288. doi: 10.1038/onc.2012.595. [DOI] [PubMed] [Google Scholar]
- Tuveson D.A., Neoptolemos J.P. Understanding metastasis in pancreatic cancer: a call for new clinical approaches. Cell. 2012;148:21–23. doi: 10.1016/j.cell.2011.12.021. [DOI] [PubMed] [Google Scholar]
- Voulgari A., Pintzas A. Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim. Biophysica Acta. 2009;1796:75–90. doi: 10.1016/j.bbcan.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Wei S., Gao X., Du J., Su J., Xu Z. Angiogenin enhances cell migration by regulating stress fiber assembly and focal adhesion dynamics. PLoS One. 2011;6:e28797. doi: 10.1371/journal.pone.0028797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski J.R., Zougman A., Nagaraj N., Mann M. Universal sample preparation method for proteome analysis. Nat. Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Wisniewski J.R., Ostasiewicz P., Mann M. High recovery FASP applied to the proteomic analysis of microdissected formalin fixed paraffin embedded cancer tissues retrieves known colon cancer markers. J. Proteome Res. 2011;10:3040–3049. doi: 10.1021/pr200019m. [DOI] [PubMed] [Google Scholar]
- Yu K.H., Barry C.G., Austin D., Busch C.M., Sangar V., Rustgi A.K., Blair I.A. Stable isotope dilution multidimensional liquid chromatography-tandem mass spectrometry for pancreatic cancer serum biomarker discovery. J. Proteome Res. 2009;8:1565–1576. doi: 10.1021/pr800904z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Wen Z., Washburn M.P., Florens L. Refinements to label free proteome quantitation: how to deal with peptides shared by multiple proteins. Anal. Chem. 2010;82:2272–2281. doi: 10.1021/ac9023999. [DOI] [PubMed] [Google Scholar]
- Zhou J., Du Y. Acquisition of resistance of pancreatic cancer cells to 2-methoxyestradiol is associated with the upregulation of manganese superoxide dismutase. Mol. Cancer Res. 2012;10:768–777. doi: 10.1158/1541-7786.MCR-11-0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann G., Papke B., Ismail S., Vartak N., Chandra A., Hoffmann M., Hahn S.A., Triola G., Wittinghofer A., Bastiaens P.I., et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.