

Abstract
Prostaglandin E2 (PGE2) promotes tumor-persistent inflammation, frequently resulting in cancer. Curcumin is a diphenolic turmeric that inhibits carcinogenesis and induces apoptosis. PGE2 inhibits curcumin-induced apoptosis; however, the underlying inhibitory mechanisms in colon cancer cells remain unknown. The aim of the present study is to investigate the survival role of PGE2 and whether addition of exogenous PGE2 affects curcumin-induced cell death. HCT-15 cells were treated with curcumin and PGE2, and protein expression levels were investigated via Western blot. Reactive oxygen species (ROS) generation, lipid peroxidation, and intracellular glutathione (GSH) levels were confirmed using specific dyes. The nuclear factor-kappa B (NF-κB) DNA-binding was measured by electrophoretic mobility shift assay (EMSA). PGE2 inhibited curcumin-induced apoptosis by suppressing oxidative stress and degradation of PARP and lamin B. However, exposure of cells to the EP2 receptor antagonist, AH6809, and the PKA inhibitor, H89, before treatment with PGE2 or curcumin abolished the protective effect of PGE2 and enhanced curcumin-induced cell death. PGE2 activates PKA, which is required for cAMP-mediated transcriptional activation of CREB. PGE2 also activated the Ras/Raf/Erk pathway, and pretreatment with PD98059 abolished the protective effect of PGE2. Furthermore, curcumin treatment greatly reduced phosphorylation of CREB, followed by a concomitant reduction of NF-κB (p50 and p65) subunit activation. PGE2 markedly activated nuclear translocation of NF-κB. EMSA confirmed the DNA-binding activities of NF-κB subunits. These results suggest that inhibition of curcumin-induced apoptosis by PGE2 through activation of PKA, Ras, and NF-κB signaling pathways may provide a molecular basis for the reversal of curcumin-induced colon carcinoma cell death.
Keywords: apoptosis, curcumin, prostaglandin E2, survival signal
INTRODUCTION
Prostaglandins (PGs) such as PGE2 are produced de novo from arachidonic acid, a polyunsaturated fatty acid, upon external or internal stimulus. The cytosolic phospholipase A2 (cPLA2) group of enzymes precisely controls cellular levels of arachidonic acid until mobilized by PGH synthase and PGH2 (Six and Dennis, 2000). PGH synthase exists in two isoforms, known as cyclooxygenase-1 and -2 (COX-1 and COX-2) (Funk, 2001). It has been shown that COX-1 is constitutively expressed and is responsible for prostaglandin synthesis, whereas COX-2 is inducible and is responsible for various pro-inflammatory activities. Based on the presence of a divergent carboxy-terminus, nine PG receptors have been identified in pre-clinical and clinical studies; four of which (EP1–EP4) bind to PGE2 (Funk, 2001; Sonoshita et al., 2001; Wang et al., 2004). Hence, numerous studies have established that COX-2 expression and up-regulation of its moderator PGE2 promote the development of colorectal tumorigenesis through the prostanoid EP2 receptor (Castellone et al., 2005). Mechanisms often overlapping PGE2 activation in colorectal cancer remain unknown. Thus, inhibition of inflammatory PGE2 using phytochemicals or by alteration of its regulation can prevent carcinogenesis.
The Ras/Raf/Erk cascades are important signal transduction pathways involved in the regulation of cell growth, proliferation, survival, and differentiation (Santarpia et al., 2012). Mutation and aberrant expression of the components of these pathways can deregulate signal transduction, resulting in mitogenic signaling and cancer progression (Roberts and Der, 2007). Ras is a small GTPase that induces Raf, ultimately activating MEK-associated extracellular signal-regulated kinases (Erk) by serial phosphorylation. Erk activation has been reported to prevent apoptosis in cancer cells (Fernando and Wimalasena, 2004). On the other hand, nuclear factor-kappa B (NF-κB) is a ubiquitous inflammatory transcription factor with anti-apoptotic effects that is involved in cell survival, proliferation, apoptosis, and cell differentiation (Sakamoto et al., 2009; Wang et al., 2009). NF-κB is constitutively expressed in various human cancers, including colorectal cancer, and is one of the major contributing factors to chemotherapy failure when attempting to induce apoptosis in cancer cells (Barnes and Karin, 1997). Therefore, inhibition of NF-κB in human malignancies could be a potential therapeutic strategy for colorectal cancer prevention (Baud and Karin, 2009). NF-κB consists of five interrelated subunits, of which p50 and p65 are the most common heterodimer forms (Seufert et al., 2013). In response to inflammatory stimuli, NF-κB is translocated to the nucleus where it encodes a large number of inflammatory genes that may be, directly or indirectly, responsible for cancer progression and development (Sakamoto et al., 2009; Wang et al., 2009). Thus, the Ras and NF-κB signaling network has been the focus of pharmaceutical research to discover novel approaches for cancer treatment.
Despite recent advancements in cancer prevention, diagnosis, and treatment, colorectal cancer remains the second leading cause of cancer-related deaths in both men and women in the United States (Shehzad et al., 2013b). Previously, it has been reported that curcumin efficiently reduced arachidonic acid metabolism by blocking the phosphorylation of cPLA2, decreasing the expression of COX-2 and the activation of 5-lipoxygenase (LOX) in RAW and HT-29 cells (Hong et al., 2004). Therefore, we selected human colorectal carcinoma (HCT-15) cells to investigate the mechanisms of curcumin-induced apoptosis as well as the effect of exogenous addition of PGE2. Curcumin induced oxidative-stress apoptosis through caspase-3 cleavage as well as through poly (ADP-ribose) polymerase (PARP) and lamin B degradation in HCT-15 cells. However, pretreatment with PGE2 inhibited curcumin-induced cell death through the EP2 receptor, as the specific EP2 antagonist, AH6809, abrogated the survival effect in HCT-15 cells. Furthermore, PGE2 reversed curcumin-induced apoptosis by activating protein kinase A (PKA), Ras, and NF-κB signaling pathways. We hope that this study provides new insights into the protection of cancer cells by PGE2 as well as the clinical application of curcumin for colorectal cancer treatment.
MATERIALS AND METHODS
Chemicals and reagents
Curcumin and propidium iodide (PI) were purchased from Sigma-Aldrich (Germany). All antibodies (PARP, lamin B, caspase-3, pCREB, catalytic PKA subunits, Ras GAP, pRaf, pErk, NF-κB subunits p50 and p65, RelB, IκBα, actin, and proliferating cell nuclear antigen (PCNA) were obtained from Santa Cruz Biotechnology (USA) and Cell Signaling (USA). Electrophoresis reagents and Bio-Rad protein assay kit were purchased from Bio-Rad Laboratories (USA). The 22-mer double-stranded NF-κB oligonucleotides were obtained from Promega (USA). Dichlorofluorescein diacetate (DCFHDA) was obtained from Molecular Probes (USA). PGE2, hydroxy PGE2, and butaprost were purchased from Cayman Chemicals (USA). H89 and PD98059 were obtained from Sigma Aldrich and Cell Signaling. Nitrocellulose membrane and X-ray reagents were purchased from (Amersham Pharmacia Biotech, UK). These chemicals were used according to the manufacturer’s instructions.
Cell culture and treatment
HCT-15 colorectal carcinoma cells were purchased from American Type Culture Collection, CCL-225 (USA). These cells were maintained at subconfluence in a 95% air and 5% CO2 humidified atmosphere at 37°C. For subculturing, RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), L-glutamine, and 1% (v/v) antibiotic-penicillin streptomycin (USA) was used. HCT-15 cells were seeded at 5 × 105 cells/ml and were subcultured when ∼70% confluent. PGE2 and hydroxy PGE2 were dissolved in ethanol, whereas curcumin, PD98059, AH6809, and H89 were dissolved in DMSO. HCT-15 cells were treated with 1 μM PGE2 for 2 h, followed by 20 μM curcumin. For further experiments, cells were exposed to PD98059, AH6809, and H89 before treatment with PGE2 and curcumin.
Apoptosis detection
To measure the level of apoptosis, HCT-15 cells were trypsinized and fixed with 70% ethanol. Cells then were stained with PI solution, and fluorescence staining of individual cells was analyzed using flow cytometry (Becton Dickinson). For cell counting, at least 20,000 events were stored. Each experiment was repeated at least six times.
Measurement of reactive oxygen species generation
To detect reactive oxygen species (ROS) production after treatment with 1 μM PGE2 followed by treatment with 30 μM curcumin, 5 μM DCFHDA solubilized in ethanol was added to the cell culture medium for 30 min in the dark. Cells were also exposed to 10 mM n-acetylcysteine (NAC), a known inhibitor of ROS generation. DCFH is a non-fluorescent dye but is converted to highly fluorescent DCF when oxidized by intracellular ROS and peroxides, and it has an excitation wavelength of 480 nm and an emission wavelength of 520 nm. HCT-15 cells were washed with 1 × PBS and then subjected to laser confocal scanning microscope (DM/R-TCS, Leica) coupled to a microscope (Leitz DM REB).
Measurement of lipid peroxidation
Lipid peroxidation was measured using the fluorescent probe diphenyl-1-pyrenylphosphine (DPPP) as previously described (Okimotoa et al., 2000). HCT-15 cells were incubated with 5 μM DPPP for 15 min in the dark and further exposed to 1 μM PGE2 or 20 μM curcumin for 30 min. DPPP is known to react with hydroperoxide, resulting in fluorescence. Images of DPPP fluorescence by ROS were measured by inverted light microscopy (Zeiss Axiovert 200) at an excitation wavelength of 351 nm and an emission wavelength of 380 nm.
Measurement of intracellular glutathione (GSH) level
HCT-15 cells were exposed to 1 μM PGE2 or 20 μM curcumin for 30 min after which cells were incubated with 5 μM 7-amino-4-chloromethylcoumarin (CMAC; fluorescent dye) for 30 min. CMAC forms an adduct with GSH, which is catalyzed by glutathione S-transferase. Images of CMAC cell tracker fluorescence by GSH were analyzed by fluorescence microscopy (Zeiss Axiovert, 200) at an excitation wavelength of 351 nm and an emission wavelength of 380 nm (Sebastia et al., 2003).
Fractionation of cytosolic and nuclear proteins
HCT-15 cells were harvested by trypsin-EDTA, collected by centrifugation, and washed two times with cold 1 × PBS. Fractions were prepared according to a previously described method (Rosner and Hengstschläger, 2008). Briefly, cell pellets were suspended in buffer A containing 100 mM HEPES, 2 M KCl, 0.1 M ethyleneglycol tetra-acetic acid (EGTA), 0.2 M ethylenediaminetetraacetic acid (EDTA), 1 M dithiothreitol (DTT), 1 mM sodium orthovanadate (Na3VO4), 100 mM phenylmethylsulfonyl fluoride (PMSF), and 6% IGEPAL (NP-40), followed by incubation for 2 min at room temperature and then 10 min on ice. The resuspended cell pellets were centrifuged at 600 × g for 5 min at 4°C, and supernatants containing cytoplasmic proteins were collected in a new tube. The remaining pellets were resuspended in buffer C containing 100 mM HEPES, 5 M NaCl, 0.1 M EGTA, 0.2 M EDTA, 1 M DTT, 1 mM Na3VO4, 100 mM PMSF, and protease inhibitors. The resuspended pellets were centrifuged at 20,000 × g for 15 min at 4°C, and supernatants containing nuclear proteins were collected in a new tube. Both cytoplasmic and nuclear fractions were stored at −80°C.
Western blot analysis
HCT-15 cells were washed twice with ice cold PBS and then suspended at 4°C in lysis buffer (50 mM Tris, pH 7.4, 0.5% NP40, 0.01% SDS) containing protease inhibitor cocktail (Roche, Germany). After scraping and centrifuging the cells at 12,000 × g for 15 min at 4°C, the supernatants were collected, and the protein concentration in cell lysates was determined using the Bio-Rad Protein Assay. Proteins were separated by 10% SDS-PAGE and transferred to a nitrocellulose membrane. After blocking with 5% non-fat milk, the membranes were incubated with the respective primary antibody overnight at 4°C. Membranes then were washed and further incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibodies. The protein complex bands were detected by an enhanced chemiluminescence system.
Electrophoretic mobility shift assay (EMSA)
EMSA was performed to study the inhibitory effect of curcumin on NF-κB DNA binding according to a previously described method, with partial modification (Khan et al., 2013). In brief, nuclear extracts prepared from PGE2 and curcumin treated-cells were incubated with 32P-end-labeled 22-mer double-stranded NF-κB consensus oligonucleotides (Promega, sequence: 5′-AGT TGA GGG GAC TTT CCC AGG C-3′) for 40 min at room temperature. To confirm specificity for NF-κB, a 50-fold excess of unlabeled NF-κB oligonucleotide was added to the reaction mixture as a competitor. For the supershift assay, 5 μg of p65 and p50 antibodies was added, followed by incubation for 40 min at room temperature. DNA protein complexes then were separated from free oligonucleotides on 6% polyacrylamide gels. The gel was dried, and the signals obtained were quantified with an FLA-3000 apparatus (Fuji) using BAS reader version 3.14 and Aida version 3.22 software (Amersham Biosciences, USA).
Statistical analysis
Unless otherwise stated, statistical comparisons were performed by one-way analysis of variance (ANOVA) from three separate experiments. A value of p* < 0.05 was chosen as statistically significant.
RESULTS
Curcumin induces oxidative-stress apoptosis in HCT-15 cells
To investigate the underlying mechanism of curcumin-induced cell death, HCT-15 cells were treated with curcumin for the indicated times. As shown in Fig. 1A, lamin B and PARP were cleaved with curcumin treatment yielding cleavage bands. As both lamin B and PARP are substrates for caspase-3, activation of caspase-3 in whole cell lysates of curcumin-treated cells was confirmed by Western blot. Disappearance of 32 KDa caspase zymogen was used to measure caspase-3 activation (Fig. 1A). These results confirm the role of caspase-3 activation in curcumin-induced apoptosis.
Fig. 1.
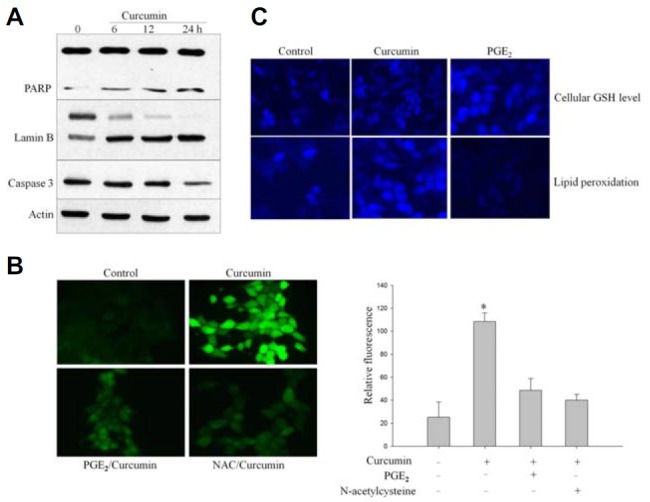
Curcumin induces oxidative-stress apoptosis. (A) HCT-15 cells were cultured in RPMI-1640 medium and treated with 20 μM curcumin. Western immunoblot analysis was performed on protein lysates using antibodies specific for PARP, lamin B, and caspase-3 cleavage. Lamin B and PARP were cleaved as a result of caspase-3 activation in apoptotic cells after treatment with curcumin. (B) HCT-15 cells were pretreated with 10 mM N-acetyl cysteine (NAC) for 1 h and exposed to curcumin. Cells were incubated further with DCFHDA, a cell-permeable n-dichlorofluorescein used as an intracellular probe for oxidative stress, for 30 min. Accumulation of probe in cells was measured based on an absorbance increase at 530 nm when the sample was excited at 485 nm. Fluorescence images of DCF-loaded cells were obtained under a microscope. The average fluorescence intensity was calculated as described by Sundaresan et al. (1995). NAC was used as an inhibitor of ROS generation. (C) HCT-15 cells were stained with 5 μM CMAC for 15 min in the dark after treatment with 1 μM PGE2 or 20 μM curcumin. Intracellular GSH levels were expressed relative to levels of FBS-treated cells that served as control. Curcumin treatment reduced cellular GSH levels, which was reversed by pretreatment with PGE2. After HCT cells were incubated with 5 μM DPPP for 15 min, cells were treated with PGE2 or curcumin for 30 min. DPPP fluorescence intensity was reduced markedly in PGE2-treated cells, whereas curcumin treatment increased lipid peroxidation as evidenced by fluorescence intensity under microscopy.
We have previously shown that curcumin-induced apoptosis is directly mediated by ROS generation in HCT-15 cells (Shehzad et al., 2013a). We used a cell permeable redox sensitive dye, DCFHDA, as an intracellular marker to measure curcumin-induced oxidative stress. Herein, we showed again that curcumin treatment caused production of ROS (Fig. 1B). However, we exposed HCT-15 cells to PGE2 before treatment with curcumin and found that PGE2 partially reversed curcumin-induced ROS generation (Fig. 1B). Consistently, NAC also reversed curcumin-induced apoptosis, based on reduced levels of DCFDA fluorescence (Fig. 1B).
Curcumin has been shown to induce oxidative stress-mediated cell death in a variety of cancer cells, including colon cancer cells. Glutathione (GSH) is a sulfhydryl-containing anti-oxidant product that is utilized mainly in cellular oxidation-reduction homeostasis by various glutathione peroxidases (Sebastia et al., 2003). Alteration of GSH to oxidized GSH (GSSG) can be measured as an indicator of oxidative stress in cells (Tauskela et al., 2001). In this study, the intracellular GSH concentration level was measured using the GSH-sensitive fluorescent dye CMAC as a probe. As shown in Fig. 1C, curcumin treatment reduced cellular antioxidants GSH levels in HCT-15 cells compared with control and PGE2-treated cells. In contrast, 1 μM PGE2 treatment increased the blue fluorescence intensity of cellular GSH observed by confocal microscopy (Fig. 1C). Furthermore, lipid peroxidation has been suggested as an alternative mechanism of cell death. Therefore, to investigate the effect of curcumin-induced oxidative damage to cellular components, membrane lipid peroxidation was measured using DPPP oxide, which is produced by the reaction of DPPA with hydroperoxides (Okimotoa et al., 2000). Exposure of HCT-15 cells to curcumin increased the amount of cell-associated DPPP fluorescence as compared with control (Fig. 1C). However, there was little difference between PGE2 treatment and control lipid peroxidation levels, indicating that PGE2 alone can inhibit membrane lipid peroxidation. DPPP fluorescence intensity was markedly lower in PGE2-treated cells (Fig. 1C). These results suggest that curcumin-induced lipid peroxidation resulted in perturbation of cellular antioxidant mechanisms through inactivation of antioxidant enzymes and depletion of GSH.
EP2receptor participates in PGE2-mediated anti-apoptosis in HCT-15 cells
Compelling evidence has shown that PGE2 regulates cellular proliferation, apoptosis, and angiogenesis through its receptors in various cancers, including colon tumorigenesis (Castellone et al., 2005). However, the functional receptor that mediates PGE2 anti-apoptotic effects remains unclear. Therefore, we treated the cells with 1 μM PGE2 and assessed the expression of four EP (EP1, EP2, EP3, and EP4) receptors by Western blot. Interestingly, the expression of EP1, EP3, and EP4 was unaffected by PGE2, whereas the expression of EP2 was considerably up-regulated in HCT-15 cells stimulated with PGE2 (Fig. 2A).
Fig. 2.
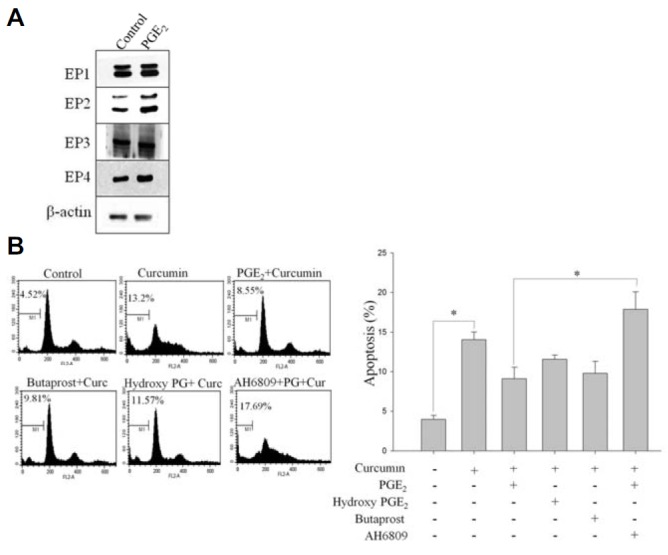
EP2 receptor is involved in PGE2-induced protection of HCT-15 cells against curcumin-induced cell death. (A) Cells were treated with 1 μM PGE2 for 8 h, and Western immunoblot analysis was performed on protein lysates using antibodies specific for EP1, EP2, EP3, and EP4 (B) HCT-15 cells were incubated with or without butaprost, AH6809, hydroxy PGE2, and PGE2 for 2 h, followed by curcumin treatment for 24 h. Cells were stained with PI, and their DNA content was analyzed by flow cytometry. The EP2 receptor agonist, butaprost, displayed the same cell death features as observed with PGE2. On the other hand, the EP2 antagonist, AH6809, enhanced curcumin-induced apoptosis in HCT-15 cells, suggesting the involvement of EP2 receptor in PGE2-induced cell survival.
To study further that EP2 receptor controls the response of HCT-15 cells to curcumin, cells were treated with the EP2 agonist, butaprost, and the antagonist, AH6809, prior to treatment with curcumin or PGE2 and then subjected to flow cytometry. As shown in Fig. 2B, when the EP2 agonist butaprost was added to HCT-15 cells, it produced similar effects to PGE2-induced cell survival against curcumin treatment. However, exposure of cells to the EP3 and EP4 agonist, PGE1 alcohol, displayed no significant effect on curcumin-induced cell death (Fig. 2B). In line with this, AH6809 is widely used as an EP2 receptor antagonist (Lee et al., 2009), and therefore, we treated HCT-15 cells with this antagonist to further determine the involvement of EP2 role in PGE2-induced cell survival. We noted that administration of the EP2 receptor antagonist, AH6809, prevented PGE2 from inducing EP2 expression in HCT-15 cells, which suggests that EP2 receptor activation is necessary for PGE2-induced cell survival (Fig. 2B). These results showed the anti-apoptotic activity of PGE2 mediated by EP2 activation, as the EP2 antagonist, AH-6809, abrogated the inhibitory effects of PGE2 on curcumin-induced apoptosis.
PGE2 activates PKA signaling pathway
Previous studies on PGE2 have shown that EP2 has the ability to activate PKA by stimulating the cAMP signaling pathway (Leone et al., 2007). It is well known that the cAMP-mediated transcriptional response involves the phosphorylation of cAMP response element-binding protein (CREB) by PKA. As shown in Fig. 3A, curcumin treatment caused HCT-15 cell death, whereas exposure of cells to PGE2 blocked curcumin-induced cell death. Co-incubation of HCT-15 cells with the PKA inhibitor, H89, for 15 min suppressed the anti-apoptotic activity of PGE2 compared with cells treated with PGE2 alone. We also studied the effect of cell-permeable dibutyryl cAMP, a known cAMP analog, on curcumin-induced cell death. PGE2 may mediate protection of cells through a cAMP-dependent mechanism (Leone et al., 2007). Addition of dibutyryl cAMP blocked curcumin-induced apoptotic cell death. As expected, treatment of HCT-15 cells with 1 μM PGE2 also effectively suppressed curcumin-induced apoptosis.
Fig. 3.
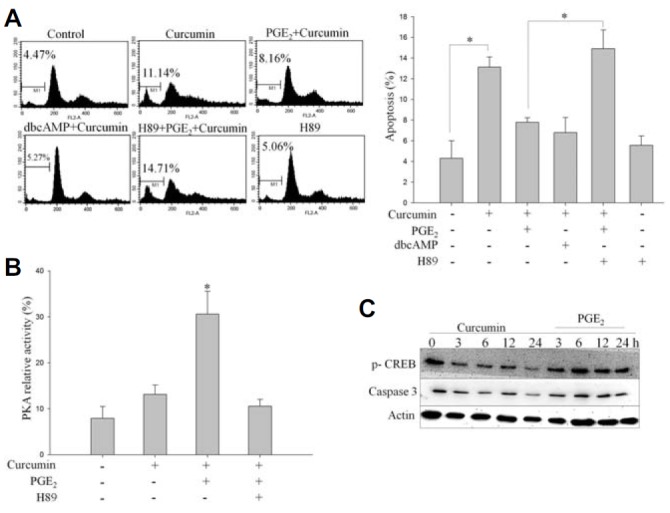
PGE2 treatment activates PKA in HCT-15 cells. (A) HCT-15 cells were exposed to 1 μM PGE2 as well as to 100 μM dibutyryl cAMP (dbcAMP) or the PKA inhibitor, H89, prior to treatment with 20 μM curcumin. Cells were harvested and stained with PI, and their DNA content was analyzed by flow cytometry. (B) HCT-15 cells were pretreated with H89 for 15 min prior to PGE2 exposure and incubated with 20 μM curcumin for 4 h. PKA activity was measured by an in vitro kinase assay as detailed in “Materials and Methods”. (C) HCT-15 cells were treated with 1 μM PGE2, followed by curcumin treatment for the indicated time. In the immunoblot analysis, curcumin inhibited CREB phosphorylation and induced apoptosis by activating caspase-3. However, pretreatment with PGE2 enhanced CREB phosphorylation and blocked curcumin-induced activation of caspase-3, thereby inhibiting apoptosis.
To further elucidate the survival role of PGE2, we measured PKA activity in HCT-15 cells using a radioisotope method that is based on phosphorylation of the synthetic substrate kemptide, a well-known substrate of PKA (Fig. 3B). We found that curcumin treatment blocked PKA substrate phosphorylation and, thus, suppressed PKA activation. However, prior treatment with PGE2 elevated PKA activity six-fold while reversing curcumin-induced inhibition of PKA. Further, the increase in PKA activity in response to PGE2 treatment was inhibited significantly by prior treatment with the PKA inhibitor, H89 (Fig. 3B).
To examine whether CREB can be activated by PGE2, we investigated the effect of PGE2 treatment on CREB phosphorylation. We measured CREB phosphorylation by detecting phosphorylation of CREB at Ser133 (p-CREB) by Western blotting. As shown in Fig. 3C, PGE2 treatment induced CREB activation. Additionally, exogenously added PGE2 to cells resulted in phosphorylation of CREB. Taken together, these results demonstrate that activation of PKA plays an important role in the regulation of PGE2-induced anti-apoptosis, and phosphorylation of CREB further potentiates the anti-apoptotic effect of PGE2 in HCT-15 cells.
PGE2 activates Ras/Ras/Erk pathway
Ras/Raf/Erk cascades reportedly are dysregulated in human cancers. To address the role of the Ras pathway in PGE2-induced anti-apoptosis, we measured the expression levels of Ras, p-Raf, and p-Erk after specific treatments with 1 μM PGE2 for the indicated times. Challenge of HCT-15 cells with PGE2 resulted in increased Ras activity as confirmed by immunoblotting with Ras GAP antibody, which detects activation of Ras GTPase-activating protein (Fig. 4A). Moreover, PGE2 treatment increased the phosphorylation levels of Raf and ERK in HCT-15 cells (Fig. 4A). To further investigate the functional role of the Ras pathway in PGE2-induced anti-apoptosis, cells were pretreated with PD98059, an inhibitor of MEK. Western blot analysis revealed that specific inhibition of MEK with PD98059 attenuated PGE2-induced phosphorylation of Erk (Fig. 4B). Further, flow cytometry was performed to investigate the effect of MEK pathway inhibition following PGE2 and curcumin treatment. As shown in Fig. 4C, PD98059 potentiated curcumin-induced cell death as confirmed by the accumulation of cells in sub-G1 phase. Pretreatment with PD98059 also prevented PGE2-induced reversal of cell death upon curcumin treatment.
Fig. 4.
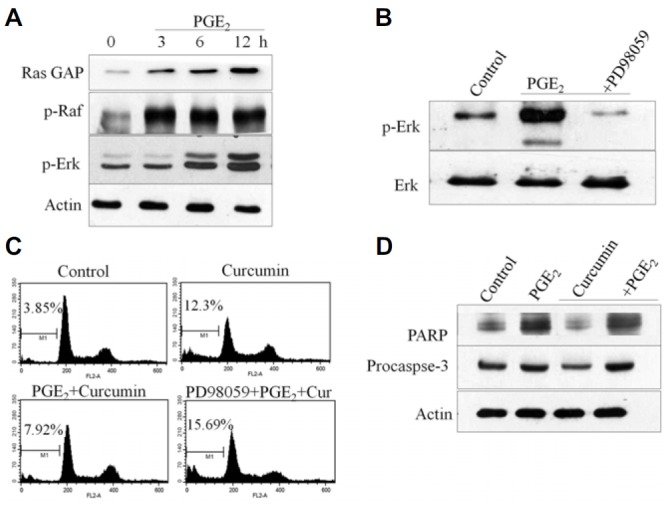
PGE2 treatment activates the Ras/Raf/Erk signaling pathway. (A) Ras GAP, Raf-1, and Erk phosphorylation were detected by immunoblot analysis with specific antibodies. Results are representative of three independent experiments. (B) Cells were treated with PGE2 or the MEK inhibitor, PD98059, and Erk phosphorylation was detected by immunoblot analysis with an antibody specific for phosphorylated Erk. PD98059 inhibited PGE2-induced phosphorylation of Erk in HCT-15 cells. (C) HCT-15 cells were pretreated with PD98095 for 1 h before treatment with PGE2. Cells were harvested and stained with PI, and their DNA content was analyzed by flow cytometry. PD98059 abolished the protective effects of PGE2 against curcumin-induced apoptosis. (D) HCT-15 cells were treated with PGE2 and curcumin. Western blot analysis was performed on protein lysates using antibodies specific for PARP and caspase-3. PGE2 protected against curcumin-induced apoptosis by reversing the effect of curcumin on PARP and caspase-3. Results were obtained from three independent experiments.
To further determine the anti-apoptotic effect of PGE2, HCT-15 cells were incubated with PGE2 or curcumin. As shown in Fig. 4D, curcumin treatment decreased PARP and caspase-3, indicating curcumin-induced apoptosis. However, following prior treatment with PGE2, curcumin failed to induce apoptosis in HCT-15 cells. PGE2 induced cell survival and protected against curcumin-induced apoptosis (Fig. 4D). These results demonstrate that activation of Ras/Raf/Erk is at least partially required for the anti-apoptotic effect of PGE2 in HCT-15 cells.
PGE2 activates NF-κB pathway in HCT-15 cells
Many reports have shown the involvement of NF-κB in cell proliferation and cell survival. Curcumin has been shown to modulate cellular functions through inhibition of NF-κB signaling in various cancer cells (Shehzad and Lee, 2013). To examine whether NF-κB activation is required for PGE2-induced anti-apoptosis and cell survival, cells were exposed to curcumin with or without PGE2 treatment. As shown in Fig. 5A, curcumin treatment for 24 h time-dependently reduced the activation of different NF-κB subunits (p50 and p65). However, pretreatment with PGE2 attenuated curcumin-induced inhibition of p50, p65, and RelB of NF-κB signaling (Fig. 5A). To determine whether the survival effect of PGE2 is mediated through NF-κB (p50 and p65) translocation, we prepared cytosolic and nuclear cell fractions. Curcumin treatment blocked activation of p50 and p65, which was significantly activated by pretreatment with 1 μM PGE2 for 2 h (Fig. 5B). Moreover, PGE2 enhanced translocation of NF-κB subunits (p50 and p65) in nuclear extracts compared with non-treated cells, as determined by Western blot (Fig. 5B).
Fig. 5.
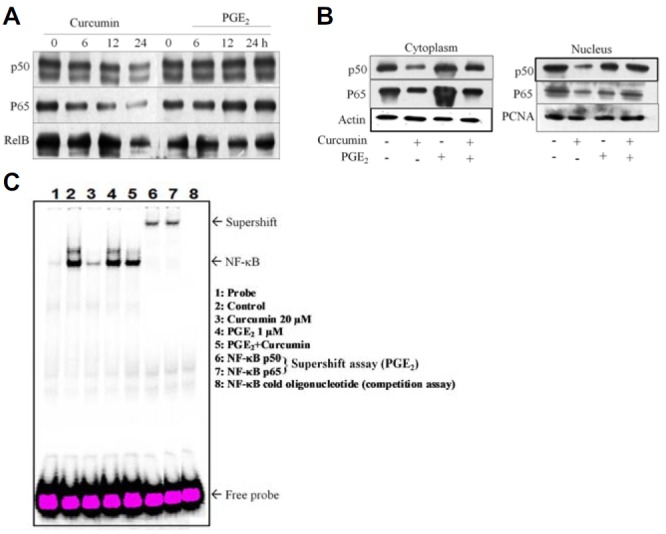
PGE2 blocks curcumin-induced cell death through the NF-κB pathway. (A) HCT-15 cells were exposed to curcumin with or without PGE2. Curcumin decreased expression of NF-κB subunits (p50, p65, and RelB) as detected by immunoblotting. Prior treatment with PGE2 reversed curcumin-induced inhibition of NF-κB subunit expression. (B) Cytoplasmic and nuclear fractions were separated from HCT-15 cells treated with PGE2 and/or curcumin. NF-κB subunits (p50 and p65) were detected by immunoblot analysis using antibodies specific for their expression in the cytoplasm and nucleus. (C) Effects of curcumin and PGE2 on DNA binding activity of NF-κB. HCT-15 cells were pretreated with PGE2 for 2 h, followed by curcumin treatment for 24 h. Nuclear extracts (5 μg) were incubated with 32P-labeled oligonucleotide specific to NF-κB and then electrophoresed on a 6% gel. EMSA results are represented, and NF-κB complexes and excessive probe are indicated by arrows. Competition and supershift assays for p50 and p65 DNA-binding are shown in lanes 6 and 7.
To further elucidate the mechanism of curcumin-mediated inhibition of NF-κB, translocation and DNA-binding activity of NF-κB were investigated by EMSA. As shown in Fig. 5C, the DNA-binding activity of NF-κB was reduced remarkably in nuclear extracts obtained from curcumin-treated cells. However, clear expression of NF-κB was observed upon pretreatment with PGE2. To identify the specific subunit of NF-κB in HCT-15 cells, we performed an EMSA competition assay using excess amounts of unlabeled NF-κB oligonucleotide as well as a supershift assay using p50 and p65 antibodies. A slow migration band of NF-κB was observed in EMSA (Fig. 5C). Additionally, nuclear extracts from PGE2-stimulated HCT-15 cells treated with antibodies against p65 and p50 were considerably supershifted. These results further confirm that PGE2 activates NF-κB translocation to the nucleus and prevents curcumin-induced cell death.
DISCUSSION
PGE2 synthesis has been reported to play a pivotal role in many pathogenic states, including chronic inflammation and oncogenesis (Castellone et al., 2005). Inflammatory function of PGE2 also promotes colorectal carcinogenesis (Zhang et al., 1997). It has been shown that production of PGE2 is higher in human as well as in experimental colon xenograft models (Ricchi et al., 2003). Studies also have demonstrated that absence of the PGE2 receptors EP2 and EP4 is associated with reduced colon cancer incidence in a xenograft mice model (Sonoshita et al., 2001). Inflammatory PGE2 promotes tumor development and activates signaling pathways that regulate cell proliferation, apoptosis, and angiogenesis. However, the molecular mechanism behind the potential anti-cancer role of phytochemicals such as curcumin has not been characterized in terms of PGE2 synthesis.
PGE2 is a pleiotropic molecule involved in tumor progression in a time- and concentration-dependent manner (Zhang et al., 1997). Curcumin-induced apoptosis was directly mediated by ROS generation, which was blocked by pretreatment with PGE2 treatment (Fig. 1B). PGE2 inhibits the apoptotic effect of curcumin, at least in part, through blocking of ROS production. PGE2 also increased the glutathione level, supporting the interpretation that PGE2 diminished curcumin-induced cell death by reducing the levels of ROS production (Eibl et al., 2003). Furthermore, PGE2 facilitates cancer cell survival by binding to its cognate receptors, resulting in enhanced cellular proliferation, invasiveness, and angiogenesis as well as inhibition of apoptosis. Here, we found that PGE2 protected HCT-15 cells against curcumin-induced oxidative stress, possibly through the increased expression of the transmembrane receptor EP2 (Eibl et al., 2003; Kamiyama et al., 2006). EP2 mediates its activity by elevating cAMP production, which has been reported to stimulate the tumor microenvironment (Wang and Klein, 2007). In our results, treatment with the EP2 agonist butaprost significantly inhibited curcumin-induced cell death, suggesting that no other receptors are involved in the PGE2-induced anti-apoptotic effect. Conversely, the EP2 antagonist, AH-6809, abrogated the inhibitory effects of PGE2 on curcumin-induced cell death.
Activation of the PKA/cAMP pathway is another possible mechanism for PGE2-induced inhibition of apoptosis (Wang and Klein, 2007). PKA is a cytosolic holoenzyme regulating cell physiology through phosphorylation of cytoplasmic and nuclear protein substrates (Chowdhury et al., 2011). However, a selective PKA inhibitor, H89, was able to reduce PGE2-induced PKA activation and restored curcumin-induced cell death. Previous observations regarding cAMP-mediated suppression of apoptosis in transformed colon epithelial cells suggested that cAMP second messenger might be involved in the development of cancer including colorectal carcinoma (Kisslov et al., 2012). Furthermore, cAMP-mediated transcriptional activation involves the phosphorylation of CREB by PKA. It has been noted that colon cancer cell proliferation is regulated by cPLA2, which is dependent on PGE2-induced activation of both PKA and PKB pathways (Kisslov et al., 2012). We observed that exogenously added PGE2 induced proliferation in colorectal carcinoma HCT-15 cells, as well as protected against curcumin-induced apoptosis, resulting in increased PKA/cAMP association.
Both PKA and cAMP regulate growth factor signaling as well as activation of the Ras/Raf/Erk pathway (Pursiheimo et al., 2002). As to the signal pathway involved in the mechanism by which curcumin inhibits colon cancer cells proliferation, we show that Ras/Raf/Erk and NF-κB pathways are both activated in HCT-15 cancer cells in response to PGE2, while inhibition of these pathways contributes to the suppressive effect of curcumin. Previously, it has been shown that Ras/Raf/Erk induced phosphorylation of cPLA2, either directly or indirectly, and that blocking of this pathway inhibited arachidonic acid release as well as the arachidonic metabolite, PGE2 (Hong et al., 2004). Our results showed that curcumin (20 μM) inhibited the activation of the Ras/Raf/Erk pathway, but that exogenous addition of PGE2 reversed curcumin-induced inhibition of this pathway (Fig. 4). The mechanism involved in the reversal of curcumin has been suggested to be due to autocrine signaling of PGE2 that promotes several inflammatory processes in cancer cells (Shehzad et al., 2014). Furthermore, activation of NF-κB pathways has been reported to be involved in cell survival and in inhibition of apoptosis (Krysan et al., 2005; Yu et al., 2009). Studies have shown that phosphorylation of p65 is involved in DNA-binding and in transactivation of NF-κB (Chen et al., 2001). PGE2 blocked curcumin-inhibition as well as activated the DNA-binding activity and nuclear translocation of NF-κB. It was shown that the NF-κB pathway mediated a positive feedback loop for amplification of Ras activity by PGE2 (Daniluk et al., 2012). Our results show that Erk and NF-κB activation by PGE2 contributes to the inhibition of curcumin-triggered tumor cell death.
In conclusion, our study suggests that PGE2 can protect HCT-15 cells against curcumin-induced cell death by activating PKA/cAMP as well as the Ras/Raf/Erk pathway. PGE2 also has the ability to inhibit curcumin-induced cell death through inhibition of inflammatory transcription factor NF-κB (p50 and p65). Further studies are necessary to investigate the underlying mechanism of PGE2 in xenograft cancer models with regard to curcumin treatment. Collectively, our results show that development of compounds that reduce cellular levels of PGE2 and/or directly inhibit the PGE2-activated signaling pathway might be a useful strategy for cancer therapy.
Acknowledgments
This research was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (NRF-2009-0078234) and by the Basic Science Research Program through the NRF funded by the Ministry of Science, ICT, and Future Planning (NRF-2013R1A1A2063612).
REFERENCES
- Barnes P.J., Karin M. Nuclear factor-κB: a pivotal transcription factor in chronic inflammatory diseases. New Eng. J. Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- Baud V., Karin M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009;8:33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellone M.D., Teramoto H., Williams B.O., Druey K.M., Gutkind J.S. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- Chen L., Fischle W., Verdin E., Greene W.C. Duration of nuclear NF-κB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- Chowdhury S., Howell G.M, Rajput A., Teggart C.A., Brattain L.E., Weber H.R., Chowdhury A., Brattain M.G. Identification of a novel TGFβ/PKA signaling transduceome in mediating control of cell survival and metastasis in colon cancer. PLoS One. 2011;6:e19335. doi: 10.1371/journal.pone.0019335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniluk J., Liu Y., Deng D., Chu J, Huang H., Gaiser S., Cruz-Monserrate Z., Wang H., Ji B., Logsdon C.D. An NF-κB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J. Clin. Invest. 2012;122:1519–1528. doi: 10.1172/JCI59743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eibl G., Bruemmer D., Okada Y., Duffy J.P., Law R.E., Reber H.A., Hines O.J. PGE2 is generated by specific COX-2 activity and increases VEGF production in COX-2-expressing human pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2003;306:887–897. doi: 10.1016/s0006-291x(03)01079-9. [DOI] [PubMed] [Google Scholar]
- Fernando R.I., Wimalasena J. Estradiol abrogates apoptosis in breast cancer cells through inactivation of BAD: Ras-dependent nongenomic pathways requiring signaling through ERK and Akt. Mol. Biol. Cell. 2004;15:3266–3284. doi: 10.1091/mbc.E03-11-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk C.D. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- Hong J., Bose M., Ju J., Ryu J.H., Chen X., Sang S., Lee M.J., Yang C.S. Modulation of arachidonic acid metabolism by curcumin and related β-diketone derivatives: effects on cytosolic phospholipase A2, cyclooxygenases and 5-lipoxygenase. Carcinogenesis. 2004;25:1671–1679. doi: 10.1093/carcin/bgh165. [DOI] [PubMed] [Google Scholar]
- Kamiyama M., Pozzi A., Yang L., DeBusk L.M., Breyer R.M., Lin P.C. EP2, a receptor for PGE2, regulates tumor angiogenesis through direct effects on endothelial cell motility and survival. Oncogene. 2006;25:7019–7028. doi: 10.1038/sj.onc.1209694. [DOI] [PubMed] [Google Scholar]
- Khan S., Choi R.J., Shehzad O., Kim H.P., Islam M.N., Choi J.S., Kim Y.S. Molecular mechanism of capillarisin-mediated inhibition of MyD88/TIRAP inflammatory signaling in in vitro and in vivo experimental models. J. Ethnopharmacol. 2013;145:626–637. doi: 10.1016/j.jep.2012.12.001. [DOI] [PubMed] [Google Scholar]
- Kisslov L., Hadad N., Rosengraten M., Levy R. HT-29 human colon cancer cell proliferation is regulated by cytosolic phospholipase A2α dependent PGE2 via both PKA and PKB pathways. Biochim. Biophys. Acta. 2012;1821:1224–1234. doi: 10.1016/j.bbalip.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Krysan K., Reckamp K.L., Dalwadi H., Sharma S., Rozengurt E., Dohadwala M., Dubinett S.M. Prostaglandin E2 activates mitogen-activated protein kinase/Erk pathway signaling and cell proliferation in non-small cell lung cancer cells in an epidermal growth factor receptor-independent manner. Cancer Res. 2005;65:6275–6281. doi: 10.1158/0008-5472.CAN-05-0216. [DOI] [PubMed] [Google Scholar]
- Lee B.P., Juvet S.C., Zhang L. Prostaglandin E2 signaling through E prostanoid receptor 2 impairs proliferative response of double negative regulatory T cells. Int. Immunopharmacol. 2009;9:534–539. doi: 10.1016/j.intimp.2009.01.023. [DOI] [PubMed] [Google Scholar]
- Leone V., di Palma A, Ricchi P., Acquaviva F., Giannouli M., Prisco A.M., Iuliano F., Acquaviva A.M. PGE2 inhibits apoptosis in human adenocarcinoma Caco-2 cell line through Ras-PI3K association and cAMP-dependent kinase A activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2007;293:673–681. doi: 10.1152/ajpgi.00584.2006. [DOI] [PubMed] [Google Scholar]
- Okimotoa Y., Watanabea A., Nikia E., Yamashitab T., Noguchi N. A novel fluorescent probe diphenyl-1-pyrenylphosphine to follow lipid peroxidation in cell membranes. FEBS Lett. 2000;474:137–140. doi: 10.1016/s0014-5793(00)01587-8. [DOI] [PubMed] [Google Scholar]
- Pursiheimo J.P., Kieksi A., Jalkanen M., Salmivirta M. Protein kinase A balances the growth factor-induced Ras/ERK signaling. FEBS Lett. 2002;521:157–164. doi: 10.1016/s0014-5793(02)02864-8. [DOI] [PubMed] [Google Scholar]
- Ricchi P., di Palma A.D., Di Matola T.D., Apicella A., Fortunato R., Zarrilli R., Acquaviva A.M. Aspirin protects Caco-2 cells from apoptosis after serum deprivation through the activation of a phosphatidylinositol 3-kinase/AKT/p21Cip/WAF1 pathway. Mol. Pharmacol. 2003;64:407–414. doi: 10.1124/mol.64.2.407. [DOI] [PubMed] [Google Scholar]
- Roberts P.J., Der C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- Rosner M., Hengstschläger M. Cytoplasmic and nuclear distribution of the protein complexes mTORC1 and mTORC2: rapamycin triggers dephosphorylation and delocalization of the mTORC2 components rictor and sin1. Hum. Mol. Genet. 2008;17:2934–248. doi: 10.1093/hmg/ddn192. [DOI] [PubMed] [Google Scholar]
- Sakamoto K., Maeda S., Hikiba Y., Nakagawa H., Hayakawa Y., Shibata W., Yanai A., Ogura K., Omata M. Constitutive NF-κB activation in colorectal carcinoma plays a key role in angiogenesis, promoting tumor growth. Clin. Cancer Res. 2009;15:2248–2258. doi: 10.1158/1078-0432.CCR-08-1383. [DOI] [PubMed] [Google Scholar]
- Santarpia L., Lippman S.M., El-Naggar A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Exp. Opin. Ther. Targets. 2012;16:103–119. doi: 10.1517/14728222.2011.645805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastià J., Cristòfol R., Martín M., Rodríguez-Farré E., Sanfeliu C. Evaluation of fluorescent dyes for measuring intracellular glutathione content in primary cultures of human neurons and neuroblastoma SH-SY5Y. Cytometry A. 2003;51:16–25. doi: 10.1002/cyto.a.10003. [DOI] [PubMed] [Google Scholar]
- Seufert B.L., Poole E.M., Whitton J., Xiao L., Makar K.W., Campbell P.T., Kulmacz R.J., Baron J.A., Newcomb P.A., Slattery M.L., et al. IκBKβ and NFκB1, NSAID use and risk of colorectal cancer in the colon cancer family registry. Carcino-genesis. 2013;34:79–85. doi: 10.1093/carcin/bgs296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Six D.A., Dennis E.A. The expanding superfamily of phospholipase A2 enzymes: classification and characterization. Biochim. Biophys. Acta. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- Shehzad A., Lee Y.S. Molecular mechanisms of curcumin action: signal transduction. Biofactors. 2013;39:27–36. doi: 10.1002/biof.1065. [DOI] [PubMed] [Google Scholar]
- Shehzad A., Lee J., Huh T.L., Lee Y.S. Curcumin induces apoptosis in human colorectal carcinoma (HCT-15) cells by regulating expression of Prp4 and p53. Mol. Cells. 2013a;35:526–532. doi: 10.1007/s10059-013-0038-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehzad A., Lee J., Lee Y.S. Curcumin in various cancers. Biofactors. 2013b;39:56–68. doi: 10.1002/biof.1068. [DOI] [PubMed] [Google Scholar]
- Shehzad A., Lee J., Lee Y.S. Autocrine prostaglandin E2 signaling promotes promonocytic leukemia cell survival via COX-2 expression and MAPK pathway. BMB Rep. 2014. Jun 26, pii: 2794. [DOI] [PMC free article] [PubMed]
- Sonoshita M., Takaku K., Sasaki N., Sugimoto Y., Ushikubi F., Narumiya S., Oshima M., Yaketo M.M. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in ApcDelta716 knockout mice. Nat. Med. 2001;7:1048–1051. doi: 10.1038/nm0901-1048. [DOI] [PubMed] [Google Scholar]
- Sundaresan M., Yu Z.X., Ferrans C.J., Irani K., Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- Tauskela J.S., Hewitt K., Kang L.P., Comas T., Gendron T., Hakim A., Hogan M., Durkin J., Morley P. Evaluation of glutathione-sensitive fluorescent dyes in conical culture. Glia. 2001;30:329–341. [PubMed] [Google Scholar]
- Wang X., Klein R.D. Prostaglandin E2 induces vascular endothelial growth factor secretion in prostate cancer cells through EP2 receptor-mediated cAMP pathway. Mol. Carcinogen. 2007;46:912–923. doi: 10.1002/mc.20320. [DOI] [PubMed] [Google Scholar]
- Wang D., Wang H., Shi Q., Katkuri S., Walhi W., Desvergne B., Das S.K., DuBois R.N. Prostaglandin E2 promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor δ. Cancer Cell. 2004;6:285–295. doi: 10.1016/j.ccr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Wang S., Liu Z., Wang L., Zhang X. NF-κB signaling pathway, inflammation and colorectal cancer. Cell. Mol. Immunol. 2009;6:327–334. doi: 10.1038/cmi.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L., Wu W.K.K., Li Z.J., Li H.T., Wu Y.C., Cho C.H. Prostaglandin E2 promotes cell proliferation via protein kinase C/extracellular signal regulated kinase pathway-dependent induction of c-Myc expression in human esophageal squamous cell carcinoma cells. Int. J. Cancer. 2009;125:2540–2546. doi: 10.1002/ijc.24607. [DOI] [PubMed] [Google Scholar]
- Zhang L., Zhou W., Velculescu. V.E., Kern S.E., Hruban R.H., Hamilton S.R., Vogelstein B., Kinzler K.W. Gene expression profiles in normal and cancer cells. Science. 1997;276:1268–1272. doi: 10.1126/science.276.5316.1268. [DOI] [PubMed] [Google Scholar]