

Abstract
Tuberous sclerosis complex (TSC) is an autosomal dominant condition characterised by the presence of multiple hamartomas in various organ systems in the body. The kidneys are affected in 80% of patients, usually in the form of renal angiomyolipomas, renal cysts or renal cell carcinoma. Although extremely rare, TSC and autosomal dominant polycystic kidney disease (ADPKD) can co-exist in the same patient as a result of concurrent deletion of both polycystic kidney disease (PKD) 1 and TSC2 genes present on the chromosome 16p13.3. These patients develop end-stage renal disease at an earlier age and have an increased risk of malignancy. We present a case of a 30-year-old man with a history of tuberous sclerosis, presenting with loin pain and subsequently diagnosed to have ADPKD.
Background
Tuberous sclerosis complex (TSC) is an autosomal dominant condition with an estimated incidence between 1 in 6000 and 1 in 11 000 births.1 2 It results from a mutation of either TSC1 or TSC2 gene; TSC1 gene codes for hamartin and is located on chromosome 9q34 while TSC2 gene codes for tubulin and is located on chromosome 16p13. These mutations lead to multiple, uncontrolled proliferations in the brain, skin, heart, lungs and kidneys. Common renal manifestations of TSC include angiomyolipomas (85.4%), cysts (44.8%) and renal cell carcinomas (4.2%).3 Autosomal dominant polycystic kidney disease (ADPKD) is rarely seen; it is found in less than 2% of patients with TSC.4
ADPKD is a systemic illness that often presents with hypertension, abdominal pain, haematuria, urinary tract infection and abdominal mass. Mutations of two genes are known to cause ADPKD; these are polycystic kidney disease (PKD) 1, located on chromosome 16p13.3 coding for polycystin 1, and PKD2, located on chromosome 4q21-q23 coding for the protein polycystin 2.5
Patients with TSC who have ADPKD are known to have a contiguous gene syndrome involving deletion of all or part of the TSC2 and PKD1 genes.6 7 In these cases, early-onset renal failure, usually in 30 s, develops due to PKD and there is greater risk of malignancy at an early age.8
We report a case of a 30-year-old man with a history of TSC who presented to us with acute loin pain. Further investigation led us to a diagnosis of ADKPD.
Case presentation
A 30-year-old white man presented to the emergency department with 1 day history of right loin pain associated with vomiting. His medical history was significant for tuberous sclerosis along with seizure disorder, hypertension, sensori-neural hearing loss, asthma and mental retardation. There was no significant family history. His home medications included amlodipine, albuterol, phenobarbital and fluticasone/salmeterol.
On examination, the patient's vitals were stable. He had obvious speech and hearing impairment. We noted multiple angiofibromas on the nasolabial fold and forehead, and hypomelanotic macules on bilateral upper and lower extremities. There was tenderness on the right lumbar region but no tenderness on bilateral costovertebral angles. The rest of the physical examination was within normal limits. The patient was oliguric with a urinary output of less than 0.5 mL/kg/h.
Investigations
Laboratory investigations were significant for elevated blood urea nitrogen (BUN) of 22 mg/dL and creatinine of 3.59 mg/dL, suggesting acute kidney injury (AKI). The patient’s last known creatinine was 1.1 mg/dL, as seen in records from 2 years prior. Urine analysis revealed a specific gravity of 1.011 and microscopy was unremarkable with no mention of the presence of abnormal casts. There were innumerable cysts within both kidneys shown by CT of the abdomen and pelvis, consistent with adult PKD (figure 1). Fatty soft tissue masses were seen in the liver and right kidney, suggestive of angiomyolipoma, and there were diffuse sclerotic lesions throughout the bones. CT of the brain revealed multiple calcified tubers within the periventricular area, a feature consistent with tuberous sclerosis (figure 2). TSC and ADPKD were clinically diagnosed and no genetic testing was performed.
Figure 1.
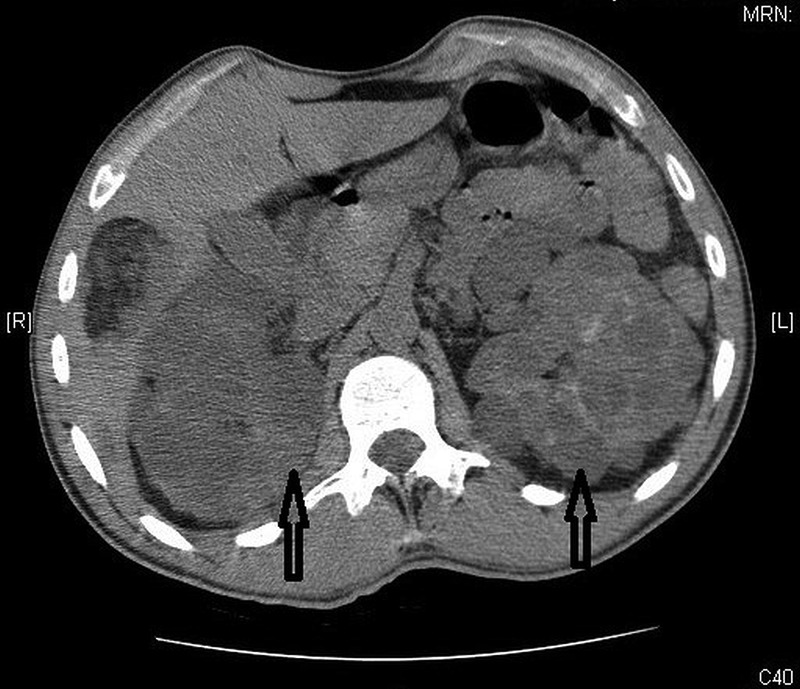
CT of the abdomen showing numerous bilateral renal cysts suggestive of polycystic kidney disease.
Figure 2.
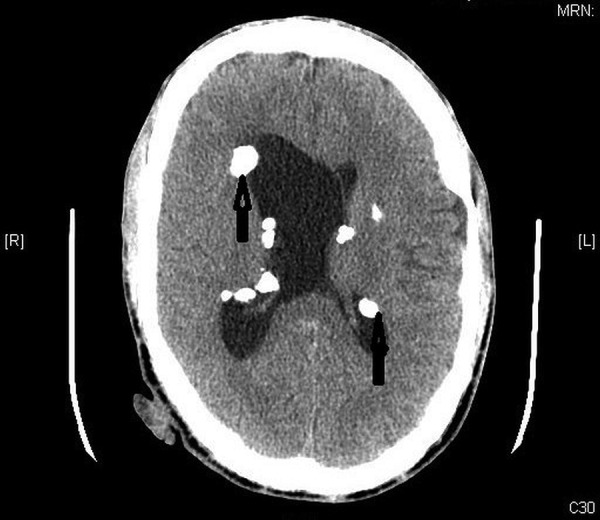
CT of the brain showing numerous calcified tubers in the periventricular area suggestive of tuberous sclerosis.
Treatment
The patient was treated with normal saline infusion and was given hydromorphone for pain. He felt significantly better over the subsequent 3 days. His renal function improved with BUN of 18 mg/dL and creatinine of 2.13 mg/dL.
Outcome and follow-up
The patient was discharged on the fourth day of hospitalisation in stable clinical condition. His clinical condition and laboratory tests were found to be stable at 2-month and 6-month follow-up (serum creatinine of 2.1 mg/dL). The patient is undergoing a biannual follow-up with renal and liver function tests, and renal ultrasonography (USG). Screening of all first-degree family members was strongly recommended as well.
Discussion
TSC is an autosomal dominant condition with multiple hamartomas in various organs of the body. Epilepsy and mental retardation, characteristic cutaneous manifestations, intracerebral hamartoma, renal angiomyolipoma and pulmonary lymphangioleiomyomatosis are among the major clinical features of TSC.9 The majority of patients (more than 80%) with TSC develop some form of kidney disease during their lifetime.10 Angiomyolipoma and cystic kidney diseases are among the common renal manifestations of TSC, with ADPKD occurring only in about 2% of TSC cases.4 The classic ADPKD renal phenotype may occur in the context of TSC disease as a result of large deletions involving the PKD1 and TSC2 genes present on the chromosome 16p13. This condition is also known as TSC2/ADPKD1 contiguous gene syndrome, and is diagnosed when renal lesions typical for ADPKD phenotype are associated with TSC phenotype.6 11
The concurrence of TSC and ADPKD is rare with very few cases reported in the literature. Most of these cases belong to the paediatric age group with adult cases representing about one-fourth of the total.6 7 12–20 Abdominal distention and hypertension are common manifestations of ADKPD in these cases.6 Progression to end-stage renal disease (ESRD) has been noted in early ages, even as early as the third decade in some cases, and requires renal replacement therapy.4 11 17 21 22 According to Sampson et al, most patients with non-mosaic deletions in TSC1/PKD2 gene were found to have enlarged cystic kidneys during infancy or childhood with features of advanced ADPKD at the time of diagnosis, but other patients had variable degrees of renal involvement. Manifestations other than renal varied considerably among all the patients.7
The diagnostic criteria for TSC consist of various major and minor features in different organs. Two major features or one major and two minor are required to make a definite diagnosis.23 Major features such as angiofibromas on the face, angiomyolipoma on the liver and right kidney, calcified tubers within the periventricular area and hypomelanotic macules on extremities, and minor feature such as multiple renal cysts, were present in our patient. In most cases, diagnosis is made by using clinical criteria but molecular genetic testing can be used for prenatal diagnosis, for screening family members of an affected individual and for cases where diagnosis is uncertain.23 Similarly, ADPKD is usually diagnosed with renal USG, although molecular genetic testing can be used for uncertain or at risk cases.24 ADPKD was diagnosed in our case with CT of the abdomen and pelvis, showing innumerable bilateral renal cysts.
The optimal surveillance protocols for renal imaging in TSC are not established, but a baseline renal USG before 5 years of age is recommended. In the presence of angiomyolipomas or cysts, renal lesions should be followed closely with imaging every 1–2 years because aggressive treatment can preserve kidney function with minimal trauma to the individual or the kidney.3 25 Patients known to have a contiguous gene syndrome should be recommended for monitoring at a younger age as most patients have early onset of ADPKD. Furthermore, AKI with subsequent acceleration in the progression of renal cystic disease can develop in TSC patients after prolonged seizure or the use of certain anticonvulsants and non-steroidal anti-inflammatory drugs.26 This requires careful drug selection and prompt management of AKI.
Currently, there is a lack of definite therapy for ADPKD and TSC, and renal failure remains one of the most common causes of death.27 Recently, therapy with sirolimus (or rapamycin), a potent inhibitor of the mammalian target of rapamycin complex 1, which is deregulated in TSC and ADPKD, has been shown in studies to slow progression of ADPKD and angiomyolipoma.28 Further understanding of such underlying molecular pathways can be gained by studying cases of co-existing ADPKD and TSC, which can hopefully help in translating targeted therapies into clinical practice.
Learning points.
Although extremely rare, tuberous sclerosis complex (TSC) and autosomal dominant polycystic kidney disease (ADPKD) can co-exist in the same patient as a result of concurrent deletion of both polycystic kidney disease (PKD) 1 and TSC2 genes present on the chromosome 16p13.3.
ADPKD associated with TSC is severe and usually presents early in life with hypertension, haematuria or renal failure.
There is no definite therapy; the goal of treatment being minimum interventions to treat the symptoms and prevent complications. Renal lesions should be followed every 1–2 years with imaging studies. Recently, targeted therapy using drugs such as sirolimus is being studied in some of these cases.
Footnotes
Contributors: JPR conceptualised the project and gathered clinical information. PD and SG wrote the first draft. KVD supervised the project. All authors agreed on the final version of the manuscript.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Lendvay TS, Marshall FF. The tuberous sclerosis complex and its highly variable manifestations. J Urol 2003;169:1635–42. [DOI] [PubMed] [Google Scholar]
- 2.Sancak O, Nellist M, Goedbloed M et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur J Hum Genet 2005;13:731–41. [DOI] [PubMed] [Google Scholar]
- 3.Rakowski SK, Winterkorn EB, Paul E et al. Renal manifestations of tuberous sclerosis complex: Incidence, prognosis, and predictive factors. Kidney Int 2006;70:1777–82. [DOI] [PubMed] [Google Scholar]
- 4.Dhakal M, Dhakal OP, Bhandari D. Polycystic kidney disease and chronic renal failure in tuberous sclerosis. BMJ Case Rep 2013;2013 pii: bcr2013200711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peters DJ, Spruit L, Saris JJ et al. Chromosome 4 localization of a second gene for autosomal dominant polycystic kidney disease. Nat Genet 1993;5:359–62. [DOI] [PubMed] [Google Scholar]
- 6.Brook-Carter PT, Peral B, Ward CJ et al. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease—a contiguous gene syndrome. Nat Genet 1994;8:328–32. [DOI] [PubMed] [Google Scholar]
- 7.Sampson JR, Maheshwar MM, Aspinwall R et al. Renal cystic disease in tuberous sclerosis: role of the polycystic kidney disease 1 gene. Am J Hum Genet 1997;61:843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonsib SM. Renal cystic diseases and renal neoplasms: a mini-review. Clin J Am Soc Nephrol 2009;4:1998–2007. [DOI] [PubMed] [Google Scholar]
- 9.Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 1998;13:624–8. [DOI] [PubMed] [Google Scholar]
- 10.Ewalt DH, Sheffield E, Sparagana SP et al. Renal lesion growth in children with tuberous sclerosis complex. J Urol 1998;160:141–5. [PubMed] [Google Scholar]
- 11.Bisceglia M, Galliani C, Carosi I et al. Tuberous sclerosis complex with polycystic kidney disease of the adult type: the TSC2/ADPKD1 contiguous gene syndrome. Int J Surg Pathol 2008;16:375–85. [DOI] [PubMed] [Google Scholar]
- 12.Boehm D, Bacher J, Neumann HP. Gross genomic rearrangement involving the TSC2-PKD1 contiguous deletion syndrome: characterization of the deletion event by quantitative polymerase chain reaction deletion assay. Am J Kidney Dis 2007;49:e11–21. [DOI] [PubMed] [Google Scholar]
- 13.Culty T, Molinie V, Lebret T et al. TSC2/PKD1 contiguous gene syndrome in an adult. Minerva Urol Nefrol 2006;58:351–4. [PubMed] [Google Scholar]
- 14.Dauwerse JG, Bouman K, van Essen AJ et al. Acrofacial dysostosis in a patient with the TSC2-PKD1 contiguous gene syndrome. J Med Genet 2002;39:136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris PC. The TSC2/PKD1 contiguous gene syndrome. Contrib Nephrol 1997;122:76–82. [DOI] [PubMed] [Google Scholar]
- 16.Laass MW, Spiegel M, Jauch A et al. Tuberous sclerosis and polycystic kidney disease in a 3-month-old infant. Pediatr Nephrol 2004;19:602–8. [DOI] [PubMed] [Google Scholar]
- 17.Longa L, Brusco A, Carbonara C et al. A tuberous sclerosis patient with a large TSC2 and PKD1 gene deletion shows extrarenal signs of autosomal dominant polycystic kidney disease. Contrib Nephrol 1997;122:91–5. [DOI] [PubMed] [Google Scholar]
- 18.Mancino C, Balducci A. Tuberous sclerosis complex and early-onset autosomal dominant polycystic kidney disease as a ‘contiguous gene’ syndrome: report of a case. Contrib Nephrol 1997;122:96–7. [DOI] [PubMed] [Google Scholar]
- 19.Martignoni G, Bonetti F, Pea M et al. Renal disease in adults with TSC2/PKD1 contiguous gene syndrome. Am J Surg Pathol 2002;26:198–205. [DOI] [PubMed] [Google Scholar]
- 20.Torra R, Badenas C, Darnell A et al. Facilitated diagnosis of the contiguous gene syndrome: tuberous sclerosis and polycystic kidneys by means of haplotype studies. Am J Kidney Dis 1998;31:1038–43. [DOI] [PubMed] [Google Scholar]
- 21.Kacerovska D, Vrtel R, Michal M et al. TSC2/PKD1 contiguous gene syndrome: a report of 2 cases with emphasis on dermatopathologic findings. Am J Dermatopathol 2009;31:532–41. [DOI] [PubMed] [Google Scholar]
- 22.Rosado C, Garcia-Cosmes P, Fraile P et al. Tuberous sclerosis associated with polycystic kidney disease: effects of rapamycin after renal transplantation. Case Rep Transplant 2013;2013:397087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Northrup H, Krueger DA. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 2013;49:243–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pei Y. Diagnostic approach in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 2006;1:1108–14. [DOI] [PubMed] [Google Scholar]
- 25.Roach ES, DiMario FJ, Kandt RS et al. Tuberous Sclerosis Consensus Conference: recommendations for diagnostic evaluation. National Tuberous Sclerosis Association. J Child Neurol 1999;14:401–7. [DOI] [PubMed] [Google Scholar]
- 26.de Chadarevian JP, Legido A, Miles DK et al. Epilepsy, atherosclerosis, myocardial infarction, and carbamazepine. J Child Neurol 2003;18:150–1. [DOI] [PubMed] [Google Scholar]
- 27.Shepherd CW, Gomez MR, Lie JT et al. Causes of death in patients with tuberous sclerosis. Mayo Clin Proc 1991;66:792–6. [DOI] [PubMed] [Google Scholar]
- 28.Bissler JJ, McCormack FX, Young LR et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 2008;358:140–51. [DOI] [PMC free article] [PubMed] [Google Scholar]