

Abstract
Thioredoxin plays a crucial role in a wide number of physiological processes, which span from reduction of nucleotides to deoxyriboucleotides to the detoxification from xenobiotics, oxidants and radicals. The redox function of Thioredoxin is critically dependent on the enzyme Thioredoxin NADPH Reductase (TrxR). In view of its indirect involvement in the above mentioned physio/pathological processes, inhibition of TrxR is an important clinical goal. As a general rule, the affinities and mechanisms of binding of TrxR inhibitors to the target enzyme are known with scarce precision and conflicting results abound in the literature. A relevant analysis of published results as well as the experimental procedures is therefore needed, also in view of the critical interest of TrxR inhibitors. We review the inhibitors of TrxR and related flavoreductases and the classical treatment of reversible, competitive, non competitive and uncompetitive inhibition with respect to TrxR, and in some cases we are able to reconcile contradictory results generated by oversimplified data analysis.
Keywords: Thioredoxin reductase, mechanism of inhibition, competitive inhibitor, uncompetitive inhibitor, irreversible inhibitor, suicide substrates, pseudo-irreversible inhibition, double-substrate enzyme.
1. INTRODUCTION
Thioredoxin Reductase (TrxR) is a ubiquitous homodimeric flavoenzyme whose physiological role is the transfer of reducing equivalents from NADPH to thioredoxin [1, 2]. Two variants of TrxR have evolved independently: the High molecular weight TrxR (H-TrxR) found exclusively in higher eukarya and the Low molecular weight TrxR (L-TrxR) found in procarya and some eukarya including fungi and plants [3]. H-TrxR is a member of a family of closely related flavoenzymes that includes Glutathione Reductase (GR), Trypanothione Reductase (TryR), Mercuric Ion Reductase (MeR) and Lipoamide Dehydrogenase (LipD). In mammals three H-TrxR isoforms are present: TrxR1 found in the cytoplasm, TrxR2 in mitochondria and TrxR3 (also called Thioredoxin Glutathione Reductase, TGR) expressed only in specialized tissues (e.g. the testis) [2]. TGR is a unique fusion of a TrxR domain (at the C terminus) and a Glutaredoxin domain (at the N-terminus; see [4]) and, interestingly, in parasitic platyhelminths, it may entirely replace both TrxR and GR [5, 6].
TrxR is necessary to all biochemical pathways in which thioredoxin is involved as a reducing substrate. These span from the reduction of ribonucleotides to deoxyribonucleotides to the detoxification of oxidants and radicals. Not surprisingly, it has been suggested to play roles in such diverse physiological and pathological conditions as apoptosis [7], cancer [8], parasitoses [4, 9, 10], chronic inflammatory and autoimmune diseases [1, 11] and neurodegenerative disorders [12].
In view of its involvement in the above mentioned pathological processes, inhibition of TrxR is an important clinical goal [1, 8, 11, 13]. Several drugs, used either currently or in the past, are proven TrxR inhibitors: e.g. gold compounds such as aurothiomalate and auranofin (used in rheumatoid arthritis), or emetic tartar (antimony tartrate) [14]. It has been demonstrated that inhibitors of TrxR (or TGR) have antimalarial and schistosomicidal activity [15, 16], and may inhibit the proliferation of tumour cells in culture [17].
As a general rule, the affinities and mechanisms of binding of TrxR inhibitors to the target enzyme are known with scarce precision and conflicting results abound in the literature. This may be partly due to the complex structure and catalytic mechanism of the enzyme. Indeed TrxR has at least two catalytically important redox sites: one is constituted by the FAD and by a couple of Cys residues next to it that receives electrons from NADPH; the other is a Sec(Selenocysteine)-Cys couple (found in H-TrxR of higher eukarya, see below) at the C-terminal end of the polypeptide chain that picks up the electrons from the FAD/Cys redox site and transfers them to transiently bound Trx [18]. Only in rare cases (e.g., the TrxR from insects and that from Apicomplexa [9]) the Sec residue is replaced by a Cys; in these cases the catalytically active Cys has a low pKa value (see below, section 2.1).
Inhibitors of TrxR may bind to the NADPH binding site, or to the Cys or Sec residues of the two redox sites of the enzyme or to other sites, unrelated with either substrate, e.g. the monomer-monomer interface of the homodimer. Some promising TrxR inhibitors are suicide substrates that alkylate or otherwise covalently modify the reactive Cys (or Sec) residues of the two main redox sites of the enzyme (see [19]). Other TrxR inhibitors are metal containing compounds capable to transfer the metal ion to the catalytic Cys (or Sec) residues [20, 21]. While reduction of the physiological substrate Trx occurs at the C-terminal Sec-Cys couple and requires the full functionality of both the redox sites of the enzyme, reduction of artificial oxidized substrates may occur at either or both couples of Cys (or Sec-Cys), and inhibitors that only or preferentially bind to the C terminal Sec-Cys couple may not prevent the conversion of artificial substrates, even when the enzyme is unreactive versus Trx. However, in the literature, there are inconsistencies that cannot be due to the complexity of the enzyme and a critical analysis of published results as well as the experimental procedures is advisable. Comprehensive reviews of TrxR inhibitors have been published [13, 22]; they have been a precious source of information for the present study. However, the incomplete recognition of the peculiarities of TrxR, coupled with the diverse possible mechanisms of inhibition of the enzyme, have resulted in a large amount of undigested data. In particular we noticed that: (i) numerous inhibitors are declared as irreversible, and yet have been assigned non-zero values of IC50 and KI, in spite of the fact that the enzyme remains inhibited after complete removal of the free inhibitor; (ii) in several cases two different KI values have been assigned to the same inhibitor, depending on which substrate was kept constant during the experiments; and (iii) a puzzling number of “atypical” inhibitors have been attributed to the enzyme. We are confident that many of these inconsistencies and peculiarities can be explained by a thorough analysis of the published data and a thermodynamically rigorous (even though simplified) modelling of the catalytic cycle. Although it is not possible to give a general explanation of the inconsistencies, it is evident that at least two factors contribute, namely: (i) analysis of experimental data is often carried out according to models developed for single-substrate enzymes, therefore in the case of two-substrates enzymes it yields apparent, rather than absolute, parameters; and (ii) the incubation of the enzyme with slow-binding irreversible inhibitors has often been insufficient or inadequate.
2. THIOREDOXIN REDUCTASE AND RELATED FLAVOENZYMES
2.1. Structure
H-TrxR, GR and related enzymes show a homodimeric quaternary arrangement, with a two-fold symmetry axis lying on the dimerization interface between the two monomers (see Fig. 1, panel A). In the case of H-TrxR, each monomer displays two major domains: (i) one N-terminal domain containing one FAD molecule inside a huge cleft (residues 1–367 numbering, according to the crystal structure of rat H-TrxR1, [23]); this domain includes the NADPH binding sub-domain (residues 164 –296 for rat H-TrxR1); and (ii) the interface domain (residues 368 – 499) which includes the highly motile C-terminal arm. Both the FAD binding domain and the NADPH binding sub-domain are variants of a Rossmann’s fold [24]. Dimerization confers a particular structural arrangement to the enzyme: the C-terminal arm of one subunit is in proximity of and receives electrons from the FAD domain of the partner subunit. The chemistry and the structure of the C-terminal sequence may vary between eukaria, being X-C-Sec-X in mammals (called Type Ia), X-C-C-X in insects (Type Ib) and C-X-X-X-X-C-X in apicomplexa (Type II) [25, 26]. The electrons transferred from NADPH to the substrates follow a precise path within the protein domains/sub-domains, being first donated to the FAD binding domain, whose active site is constituted by the FAD itself and a couple of Cys residues on the si-face of the isoalloxazine ring, and from these to the C-terminal arm.
Fig. (1).

Panel A: cartoon representation of human TrxR (hTrxR1 – PDB entry 2ZZC). Each monomer is depicted in such a way to differentiate the domains/sub-domains, according to the colour code reported on the right of the frame. The overall structure is a homodimer where a 2-fold symmetry axis was across the dimerization domain. The FAD and NADPH moieties interact at the level of the FAD binding site, where the isoalloxazine ring of flavine stacks onto the nicotinamide ring of NADPH. A peculiarity of TrxR and TrxR-like enzymes (e.g. TGR) is the presence of a second couple of reactive residues (Sec/Cys or Cys/Cys) located on the C-terminal tail. This element of the structure lies in true proximity of the first couple of Cys residues on the si-face of the FAD of the partner subunit in the homodimer. In this way electrons are transferred from the reduced FAD to the couple on the C-terminus belonging to the partner subunit. Panel B: Cartoon representation of human TrxR (PDB entry 2ZZC) in which the red circles highlight the sites where inhibitors have been found for all related flavoenzyme, namely TrxR, GR, TryR and TGR. Examples of inhibitors binding to different sites are depicted in Figs. 2 and 5.
In the oxidized state, the phenolic group of a conserved tyrosine (Y200 in rat H-TrxR) points towards the re-face of the isoalloxazine ring of FAD and the Cys on the si-face are linked by a disulfide bridge. In the presence of NADPH the Tyr side chain swings out in order to accommodate the nicotinamide ring of NADPH. As a consequence of this structural rearrangement, a п-stacking interaction is formed by the phenol ring of Tyr, the nicotinamide ring of NADPH and the isoalloxazine ring of FAD. This facilitates a hydride ion transfer from NADPH to FAD; the reducing equivalent is then transferred from the reduced flavin to the active site disulfide. In the absence of the oxidizing substrate, a thiolate-flavin charge transfer complex is stabilized, resulting in an absorption band detected at 540 nm [27]. Subsequently, a dithiol-disulfide interchange occurs between the reduced cysteines of the FAD domain and the C-terminal redox centre of the partner subunit. This domain swapping electron transfer process makes the dimeric structure mandatory for catalysis. Electrons are then transferred to Trx via the reduced C-terminus [28]. The redox center of the FAD domain could be regarded as a reservoir of electrons from which the redox active C-terminal of the other subunit takes up the reducing equivalents to reduce the incoming substrate.
The first mammalian TrxR to be cloned, TrxR1 from human placenta, was found to have only 31% sequence identity with prokaryotic TrxRs, but to have 44% identity with eukaryotic and prokaryotic glutathione reductases (GR) [29]. A comparison between TrxR and GR is not only desirable but also useful to understand the structure, the catalytic mechanism and the substrate specificity. The amino acid sequence in the catalytic site of human TrxR, -Cys-Val-Asn-Val-Gly-Cys-, is also found in human GR and is located in the FAD domain of both enzymes [29], whereas in the TrxR of Escherichia coli the catalytic site, -Cys-Ala-Thr-Cys-, is part of the NADPH-binding domain [30]. A comparison between TrxR and GR or TryR (the latter two being dissimilar because of a handful of amino acid substitutions, [31]) is useful to understand the catalytic mechanism and the mechanism of inhibition of some classes of compounds that bind to NADPH-dependent flavoreductases of this family [20, 32]. Indeed, structural superposition of human TrxR and GR shows that the same basic fold is shared among all the family members, with major differences located at the C-terminal arm, in some loops exposed to solvent and at the dimerization interface [33]. In GR, the electron transfer from NADPH to the FAD domain takes place in the same way as already seen for TrxR. However, oxidized glutahione (GSSG) is able to bind to the solvent exposed FAD domain Cys couple and the dithiol-disulfide exchange occurs without the C-terminus acting as a mediator. As a consequence, GRs have shorter C-terminal domains with respect to TrxR, and lack the Sec-Cys couple.
Despite the importance of TrxR in several pathological conditions, very few 3D structures of enzyme-inhibitor complexes, that may guide rational drug-design, are deposited in the protein data bank (PDB). In the case of mammalian TrxR, this is difficult to explain, in view of the fact that many of the known inhibitors are irreversible because of covalent binding to the catalytic nucleophilic Cys or Sec residues, and thus they should easily appear in the electron density maps. One possible explanation is that heterologous expression of the protein yields a mixture of Sec-lacking and Sec-containing species (the best incorporation of the Sec observed in E. coli was found to be 50%, [34]). Moreover the presence of two Cys (and Sec) containing active sites and the high concentration of enzyme used for crystallization may increase the number of possible adducts between the reduced protein and the inhibitor (not necessarily present in the conditions of the steady state experiments), yielding a non-homogenous protein preparation. This may have effects at several levels from hampering the crystallization process to lowering the quality of the obtained crystals or to confounding the interpretation of the electron density maps, if any is obtained. Finally, in many structures of TrxR the C-terminal arm, which is an important potential binding site for inhibitors, is disordered. Therefore, it is impossible to ascertain whether the inhibitor is present or not.
Within this scenario, the more abundant 3D structures of inhibitor complexes of GR and TryR present in the PDB (reported in Tables 1 and 2) with compounds similar to some TrxR inhibitors are of great importance to rationalize their mechanism of inhibition against TrxR. Indeed, GR (or TryR) can be considered a simplified version of TrxR, due to the presence of one single redox active site, which uses the same chemistry and stereochemistry of that of TrxR. (Fig. 1, panel B) reports the inhibitors binding sites identified in any of the of closely related flavoreductase family members, TrxR, GR, TryR and TGR, while selected examples of enzyme-inhibitor complexes whose 3D structures have been solved by X-ray crystallography are reported in (Fig. 2). The great majority of known inhibitors bind in proximity of active site Cys, as reported in (Fig. 2, panel A) for a 3,4-dihydroquinazoline derivative inhibitor to Tripanosoma brucei TryR [35]. (Fig. 2, panel B) reports the structure of human GR in complex with a xanthene derivative bound at the dimerization interface, one of the few inhibitors effectively proved to bind at this site [36].
Table 1.
Competitive, rapidly equilibrating inhibitors of TrxR.
| Inhibitor | Enzyme | Competes with | Apparent Ki | Reference | Remarks |
|---|---|---|---|---|---|
| CH3AsI2 | Mouse TrxR | DTNB, Trx | 100 nM | [45] | (1) |
| PpIX (protoporphyrin IX) | Rat TrxR1 | Trx | 2.7 μM | [43] | (1,2) |
| EGCG (Epigallocatechin-3-O-Gallate) | Mammalian TrxR1 | DTNB | 64 μM | [44] | (1) |
| n-butyl 2-imidazolyl disulfide | Human placental TrxR | DTNB | 3.3 μM | [50] | (3) |
| 1-methylpropyl 2-imidazolyl disulfide | Human placental TrxR | DTNB | 13 μM | [50] | (3) |
| n-decyl 2-imidazolyl disulfide | Human placental TrxR | DTNB | 8.6 μM | [50] | (3) |
| 9,10 phenantrene-quinone | Rat TrxR1 | DTNB | 6.3 μM | [51] | (4) |
| Diarylpirrole (4-((1-(4-ethylphenyl)-2-methyl-5-(4-(methylthio)phenyl)-1H-pyrrol-3-yl)methyl)thiomorpholine) | Leishmania infantum TryR | NADPH | 4.6 μM | [52] | (5) |
| 3,4-dihydroquinazoline derivatives | Trypanosoma brucei TryR | DTNB | 0.19-1.5 μM | [35] | (6) |
Notes:
(1) On long incubation times binding becomes irreversible.
(2) Inhibition is only observed in the assay in which [NADPH] concentration is kept constant, while varying the concentration of [Trx].
(3) Inhibition was non-competitive vs NADPH.
(4) The quinone can be reduced producing oxygen superoxides which could lead to the real inhibition of the enzyme.
(5) The structure of the enzyme-inhibitor complex has been determined: PDB entry 4APN.
(6) The structures of the enzyme-non-covalent-inhibitor complexes have been determined: PDB entry 2WP6, 2WPF, 2WPS, 2WPC, 2WPE.
Table 2.
Collection of non competitive inhibitors. The inhibition constants have been taken from the cited references.
| Inhibitor | Enzyme | PDB Entry | Binding Site | Apparent KI | Reference |
|---|---|---|---|---|---|
| Xanthene (6-hydroxy-3-oxo-3H-xanthene-9-propionic acid) | Human cytosolic GR | 1XAN | dimerization interface | 27-48 μM (vs GSSG) 144-176 μM (vs NADPH) |
[36] |
| Safranin (3,7-diamino- 2,8-dimethyl-5-phenyl-phenazinium chloride) | Human cytosolic GR | (coordinates will be deposited) | dimerization interface | 453-586 μM (vs GSSG) | [90] |
| Safranin | Yeast GR | n.d. | n.d. | 500 μM (vs GSSG) | [91] |
| Pyocyanin (5-methylphenazin-1(5H)-one) | Human mitochondrial GR | 3SQP | dimerization interface | given as IC50, 7 μM (vs NADPH) |
[53] |
| EGCG (Epigallocatechin-3-O-Gallate) | Mammalian TrxR1 | n.d | Possibly the C-terminal Cys-Sec | 92μM (vs NADPH) | [44] |
Fig. (2).

Panel A: Cartoon representation of T.brucei TryR (PDB entry 2WPF) in complex with 3-[(4S)-6-chloro-2-methyl-4-(4-methylphenyl)quinazolin-3(4H)-yl]-N,N-dimethylpropan-1-amine, a 3,4-dihydroquinazoline derivative (reported in [35]). The molecule is representative of competitive inhibitors binding near the active site, in true proximity of the active site Cys on the si-face of FAD. The inhibitor moiety is highlighted in green stick. The FAD and active site Cys are depicted in stick. Panel B: Cartoon representation of human GR (PDB entry 1XAN) in complex with 6-hydroxy-3-oxo-3H-xanthene-9-propionic acid (XAN), a xanthene derivative (reported in [36]). The inhibitor is highlighted in blue stick; the FAD and active site Cys are also in stick. XAN binds in a cleft at the dimerization interface, resulting in the noncompetitive inhibition of the enzyme. Panel C: Cartoon representation of SmTGR (PDB entry 3H4K) in complex with two Au atoms, once the enzyme was incubated with gold-containing compund auranofin [20]. The gold atoms were located in two distinct position within the protein scaffold (see inset); Angelucci and coworkers referred to them as Site 1 and Site 2.
2.2. Catalytic Mechanism
Theory
The interpretation of enzyme kinetics needs some hypothesis on the catalytic cycle of the enzyme. As it is the case for many redox enzymes, the catalytic mechanism of TrxR and related reductases may be separated into a reductive and an oxidative half cycle:
| (2.1) |
| (2.2) |
This mechanism is oversimplified and cannot explain all the experiments one may carry out on these enzymes: in particular it overlooks the kinetics of the internal electron transfer between the two (or more) redox sites of the enzyme, and the two (or more) reduced states of the enzyme called EH2 and EH4 in the literature. A more realistic reaction mechanism is reported in (Scheme 2) of [37]. However, the ambition of having a comprehensive and realistic reaction scheme for such a complex enzyme produces a kinetic mechanism that is too complicated to be fully tested and effectively obscures the phenomena that it aims to clarify. Thus, unless specifically needed by very refined kinetic experiments, we shall prefer the simplified mechanism reported above, and shall introduce the further simplification that the first step of both the reductive and oxidative reactions is fast with respect to the second step. This simplification entails that each half cycle is fully described by only two constants, one for the equilibrium with the substrate, the other for the electron transfer reaction. Under most experimental conditions it is unnecessary to consider the backwards reactions (i.e. the electrons transfer from reduced Trx to TrxR to NADP+) given the redox potentials of the oxidizing and reducing substrates: thus both half cycles are assumed to be irreversible.
We define two equilibrium constants and two kinetic rate constants:
| (2.3) |
| (2.4) |
| (2.5) |
| (2.6) |
where k1 is the kinetic rate constant assigned to the irreversible reduction of the enzyme by NADPH and k2 the one assigned to the oxidation of the reduced enzyme by the oxidizing substrate. Strictly speaking the latter reaction should be reversible, but we assume that, under the most commonly employed experimental conditions, reversibility may be neglected, due to the high concentration of the oxidizing substrate and the large difference in the redox potentials of NADPH and the oxidizing substrate(s).
Under steady-state conditions one may equate eqs. 2.5 and 2.6, i.e.,
| (2.7) |
This allows one to calculate the partition of the total enzyme among the four possible catalytic intermediates at any given concentration of its substrates, using the concentration of EO as the reference species:
| (2.8) |
| (2.9) |
| (2.10) |
The above equations allow us to calculate partition function of the total enzyme:
| (2.11) |
The velocity of the reaction catalyzed by the enzyme is usually measured from the oxidation of NADPH, that has a convenient absorbance change (Ó340 nm = 6220 M-1cm-1):
| (2.12) |
In a typical experiment of steady state one keeps constant the concentration of one substrate at a time, while systematically varying the other. This yields two Michaelis-like hyperbolas from which the kcat and KM of each substrate can be derived. Unfortunately these parameters are not absolute, but relative to the concentration of the fixed substrate. The equations above allow us to define the apparent Michaelis parameters as follows.
If the concentration of NADPH is kept constant, the values of KM and kcat for the oxidizing substrate SO (be it Trx or DTNB or any other artificial substrate) may be determined:
| (2.13) |
| (2.14) |
As expected: (i) the calculated parameters depend on the concentration of the fixed substrate (NADPH); (ii) KM So is a function of kcat So and of the affinity of the reduced enzyme for SO.
If the concentration of SO is kept constant, KM and kcat for NADPH may be determined:
| (2.15) |
| (2.16) |
We remark that since the reductive and oxidative half cycles obey similar laws, the formulas that define kcat and KM for the two substrates are identical except that they invert [NADPH] and [So], KN and KS, and k1 and k2.
A considerable simplification could be introduced by using “saturating” concentrations of the fixed substrate. E.g. if NADPH concentration is kept constant, at a value much higher than KN, so that KN +[NADPH]≈[NADPH], one obtains kcat So=k1k2/(k1+k2). However, unless KN has been carefully determined and the concentration of NADPH has been demonstrated to be saturating, the use of this simplification in the analysis of experimental data may lead to wrong results. Indeed, one is not justified in assuming that KN ≈ KM NADPH, since eq. 2.15 and 2.16 make it clear that KN › KM NADPH.
3. GENERAL CONSIDERATIONS ON THE INHIBITION OF TrxR AND RELATED THIOL-BASED REDUCTASES
Inhibitors of TrxR and its homologues are known to act via every possible mechanism of inhibition: competitive, non competitive, irreversible, and mechanism based. In view of the peculiarities of this class of enzymes, however, it is important to make some specific considerations on these mechanisms of inhibition.
3.1. Rapidly Binding Reversible Inhibitors
The textbook treatment of classical reversible inhibitors, competitive, non competitive and uncompetitive, is based on the assumption that they rapidly equilibrate with the enzyme, i.e. that they bind to and dissociate from the enzyme at a rate faster than that of the transformation of the substrates to the products. Inhibitors that satisfy this condition do not require an incubation with the enzyme before the addition of the substrates, and reach a pseudo-equilibrium condition throughout the time of the measurement of the enzyme activity, which usually lasts for less than one minute. Conversely, if an inhibitor requires pre-incubation with the enzyme, its behaviour cannot be analyzed using the classical textbook equations for reversible inhibitors. Unfortunately rapidly binding reversible inhibitors do not constitute the majority of TrxR inhibitors, and some of the confusions present in the literature are due to the incomplete recognition of this fact, and therefore to insufficient incubation time. It would be highly advisable to systematically test, for any new inhibitor, the effect of variable incubation times, as well as any possible recovery of activity after dialysis.
As a general rule, the apparent affinity of the chosen inhibitor for the enzyme, measured at a fixed concentration of one substrate while systematically varying the concentration of the other, and repeating the experiment at several concentrations of the inhibitor, will depend on the concentration of the fixed substrate. The exact relationship between the observed KI and the concentration of the fixed substrate depends on the mechanism of binding of the inhibitor and the details of the catalytic cycle; several pertinent examples are analyzed in detail below. The same applies to the value of the IC50, defined as the concentration of the free inhibitor required to reduce the rate of the catalyzed reaction to half the value observed in the absence of the inhibitor (for a general treatment of this subject see [38]). Consideration of the concentration of the constant substrate is often neglected in the experiments published in the literature that are analyzed using the standard equations developed for single-substrate enzymes. As a result, the meaning of the values of KI that appear in the literature is often unclear, and sometimes frankly perplexing. Indeed there are instances in which the same reaction is assigned two different values of KI, one obtained at fixed concentration of NADPH and variable concentration of Trx (or DTNB), the other obtained in the opposite way round (see below).
In order to determine the value(s) of KI for single-substrate enzymes, the dependence of the apparent values of kcat, KM or kcat/KM on the concentration of the inhibitor is exploited, and it seems worthwhile to summarize the possible cases, in order to facilitate the comparison with the corresponding equations for a two-substrate enzyme like TrxR. The general scheme for a single-substrate enzyme is as follows:
| (3.1) |
A competitive inhibitor has KIS → ∞; a classical non competitive inhibitor has KI=KIS (and KS=KSI); an uncompetitive inhibitor has KI ≠ KIS (and KS ≠ KSI); a special case of uncompetitive inhibitor is obtained if KI→ ∞ (and KSI → 0). The standard relationships between KI (or KIS) and the apparent Michaelis parameters are as follows:
(i) in the case of competitive inhibitors KM app exhibits a linear dependence on the concentration of the inhibitor:
| (3.2) |
and KI = intercept / slope. Since the classical competitive inhibitor shares the same binding site as the substrate, this type of inhibition provides structural information, also in the absence of direct structural data.
(ii) in the case of non competitive inhibitors 1/kcat app exhibits a linear dependence on the concentration of the inhibitor:
| (3.3) |
and KI=intercept/slope
(iii) in the case of uncompetitive inhibitors 1/kcat app and KM app / kcat app both exhibit a linear dependence on the concentration of the inhibitor:
| (3.4) |
and the equilibrium dissociation constant of the complex ESI results KIS = intercept / slope;
| (3.5) |
and the equilibrium dissociation constant of the complex EI results KI = intercept / slope;
(iv) in the special case of uncompetitive inhibition in which KI → ∞ (i.e., where the inhibitor only binds to the enzyme-substrate complex):
| (3.6) |
| (3.7) |
and kcat app/KM app=kcat/KS=constant. In this case KIS can be determined from either kcat app or KM app:
| (3.8) |
and KIS = intercept/slope.
We shall refer to these equations in the text below, in order to make explicit their relationship with the corresponding ones for a two-substrate enzyme like TrxR and related flavoreductases.
Estimating the values of KI from linear regressions of KM app or kcat app is not a statistically sound procedure and should be avoided: indeed the algebraic operations necessary to linearize the Michaelis hyperbolas or the relationships between their parameters and the inhibitor concentration cause a distortion of the experimental error associated with the single measurements, so that small uncertainties in the measured values of the initial velocity, or the parameters are amplified by the mathematical transformation. In a steady-state experiment, the measures of initial velocities obtained at low concentrations of substrate should be regarded as the more error-prone. As stated by Copeland, linear transformations are now of limited usefulness as computer programs that allow non-linear curve fitting can be used in order to estimate the kinetic parameters from the raw data without distorting their experimental errors [39]. However, analyzing the equations 3.2 to 3.8 in themselves is perfectly admissible, since in doing so we are not distorting in any way the experimental error and its statistical distribution. Thus, by writing down eqs. 3.2 to 3.8 we are in no way advocating that they should be used in practice to determine the KI values of any enzyme inhibitor; our only goal is to make explicit which set of relationships our system obeys to.
3.2. Slowly Binding Inhibitors; Irreversible Inhibitors
Slowly binding inhibitors require prior incubation with the enzyme before the addition of substrates; these inhibitors do not reach a pseudo-equilibrium condition during the activity assay and when their effect is analyzed in terms of the textbook equations, they always appear non competitive, even if they bind at the same site of either substrate. This occurs because competition with the substrate requires a time scale longer than that of the assay, thus the fraction of the bound inhibitor seems not to respond to changes in the substrate concentration. To identify this type of inhibitors one should record the enzyme activity in the presence of the inhibitor after different incubation times, and one should also check whether or not the inhibited enzyme recovers activity after extensive dialysis. Indeed differentiating a slow binding inhibitor from an irreversible one may not be an easy task.
Irreversible and mechanism based inhibitors may bind rapidly or slowly and when their reaction with the enzyme is complete, they always mimic the behaviour of non competitive inhibitors, even if they bind at the same site of either substrate. These inhibitors are relatively common for this class of enzymes, which have reactive Cys residues, prone to selective covalent modification by inhibitors. Two conditions are often encountered in the literature, namely (i) the inhibitor requires some incubation with the reduced enzyme, the oxidized Cys being usually unreactive: i.e. for full development of the inhibition TrxR must be incubated with the inhibitor in the presence of the reducing substrate NADPH, and possibly in the absence of oxygen; the oxidizing substrate must not be added before the end of the incubation time. Moreover (ii) the inhibitor may initially behave as a reversible, rapidly binding competitive inhibitor, to become only slowly irreversibly bound. The reaction mechanism may be imagined to proceed via two steps:
| (3.9) |
where the first step is fast (in both directions) and fully reversible, whereas the second one is slow and irreversible. In these cases it is reasonable to determine the KI of the initial, reversible step, and the mechanism and rate constant of the formation of the irreversible end complex.
The redox-active Cys or Sec of TrxR active sites are exquisitely sensitive to irreversible combination with inhibitors; and as a consequence the majority of irreversible inhibitors of TrxR bind in the proximity of the binding sites of oxidizing substrates (Trx, DTNB, etc.), i.e. either at the Cys couple that lies on the si-face of FAD or at the Sec-Cys couple at the C-terminus. By contrast, the binding site of NADPH is not crowded with reactive amino acids, thus inhibitors that compete with NADPH are often reversible. Due to this structural peculiarity, in the initial reversible process, the inhibitor often competes with the oxidizing substrate.
An almost unique peculiarity of thiol-NADPH oxidoreductases is that the oxidizing substrate (e.g. Trx) is chemically similar to the active site of the enzyme, since both contain a redox active Cys couple. Thus, the reduced form of the oxidizing substrate may often compete with the active site of the enzyme for the “irreversible” inhibitor and effectively restore the enzyme activity [40]. The overall reaction is as follows:
TrxR-S-I + Trx-SH → TrxR-SH + Trx-S-I (3.10)
This is not ordinary reversibility: rather, it is similar to ligand exchange between two proteins (e.g. the transfer of O2 from hemoglobin to myoglobin), and has the paradoxical effect of reverting an otherwise irreversible inhibition, under physiological conditions.
4. RAPIDLY BINDING REVERSIBLE COMPETITIVE INHIBITORS OF TrxR AND RELATED ENZYMES
Rapidly binding inhibitors reach the equilibrium with the enzyme over a time scale which is significantly faster than that of the enzyme activity assay. They do not require pre-incubation with the enzyme. If competition of the inhibitor with either substrate is effectively observed in the assay, this is sound proof that the inhibitor equilibrates rapidly with the enzyme. It should be noticed that the opposite is not true: if competition is not observed in standard steady-state experiments this may be due to the inhibitor being competitive but slow to equilibrate with the enzyme, or to the inhibitor being non competitive or uncompetitive, or to the inhibitor being irreversible.
For a two-substrate enzyme, the measurement of the steady state parameters is usually carried out by keeping fixed the concentration of one substrate, while systematically varying the concentration of the other, and that of the inhibitor; the measurement is then repeated by inverting the fixed and variable substrate. Competition will be observed in the Michaelis curves recorded while varying the competing substrate, whereas atypical non competitive curves will be recorded while varying the other substrate.
4.1. Experimental Data
True competitive inhibition of TrxR may occur in the case of compounds that mimic the reducing substrate (NADPH) or in the case of compounds that prevent binding of the oxidizing substrate (Trx), provided that the enzyme and inhibitor rapidly equilibrate. A partial list of inhibitors of this class is reported in (Table 1).
Inhibitors competing with NADPH are rare in the literature; an example is indomethacin that is reported to compete with NADPH in GR [41]. Possibly, these inhibitors are pharmacologically less interesting than competitive inhibitors of the oxidizing substrate because many enzymes use NADPH and thus target specificity is unlikely: indeed indomethacin inhibited all five flavoreductases tested by Chen and coworkers, with KI about 170-500 μM [41]. Another generic inhibitor of NADPH dependent reductases is the dye cibachron blue [42], that has the advantage of undergoing an absorbance change upon binding to the free enzyme. This allows one to directly measure the affinity of this compound for the enzyme in the absence of substrates.
Examples of inhibitors that compete with Trx are Protoporphyrin IX [43] or catechins [44]. They may present two modes of action, being competitive and reversible on short incubation time scales, but becoming irreversibly bound after longer incubation times (see eq. 3.9). This is also the case of arsenic derivatives [45] and of the slowly reacting substrate chaetocin [46]. This phenomenon occurs because the inhibitor initially forms a weak, non-covalent complex with the binding site of the oxidizing substrate, and then slowly forms a covalent complex, presumably with the reactive Cys residues. It is interesting to remark that inhibitors of GR that compete with GSSG and of TryR that compete with trypanothione (e.g., phenothiazines, [47, 48]; synthetic substrate analogues, [49]) are known and present similar properties.
Arsenic derivatives are powerful inhibitors of TrxR. These compounds exhibit a complex inhibition mechanism that essentially corresponds to eq. 3.9: if they are not pre-incubated with TrxR they behave as reversible inhibitors competing with the oxidizing substrate; after some incubation their binding becomes irreversible. Lin and coworkers [45] measured the steady state consumption of substrate by mouse liver TrxR while keeping constant the concentrations of NADPH (the non-competing substrate), systematically varying the concentration of DTNB (a competing chromogenic substrate) and that of the inhibitor methylarsonous diiodide. They obtained Michaelis or Lineweaver plots showing the usual features of competitive inhibition: the inhibitor increases the apparent KM and does not change the Vmax (or kcat; see Fig. 3, upper panel). From these experiments a KI of 100 nM was estimated, using eq. 3.2.
Fig. (3).

Inhibition of mouse liver TrxR by methylarsonous diiodide, CH3AsI2 (redrawn and modified from Figs. 2 and 3 of [45]). Arsenicals are inhibitors of TrxR which on short time scales compete with either Trx or the chromogenic substrate DTNB; on longer time scales they become irreversibly bound to the Cys residues of the enzyme. The data above were taken immediately after mixing the enzyme with the inhibitor and the substrates, hence before covalent binding of CH3AsI2 to TrxR. Upper panel reports an experiment in which the concentration of NADPH is kept constant while that of DTNB is varied. The Lineweaver data plot obtained is typical of competitive inhibition. Lower panel: same experiment as in the upper panel but the concentration of DTNB is kept constant and that of NADPH is varied. An atypical Linweaver plot is recorded in which the lines run more or less parallel to each other (see text).
When the authors run the opposite experiment, i.e. when they measured the steady state substrate consumption at constant DTNB and variable NADPH, they recorded highly atypical Lineweaver plots (Fig. 3, lower panel). Indeed, the lines obtained at different inhibitor concentrations run parallel to each other or almost so, reminiscent of those one would obtain for an inhibitor that only binds to the Michaelis complex of a single-substrate enzyme (eqs. 3.6-3.8). The KI estimated from replot of Vmax values (eq. 3.8) yielded a value of 250 nM, significantly higher than the one measured at constant [NADPH].
These data are perplexing because of at least two reasons: (i) the KI is by definition the affinity of the enzyme for the inhibitor in the absence of substrates, and an inhibitor of this type is expected to have one and only one KI. How is it possible that different methods lead to two so different estimates? And which one is closer to the true value? (ii) The parallel Lineweaver plots in the lower panel of (Fig. 3) imply that this inhibitor, under these experimental conditions lowers the kcat app but leaves (approximately) constant the specificity constant kcat app / KM app. This in turn implies that the inhibitor increases the apparent affinity of the enzyme for the non competing substrate (as evident from the intercepts on abscissa, Fig. 3, lower panel). This fact may be described as an apparent, indirect cooperativity between the inhibitor and the non-competing substrate: binding of the inhibitor seems to favor the binding of NADPH and vice versa. As a consequence of this fact the IC50 (not reported by the authors) is expected to increase with increasing DTNB concentration and to decrease with increasing the NADPH concentration.
A quantitative explanation of these perplexing observations is deferred to next chapter, 4.2, where we shall demonstrate that the lower value is the actual parameter, while the higher value is an apparent, uncorrected estimate of the same. Unfortunately, the authors do not report information enough to prove this point beyond confirming the expectation that KI app > KI. Before turning to this we shall examine other competitive inhibitors to demonstrate that the problems highlighted above are a general occurrence. However, we want to firmly state one point: these results are not the consequence of faulty experimental methods, nor are they due to the inhibitor binding irreversibly during the assay. Indeed, if it is true that upon long incubation times under reducing conditions irreversible binding occurs and the enzyme loses its activity, Lin and coworkers [45] carefully demonstrated the reversibility of inhibition after short incubation.
Protoporphyrin IX is a very peculiar inhibitor of rat TrxR1 [43]. When this molecule is added to a TrxR assay in which the concentration of [NADPH] is kept constant while that of Trx is varied, a typical competitive inhibition is observed with an apparent KI of 2.7 µM. After long incubation time, however, the inhibitor becomes irreversibly bound with an apparent second order rate constant k = 0.73x103 M-1s-1. By contrast when the concentration of Trx is kept constant while varying that of NADPH little inhibition is observed. This result is paradoxical and at present unexplained. A similar irreversible inactivation was obtained with rottlerin (k = 0.3x103 M-1s-1) [43].
Green tea extracts inhibit rat and calf TrxR1 by competing with Trx and other oxidizing substrates. The most important identified inhibitors from this source are (-)-epicatechin-gallate (ECG) and (-)-epigallocatechin-3-gallate (EGCG), but other catechin derivatives may also contribute to the observed effect. The IC50 values, under the experimental conditions used by Wang and coworkers are 17 µM for ECG and 26 µM for EGCG [44]. On long incubation times under reducing conditions, these compounds become irreversibly bound to the enzyme. The authors report a KI of 64 µM in the assay at constant [NADPH] and variable [DTNB] (where competition is observed) and of 92 µM in the assay at variable [NADPH] and constant [DTNB] (where competition is not observed). This is the same type of discrepancy we noticed in the case of methylarsonous diiodide (see above), on a smaller scale.
Alkyl 2-imidazolyl disulfide analogues are reported to be reversible competitive inhibitors of human placental TrxR [50]. These inhibitors compete with the chromogenic substrate DTNB, with KI values in the order of 5-10 µM (see Table 1); inhibition was found to be non competitive with NADPH. The chemical structure of these compounds, and their ability to react with reduced glutathione suggest that they might be slowly reacting substrates.
As a general rule, quinones are substrates, rather than inhibitors, of TrxR. However, 9,10 phenantrene-quinone is reported to be a competitive inhibitor (KI = 6.3 μM) of the reduction of the chromogenic substrate DTNB [51]. This is probably due to the fact that this compound effectively binds to the active site of TrxR with higher affinity than the substrate, but is more slowly transformed. Indeed Cenas and coworkers demonstrated that 9,10-phenantrene quinone is slowly oxidized by the enzyme [51].
Phenothiazines are tricyclic compounds acting as inhibitors of trypanosome TryR [47, 48]. Trifluoperazine competes with the oxidizing substrate trypanothione and has KI = 22 μM when measured at variable trypanothione concentration and KI = 31μM at variable NADPH concentration. Here again we notice that the KI value recorded in experiments carried out at variable concentration of the competing substrate (trypanothione) is lower than that recorded when variable concentration of the non-competing substrate are used, as already observed for dimethylarsonous diiodide and catechins (see above).
Unfortunately, 3D complexes between TrxR and competitive inhibitors are not present in the PDB; the only structures available are those of the related TryR from Leishmania infantum and Trypanosoma brucei in complex with diarylpirrole [52] and 3,4-dihydroquinazoline [35], respectively. Both these inhibitors interact with key residues of the trypanothione binding site, in particular with those that define the hydrophobic pocket necessary to recognize the spermidine moiety of the substrate (Fig. 2, panel A). The steady state experiments recorded at variable concentration of the competitive substrate confirm the expected competition with trypanothione with KI values in the micromolar range.
4.2. Theory
When an enzyme sequentially reacts with two substrates in the presence of a competitive inhibitor of one of these, classical Michaelis plots can be obtained by keeping constant the concentration of either substrate and varying the other. Referring to the simplified catalytic model described above (eq. 2.1 and 2.2), and adding a rapidly binding, reversible inhibitor that competes with the oxidizing substrate, we may schematize:
| (4.1) |
| (4.2) |
In the above scheme, the inhibitor binds to the same site as the oxidizing substrate with the equilibrium dissociation constant KI, and in principle is insensitive to the oxidation state of the enzyme; thus it may also populate the species EOI. This species can be safely neglected in our simplified scheme, under the assumption that the inhibitor does not perturb the binding site of NADPH: thus EOI can bind NADPH and be reduced to ERI at the same rate of EO. Again, this assumption is an oversimplification and the inhibitor might indeed affect the rate of enzyme reduction, but unless one is compelled by strong experimental evidence, Occam’s razor dictates that this possibility is ignored.
Under the assumption that the equilibration of the inhibitor with ER is fast with respect to the time of the assay, the pseudo-equilibrium approximation applies and the steady state consumption of NADPH is described by the following equation:
 (4.3)
(4.3)
On the one hand, in an experiment in which the concentration of [NADPH] is kept constant while systematically varying that of the oxidant (Trx or any artificial substrate) one would obtain a classical Michaelis plot for competitive inhibition, with the following apparent steady state parameters (to be compared with 2.12 and 2.13):
 (4.4)
(4.4)
 (4.5)
(4.5)
Since the competitive inhibitor does not change the kcat So of the enzyme, we do not need to call this an “apparent” value; by contrast the competitive inhibitor changes the apparent KM (KM app So) of the enzyme and we use eq. 4.5 to correlate the KM app So, measured in the presence of the inhibitor, with the true KM So, measured in the absence of the inhibitor (eq. 2.14). Comparison of eqs. 4.5 and 3.2 shows that application of the analysis devised for single-substrate enzymes in this case yields the correct estimate of KI.
On the other hand, in an experiment in which the oxidant is kept constant while systematically varying NADPH one would obtain an “atypical” case of non-competitive inhibition, with the following apparent steady state parameters (to be compared with eq. 2.15 and 2.16):
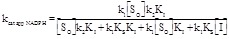 (4.6)
(4.6)
 (4.7)
(4.7)
We remark that in this type of experiment the inhibitor decreases the kcat:
 (4.8)
(4.8)
that is kcat app NADPH < kcat NADPH, as expected. Quite surprisingly this inhibitor also decreases the apparent KM for the reducing substrate (i.e. KM app NADPH < KM NADPH), given that KM app NADPH is directly proportional to kcat app NADPH. (eq. 4.7). This effect has been often observed in the experiments (see point (ii) in section 4.1 and Fig. 3, lower panel). We also remark that the specificity constant, kcat app NADPH / KM app NADPH does not vary, irrespective of the concentration of the inhibitor, and equals kcat NADPH / KM NADPH. Since the slope of the Lineweaver and Burk plot is the reciprocal of this parameter, the straight lines one obtains at different concentrations of the inhibitor run parallel to each other (Fig. 3, lower panel).
Even though equations 4.6 through 4.8 do not require an explanation, one may want to visualize the reason why the inhibitor decreases the apparent KM of the non-competing substrate. This happens because, in the simplified reaction scheme considered above, the oxidized TrxR is catalytically active irrespective of the presence or absence of the inhibitor in the Trx binding site (and indeed the species EOI has not been explicitly taken into account since it is assumed to be functionally indistinguishable from EO). The only inhibited intermediate is ERI, which, in an experiment where the concentration of SO is kept constant, constitutes a fixed fraction of the total amount of the reduced enzyme. Thus, the higher the concentration of NADPH, the greater the amount of the reduced enzyme, and the greater the fraction of the total enzyme which is inhibited. In an experiment in which the concentration of NADPH is varied, the velocity of the reaction reaches a plateau at relatively low concentrations of NADPH, because the reducing substrate indirectly favors the binding of the inhibitor. Hence the apparent KM for the reducing substrate is lower. This reasoning also explains the similarity of the Lineweaver plots obtained by Lin and coworkers ([45], Fig. 3, lower panel) with those expected for an inhibitor that binds to the Michaelis complex of a single-substrate enzyme (eqs. 3.6-3.8), since in both cases the inhibitor promotes the binding of the variable substrate.
The IC50 is defined as the free inhibitor concentration required to reduce the rate of the catalyzed reaction to half its value in the absence of the inhibitor [38]:
 (4.9)
(4.9)
We remark that if SO is increased (at constant [NADPH]) the IC50 also increases, as expected since the inhibitor and the oxidized substrate compete for the same binding site on the macromolecule. By contrast, if [NADPH] is increased while keeping constant SO the IC50 decreases as a hyperbolical function of [NADPH].
These observations lead us to an important conclusion: if the steady state experiments are interpreted using the textbook equations for single
substrate enzymes, one obtains two different apparent values of KI (see section 4.1 above for some examples). On the one hand, if the
experiment is carried out at constant [NADPH] and variable [SO], replot of KM app So vs. [I] yields a straight line with slope K M So / KI and intercept KM So (eq. 4.5), as in the textbook treatment of competitive inhibitors of single substrate
enzymes (eq. 3.2). On the other hand, if the experiment is carried out at constant [SO] and variable [NADPH], replot of 1/k cat app NADPH vs. [I] yields a straight line with slope
 (eq. 4.8), much different from the slope
(eq. 4.8), much different from the slope
 obtained in the case of single substrate enzymes (eqs. 3.3 or 3.4 or 3.8). If an apparent KI value is erroneously calculated as in the case
of single substrate enzymes, one obtains KI app = intercept/slope, the relationship between KI app and KI being
obtained in the case of single substrate enzymes (eqs. 3.3 or 3.4 or 3.8). If an apparent KI value is erroneously calculated as in the case
of single substrate enzymes, one obtains KI app = intercept/slope, the relationship between KI app and KI being
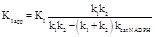 (4.10)
(4.10)
We remark that KI,app > KI, i.e. in the experiments carried out at constant NADPH concentration and analyzed using the equations for single-substrate enzymes (eq. 3.3 or 3.4 or 3.8), KI is overestimated (see point (i) of section 4.1, above). Moreover, KI app depends on kcat NADPH, which in turn is directly proportional to the concentration of the constant substrate SO. Obviously it is always advisable to fit the experimental data with the rate equations, rather than to rely on the replot of apparent equilibrium and kinetic parameters, since the former procedure is statistically more sound and avoids the problem of converting apparent into real parameters. However in this case the problem arises because of the use of equations developed for single-substrate enzymes and would not be solved by the mere application of non-linear regressions instead of linearizations: there is no substitute for the application of the correct equations.
In conclusion, it is our opinion that the discrepancies present in the literature are due to incomplete realization of the complexities inherent in the application of the textbook equations for inhibition of single-substrate enzymes to a two-substrates enzyme. Unfortunately, even under the most accurate analysis, the so called real parameters are model dependent, and two authors using different models might still come out with different values of KI for the same inhibitor.
5. NON COMPETITIVE AND UNCOMPETITIVE INHIBITORS
Classical, fully reversible non competitive and uncompetitive inhibitors of TrxR and its homologues are known, as listed in (Table 2). Only a few of them have been satisfactorily characterized, and are described here, whereas many others are grouped further below as dubious cases. The reason behind this decision is simple: inhibitors that bind slowly to the enzyme, and irreversible inhibitors, at a first screening may behave like reversible non competitive inhibitors; thus if reversibility has not been specifically looked for, an apparently non competitive inhibitor may actually belong to a different group. The most obvious test of reversibility is the full recover of the enzyme activity after dialysis; the most obvious test of rapid equilibration between the enzyme and the inhibitor is that no incubation is needed for inhibition to develop. Doubts may arise in the case of high affinity inhibitors, when the bound inhibitor accounts for a significant fraction of the total inhibitor concentration. In the case of non competitive or uncompetitive inhibitors the usual enzyme assays give no obvious indication whether chemical equilibrium of the enzyme-inhibitor complex has been reached, and the effect of pre-incubation of the enzyme with the inhibitor should be systematically tested. A good experimental practice is as follows: the mixture of the enzyme plus either of its substrates is incubated with the chosen inhibitor for variable times, then the other substrate is added and the activity is recorded. In the case of rapidly binding inhibitors, the activity of the enzyme is independent of the incubation time. The same experiment should then be repeated exchanging the substrates.
5.1. Experimental Data
Even though control experiments to recognize a non-competitive from a slow and/or irreversible inhibitor were not always run, inhibitors whose complex with the enzyme has been characterized by X-ray crystallography to demonstrate that binding is non-covalent and that the binding site does not overlap with that of NADPH or the reducing substrates are likely to be true non-competitive inhibitors. In the majority of the cases that have been subjected to detailed structural characterization, the binding site lies at the monomer-monomer interface. Three fused rings differently derivatized and oriented in an anthracene-like structure characterize the architecture of some such compounds. The 3D complexes found in the PDB (see Table 2) are those between human GR in complex with 6-hydroxy-3-oxo-3H-xanthene-9-propionic acid (XAN, a xanthene derivative; [36], PDB entry 1XAN); safranin (a protein-staining dye); and pyocyanin (a toxin produced and secreted by the Gram negative bacterium Pseudomonas aeruginosa; [53]). The binding pocket at the dimerization interface contains the two-fold symmetry axis of the homodimer and interconnects the two GSSG binding sites. In the case of human GR-safranin complex, it is evident how the enzyme symmetry axis corresponds to that of the inhibitor, which is generated by the planarity and the linear disposition of the three rings [36]. Inhibitors of TrxR which share similar stereochemical characteristic are known [14, 54] but due to incomplete characterization of the steady state parameters and to the lack of 3D structures one cannot be confident that their mode of action is the same as that observed in GR (Fig. 2, panel B).
Savvides and Karplus gave important information on the mechanism of enzyme inhibition while describing two interesting inhibitors of human GR, safranin and XAN [36]. They reported that the former behaves as an uncompetitive inhibitor when the enzyme activity is tested at variable [GSSG] and constant [NADPH] with KI = 453 μM, KIS = 586 μM (in the original work the authors name these parameters KIS and KII respectively) and that XAN behaves like an uncompetitive inhibitor with respect to both substrates with KI = 27 μM and KIS = 48 μM when tested at constant [NADPH] and variable [GSSG] (Fig. 4), and KI = 144 μM, KIS = 176 μM at constant [GSSG] and variable [NADPH]. The authors analyze their functional data using the equations of single-substrate enzymes, even though they acknowledge that the procedure is not ideal in the case of a two-substrate enzyme. An intriguing observation is that XAN significantly lowers the affinity of the enzyme for GSSG, as this substrate cannot be observed in the electron density maps; this effect is more marked than one would expect from the KI and KIS values. To minimize the difficulties inherent in the analysis of a two-substrate enzyme with equations developed for one-substrate ones, the authors in their experiments used “saturating” concentrations of the fixed substrate. This procedure is sound but does not yield real values of the KI and KIS, which should in any case be considered apparent parameters (see below).
Fig. (4).

Inhibition of hGR by XAN. Kinetic measurements reported as a Lineweaver-Burk plot showing the effect of XAN on hGR activity measured at variable GSSG concentrations (30 – 1000 μM) and at constant, saturating 100 μM NADPH concentration [36]. The concentrations of XAN used were 10μM (◊), 20μM (+), and 30 μM (♦). The control measurements in absence of inhibitor were marked as (●). (Modified from a research originally published in Journal of Biological Chemistry by Savvides and Karplus [36]).
The redox active cofactor of bacterial quinoproteins, pirroloquinoline quinone (PQQ), seems to act as a reversible inhibitor of Trx reduction by TrxR [55] while increasing the enzyme activity versus naphthoquinone substrates. The mechanisms of inhibition of Trx reduction, whether competitive or non competitive, is unclear. PQQ was also demonstrated by the same authors to inhibit GR, with a mechanism that cannot be purely competitive, and seems to be uncompetitive, since both kcat and KM are affected. The affinity of PQQ for yeast GR is in the micromolar range.
In an extensive search of antimalarial drugs Theobald and coworkers [56] found seven new non competitive apparently reversible inhibitors of Plasmodium falciparum TrxR, with apparent KI values in the range of 1.3-4.1μM. These compounds have a similar and quite complex polycyclic chemical structure that the authors grouped into two novel chemical families not previously described to have antimalarial activity. All seven inhibitors are potent and selective, and contain electrophilic moieties. No structural data were presented, but the authors ascribed the loss of catalytic activity of the enzyme to a non competitive inhibition with respect Trx and NADPH, and indicated the dimer interface as the possible binding site of the molecule, further alluding to possible allosteric mechanism.
Andricopulo and coworkers described a similar behaviour for three P. falciparum TrxR inhibitors [57]. The IC50 of these compounds is in the 0.5-2 μM range for P. falciparum TrxR and 4-140 μM for human TrxR. The authors suggest that these inhibitors could interfere with dithiol−disulfide exchange, thus preventing the reduction of the C- terminal Cys pair (it should be remembered that in P. falciparum TrxR Sec is replaced by Cys) and their solvent exposure, required to reduce the Trx substrate.
5.2. Theory
A complete reaction scheme for an uncompetitive inhibitor is depicted below:
 (5.1)
(5.1)
In this scheme the inhibitor can bind to both the reduced and oxidized state of the enzyme, and in the presence or in the absence of either or both substrates, in principle with four different affinity constants. Moreover either the oxidation or reduction of the enzyme may still be possible (not indicated in the scheme).
Developing the complete treatment of this reaction mechanism is complex, since eight constants are involved; the experimental measurements, to be performed in order to evaluate these constants, are even more complex and error-prone. Thus, it is reasonable to try to simplify the scheme as much as possible to see whether the experimental data really require the entire scheme or are compatible with a simplified version of it.
The first possible simplification is to assume that KOI = KOI N = KRI = KRI S. Under this assumption we have the perfect non competitive mechanism governed by just one KI and the total enzyme is divided into two fractions: the inhibitor-bound and inactive, that amounts to [I] / ([I] + KI) and the inhibitor-free and active, amounting to KI / ([I] + KI). Substrates and redox state do not influence the fraction of inactive enzyme. The kinetic parameters KM and kcat are identical to those calculated in the absence of the inhibitor (eq. 2.12-2.15) but the Vmax, that equals the product of the total enzyme concentration times the kcat, should be further multiplied by KI / ([I] + KI) to take into account that only a fraction of the enzyme is active:
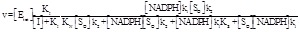 (5.2)
(5.2)
The dependence of 1/Vmax app on [I] is linear, with slope 1/KI Vmax, irrespective of the concentration of the constant substrate used in the experiment (see eq. 3.3). The above simplification is most often too radical to account for the actual experimental data: most non-competitive inhibitors behave in a more complex way.
The next possible step is to assume that the inhibitor’s affinity depends on the redox state of the enzyme but not on the presence of either substrate, i.e.: KOI = KOIN and KRI = KRIS. Under this simplification we obtain that the fractions of inhibitor-bound oxidized and reduced enzyme are independent of either substrate, but different from each other:
 (5.3)
(5.3)
 (5.4)
(5.4)
Contrary to the simple non competitive scheme considered above, in this scheme the relative concentrations of the two substrates influence the total amount of inhibited enzyme. This happens because the relative concentrations of the substrates influence the fractions of the oxidized and reduced enzyme, which differ in their affinity for the inhibitor. In particular, if KRI > KOI (i.e. the oxidized enzyme has greater affinity for the inhibitor than the reduced form) then an increase in the concentration of the oxidizing substrate SO results in greater inhibition, and an increase in the concentration of NADPH results in a decrease in inhibition. The opposite holds if KRI < KOI.
The partition of the total enzyme among the catalytic intermediates at any given concentration of its substrates and of the inhibitor, using the concentration of EO as the reference species is as follows:
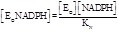 (5.5)
(5.5)
 (5.6)
(5.6)
 (5.7)
(5.7)
 (5.8)
(5.8)
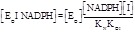 (5.9)
(5.9)
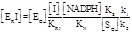 (5.10)
(5.10)
 (5.11)
(5.11)
The rate of NADPH consumption results:
 (5.12)
(5.12)
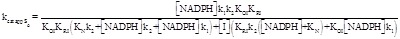 (5.13)
(5.13)
If the concentration of NADPH is kept constant and a family of steady state curves is recorded by systematically varying [SO] and [I], one obtains:
 (5.14)
(5.14)
and
 (5.15)
(5.15)
These equations clearly show that the KM app So depends on [I], with a law different from that of kcat app So; as a consequence an inhibitor of this type will simulate the behaviour of an uncompetitive inhibitor of a single-substrate enzyme in that it will affect both the apparent Michaelis parameters (see eqs. 3.4 and 3.5), and will do so with both substrates.
It is easily appreciated that the replot of 1/kcat app So vs. 1/[I] in this case yields a straight line whose slope is a complex convolution of KRI and KOI. Even more interesting, replot of KM app So/kcat app So vs. [I] yields a straight line with intercept KS/k2 and slope KS/k2 KRI; thus KRI = intercept / slope, a further apparent similarity to the uncompetitive inhibitor (see eq. 3.4). Since the system is symmetric with respect to its two substrates, the experiment in which the effect of the inhibitor is tested at fixed concentration of So and a variable concentration of [NADPH] will yield 1/kcat app NADPH vs. [I] and KM app So/kcat app So vs. [I] plots whose slopes will be a different convolution of the same parameters.
Given that this type of inhibitor changes both the apparent KM and kcat (for each substrate), and that the above relationships bear a rough similarity with those observed in the case of the uncompetitive inhibitor binding to a single-substrate enzyme, one may easily be mislead and analyze the Lineweaver plots of an inhibitor of this type to obtain four apparent KI values, while in the above (Scheme 5.1-5.4) only two are needed. We are not in the position of saying that this analysis would give an acceptable description of the data on either GR and safranin or GR and XAN (or of any other data set in the literature), but we are tempted to say that at least some of the data of this kind may hardly require more than two values of KI. Unfortunately an enzyme may be more complex than our experiments can resolve, and it is perfectly plausible that the chemical equilibria between GR and XAN effectively entail four constants, even if they might be acceptably described by only two.
6. IRREVERSIBLE AND SLOW-BINDING INHIBITORS
Irreversible inhibitors of thiol reductases are common. Most usually these compounds covalently bind to reactive Cys or Sec residues of the catalytic site(s) of the enzyme. This mode of binding requires these residues to be reduced, thus some incubation with the enzyme in the presence of both NADPH and the chosen inhibitor is usually necessary. This peculiarity blurs the distinction between irreversible inhibitors and mechanism-based inhibitors. The enzyme remains inhibited in the presence of all substrates and dialysis or gel filtration do not recover its activity. In some cases, however, activity can be restored by treating the enzyme with reduced organic sulfides (e.g. cysteine, glutathione, mercaptoethanol, dimercaptopropanol or Trx) that effectively remove the inhibition by a ligand exchange reaction ascribed to the sulfur atoms of thiol residues in the active site (eq. 3.10). Since reduced thiols are the product of the reaction catalyzed by these enzymes, if the enzyme is not completely inhibited and its substrates are present, its residual activity reduces some thiols that may in turn partially restore the fraction of the active enzyme, yielding an autocatalytic time course.
Irreversible inhibition may or may not be preceded in time by a condition of measurable reversible inhibition, and may require considerable time to fully develop.
The chemical properties of inhibitors that covalently bind to the sulfur atom of Cys (or the selenium atom of Sec) should be carefully studied, because these enzyme-inhibitor couples may have a peculiar and very dynamic reactivity. Indeed, not only an irreversible inhibitor may often be removed by incubation with a low molecular weight thiol or by chemical reduction, but an enzyme evolutionarily related to TrxR and GR, Mercuric Reductase, specifically exploits this property, using NADPH to reduce mercury bound to a Cys residue of the protein, which is released as metallic Hg0 ([3] and references therein).
6.1. Experimental Data
Since irreversible inhibitors of TrxR and related flavoreductases are numerous (see Table 3), it seems convenient to classify them under separate headings, depending on their chemical structure. All or almost all of them bind to the catalytic Cys or Sec residues.
Table 3.
A partial list of families of irreversible inhibitors of TrxR and related flavoreductases.
| Family of Inhibitor | Enzyme | References | Notes | PDB Entry |
|---|---|---|---|---|
| Metals | ||||
| Gold-based | ||||
| auranofin | Schistosoma mansoni TGR | [20] | (1) | 3H4K |
| auranofin | Leishmania infantum TryR | [92] | (1) | 2YAU |
| aurothiomalate | Rat cytosolic TrxR1 | [93] | ||
| Phosphine Gold(I) | Human cytosolic TrxR1 | [94] | ||
| GoPI ([{1-phenyl-2,5-di(2-pyridyl)phosphole}AuCl]) | Human GR | [95] | (1) | 2AAQ |
| Gold cyanide | Entamoeba histolytica TrxR | Parsonage et al. | (2) | 4A65 |
| Et3PAuCl [N-(N’,N’-dimethylaminoethyl)-1,8-naphthalimide-4 sulfide]triethylphosphineAu(I)] |
Rat liver TrxR | [96] | (1) | |
| Gold(I)carbene complexes | Human TrxR1 Human mitochondrial TrxR2 Human GR |
[97] | (1) | |
| Gold(III)-dithiocarbamato complexes | Human TrxR1 | [98] | (1) | |
| AuBiPy AuXil AuPy |
Human TrxR2 | [99] | ||
| Silver-based | ||||
| Silver-nanoparticles | L. infantum TryR | [78] | (1) | 2X50 |
| Antimony-based | ||||
| Tartar emetic | L. infantum TryR | [78] | (1) | 2W0H |
| Platinum-based | ||||
| Terpyridine-Pt(II) | Human TrxR1 | [100, 101] | 2ZZB | |
| Cisplatin [cis-diaminodichloroPt(II)] | Human placental TrxR | [102] | ||
| Carboplatin | Human placental TrxR | [102] | ||
| Oxaliplatin | Rat TrxR | [103] | ||
| Terpyridine-platinum(II) | Human TrxR1 | [100, 103] | ||
| Ruthenium | ||||
| trans-[bis(2-amino-5-methylthiazole)tetrachloro ruthenate(III)] |
Rat TrxR | [63] | ||
| Mercury | ||||
| Monomethylmercury | Rat TrxR | [68] | (1) | |
| Mercuric chloride | Rat TrxR | [68] | (1) | |
| Gadolinium | ||||
| Motexafin Gd | Rat TrxR1 | [104] | ||
| Arsenic | ||||
| Arsenic trioxide | Human TrxR1 | [105] | ||
| Methyl As(III) | Human TrxR1 | [106] | ||
| Organic compounds | ||||
| 13-cis retinoic acid | [107] | (1) | ||
| Curcumin | Rat TrxR | [73] | (3) | |
| 2,4-Dihydroxybenzylamine | Human GR | [74] | ||
| nitrosoureas | Human GR | [40] | (3) | 1GRH 1GRG |
| Dinitrohalobenzenes | Human TrxR1 | [71, 72] | ||
| Quinacrine mustard | T. brucei TryR | [108] | (4) | 1GXF |
| Ajoene ((E,Z)-4,5,9-trithiadodeca-1,6,11-triene 9-oxide) | Human GR T. brucei TryR |
[109] | (3) | 1BWC |
| fluoro-analogue of a menadione derivative | Human mitochondrial GR | [79] | (5) | 2GH5 |
| bromo-isophosphoramide | Human GR, human TrxR1 | [110] | ||
| Benzisothiazolone | T. brucei TryR | [111] | ||
| Nitrogen Reactive Species | ||||
| Peroxynitrite | HumanGR | [112] | (6) | 1K4Q |
| Dinitrosoglutathione | HumanGR | [113] | (7) | 1GSN (hydroxyCys) 1DNC (sulfinoCys) |
| S-nitrosoglutathione | [75] | (8) | ||
Notes
(1) Coordination of catalytic Cys residues adjacent to the FAD (possibly also other Cys or Sec)
(2) structure deposited, paper to be published
(3) Alkylation of the FAD catalytic cysteine
(4) Modification of the FAD catalytic cysteine
(5) Alkylation of the FAD catalytic cysteine
(6) Nitration of Tyr residues at the glutathione disulfide-binding site
6.1.1. Metal Ions and Their Complexes
Gold complexes are excellent irreversible inhibitors of thiol-reductases. Cai and coworkers report quite an extensive list of Au(I) and Au(III) complexes that are capable of inhibiting TrxR and related thiol reductases, and in many cases they list the IC50 or KI values found in the literature [13]. It is not always clear whether or not the reversibility of the enzyme-inhibitor complex has been tested by dialysis, thus these values should be regarded with caution. Indeed, if an inhibitor irreversibly binds to the enzyme its KI and IC50 should be both zero [38, 58], given that the complex does not dissociate under conditions in which the free inhibitor has been removed (usually by dialysis). An explanation of the possible reason(s) why non-zero values of these parameters are so often measured will be provided in the following chapter 6.4.
The typical mechanism of Au(I) binding to TrxR and TGR was reviewed by us [21] for the case of auranofin (Figs. 7 to 9). The coordination geometry of Au is linear bidentate and both sulfur and selenium are excellent gold ligands. We could demonstrate by means of X-ray crystallography that in Schistosoma mansoni TGR treated with auranofin (AF), at least two gold ions are bound to the proteins: one is coordinated between the two Cys residues on the si-face of FAD co-factor, and the other between two other Cys residues (at positions 520 and 574; Fig. 2, panel C). The C-terminus is disordered in our structures, thus we cannot state, nor exclude, binding to the Cys 596 – Sec 597 couple. AF is a bidentate gold complex in which the metal is coordinated by acetoxythioglucose and triethylphosphine, but the molecule is not found in the electron density maps of the inhibited enzyme(s): most probably the metal is freed first of thioglucose, which is replaced by the Se atom of Sec597, and then of triethylphosphine, replaced by a Cys residue. The metal would then be transferred to other Cys couples by thiol exchange. We hypothesize that both reactions occur via a transient three-coordinate intermediate (Fig. 5).
Fig. (7).
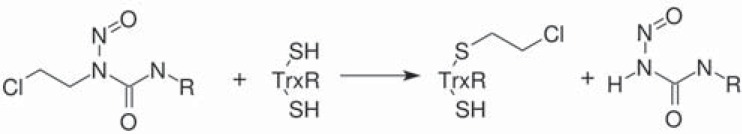
The proposed reaction of nitrosourea derivatives with TrxR active site Cys residues.
Fig. (9).

Mitomycin C and its reaction with Sec-containing TrxR.
Fig. (5).

Possible reaction path for AF and SmTGR (this research was originally published in Journal of Biological Chemistry by the authors. [20]. © the American Society for Biochemistry and Molecular Biology).
Gromer and coworkers demonstrated that pre-incubation of AF with NADPH-reduced TrxR from human placenta is necessary to observe significant inhibition and reported that AF is a much more efficient inhibitor of TrxR than of GR, with estimated KI values of 2-3nM and in the μM range respectively [59]. However GoPI, a different gold-containing compound, is a strong inhibitor of GR and the structure of the GR-Au complex reveals a gold ion coordinated to the two active site Cys residues, with exactly the same geometry observed for the TGR-Au couple in the case of AF [60]. GoPI forms an initial reversible complex with human GR in which it behaves as a competitive inhibitor (competing with GSSG), with KI = 460 nM; gold is then transferred to the Cys residues of the active site and inhibition becomes irreversible. Comparison of the results obtained on GR using AF and GoPI is illuminating. Indeed if the end complex were the same, both inhibitors should be irreversibly bound and no difference in affinity should exist between the two (except for the possible initial reversible complex, if formed to any measurable extent in the case of AF). We searched for a possible alternative explanation of the postulated difference in the KI of GR and TrxR for AF and demonstrated that the reaction of AF with GR (or with a truncated variant of TGR from S. mansoni, lacking the Sec residue) is much slower than with wild type, Sec containing, TGR [10, 20]. Moreover, we demonstrated that the reaction of gold incorporation is greatly accelerated by the addition of an external source of Se (benzene-selenol; Fig. 6).
Fig. (6).

A, time course of baker’s yeast GR inactivation by AF – concentrations of 1 μM (■), 4 μM (●), 10 μM (○), and 50 μM (∆). B, time course of baker’s yeast GR inactivation by AF after adding BzSe – 4 μM AF (●) or 4 μM AF plus 2 μM BzSe (▲); GR exposed to a mixture in which 4 μM AF and 2 μM BzSe were preincubated for 2 h before the assay (closed pentagon). C, time courses of wild type SmTGR and truncated SmTGR inactivation by AF, and the effect of BzSe – 8 μM AF (●) or 8 μM AF plus 3 μM BzSe (▲). Truncated SmTGR exposed to a mixture in which 8 μM AF and 3 μM BzSe were preincubated for 2 h before the assay (closed pentagon); time course of wild type 20 nM SmTGR incubated with 50 nM AF (○).(this research was originally published in Journal of Biological Chemistry by the authors. [20]. © the American Society for Biochemistry and Molecular Biology).
We suggest that, at least in some instances, the apparent “low affinities” of thiol-reductases for gold are artifacts due to three possible reasons, namely: (i) slow reactivity of the chosen enzyme and insufficient incubation time (see below, section 6.4); (ii) measurement of the effective KI of an initial reversible complex, possibly mixed with a fraction of the irreversible one; and (iii) pseudo reversibility due to transfer of the metal to the substrate (see below, section 6.2). It is highly unlikely that there is any real difference in the affinity of gold based inhibitors and thiol-reductases, given that these inhibitors only act as gold carriers and once the metal has been transferred to the enzyme, the complex has exactly the same coordination geometry. It is also unlikely that the values of KI thus measured are reliable. Indeed the linear S-Au-S geometry allowed by the redox-active Cys couples of the enzyme is expected to be similar to that of the corresponding inorganic complex, whose estimated stability is in the order of 55 kcal /mol [61]. This value is inconsistent with the estimated KI values in the nanomolar range (corresponding to approximately 11 kcal /mol), let alone the μM values reported for GR. There is no doubt that, as a general rule, metal based inhibitors strongly prefer Se containing TrxR to Se-lacking GR, an observation that needs to be reconciled with the practically irreversible nature of both complexes. Our best explanation of this apparent inconsistency is based on kinetic, rather than thermodynamic, properties of S and Se; indeed we demonstrated that under common experimental conditions Sec is a much more efficient ligand than Cys to undress gold and to release thioglucose [20], possibly due to its lower pKa that makes it negatively charged at neutral pH, where the sulfydryl of Cys would be almost completely protonated.
Silver interferes with selenium metabolism and inhibits TrxR, presumably by coordination to the Cys or Sec-Cys redox couples [62]. Since the Ag+ ion is more soluble in water than Au+, it is effective as a TrxR inhibitor in free form as well as in the form of its coordination complexes or nanoparticles.
Some ruthenium-containing compounds inhibit TrxR [63, 64]. Selectivity of ruthenium-containing compounds for selenoenzymes and/or thiol reductases suggests that Sec-Cys or Cys-Cys couples may be the binding site of this metal [65].
Inorganic mercury (in the oxidation states Hg+ and Hg2+) and organic mercurials (e.g., methylmercury) are strong inhibitors of the selenoproteins glutathione peroxidase and TrxR in animal models [66, 67]. This result is not surprising given the high affinity of mercury for S and Se. Contrary to gold (and probably silver), Hg forms a stable complex also with single Cys or Sec residues and thus it lacks the specificity of Au for the redox active Cys or Sec-Cys couples. Inhibition of TrxR by Hg can be prevented or removed by selenites or thiols (e.g., GSH, BAL, DMSA or lipoic acid, see [68]). Holmgren and coworkers measured IC50 values of rat recombinant TrxR for HgCl2 and CH3HgCl; however the values obtained by these authors (7.2 and 19.7 nM respectively) are lower than the enzyme concentration in the incubation mixture (50 nM) [69]. Although this is in principle possible, it should be recalled that the IC50 is defined as the free concentration of the inhibitor required to reduce the catalytic efficiency of the enzyme to 50% of its value in the absence of the inhibitor [38]. Thus, the measurement of IC50 values lower than the enzyme concentration would require either direct measurement of the free inhibitor or extreme precision in the measurement of the total inhibitor concentration and correction for the amount of bound inhibitor (a procedure that is not described in the original paper). It is our opinion that such low IC50 values indicate irreversible binding, at least under the experimental conditions used by the authors.
6.1.2. Alkylating Agents
Several alkylating agents are able to selectively modify the redox active Cys or Sec in the active site(s) of TrxR, TGR and GR, causing irreversible inhibition. The reactive species are either the thiolate or selenolate anions; thus, as a general rule, Sec (pKa ~ 5.5, [70]) and low pKa Cys are preferred over a non-polarized Cys (pKa ~ 8.5).
Examples of this class of inhibitors are nitrosoureas [40]; iodoacetate and bromoacetate; chlorodinitrobenzene and its derivatives [71, 72]; curcumin and its derivatives [73]; the anticancer drug mitomycin C [19] and other compounds (see Table 3). Alkylating agents are always mechanism based inhibitors that are activated by the enzyme and form a reactive intermediate during the catalytic cycle, capable to transfer a portion of their molecule to the active site Sec or Cys residues: as such they share many features with other suicide substrates (see below).
Curcumin was shown to be an irreversible inhibitor of rat TrxR [73]. Its reported IC50 = 3.6 μM is inconsistent with the mode of binding and should be regarded with caution: it may refer to the formation of an initial reversible complex or it may have been measured after insufficient incubation time (see below, section 6.4). Upon two hours incubation in the presence of NADPH the enzyme is irreversibly inhibited and resulted to be alkylated at the Sec-Cys couple. As observed in the case of other mechanism based inhibitors (e.g., naphthoquinones, see below), curcumin induces a strong NADPH oxidase activity.
Mitomycin C is an anticancer drug that alkylates and irreversibly inhibits reduced TrxR. Full inhibition requires several hours of incubation at 60 μM mitomycin C and 200 μM NADPH, and the apparent half time of the reaction is of approx. 1 hour [19].
2,4-Dihydroxybenzylamine (2,4-DHBA) and possibly 1,3-bischloroethyl-nitrosourea (BCNU) are irreversible mechanism based inhibitors of GR and TrxR. Competition between 2,4-DHBA and the oxidizing substrate glutathione indicates the active site as the inhibitor binding site. The observation that reducing agents (e.g. glutathione or dithioerythritol) protect the enzyme from the inhibitory effect of 2,4-DHBA lead FitzGerald and coworkers to suggest that the inhibition may require the formation of a free radical at or near the active site [74].
6.1.3. Elements of the V Group of the Periodic Table
Elements of the V and VI group react readily with Cys or Sec residues and many of their compounds behave as inhibitors of TrxR and related thiol reductases.
Nitrogen in the form of nitric oxide (NO) or nitrosonium ion (NO+) is a good ligand of Cys, able to form the S-nitroso Cys derivative. Nitrosoglutathione (GSNO; [75]) was shown to be able to transfer the NO group to free cysteine; it should be regarded as an aspecific inhibitor of TrxR, given that transnitrosylation is possible for virtually any reactive Cys residue in any enzyme. Other NO donors (e.g., furoxane derivatives) are excellent inhibitors of TrxR [14, 76] and at least in some cases the S-nitroso derivative of active site Cys could be directly demonstrated (unpublished observations from our Laboratory). However S-nitrosylation may not be the only mechanism of action of furoxane derived inhibitors of TrxR and compounds of this class can also act as suicide substrates (see below).
Although phosphorous can form high affinity complexes with sulfur, and P-based inhibitors are known to act on tyrosine phosphatases [77], no references about phosphorous-based inhibitors of TrxR can be found in the literature.
Some arsenic derivatives are potent irreversible inhibitors of TrxR. Among the best characterized such compounds is dimethyl arsonous diiodide [45] which forms an initial reversible complex with the enzyme, acting as a competitive inhibitor (as already described, section 4.1), and becomes irreversibly bound on longer incubation times.
Antimony derivatives include emetic tartar, a drug used since the XV century or even before. Emetic tartar probably acts on several targets, only one of which being TrxR and related thiol reductases. The drug was used to treat several pathological conditions including parasitoses and infections and indeed it was demonstrated to inhibit TGR from S. mansoni [14] and TryR from L. infantum [78]; the 3D structure of the complex between TryR from L. infantum and Sb released by emetic tartar was solved by means of X-ray crystallography (PDB entry 2W0H) and antimony (but not tartrate ion) was found complexed to the Cys residues on the si-face of FAD.
6.1.4. Suicide Substrates
A suicide substrate is a specific, irreversible, mechanism-based inhibitor that requires to be processed by the enzyme and that can possibly be released as a product. During its transformation, a highly reactive intermediate is produced that may covalently bind to active site residues. The distinction between “common” irreversible inhibitors, mechanism based inhibitors, and suicide substrates is not always obvious. A specific feature of suicide substrates is that the apparent stoichiometry of binding may be much higher than 1:1, given that the reactive intermediate is usually short lived and has a lower than 1 probability of reacting with the enzyme (see below, section 6.4).
Several naphthoquinone derivatives are substrates of TrxR and related thiol oxidases (e. g., juglone [51, 55]). These compounds may become covalently bound to the enzyme active site, possibly because of transient formation of a reactive intermediate. So far, an unexplained feature of these inhibitors is that they increase the NADPH oxidase activity of TrxR and stimulate the reduction of O2 to H2O2, as shown by the observation that NADPH consumption occurs in the presence of O2, stops upon exhaustion of the gas and resumes upon addition of catalase (unpublished data by F. Saccoccia and A. Bellelli). No high resolution 3D structures of enzyme-inhibitor complexes are available in the literature, and in spite of our efforts we never succeeded in obtaining crystals of TrxR irreversibly inhibited by naphthoquinone derivatives, or containing a covalently bound naphthoquinone product. It is possible that these suicide substrates should be classed as pseudo-irreversible (see below).
An interesting example of suicide substrate of GR is the fluoro-menadione derivative 6-2’-(3’-methyl)-1’,4’-naphthoquinolylhexanoic acid [79]. Bauer and coworkers took advantage on the capability of GR to use 1,4-naphthoquinone as substrate and on the possibility to derivatize the 1,4-naphthoquinone moiety. They added a fluorine atom to the quinone scaffold in order to generate, upon enzymatic reduction, an electron vacancy near the nucleophilic cysteine of the active site; the consequent formation of a covalent bond, also demonstrated by the 3D structure of the complex, irreversibly alkylated the catalytic residue, thus inhibiting the enzyme (see Table 3).
Furoxanes (oxadiazole-2-oxide derivatives) behave as suicide substrates of SmTGR [14, 76]. The mechanism of inhibition is not completely understood: TGR has the ability to undergo a nucleophilic attack by furoxan which reacts with reactive Cys or Sec, to effect molecular rearrangements of the compound, and to release from the heterocycle scaffold NO or other reactive species which in turn are able to nitrosylate crucial Cys residues, thus inactivating the enzyme (see above). The authors calculated that 15 nM reduced GR in the presence of 10μM inhibitor produces 1.5μM NO, thus only after many redox cycles the enzyme is inactivated by furoxane.
6.2. Pseudo-irreversible Inhibition
TrxR and related reductases are peculiar as the catalytic residues of the active site share the same chemical structure and reactivity of the oxidizing substrate. As a consequence and thanks to the reactivity of these residues, at least in some cases, the substrate can effectively compete with the enzyme and remove an otherwise irreversibly bound inhibitor. The most clear-cut example is provided by metal-based inhibitors, thus it will be described as paradigmatic; other cases, however will be mentioned.
A typical metal-based inhibitor, like AF or GoPI, reacts with the reduced Cys (or Sec) enzyme in a sequence of reactions, as depicted in (Fig. 5). True thermodynamic reversibility would imply not only the dissociation of the bound metal, but also the re-synthesis of the original drug, with the two gold ligands (e.g. thioglucose and triethylphosphine in the case of auranofin). This event, however, is unlikely in the case of thiol reductases, given that the geometry of the two Cys residues required to form a disulfide bridge is also very favourable for gold coordination. An interesting consideration is that, if the equilibrium concentration of the presumably unstable intermediate X-Au-S-TrxR-SH can be neglected, the hypothetical dissociation equilibrium constant (KI) for the above reaction scheme would have the measure units of M-1, rather than M as in the usual cases, and dilution would favor the formation of the EI complex, rather than its dissociation.
Once gold has been bound it will not appreciably dissociate in water and inhibition may be considered irreversible, especially if the original gold ligands are removed by dialysis or diluted. However, the metal can be transferred from a reduced Cys (or Sec-Cys) couple to another, if available. We demonstrated that in SmTGR, AF transfers gold to the C-terminal Cys596-Sec597 couple of the enzyme, which in turn releases the metal to at least two other Cys couples of the enzyme (at positions 154 and 159, 520 and 574), and that the role of Sec is catalytic, rather than thermodynamic (see above section 6.1.1 [20, 80, 81]).
It is perfectly reasonable, and indeed expected, that gold can also be transferred to reduced thioredoxin via the C terminal Sec-Cys couple; thus an excess of thioredoxin may be able to restore the activity of a sample of TrxR “irreversibly” inhibited by gold. The same applies to GR and glutathione. Indeed it is a common observation of our and other laboratories that “irreversible” inhibitors often do not appear in the X-ray crystallography maps of TrxR or GR, presumably because they have been removed by reductants present in the crystallization reservoir during the long incubation required to obtain protein crystals. Recovery of enzymatic activity upon incubation with Trx (or GSH) is a slow process and can often be directly followed; it usually takes hours or days.
Nitrosoureas represent another class of covalent inhibitors of TrxR that can be removed by Trx; Schallreuter and coworkers indicated an optimal TrxR:Trx ratio of 12:1 for the reaction [40]. These inhibitors are of special interest in the present discussion because of their mechanism of action. Indeed nitrosoureas are mechanism based inhibitors that alkylate the reduced Cys (or Sec) residues of the active site and hence the entire inhibitor molecule is not present in the (pseudo-) irreversibly inhibited enzyme. In this case, the initial mechanism of binding, which requires the reduced enzyme and the parent compound(s), is different from the subsequent mechanism of transfer of alkyl moiety of the inhibitor from the enzyme to reduced Trx, and it may be expected that Trx might also be able to remove from TrxR alkyl moieties transferred to the enzyme by other, unrelated mechanism-based inhibitors.
6.3. Slow Substrates and Other Atypical Covalent Inhibitors
TrxR and related flavoreductases are also able to combine with several molecules that may or may not be subsequently released, and behave as inhibitors or as slow substrates. On the short time scale of the activity assay these compounds resemble irreversible inhibitors; however their mode of binding suggests that they be classed as atypical inhibitors. Often atypical inhibition may be reversed by the reduced form of the oxidizing substrate (see section 6.2), or by exposure to acidic conditions, or by prolonged incubation with excess NADPH.
Azelaic acid is reported to be a reversible competitive inhibitor of TrxR [82]. However, this compound seems to exert its activity by forming a thioesther with a Cys residue of the active site; by transacylation the azelaic acid residue would then be transferred to a basic residue of the active site. Such a complex mechanism of action cannot be considered typical of a reversible competitive inhibitor, even though the thioester bond may be easily hydrolyzed, thus accounting for reversibility. Clearly this interesting compound would deserve a thorough biochemical characterization.
Several organic compounds containing calchogens behave as artificial substrates, slow substrates or inhibitors of TrxR. In these cases, the distinction between an artificial substrate and an irreversible or slowly reacting inhibitor is blurred, since a substrate with turnover number in the range of several hours behaves as an irreversible inhibitor in any conventional activity assay. Methylseleninate, previously thought to be an inhibitor of TrxR and other thiol reductases was demonstrated to be an efficient substrate of Sec-containing TxrRs, with kcat = 23 s-1 and KM = 18 μM, measured at constant concentration of NADPH (100 μM) [83]; Sec-lacking thiol reductases showed poor catalytic activity versus this substrate. A wide array of experimental data about sulfur-, selenium- and tellurium-containing compounds has been reviewed by Cai and coworkers [13]. Organoselenium compund BBSKE (1,2-bis(1,2-Benzisoselenazolone-3(2H)-ketone)ethane) was reported to inhibit TrxR in some cancer line cells [84]. The structure and binding mechanism of this and similar selenium-containing /compounds inhibitors are closely related to that of the artificial substrate ebselen, able to compete with Trx, with an apparent KM of 2.5 μM and a kcat of 588 min-1, measured at 100μM NADPH [85].
An interesting class of compounds are the 2-imidazolyl disulfide antitumor compounds which demostrated to be specific substrates or inhibitors of TrxR, with KI ranging from 3 to 13 μM [50], while glutathione reductase was not appreciably inhibited by any of these compounds.
Organotellurium compounds were synthesized by Engman and coworkers and some water-soluble molecules displayed IC50 in the submicromolar range. The cyclodextrin-derived diorganyl tellurides displayed efficient inhibition of TrxR, with IC50 in the low micromolar range. Sulfur mustard vescicants as 2-chloroethyl ethyl sulfide (CEES) target Sec-containing TrxR with IC50 about 5 μM [86]. The authors argued that the inhibitor acts in order to give a selenium-ethylthioethyl adduct on the C-terminus.
6.4. Theory
The effect of an irreversible inhibitor is to lower the concentration of the active enzyme in the assay; thus the KM and kcat are unaffected, whereas the Vmax, that equals the product of kcat times the active enzyme concentration, is reduced. This is reminiscent of what we already observed for pure reversible non-competitive inhibition (eq. 5.2) except that the fraction of the irreversibly inhibited enzyme does not depend on KI but only on the concentration of the inhibitor and the incubation time. If the inhibitor concentration is higher than the reaction stoichiometry and the incubation time is sufficient, the enzyme will be 100% inhibited. The IC50 of an irreversible enzyme inhibitor is zero and the total amount of inhibitor required to inhibit half of the enzyme is a function of the amount of the enzyme present in the sample and of the stoichiometry and mechanism of the reaction. No true KI can be determined (see [58]), except for the case in which the formation of a reversible complex precedes in time that of the irreversible one; in this case the KI value is determined for the reversible complex, not for the irreversible one. In the simplest case, the inhibitor binds to the enzyme with 1:1 stoichiometry, but more complex events may occur (e.g. the inhibitor may be a suicide substrate that needs to be activated and partially transformed by the enzyme and has a less than unity probability of forming a covalent bond with the enzyme). It is important to state that irreversible inhibition may be confused with reversible non competitive inhibition, given that both are insensitive to the concentration of the substrate(s): therefore reversibility should be specifically checked (e.g. by dialysis).
Almost all thiol reagents are potential irreversible inhibitors of GR, TrxR and related oxidoreductases, such as organic mercurials, NO donors, iodoacetate and iodoacetamide. Many of these compounds, however, are unspecific and highly toxic, hence their practical interest is limited.
Since the combination of irreversible inhibitors with the target enzyme may be slow with respect to the time required to measure the enzyme activity, some pre-incubation of the enzyme with the inhibitor may be required, and it is necessary to measure the time course of the inhibitor-enzyme combination in order to demonstrate that full combination has been achieved. In some instances, values of KI have been erroneously reported for irreversible inhibitors (e.g. in the case of gold containing compounds it is commonly assumed that, in spite of both complexes being irreversible, TrxR has a lower KI than GR, see the above section 6.1). These apparent KI values must be interpreted with reference to the mechanism of binding of the inhibitor and the experimental conditions used by the authors. The basic mechanism of binding of an irreversible inhibitor of TrxR (and related thiol reductases) is as follows:
 (6.1)
(6.1)
In the above scheme it is assumed that only the reduced form of the enzyme is sensitive, given that binding of irreversible inhibitors usually occurs at the reduced Sec or Cys residues of the active sites. An initial reversible ERI complex is formed in a bimolecular second order process governed by kI,1. The complex may decay unproductively to unmodified ER and I by the monomolecular first order process governed by kI,2 or proceed to the irreversible complex ER I* by the monomolecular first order process governed by kinact. Many of the observed mechanisms represent variants of this fundamental scheme (which is itself a variant of eq. 3.9).
In the case of several metal-based inhibitors the process is rate limited by kI,1 while kI,2 is often negligible. As a consequence, no reversible intermediate accumulates and the true course of inhibition follows a second order kinetics. If the inhibitor is in excess with respect to the enzyme this mechanism further simplifies to a pseudo-first order time course, in which the concentration of the active enzyme decreases exponentially during the incubation time:
 (6.2)
(6.2)
This equation implies that the fraction of uninhibited enzyme after an incubation of fixed duration t is an exponential function of the time but also of the inhibitor concentration [I], i.e. if one plots the fraction of active enzyme as a function of the inhibitor concentration (keeping fixed the incubation time), one obtains an exponential, very similar to the one obtained in the more conventional plot of the active enzyme vs. time (at fixed inhibitor concentration). If the incubation time is not long enough to allow the reaction to go to completion (with disappearance of either the uninhibited enzyme or the inhibitor), some active enzyme will be present in the assay and the velocity of the catalyzed reaction will be:
 (6.3)
(6.3)
Eq. 6.2 and 6.3 show that the enzyme activity in the presence of an irreversible inhibitor incubated for too short a time is an exponential function of the inhibitor concentration. Unfortunately an exponential at a first glance may appear more or less similar to a hyperbola from which a “KI” can be determined. Needless to say, this procedure and the parameter one obtains are wrong. Comparison of eqs. 5.2 and 6.3 and chemical kinetics tell us that the “IC50” one may derive from fitting the exponential to a hyperbola approximates “IC50” = ln (2) / kt; since the IC50 of a non competitive inhibitor equals its KI, we suspect that this is the origin of some “KI” values of irreversible inhibitors reported in the literature.
The correct procedure to deal with irreversible inhibitors is to assess reversibility or lack thereof by dialysis; to measure a time course of binding of the inhibitor by recording the enzyme activity after variable incubation times; and to choose an incubation time greater than 4 or 5 half times. Attention should be paid to the time course, that at low inhibitor concentration may not conform to a pseudo-first order reaction (and therefore may require much longer incubation times at lower inhibitor concentrations). The relevant parameters to be measured are the binding stoichiometry and the rate constant of the reaction over and above its mechanism.
Other inhibitors (curcumin and its derivatives; other alkylating agents; arsenic derivatives) may follow a more complex time course, because kI,1 and kI,2 are both faster than kinact. As a consequence, the time course of inhibition is described by two widely time resolved processes: a fast or almost instantaneous one in which the reversible ERI complex reaches its pseudo-equilibrium concentration and a slower one in which formation of the irreversible complex occurs. In this case a “true” KI can be measured upon short incubation times and the overall reaction fully obeys eq. 3.9, except that it should usually be referred to the reduced fraction of the enzyme ER only.
If the inhibitor concentration significantly exceeds that of the enzyme, and the enzyme is kept reduced during the incubation with the inhibitor, one obtains that: (i) the ratio [ERI]/[ER] is constant; and (ii) the irreversibly inhibited species EI* forms at a rate which equals the product [ERI]kinact:
 (6.4)
(6.4)
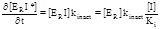 (6.5)
(6.5)
To calculate the fraction of uninhibited enzyme after an incubation time of duration t we need to further elaborate. Let us consider two populations of the total enzyme: the irreversibly inhibited ERI* and the rapidly equilibrating, non-irreversibly inhibited [ER]+[ERI] (since incubation is assumed to be carried out under reducing conditions, we may assume that the oxidized derivatives EO and EOI are not present in the mixture). The rate of decay of the non irreversibly inhibited population is:
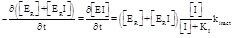 (6.6)
(6.6)
Eq. 6.6 is identical to 6.5 except that it takes into account the partial reformation of ERI from reaction ER + I ↔ ER I; if [I]>>Etot it describes an exponential time course with kapp=[I]kinact/([I]+KI). Integrating eq. 6.6 and taking into account the rapidly equilibrating reversibly inhibited species ERI we can calculate the fraction of uninhibited enzyme after an incubation with the inhibitor of duration t:
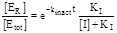 (6.7)
(6.7)
Eq. 6.7 was employed to simulate the expected time course of inhibition of TrxR by methylarsonous diiodide using the empirical data given by Lin and coworkers (i.e. KI = 100 nM and 90% inhibition after 1 hour incubation, Fig. 10). Inhibitors acting via the above mechanism may indeed be assigned a meaningful IC50, referred to the initial reversible process; this parameter however varies with the incubation time and becomes meaningless if a significant population of the irreversible complex has been formed.
Fig. (10).

Calculated time course of inhibition of TrxR by methylarsonous diiodide using eq. 6.7 with kinact = 0.004 min-1, and [I]=100nM.
Suicide substrates do not obey the general equation of irreversible inhibitors (eq. 6.2 or 6.7) and demand a different approach. In this review we have used the term “mechanism-based inhibitors” to indicate a class of inhibitors capable of forming a covalent complex with the enzyme because of specific and selective activation by the enzyme itself. TrxR and related enzymes are prone to this type of inhibition. The term “suicide substrates” has often been used as synonymous of “mechanism-based inhibitors”; in this work, however we have used this term in a more limited sense to refer only to those mechanism-based inhibitors that are fully processed by the enzyme, with oxidation of NADPH and formation of a product. Suicide substrates thus defined have a less than unity probability of forming a covalent complex with the enzyme; their general reaction mechanism is as follows:
 (6.8)
(6.8)
In (Scheme 6.8) it is assumed that the reaction is limited to the reduced enzyme and that the inhibitor is a substrate that transiently populates a reactive intermediate (ERI) that has a defined probability p = kinact/(kinact+kI,3) of converting to the irreversibly inhibited species (EI*). A true suicide substrate can oxidize the enzyme and be released as the product(s): thus full inhibition requires several cycles and, given that the enzyme is oxidized in the process, NADPH is consumed during the incubation time. As previously stated [87] for a suicide substrate mechanism, the rates of consumption of substrate (inhibitor) and appearance of product differ; one should expect that for a NADPH-dependent enzyme, like thiol reductases, more NADPH is consumed than it would be required for the product formation, since some is used to reduce a fraction of the inhibitor that will ultimately remain covalently bound to the enzyme. By contrast, not all mechanism based inhibitors do necessarily cycle and yield a product (as in the case with alkylating agents) or consume NADPH. In both cases, if the enzyme is simultaneously incubated with the inhibitor and the substrate, the conversion of the physiological substrate is inhibited because of competition with the inhibitor, but NADPH consumption may be scarcely affected (as in the case of TrxR and naphthoquinones).
A clear distinction between enzyme-activated irreversible inhibitors and suicide substrates was not always clearly drawn and many authors use these terms as synonymous [88]; other authors, however, have drawn a distinction [89] and we feel that the latter option is preferable. The most general approach is to consider suicide substrates as a subgroup of the larger class of mechanism-based inhibitors. Loosely speaking, the “common” mechanism-based inhibitor is not prone to release a product even though it specifically binds to target enzyme at the active site and is activated during the process; on the contrary, suicide substrate has a certain probability to be processed in order to be released as a product.
7. CONCLUSIONS
The aim of the present analysis was to carry out a systematic reappraisal of the literature data on the inhibitors of TrxR and related enzymes. It was prompted by the authors noticing inconsistencies in several published works. In spite of its obvious shortcomings and oversimplifications, this work demonstrates that (i) rapidly binding reversible inhibitors have often been attributed a behaviour more atypical than it was probably the case, due to the fact that the experimental data were usually analyzed using equations developed for single-substrate enzymes; and (ii) slowly binding or irreversible inhibitors were often misunderstood as rapidly equilibrating reversible inhibitors because of insufficient incubation times, incubation under unfavorable experimental conditions, or incomplete time separation between reversible and irreversible reactions.
While point (ii) is straightforward and calls for greater attention to experimental details when studying irreversible inhibitors, point (i) may deserve some more explanation. The usual practice of running steady-state experiments on two-substrate enzymes at constant concentration of either substrate and varying the other yields Michaelis-like plots that conform to the equations developed for single-substrate enzymes. This practice is acceptable, but yields apparent parameters that are convolutions of the desired ones with the other constants and with the concentration of the constant substrate. Thus one gets a set of parameters for each substrate and more KI values than it is actually the case: e.g. two KI values may be assigned to competitive inhibitors which have only one. Since TrxR is an important enzyme, whose inhibition has great relevance in pharmacology, it is important to be aware of the possible pitfalls in the analysis of experimental data present in the literature which by themselves are extensive and accurate.
Fig. (8).

Chemical formulas of (A) curcumin and (B) nitrosoglutathione.
ACKNOWLEDGEMENTS
This work was supported by grants from the Fondazione Roma (project ‘‘Rational Approach to the Specific Inhibition of Plasmodium falciparum and Schistosoma mansoni’’); International FIRB RBIN06E9Z8 to A. Bellelli; “Sapienza” University of Rome under Grants “Progetti di Università” 2011 and 2012 to A. Bellelli and 2013 to A. E. Miele.
We have been educated over many years by long discussions with Prof. Maurizio Brunori, Prof. Gino Amiconi, and Prof. Bruno Giardina, whose indirect intellectual contribution to the present work is invaluable.
ABBREVIATIONS
- Trx
= Thioredoxin
- TrxR
= Thioredoxin Reductase
- H-TrxR
= High molecular weight TrxR
- L-TrxR
= Low molecular weight TrxR
- GR
= Glutathione Reductase
- TryR
= Trypanothione Reductase
- MeR
= Mercuric Ion Reductase
- LipD
= Lipoamide Dehydrogenase
- TGR
= Thioredoxin Glutathione Reductase
- Sec
= Selenocysteine
- hTrxR
= Human TrxR
- GSSG
= Oxidized glutathione
- XAN
= 6-hydroxy-3-oxo-3H-xanthene-9-propionic acid
- SmTGR
= TGR from Schistosoma mansoni
- DTNB
= 5,5’-dithiobis-(2-nitrobenzoic acid)
- ECG
= (-)-epicatechin-gallate
- EGCG
= (-)-epigallocatechin-3-gallate
- PQQ
= Pirroloquinoline quinone
- AF
= Auranofin
- BzSe
= Benzene-selenol
- GoPI
= {1-phenyl-2,5-di(2-pyridyl)phosphole} AuCl
- BAL
= 2,3-dimercapto-1-propanol
- DMSA
= Meso-2,3-dimercaptosuccinic acid
- GSNO
= Nitrosoglutathione
- 2,4-DHBA
= 2,4-Dihydroxybenzylamine
- BCNU
= 1,3-bischloroethyl-nitrosourea
- BBSKE
= 1,2-bis(1,2-Benzisoselenazolone-3(2H)-ketone)ethane
- CEES
= 2-chloroethyl ethyl sulfide
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
REFERENCES
- 1.Holmgren A, Lu J. Thioredoxin and thioredoxin reductase current research with special reference to human disease. Biochem. Biophys. Res. Commun. 2010;396:120–124. doi: 10.1016/j.bbrc.2010.03.083. [DOI] [PubMed] [Google Scholar]
- 2.Arnér E S. Focus on mammalian thioredoxin reductases important selenoproteins with versatile functions. Biochim. Biophys. Acta. 2009;1790:495–526. doi: 10.1016/j.bbagen.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 3.Hirt RP, Müller S, Embley TM, Coombs GH. The diversity and evolution of thioredoxin reductase new perspectives. Trends Parasitol. 2002;18:302–308. doi: 10.1016/s1471-4922(02)02293-6. [DOI] [PubMed] [Google Scholar]
- 4.Prast-Nielsen S, Huang HH, Williams DL. Thioredoxin glutathione reductase its role in redox biology and potential as a target for drugs against neglected diseases. Biochim. Biophys. Acta. 2011;1810:1262–1271. doi: 10.1016/j.bbagen.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cioli D, Valle C, Angelucci F, Miele AE. Will new antischistosomal drugs finally emerge?. Trends Parasitol. 2008;24:379–382. doi: 10.1016/j.pt.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 6.Williams DL, Bonilla M, Gladyshev VN, Salinas G. Thioredoxin glutathione reductase-dependent redox networks in platyhelminth parasites. Antioxid Redox Signal. 2013;19:735–745. doi: 10.1089/ars.2012.4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tonissen KF, Di Trapani. G. Thioredoxin system inhibitors as mediators of apoptosis for cancer therapy. Mol. Nutr. Food Res. 2009;53:87–103. doi: 10.1002/mnfr.200700492. [DOI] [PubMed] [Google Scholar]
- 8.Selenius M, Rundlof AK, Olm E, Fernandes AP, Bjornstedt M. Selenium and the selenoprotein thioredoxin reductase in the prevention treatment and diagnostics of cancer. Antioxid. Redox Signal. 2010;12:867–880. doi: 10.1089/ars.2009.2884. [DOI] [PubMed] [Google Scholar]
- 9.Boumis G, Giardina G, Angelucci F, Bellelli A, Brunori M, Dimastrogiovanni D, Saccoccia F, Miele AE. Crystal structure of Plasmodium falciparum thioredoxin reductase a validated drug target. Biochem. Biophys. Res. Commun. 2012;425:806–811. doi: 10.1016/j.bbrc.2012.07.156. [DOI] [PubMed] [Google Scholar]
- 10.Saccoccia F, Di Micco P, Boumis G, Brunori M, Koutris I, Miele AE, Morea V, Sriratana P, Williams DL, Bellelli A, Angelucci F. Moonlighting by different stressors crystal structure of the chaperone species of a 2-Cys peroxiredoxin. Structure. 2012;20:429–439. doi: 10.1016/j.str.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Becker K, Gromer S, Schirmer RH, Muller S. Thioredoxin reductase as a pathophysiological factor and drug target. Eur. J. Biochem. 2000;267:6118–6125. doi: 10.1046/j.1432-1327.2000.01703.x. [DOI] [PubMed] [Google Scholar]
- 12.Cimini A, Gentile R, Angelucci F, Benedetti E, Pitari G, Giordano A, Ippoliti R. Neuroprotective effects of PrxI over-expression in an in vitro human Alzheimer’s disease model. J. Cell Biochem. 2013;114:708–715. doi: 10.1002/jcb.24412. [DOI] [PubMed] [Google Scholar]
- 13.Cai W, Zhang L, Song Y, Wang B, Zhang B, Cui X, Hu G, Liu Y, Wu J, Fang J. Small molecule inhibitors of mammalian thioredoxin reductase. Free Radic. Biol. Med. 2012;52:257–265. doi: 10.1016/j.freeradbiomed.2011.10.447. [DOI] [PubMed] [Google Scholar]
- 14.Kuntz AN, Davioud-Charvet E, Sayed AA, Califf LL, Dessolin J, Arnér ES, Williams DL. Thioredoxin glutathione reductase from Schistosoma mansoni an essential parasite enzyme and a key drug target. PLoS Med. 2007;4:e206. doi: 10.1371/journal.pmed.0040206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sannella AR, Casini A, Gabbiani C, Messori L, Bilia AR, Vincieri FF, Majori G, Severini C. New uses for old drugs. Auranofin a clinically established antiarthritic metallodrug exhibits potent antimalarial effects in vitro Mechanistic and pharmacological implications. FEBS Lett. 2008;582:844–847. doi: 10.1016/j.febslet.2008.02.028. [DOI] [PubMed] [Google Scholar]
- 16.Angelucci F, Miele AE, Boumis G, Brunori M, Dimastrogiovanni D, Bellelli A. Macromolecular bases of antischistosomal therapy. Curr. Top. Med. Chem. 2011;11:2012–2028. doi: 10.2174/156802611796575939. [DOI] [PubMed] [Google Scholar]
- 17.Casini A, Messori L. Molecular mechanisms and proposed targets for selected anticancer gold compounds. Curr. Top. Med. Chem. 2011;11:2647–2660. doi: 10.2174/156802611798040732. [DOI] [PubMed] [Google Scholar]
- 18.Angelucci F, Dimastrogiovanni D, Boumis G, Brunori M, Miele AE, Saccoccia F, Bellelli A. Mapping the catalytic cycle of Schistosoma mansoni thioredoxin glutathione reductase by X-ray crystallography. J. Biol. Chem. 2010;285:32557–32567. doi: 10.1074/jbc.M110.141960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paz MM, Zhang X, Lu J, Holmgren A. A new mechanism of action for the anticancer drug mitomycin C mechanism-based inhibition of thioredoxin reductase. Chem. Res. Toxicol. 2012;25:1502–1511. doi: 10.1021/tx3002065. [DOI] [PubMed] [Google Scholar]
- 20.Angelucci F, Sayed AA, Williams DL, Boumis G, Brunori M, Dimastrogiovanni D, Miele AE, Pauly F, Bellelli A. Inhibition of Schistosoma mansoni thioredoxin-glutathione reductase by auranofin structural and kinetic aspects. J. Biol. Chem. 2009;284:28977–28985. doi: 10.1074/jbc.M109.020701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saccoccia F, Angelucci F, Boumis G, Brunori M, Miele AE, Williams DL, Bellelli A. On the mechanism and rate of gold incorporation into thiol-dependent flavoreductases. J. Inorg. Biochem. 2012;108:105–111. doi: 10.1016/j.jinorgbio.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urig S, Becker K. On the potential of thioredoxin reductase inhibitors for cancer therapy. Semin. Cancer Biol. 2006;16:452–465. doi: 10.1016/j.semcancer.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 23.Sandalova T, Zhong L, Lindqvist Y, Holmgren A, Schneider G. Three-dimensional structure of a mammalian thioredoxin reductase implications for mechanism and evolution of a selenocysteine-dependent enzyme. Proc. Natl. Acad. Sci. U. S. A. 2001;98:9533–9538. doi: 10.1073/pnas.171178698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rao ST, Rossmann MG. Comparison of super-secondary structures in proteins. J. Mol. Biol. 1973;76:241–256. doi: 10.1016/0022-2836(73)90388-4. [DOI] [PubMed] [Google Scholar]
- 25.Lothrop AP, Ruggles EL, Hondal RJ. No selenium required Reactions catalyzed by mammalian thioredoxin reductase that are independent of a selenocysteine residue. Biochemistry. 2009;48:6213–6223. doi: 10.1021/bi802146w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamura T, Stadtman TC. A new selenoprotein from human lung adenocarcinoma cells purification properties and thioredoxin reductase activity. Proc. Natl. Acad. Sci. U. S. A. 1996;93:1006–1011. doi: 10.1073/pnas.93.3.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arscott LD, Gromer S, Schirmer RH, Becker K, Williams C H ., Jr The mechanism of thioredoxin reductase from human placenta is similar to the mechanisms of lipoamide dehydrogenase and glutathione reductase and is distinct from the mechanism of thioredoxin reductase from Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 1997;94:3621–3626. doi: 10.1073/pnas.94.8.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fritz-Wolf K, Urig S, Becker K. The structure of human thioredoxin reductase 1 provides insights into C-terminal rearrangements during catalysis. J. Mol. Biol. 2007;370:116–127. doi: 10.1016/j.jmb.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 29.Gasdaska PY, Gasdaska JR, Cochran S, Powis G. Cloning and sequencing of a human thioredoxin reductase. FEBS Lett. 1995;373:5–9. doi: 10.1016/0014-5793(95)01003-w. [DOI] [PubMed] [Google Scholar]
- 30.Russel M, Model P. Sequence of thioredoxin reductase from Escherichia coli.Relationship to other flavoprotein disulfide oxidoreductases. J. Biol. Chem. 1988;263:9015–9019. [PubMed] [Google Scholar]
- 31.Stoll VS, Simpson SJ, Krauth-Siegel RL, Walsh CT, Pai E F. Glutathione reductase turned into trypanothione reductase structural analysis of an engineered change in substrate specificity. Biochemistry. 1997;36:6437–6447. doi: 10.1021/bi963074p. [DOI] [PubMed] [Google Scholar]
- 32.Angelucci F, Miele AE, Boumis G, Dimastrogiovanni D, Brunori M, Bellelli A. Glutathione reductase and thioredoxin reductase at the crossroad the structure of Schistosoma mansoni thioredoxin glutathione reductase. Proteins. 2008;72:936–945. doi: 10.1002/prot.21986. [DOI] [PubMed] [Google Scholar]
- 33.Urig S, Lieske J, Fritz-Wolf K, Irmler A, Becker K. Truncated mutants of human thioredoxin reductase 1 do not exhibit glutathione reductase activity. FEBS Lett. 2006;580:3595–3600. doi: 10.1016/j.febslet.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 34.Hondal RJ. Using chemical approaches to study selenoproteins-focus on thioredoxin reductases. Biochim. Biophys. Acta. 2009;1790:1501–1512. doi: 10.1016/j.bbagen.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patterson S, Alphey MS, Jones DC, Shanks EJ, Street IP, Frearson JA, Wyatt PG, Gilbert IH, Fairlamb AH. Dihydroquinazolines as a novel class of Tripanosoma brucei trypanothione reductase inhibitors discovery synthesis and characterization of their binding mode by protein crystallography. J. Med. Chem. 2011;54:6514–6530. doi: 10.1021/jm200312v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Savvides SN, Karplus PA. Kinetics and Crystallographic Analysis of Human Glutathione Reductase in Complex with a Xanthene Inhibitor. J. Biol. Chem. 1996;271:8101–8107. doi: 10.1074/jbc.271.14.8101. [DOI] [PubMed] [Google Scholar]
- 37.Bauer H, Gromer S, Urbani A, Schnölzer M; Schirmer RH, Müller HM. Thioredoxin reductase from the malaria mosquito Anopheles gambiae. Eur. J. Biochem. 2003;270:4272–4281. doi: 10.1046/j.1432-1033.2003.03812.x. [DOI] [PubMed] [Google Scholar]
- 38.Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50): of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 39.Copeland RA, editor. 2nd ed. WileyVCH 2000.: Enzymes A Practical Introduction to Structure Mechanism and Data Analysis. [Google Scholar]
- 40.Schallreuter KU, Gleason FK, Wood JM. The mechanism of action of the nitrosourea anti-tumor drugs on thioredoxin reductase glutathione reductase and ribonucleotide reductase. Biochim. Biophys. Acta. 1990;1054:14–20. doi: 10.1016/0167-4889(90)90199-n. [DOI] [PubMed] [Google Scholar]
- 41.Chen GP, Raybuck S, Ziegler DM. Flavoenzymes inhibited by indomethacin. Drug Metabol. Drug Interact. 1994;11:153–160. doi: 10.1515/dmdi.1994.11.2.153. [DOI] [PubMed] [Google Scholar]
- 42.Streitenberger SA, López-Mas JA, Sánchez-Ferrer A, García-Carmona F. Non-linear slow-binding inhibition of Aerococcus viridans lactate oxidase by Cibacron Blue 3GA. J. Enzyme Inhib. 2001;16:301–312. [PubMed] [Google Scholar]
- 43.Prast-Nielsen S, Dexheimer TS, Schultz L, Stafford WC, Cheng Q, Xu J, Jadhav A, Arnér ES, Simeonov A. Inhibition of thioredoxin reductase 1 by porphyrins and other small molecules identified by a high-throughput screening assay. Free Radic. Biol. Med. 2011;50:1114–1123. doi: 10.1016/j.freeradbiomed.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, Zhang H, Holmgren A, Tian W, Zhong L. Inhibitory effect of green tea extract and (-)-epigallocatechin-3-gallate on mammalian thioredoxin reductase and HeLa cell viability. Oncol. Rep. 2008;20:1479–1487. [PubMed] [Google Scholar]
- 45.Lin S, Cullen WR, Thomas DJ. Methylarsenicals and arsinothiols are potent inhibitors of mouse liver thioredoxin reductase. Chem. Res. Toxicol. 1999;12:924–930. doi: 10.1021/tx9900775. [DOI] [PubMed] [Google Scholar]
- 46.Tibodeau JD, Benson LM, Isham CR, Owen WG, Bible KC. The anticancer agent chaetocin is a competitive substrate and inhibitor of thioredoxin reductase. Antioxid. Redox Signal. 2009;11:1097–1106. doi: 10.1089/ars.2008.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benson TJ, McKie JH, Garforth J, Borges A, Fairlamb AH, Douglas KT. Rationally designed selective inhibitors of trypanothione reductase.Phenothiazines and related tricyclics as lead structures. Biochem. J. 1992;286:9–11. doi: 10.1042/bj2860009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan C, Yin H, Garforth J, McKie JH, Jaouhari R, Speers P, Douglas KT, Rock PJ, Yardley V, Croft SL, Fairlamb AH. Phenothiazine inhibitors of trypanothione reductase as potential antitrypanosomal and antileishmanial drugs. J Med Chem. 1998;41:148–156. doi: 10.1021/jm960814j. [DOI] [PubMed] [Google Scholar]
- 49.Garrard EA, Borman EC, Cook BN, Pike EJ, Alberg DG. Inhibition of trypanothione reductase by substrate analogues. Org. Lett. 2000;2:3639–3642. doi: 10.1021/ol0065423. [DOI] [PubMed] [Google Scholar]
- 50.Oblong JE, Chantler EL, Gallegos A, Kirkpatrick DL, Chen T, Marshall N, Powis G. Reversible inhibition of human thioredoxin reductase activity by cytotoxic alkyl 2-imidazolyl disulfide analogues. Cancer Chemother. Pharmacol. 1994;34:434–438. [PubMed] [Google Scholar]
- 51.Cenas N, Nivinskas H, Anusevicius Z, Sarlauskas J, Lederer F, Arnér ES. Interactions of quinones with thioredoxin reductase a challenge to the antioxidant role of the mammalian selenoprotein. J. Biol. Chem. 2004;279:2583–2592. doi: 10.1074/jbc.M310292200. [DOI] [PubMed] [Google Scholar]
- 52.Baiocco P, Poce G, Alfonso S, Cocozza M, Porretta GC, Colotti G, Biava M, Moraca F, Botta M, Yardley V, Fiorillo A, Lantella A, Malatesta F, Ilari A. Inhibition of Leishmania infantum trypanothione reductase by azole-based compounds a comparative analysis with its physiological substrate by X-ray crystallography. ChemMedChem. 2013;8:1175–1183. doi: 10.1002/cmdc.201300176. [DOI] [PubMed] [Google Scholar]
- 53.Kasozi DM, Gromer S, Adler H, Zocher K, Rahlfs S, Wittlin S, Fritz-Wolf K, Schirmer RH, Becker K. The bacterial redox signaller pyocyanin as an antiplasmodial agent comparisons with its thioanalog methylene blue. Redox Rep. 2011;16:154–165. doi: 10.1179/174329211X13049558293678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simeonov A, Jadhav A, Sayed AA, Wang Y, Nelson ME, Thomas CJ, Inglese J, Williams DL, Austin CP. Quantitative high-throughput screen identifies inhibitors of the Schistosoma mansoni redox cascade. PLoS Negl. Trop. Dis. 2008;2:e127. doi: 10.1371/journal.pntd.0000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu J, Arnér E S. Pyrroloquinoline quinone modulates the kinetic parameters of the mammalian selenoprotein thioredoxin reductase 1 and is an inhibitor of glutathione reductase. Biochem. Pharmacol. 2012;83:815–820. doi: 10.1016/j.bcp.2011.12.028. [DOI] [PubMed] [Google Scholar]
- 56.Theobald AJ, Caballero I, Coma I, Colmenarejo G, Cid C, Gamo FJ, Hibbs MJ, Bass AL, Thomas DA. Discovery and biochemical characterization of Plasmodium thioredoxin reductase inhibitors from an antimalarial set. Biochemistry. 2012;51:4764–4771. doi: 10.1021/bi3005076. [DOI] [PubMed] [Google Scholar]
- 57.Andricopulo AD, Akoachere MB, Krogh R, Nickel C, McLeish MJ, Kenyon GL, Arscott LD, Williams CH , Jr, Davioud-Charvet E. B , Becker K. Specific inhibitors of Plasmodium falciparum thioredoxin reductase as potential antimalarial agents. Bioorg. Med. Chem. Lett. . 2006; 16(8):2283–2292. doi: 10.1016/j.bmcl.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 58.Palmer T, editor. 2nd ed . Horwood Ltd.: Chichester U.K.; 1985. Understanding enzymes . [Google Scholar]
- 59.Gromer S., Arscott LD, Williams CH, Jr, Schirmer RH, Becker K. Human placenta thioredoxin reductase.Isolation of the selenoenzyme steady state kinetics and inhibition by therapeutic gold compounds. J. Biol. Chem. 1998;273:20096–20101. doi: 10.1074/jbc.273.32.20096. [DOI] [PubMed] [Google Scholar]
- 60.Deponte M, Urig S, Arscott LD, Fritz-Wolf K, Réau R, Herold-Mende C, Koncarevic S, Meyer M, Davioud-Charvet E, Ballou DP, Williams CH, Jr, Becker K. Mechanistic studies on a novel highly potent gold-phosphole inhibitor of human glutathione reductase. J. Biol. Chem. 2005;280:20628–20637. doi: 10.1074/jbc.M412519200. [DOI] [PubMed] [Google Scholar]
- 61.Schroder D, Schwarz H, Hrusak J, Pyykko P. Cationic gold (I) complexes of xenon and of ligands containing the donor atoms oxygen nitrogen phosphorous and sulfur. Inorg. Chem. 1998;37:624–632. [Google Scholar]
- 62.Srivastava M, Singh S, Self WT. Exposure to silver nanoparticles inhibits selenoprotein synthesis and the activity of thioredoxin reductase. Environ. Health Perspect. 2012;120:56–61. doi: 10.1289/ehp.1103928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mura P, Camalli M, Bindoli A, Sorrentino F, Casini A, Gabbiani C, Corsini M, Zanello P, Rigobello MP, Messori L. Activity of rat cytosolic thioredoxin reductase is strongly decreased by trans-[bis(2-amino-5methylthiazole)tetrachlororuthenate (III)]: first report of relevant thioredoxin reductase inhibition for a ruthenium compound. J. Med. Chem. 2007;50:5871–5874. doi: 10.1021/jm0708578. [DOI] [PubMed] [Google Scholar]
- 64.Casini A, Gabbiani C, Sorrentino F, Rigobello MP, Bindoli A, Geldbach TJ, Marrone A, Re N, Hartinger CG, Dyson PJ, Messori L. Emerging protein targets for anticancer metallodrugs inhibition of thioredoxin reductase and cathepsin B by antitumor ruthenium(II)-arene compounds. J. Med. Chem. 2008;51:6773–6781. doi: 10.1021/jm8006678. [DOI] [PubMed] [Google Scholar]
- 65.Oehninger L, Stefanopoulou M, Alborzinia H, Schur J, Ludewig S, Namikawa K, Muñoz-Castro A, Köster RW, Baumann K, Wölfl S, Sheldrick WS, Ott I. Evaluation of arene ruthenium(II) N-heterocyclic carbene complexes as organometallics interacting with thiol and selenol containing biomolecules. Dalton Trans. 2013;42:1657–1666. doi: 10.1039/c2dt32319b. [DOI] [PubMed] [Google Scholar]
- 66.Branco V, Canário J, Lu J, Holmgren A, Carvalho C. Mercury and selenium interaction in vivo effects on thioredoxin reductase and glutathione peroxidase. Free Radic. Biol. Med. 2012;52:781–793. doi: 10.1016/j.freeradbiomed.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 67.Wagner C, Sudati JH, Nogueira CW, Rocha JB. In vivo and in vitro inhibition of mice thioredoxin reductase by methylmercury. Biometals. 2010;23:1171–1177. doi: 10.1007/s10534-010-9367-4. [DOI] [PubMed] [Google Scholar]
- 68.Carvalho CM, Lu J, Zhang X, Arnér ES, Holmgren A. Effects of selenite and chelating agents on mammalian thioredoxin reductase inhibited by mercury implications for treatment of mercury poisoning. FASEB. J. 2011;25:370–381. doi: 10.1096/fj.10-157594. [DOI] [PubMed] [Google Scholar]
- 69.Carvalho CM, Chew E.H Hashemy, S.I. Lu . Holmgren, A. Inhibition of the human thioredoxin system. A molecular mechanism of mercury toxicity. J. Biol. Chem. 2008;283:11913,–11923,. doi: 10.1074/jbc.M710133200. [DOI] [PubMed] [Google Scholar]
- 70.Hondal RJ, Ruggles E L. Differing views of the role of selenium in thioredoxin reductase. Amino Acids. 2011;41:73–89. doi: 10.1007/s00726-010-0494-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arnér ES, Björnstedt M, Holmgren A. 1-Chloro-24-dinitrobenzene is an irreversible inhibitor of human thioredoxin reductase.Loss of thioredoxin disulfide reductase activity is accompanied by a large increase in NADPH oxidase activity. J. Biol. Chem. 1995;270:3479–3482. doi: 10.1074/jbc.270.8.3479. [DOI] [PubMed] [Google Scholar]
- 72.Nordberg J, Zhong L, Holmgren A, Arnér E S. Mammalian thioredoxin reductase is irreversibly inhibited by dinitrohalobenzenes by alkylation of both the redox active selenocysteine and its neighboring cysteine residue. J. Biol. Chem. 1998;273:10835–10842. doi: 10.1074/jbc.273.18.10835. [DOI] [PubMed] [Google Scholar]
- 73.Fang J, Lu J, Holmgren A. Thioredoxin Reductase Is Irreversibly Modified by Curcumin.A novel molecular mechanism for its anticancer activity. J. Biol. Chem. 2005;280:25284–25290. doi: 10.1074/jbc.M414645200. [DOI] [PubMed] [Google Scholar]
- 74.FitzGerald GB, Bauman C, Hussoin MS, Wick MM. 24-Dihydroxybenzylamine a specific inhibitor of glutathione reductase. Biochem. Pharmacol. 1991;41:185–190. doi: 10.1016/0006-2952(91)90475-k. [DOI] [PubMed] [Google Scholar]
- 75.Becker K, Gui M, Schirmer RH. Inhibition of human glutathione reductase by S-nitrosoglutathione. Eur. J. Biochem. 1995;234:472–478. doi: 10.1111/j.1432-1033.1995.472_b.x. [DOI] [PubMed] [Google Scholar]
- 76.Rai G, Sayed AA, Lea WA, Luecke HF, Chakrapani H, Prast-Nielsen S, Jadhav A, Leister W, Shen M, Inglese J, Austin CP, Keefer L, Arnér ES, Simeonov A, Maloney DJ, Williams DL, Thomas CJ. Structure mechanism insights and the role of nitric oxide donation guide the development of oxadiazole-2-oxides as therapeutic agents against schistosomiasis. J. Med. Chem. 2009;52:6474–6483. doi: 10.1021/jm901021k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhao Z. Thiophosphate derivatives as inhibitors of tyrosine phosphatases. Biochem. Biophys. Res. Commun. 1996;218:480–484. doi: 10.1006/bbrc.1996.0085. [DOI] [PubMed] [Google Scholar]
- 78.Baiocco P, Colotti G, Franceschini S, Ilari A. Molecular basis of antimony treatment in leishmaniasis. J. Med. Chem. 2009;52:2603–2612. doi: 10.1021/jm900185q. [DOI] [PubMed] [Google Scholar]
- 79.Bauer H, Fritz-Wolf K, Winzer A, Kühner S, Little S, Yardley V, Vezin H, Palfey B, Schirmer RH, Davioud-Charvet E. A fluoro analogue of the menadione derivative 6-[2’-(3’-methyl)-1’4’-naphthoquinolyl]hexanoic acid is a suicide substrate of glutathione reductase.Crystal structure of the alkylated human enzyme. J. Am. Chem. Soc. 2006;128:10784–10794. doi: 10.1021/ja061155v. [DOI] [PubMed] [Google Scholar]
- 80.Hill DT, Isab AA, Griswold DE, DiMartino MJ, Matz ED, Figueroa AL, Wawro JE, DeBrosse C, Reiff WM, Elder RC, Jones B, Webb JW, Shaw CF. Seleno-auranofin (Et3PAuSe-tagl) synthesis spectroscopic (EXAFS 197Au Mössbauer 31P 1H 13C and 77Se NMR ESI-MS) characterization biological activity and rapid serum albumin-induced triethylphosphine oxide generation. Inorg. Chem. 2010;49:7663–7675. doi: 10.1021/ic902335z. [DOI] [PubMed] [Google Scholar]
- 81.Shaw CF. Gold-based therapeutic agents. Chem. Rev. 1999;99:2589–2600. doi: 10.1021/cr980431o. [DOI] [PubMed] [Google Scholar]
- 82.Schallreuter KU, Wood JM. Azelaic acid as a competitive inhibitor of thioredoxin reductase in human melanoma cells. Cancer Lett. 1994;36:297–305. doi: 10.1016/0304-3835(87)90023-1. [DOI] [PubMed] [Google Scholar]
- 83.Gromer S, Gross JH. Methylseleninate is a substrate rather than an inhibitor of mammalian thioredoxin reductase.Implications for the antitumor effects of selenium. J. Biol. Chem. 2002;277:9701–9706. doi: 10.1074/jbc.M109234200. [DOI] [PubMed] [Google Scholar]
- 84.Shi C, Yu L, Yang F, Yan J, Zeng H. A novel organoselenium compound induces cell cycle arrest and apoptosis in prostate cancer cell lines. Biochem. Biophys. Res. Commun. 2003;309:578–583. doi: 10.1016/j.bbrc.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 85.Zhao R, Masayasu H, Holmgren A. Ebselen a substrate for human thioredoxin reductase strongly stimulating its hydroperoxide reductase activity and a superfast thioredoxin oxidant. Proc. Natl. Acad. Sci. U. S. A. 2002;99:8579–8584. doi: 10.1073/pnas.122061399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jan YH, Heck DE, Gray JP, Zheng H, Casillas RP, Laskin DL, Laskin JD. Selective targeting of selenocysteine in thioredoxin reductase by the half mustard 2-chloroethyl ethyl sulfide in lung epithelial cells. Chem. Res. Toxicol. 2010;23:1045–1053. doi: 10.1021/tx100040k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Waley SG. Kinetics of suicide substrates. Biochem. J. 1985;227:843–849. doi: 10.1042/bj2270843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Walsh C. Suicide substrates mechanism-based enzyme inactivators recent developments. Annu. Rev. Biochem. 1984;53:493–535. doi: 10.1146/annurev.bi.53.070184.002425. [DOI] [PubMed] [Google Scholar]
- 89.Fersht A, editor. 2nd ed . New York: Freeman ; 1984. Enzyme structure and mechanism W. [Google Scholar]
- 90.Karplus PA, Pai EF, Schulz GE. A crystallographic study of the glutathione binding site of glutathione reductase at 0.-nm resolution. Eur. J. Biochem. 1989;178:693–703. doi: 10.1111/j.1432-1033.1989.tb14500.x. [DOI] [PubMed] [Google Scholar]
- 91.Lüönd RM, McKie JH, Douglas KT, Dascombe MJ, Vale J. Inhibitors of glutathione reductase as potential antimalarial drugs.Kinetic cooperativity and effect of dimethyl sulphoxide on inhibition kinetics. J. Enzyme Inhib. 1998;13:327–345. doi: 10.3109/14756369809021479. [DOI] [PubMed] [Google Scholar]
- 92.Colotti G, Ilari A, Fiorillo A, Baiocco P, Cinellu MA, Maiore L, Scaletti F, Gabbiani C, Messori L. Metal-Based Compounds as Prospective Antileishmanial Agents Inhibition of Trypanothione Reductase by Selected Gold Complexes. ChemMedChem. 2013 [Epub ahead of print]. doi: 10.1002/cmdc.201300276. [DOI] [PubMed] [Google Scholar]
- 93.Omata Y, Folan M, Shaw M, Messer RL, Lockwood PE, Hobbs D, Bouillaguet S, Sano H, Lewis JB, Wataha JC. Sublethal concentrations of diverse gold compounds inhibit mammalian cytosolic thioredoxin reductase (TrxR1). Toxicol. In Vitro. 2006;20:882–890. doi: 10.1016/j.tiv.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 94.Viry E, Battaglia E, Deborde V, Muller T, Reau R, Davioud-Charvet E, Bagrel D. A Sugar-modified phosphole gold complex with antiproliferative properties acting as a thioredoxin reductase inhibitor in MCF-7 cells. ChemMedChem. 2008;3:1667–1670. doi: 10.1002/cmdc.200800210. [DOI] [PubMed] [Google Scholar]
- 95.Urig S, Fritz-Wolf K, Réau R, Herold-Mende C, Tóth K, Davioud-Charvet E, Becker K. Undressing of phosphine gold(I) complexes as irreversible inhibitors of human disulfide reductases. Angew. Chem. Int. Ed. Engl. 2006;45:1881–1886. doi: 10.1002/anie.200502756. [DOI] [PubMed] [Google Scholar]
- 96.Ott I, Qian X, Xu Y, Vlecken DH, Marques IJ, Kubutat D, Will J, Sheldrick WS, Jesse P, Prokop A, Bagowski CP. A gold(I) phosphine complex containing a naphthalimide ligand functions as a TrxR inhibiting antiproliferative agent and angiogenesis inhibitor. J. Med. Chem. 2009;52:763–770. doi: 10.1021/jm8012135. [DOI] [PubMed] [Google Scholar]
- 97.Schuh E, Pflüger C, Citta A, Folda A, Rigobello MP, Bindoli A, Casini A, Mohr F. Gold(I) carbene complexes causing thioredoxin 1 and thioredoxin 2 oxidation as potential anticancer agents. J. Med. Chem. 2012;55:5518–5528. doi: 10.1021/jm300428v. [DOI] [PubMed] [Google Scholar]
- 98.Saggioro D, Rigobello MP, Paloschi L, Folda A, Moggach SA, Parsons S, Ronconi L, Fregona D, Bindoli A. Gold(III)-dithiocarbamato complexes induce cancer cell death triggered by thioredoxin redox system inhibition and activation of ERK pathway. Chem. Biol. 2007;14:1128–1139. doi: 10.1016/j.chembiol.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 99.Baiocco P, Ilari A, Ceci P, Orsini S, Gramiccia M, Di Muccio T, Colotti G. Inhibitory Effect of Silver Nanoparticles on Trypanothione Reductase Activity and Leishmania infantum Proliferation. Med. Chem. Lett. 2011;2:230–233. doi: 10.1021/ml1002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Becker. K, Herold-Mende C, Park JJ, Lowe G, Schirmer RH. Human thioredoxin reductase is efficiently inhibited by (22’: 6’2’ ‘-terpyridine)platinum(II) complexes.Possible implications for a novel antitumor strategy. J. Med. Chem. 2001;44:2784–2792. doi: 10.1021/jm001014i. [DOI] [PubMed] [Google Scholar]
- 101.Lo YC, Ko TP, Su WC, Su TL, Wang AH. Terpyridine-platinum(II) complexes are effective inhibitors of mammalian topoisomerases and human thioredoxin reductase 1. J. Inorg. Biochem. 2009;103:1082–1092. doi: 10.1016/j.jinorgbio.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 102.Arnér ES, Nakamura H, Sasada T, Yodoi J, Holmgren A, Spyrou G. Analysis of the inhibition of mammalian thioredoxin thioredoxin reductase and glutaredoxin by cis-diamminedichlo- roplatinum (II) and its major metabolite the glutathione-platinum complex. Free Radic. Biol. Med. 2001;31:1170–1178. doi: 10.1016/s0891-5849(01)00698-0. [DOI] [PubMed] [Google Scholar]
- 103.Witte AB, Anestål K, Jerremalm E, Ehrsson H, Arnér E S. Inhibition of thioredoxin reductase but not of glutathione reductase by the major classes of alkylating and platinum-containing anticancer compounds. Free Radic. Biol. Med. 2005;39:696–703. doi: 10.1016/j.freeradbiomed.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 104.Hashemy SI, Ungerstedt JS, Zahedi Avval F, Holmgren A. Motexafin gadolinium a tumor-selective drug targeting thioredoxin reductase and ribonucleotide reductase. J. Biol. Chem. 2006;281:10691–10697. doi: 10.1074/jbc.M511373200. [DOI] [PubMed] [Google Scholar]
- 105.Lu J, Chew EH, Holmgren A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. U. S. A. 2007;104:12288–12293. doi: 10.1073/pnas.0701549104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lin S, Del Razo LM, Styblo M, Wang C, Cullen WR, Thomas DJ. Arsenicals inhibit thioredoxin reductase in cultured rat hepatocytes. Chem. Res. Toxicol. 2001;14:305–311. doi: 10.1021/tx0001878. [DOI] [PubMed] [Google Scholar]
- 107.Schallreuter KU, Wood JM. The stereospecific suicide inhibition of human melanoma thioredoxin reductase by 13-cis retinoic acid. Biochem. Biophys. Res. Comm. 1989;160:573–579. doi: 10.1016/0006-291x(89)92471-6. [DOI] [PubMed] [Google Scholar]
- 108.Saravanamuthu A, Vickers TJ, Bond CS, Peterson MR, Hunter WN, Fairlamb AH. Two interacting binding sites for quinacrine derivatives in the active site of trypanothione reductase a template for drug design. J. Biol. Chem. 2004;279:29493–29500. doi: 10.1074/jbc.M403187200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gallwitz H, Bonse S, Martinez-Cruz A, Schlichting I, Schumacher K, Krauth-Siegel RL. Ajoene is an inhibitor and subversive substrate of human glutathione reductase and Trypanosoma cruzi trypanothione reductase crystallographic kinetic and spectroscopic studies. J. Med. Chem. 1999;42:364–372. doi: 10.1021/jm980471k. [DOI] [PubMed] [Google Scholar]
- 110.Li S, Zhang J, Li J, Chen D, Matteucci M, Curd J, Duan JX. Inhibition of both thioredoxin reductase and glutathione reductase may contribute to the anticancer mechanism of TH-302. Biol. Trace Elem. Res. 2010;136:294–301. doi: 10.1007/s12011-009-8544-1. [DOI] [PubMed] [Google Scholar]
- 111.Lu J, Chew EH, Holmgren A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. U. S. A. 2007;104:12288–12293. doi: 10.1073/pnas.0701549104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Savvides SN, Scheiwein M, Bohme CC, Arteel GE, Karplus PA, Becker K, Schirmer RH. Crystal structure of the antioxidant enzyme glutathione reductase inactivated by peroxynitrite. J. Biol. Chem. 2002;277:2779–2784. doi: 10.1074/jbc.M108190200. [DOI] [PubMed] [Google Scholar]
- 113.Becker K, Savvides SN, Keese M, Schirmer RH, Karplus PA. Enzyme inactivation through sulfhydryl oxidation by physiologic NO-carriers. Nat. Struct. Biol. 1998;5:267–271. doi: 10.1038/nsb0498-267. [DOI] [PubMed] [Google Scholar]