

Abstract
DNA capture coupled with next generation sequencing is highly suitable for the study of ancient pathogens. Screening for pathogens can, however, be meticulous when assays are restricted to the enrichment of single organisms, which is common practice. Here, we report on an array-based DNA capture screening technique for the parallel detection of nearly 100 pathogens that could have potentially left behind molecular signatures in preserved ancient tissues. We demonstrate the sensitivity of our method through evaluation of its performance with a library known to harbour ancient Mycobacterium leprae DNA. This rapid and economical technique will be highly useful for the identification of historical diseases that are difficult to characterize based on archaeological information alone.
Keywords: pathogen screening, palaeopathology, array capture, ancient DNA
1. Introduction
Next generation sequencing as applied to ancient DNA has improved our understanding of the evolutionary history of pathogenic bacteria and the phylogeographic distribution of their associated diseases [1–6]. Thus far, most genomic analyses have relied on molecular capture techniques designed to target DNA from individual organisms suspected to have left molecular traces in preserved tissues. Candidate biological specimens have been selected based on either historical documentation [1,4,6], a burial context that links them to a past pandemic [5], or diagnostic pathological changes that occur in bone as a result of prolonged infection [2,3]. Such exclusive evidence that links an individual to a specific disease, however, is rare in the archaeological record. Many modern diseases for which genetic information obtained from ancient tissues would be highly valuable are not associated with major pandemics of the past; thus, data to implicate a suspected pathogen in a given skeletal assemblage is often lacking. In addition, physical changes in bone occur in a minority of diseases and form only after prolonged active infection. The vast majority of illnesses leave no physical evidence on the skeleton [7], and even when present, morphological changes often lack the specificity needed for reliable disease classification [8].
Close on the horizon are investigations that will consider co-circulating infectious diseases in the past. Synergistic interactions between seemingly unrelated pathogens that influence transmission and evasion of host immune responses have been demonstrated for several contemporary infections [9], suggesting that facilitative relationships may play a role in pathogen adaptation and evolution. Parallel information of co-infections in historical populations will be highly valuable to investigate the changing landscape of infectious disease through time. In the absence of obvious genetic evidence for differences in virulence between ancient and modern organisms investigated thus far, these themes have been raised potentially to account for the presumed higher rates of infection and greater disease severity in the past [1].
As such, more efficient and more comprehensive screening techniques are needed to identify preserved molecular signals of pathogens in ancient tissues. DNA capture microarrays have shown phenomenal success for pathogen DNA retrieval from highly complex ancient DNA extracts [1–5]; hence we have adapted this technology to screen for not just one, but multiple pathogens at the same time. Here, we describe an ancient pathogen screening array (APSA) designed for the parallel DNA detection of almost 100 different pathogens that might be found in ancient remains including bacteria, DNA viruses, protozoa, and multicellular organisms. Capture can be performed with multiple libraries in parallel, making this both a highly powerful and economical method for pathogen screening of archaeological material. Our enrichment technique offers advantages over alternative fluorescent-based approaches [10] as captured reads can be subsequently authenticated by evaluation of DNA damage patterns [11] and in some cases by provisional phylogenetic placement. In addition, adjustments in mapping stringency can be applied in downstream analyses to diminish the threat of false-positives. Here, we demonstrate the value of our tool by evaluating its performance with an ancient sample known to harbour preserved Mycobacterium leprae DNA.
2. Capture array design
We designed probes for an Agilent 1-million feature array to accommodate unique regions in 92 different pathogens. Candidate pathogens were chosen to represent those that could reasonably be present in archival samples including bone, dental pulp, mummified tissue, or specimens from soft tissue anatomical collections. RNA viruses were omitted in our design owing to their predicted poor preservation in ancient tissues [12]. As most ancient DNA extracts are dominated by molecules derived from environmental sources that can share genomic motifs with pathogenic organisms, care was taken to ensure high specificity in the capture regions. A summary of the array design workflow is shown in figure 1. Selection of capture regions was performed based on NCBI taxonomic relationships [13]. For organisms that share pathogenicity at the species or subspecies level (such as the different pathogenic subspecies in the Mycobacterium tuberculosis complex), regions were identified that are conserved among all strains and that are unique to this group. Where only individual strains are pathogenic (such as Escherichia coli), unique regions were identified. If genomic sequences of more than one pathogenic strain were available, one was chosen at random. Candidate regions were then screened for uniqueness by BLAST searches against the NCBI nucleotide database. Regions were retained if no other organisms were represented in the search results using an e-value cut-off of e ≤ 10−3. Those chosen for probe design ranged from 84 to 3 291 871 bp.
Figure 1.

Workflow followed in probe design for the ancient pathogen screening array (APSA).
Probes 60 bp in length were generated at 6 bp tiling for each genomic region considered suitable for capture design. Oligos were subsequently filtered for: (i) complexity by removing probes with repetitive elements, (ii) removal of all probes that contained characters other than A, C, T or G, (iii) self-complementarity using mfold v. 3.5 [14], and (iv) temperature compatibility retaining only those with melting temperatures (Tm) between 60 and 85°C. A total of 974 016 probes are permitted on an Agilent 1-million feature array, and efforts were made to provide equal representation of all pathogens, regardless of genome size. For pathogens with fewer than 30 000 probes remaining after the filtering steps, all candidate probes were included on the array. The remaining probes were distributed evenly between organisms with greater than 30 000 candidates. A list of all pathogens and probe numbers is provided in the electronic supplementary material, table S1.
3. DNA capture
We used a library generated from a leprosy sample (SK8) we reported on in an earlier publication where M. leprae DNA was successfully obtained using DNA array-based capture [2]. Libraries were generated by ligating adaptors and unique index identifiers to both ends of the molecules following established protocols [15,16]. Three negative extraction controls were included, as were three library blanks generated by adding water in place of template. Libraries were subsequently amplified [15] to obtain 0.7 µg of library SK8 and 0.9 µg of each negative control, which were serially enriched over two identical capture arrays in parallel (SK8 on one array and the negative controls on another), as described elsewhere [1,17]. Enriched products for SK8 were sequenced on an Illumina MiSeq platform with 2 × 150 + 8 + 8 cycles using the MiSeq reagent kit v. 2 and the manufacturer's protocol for multiplex sequencing. For higher resolution identification of false-positives, negative controls were subsequently sequenced on an Illumina HiSeq 2500 following the manufacturer's protocol.
4. Read processing and analysis
Paired-end reads were merged as described elsewhere [2]. Reads that could not be merged were incorporated as single reads, which were subject to an additional quality filtering step that trimmed all reads from the 3′-end until a Phred score of greater than 20 was obtained for each nucleotide in the sequence. Reads shorter than 30 bp were removed and the remaining reads were mapped to the full genomes of organisms used in APSA probe design with BWA [18] using the aln/samse algorithm with the –n parameter set to 0.1 for increased mapping specificity. To eliminate artefacts, all reads with mapping quality scores below Q37 were removed. Duplicate removal was performed using an in-house modification to the samtools rmdup function [18], which allows duplicate fragments to be removed from both merged and unmerged pools. Reads were subsequently filtered based on their overlap of the template regions, with retention of only those reads that overlapped an array probe sequence by a minimum of 10 bp. Mapping results based on these parameters are reported in the electronic supplementary material, table S2. To reduce the bias of greater probe representation for certain organisms, we further normalized the reads by dividing the number of mapped reads by the number of oligos on the array for the corresponding organism. Normalized data are also reported in the electronic supplementary material, table S2.
The normalized read count tables were visualized using the R programming environment [19]. Figure 2 shows a scatter plot of the normalized read counts and the distribution of reads mapping after duplicate removal to the different pathogens on our array.
Figure 2.

Plots for all mapped APSA-captured reads post duplicate removal, normalized to show the number of hits per probe for sample SK8.
5. Analysis of negative controls
Each of our negative controls consisted of between 71 766 and 279 577 shotgun reads and between 44 761 and 991 460 reads after ASPA capture. Mapping results are presented in the electronic supplementary material, table S2. Duplicate removal statistics are not reported because no reads were removed after applying this filter. The majority of APSA-enriched organisms had no detectable reads in our shotgun data at our current sequencing depth; hence enrichment efficiencies could not be calculated. Predictably, greater manipulation of the extraction blanks contributed to an increased number of APSA-captured reads compared with the library blanks. Of those organisms for which enrichment efficiencies could be calculated, the highest enrichment was obtained for Streptococcus pneumonia (electronic supplementary material, table S2). While this bacterium can reside in the nasopharynx of asymptomatic carriers [20], this enrichment derived from only three unique fragments (electronic supplementary material, table S2); hence potential in-lab contaminants are present in very low quantities. Additional human commensal and soil-dwelling bacterial species such as Veillonella parvula, E. coli, Burkholderia cepacia and Peptostreptococcus magnus received enrichments in the extraction blanks, which indicates that analyses of commensal organisms that can become pathogenic in compromised individuals may be challenging with our DNA capture approach owing to their ubiquity in the environment. In addition, enrichments stemming from up to 25 (non-unique) reads were observed for Trypanosoma cruzi, though normalization reveals that this seemingly high capture is merely an artefact of its higher representation on the array due to its larger genome.
Our library blanks had far fewer mapped reads, with notable enrichments only of Trypanosoma brucei in one blank and V. parvula in another. In all, the low background in our negative controls suggests that our capture approach, coupled with our strict mapping parameters, yields a low number of false-positives.
6. Analysis of the Mycobacterium leprae positive control library
Library SK8 was generated from a mediaeval bone from Winchester in the United Kingdom, and was included in a previous publication that explored genetic diversity in M. leprae. This sample was confirmed to have preserved M. leprae DNA sufficient for genome assembly through array-based enrichment [2]. An analysis of multiple samples revealed that M. leprae DNA has exceptional preservation, likely owing to protection by its lipid-rich cell wall; for this reason, we chose a sample here (SK8) that contained fewer endogenous M. leprae molecules (8.4%) compared with other samples from that study (up to 40%) in order to mimic a more realistic situation whereby pathogen DNA has low representation in a highly complex DNA library. Shotgun reads generated from 20 µl of extract were obtained from a previous investigation and consisted of 3 058 969 reads. Of these, 149 (0.0049%) mapped to genomic regions used in APSA probe design for all organisms on the array (electronic supplementary material, table S2). No reads were filtered out with our duplicate removal, indicating that all mapping reads are unique. Of these 149 reads, 148 mapped to the M. leprae probe template regions.
Our APSA enrichment from the above library generated 213 611 reads, of which 4774 (2.25%) mapped to our capture regions. A visual representation of the captured reads normalized by the number of hits per probe is shown in figure 2. By far the best mapping was to M. leprae which comprised 4756 (99.6%) of the total mapped reads, thus constituting a 460-fold enrichment and 1.16 hits per probe. Duplicate removal revealed that this enrichment derived from 3656 unique reads, constituting 0.89 unique reads per probe. To authenticate our captured fragments as ancient, damage plots were constructed using MapDamage [21], yielding a pattern expected of ancient mycobacterial DNA [2], with 10% C to T damage at the 5′-ends of the molecules (figure 3). To investigate possible phylogenetic signals, the inclusion of previously defined diagnostic M. leprae SNP positions [2] in our APSA probe set was investigated. The number of SNPs between M. leprae strains is low; however, the branch 2-specific SNP position 2 654 119 was present, and our APSA-captured reads indeed showed the expected derived character for sample SK8 at that position [2], albeit at only twofold coverage. While this coverage is too low to permit a robust phylogenic placement, it provides further evidence that the captured reads are endogenous. The higher ninefold coverage of this SNP reported in our previous genomic work likely resulted from denser 3 bp tiling of the capture probes in comparison to the 6 bp tiling used here, and the richer library used for full genome assembly. While the non-enriched SK8 library had an appreciable amount of M. leprae DNA, mapping of the APSA-captured reads to the leprosy TN reference genome [22] revealed only 856 reads mapping to the 3.2 Mb region excluded in APSA probe design. Duplicate removal reduced this to only 607 reads. This indicates that the 3656 reads mapping to our M. leprae APSA probe regions derived from array enrichment as opposed to non-specific carry-over into the enriched fraction.
Figure 3.
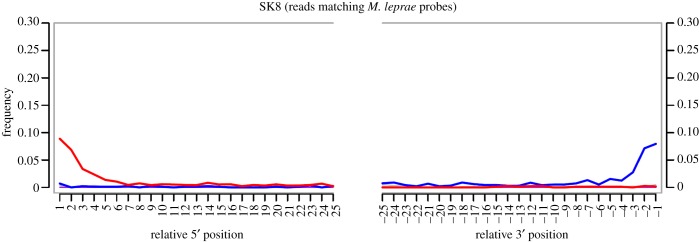
Damage plots generated using MapDamage: red indicates C to T transitions, blue indicates G to A transitions. Y-axis denotes percentage of sites containing a nucleotide change from the reference sequence, and x-axis denotes position along the DNA fragment [21]. (Online version in colour.)
Single M. leprae molecules captured in extraction blank 1, and library blanks 2 and 3 indicate that background contamination, post-capture amplification artefact formation, and/or mismapping are very low for this pathogen in our capture regions. Additional molecules were captured for organisms that also had representation in the captured extraction and library blanks, though the levels identified in the SK8 library are lower than those in the negative controls. Aside from M. leprae, the only other organism unique to this sample was Burkholderia pseudomallei, a pathogenic organism responsible for melioidosis in humans; however, this organism can also survive in soil and water [23], and without a proper control sample for comparison, an environmental origin cannot be excluded.
The efficient capture for endogenous ancient M. leprae DNA, which we had previously determined to be present in this sample, provides a good indication that our APSA technique performs well when DNA matching our probes is present, even in small amounts relative to the background.
7. Conclusion
We have successfully demonstrated the efficiency of our capture screening technique through excellent performance with a known positive control, and low background from deep sequencing of negative controls. Reads were authenticated as ancient by both damage profile and phylogenetic placement. We look forward to exploring the uses of our screening approach in future investigations of infectious diseases in the past, and further evaluating its sensitivity in identifying candidate pathogenic organisms amidst proportionately high amounts of environmental contaminants.
Supplementary Material
Supplementary Material
Acknowledgements
We thank Michael Taylor and Stewart Cole for providing us access to the leprosy bone sample, and Pushpendra Singh for his participation in library preparation. We thank Alexander Peltzer for implementing the modification to the samtools rmdup function. We thank Anne Stone and Jane Buikstra for helpful comments on an earlier version of the manuscript.
Authors' contributions
K.I.B. conceived of the investigation, designed the array, designed experiments, analysed data and drafted the manuscript. G.J. designed the array and analysed data. V.J.S. performed experiments. Å.J.V. performed experiments. M.A.S. performed experiments. A.H. analysed data. K.N. conceived of the investigation, designed experiments and analysed data. J.K. conceived of the investigation, designed experiments and analysed data. All authors contributed to and approved the final version of the manuscript.
Funding statement
This research was funded by European Research Council Starting Grant APGREID (to J.K.) and Social Sciences and Humanities Research Council of Canada (SSHRC) postdoctoral fellowship 756-2011-501 (to K.I.B.).
References
- 1.Bos KI, et al. 2011. A draft genome of Yersinia pestis from victims of the Black Death. Nature 478, 506–510. ( 10.1038/nature10549) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schuenemann VJ, et al. 2013. Genome-wide comparison of medieval and modern Mycobacterium leprae. Science 341, 179–183. ( 10.1126/science.1238286) [DOI] [PubMed] [Google Scholar]
- 3.Bos KI, et al. 2014. Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature 514, 494–497. ( 10.1038/nature13591) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Devault AM, et al. 2014. Second-pandemic of Vibrio cholerae from the Philadelphia cholera outbreak of 1849. New Engl. J. Med. 307, 334–340. ( 10.1056/NEJMoa1308663) [DOI] [PubMed] [Google Scholar]
- 5.Wagner DM, et al. 2014. Yersinia pestis and the Plague of Justinian 541–543 AD: a genomic analysis. Lancet Infect. Dis. 14, 319–326. ( 10.1016/S1473-3099(13)70323-2) [DOI] [PubMed] [Google Scholar]
- 6.Schuenemann VJ, et al. 2011. Targeted enrichment of ancient pathogens yielding the pPCP1 plasmid of Yersinia pestis from victims of the Black Death. Proc. Nat Acad. Sci. USA 108, E746–E752. ( 10.1073/pnas.1105107108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wood JW, Milner GR, Harpending HC, Weiss KM. 1992. The osteological paradox: problems of inferring prehistoric health from skeletal samples. Curr. Anthrolopol. 33, 343–370. ( 10.1086/204084) [DOI] [Google Scholar]
- 8.Lovell NC. 2000. Paleopathological description and diagnosis. In Biological anthropology of the human skeleton (eds Katzenberg MA, Saunders SR.), pp. 217–248. Hoboken, NJ: Wiley. [Google Scholar]
- 9.Singer M. 2010. Pathogen-pathogen interaction: a syndemic model of complex biosocial processes in disease. Virulence 1, 10–18. ( 10.4161/viru.1.1.9933) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Devault AM, et al. 2014. Ancient pathogen DNA in archaeological samples detected with a microbial detection array. Sci. Rep. 4, 4245 ( 10.1038/srep04245) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krause J, Briggs AW, Kircher M, Maricic T, Zwyns N, Derevianko A, Pääbo S. 2010. A complete mtDNA genome of an early modern human from Kostenki, Russia. Curr. Biol. 20, 231–236. ( 10.1016/j.cub.2009.11.068) [DOI] [PubMed] [Google Scholar]
- 12.Okello JBA, et al. 2010. Quantitative assessment of the sensitivity of various commerical reverse transcriptases based on armored HIV RNA. PLoS ONE 5, e13931 ( 10.1371/journal.pone.0013931) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Federhen S. 2012. The NCBI taxonomy database. Nucleic Acids Res. 40, D136–D143. ( 10.1093/nar/gkr1178) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415. ( 10.1093/nar/gkg595) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meyer M, Kircher M. 2010. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, pdb.prot5448 ( 10.1101/pdb.prot5448) [DOI] [PubMed] [Google Scholar]
- 16.Kircher M, Sawyer S, Meyer M. 2012. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 40, e3 ( 10.1093/nar/gkr771) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodges E, et al. 2009. Hybrid selection of discrete genomic intervals on custom-designed microarrays for massively parallel sequencing. Nat. Protoc. 4, 960–974. ( 10.1038/nprot.2009.68) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Durbin R. 2009. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. ( 10.1093/bioinformatics/btp324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.R Core Team. 2014. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; See www.R-project.org. [Google Scholar]
- 20.Falup-Pecurariu O, et al. 2011. Streptococcus pneumoniae nasopharyngeal colonization in children in Brasov, Central Romania: high antibiotic resistance and coverage by conjugate vaccines. Pediatr. Infect. Dis. J. 30, 76–78. ( 10.1097/INF.0b013e3181f42bb6) [DOI] [PubMed] [Google Scholar]
- 21.Jónsson H, Ginolhac A, Schubert M, Johnson PLF, Orlando L. 2013. MapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684. ( 10.1093/bioinformatics/btt193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monot M, et al. 2009. Comparative genomic and phylogeographic analysis of Mycobacterium leprae. Nat. Genet. 41, 1282–1289. ( 10.1038/ng.477) [DOI] [PubMed] [Google Scholar]
- 23.Brook MD, Currie B, Desmarchelier PM. 1997. Isolation and identification of Burkholderia pseudomallei from soil using selective culture techniques and the polymerase chain reaction. J. Appl. Microbiol. 82, 589–596. ( 10.1111/j.1365-2672.1997.tb02867.x) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.