

Abstract
Our understanding of the evolution of domestication has changed radically in the past 10 years, from a relatively simplistic rapid origin scenario to a protracted complex process in which plants adapted to the human environment. The adaptation of plants continued as the human environment changed with the expansion of agriculture from its centres of origin. Using archaeogenomics and computational models, we can observe genome evolution directly and understand how plants adapted to the human environment and the regional conditions to which agriculture expanded. We have applied various archaeogenomics approaches as exemplars to study local adaptation of barley to drought resistance at Qasr Ibrim, Egypt. We show the utility of DNA capture, ancient RNA, methylation patterns and DNA from charred remains of archaeobotanical samples from low latitudes where preservation conditions restrict ancient DNA research to within a Holocene timescale. The genomic level of analyses that is now possible, and the complexity of the evolutionary process of local adaptation means that plant studies are set to move to the genome level, and account for the interaction of genes under selection in systems-level approaches. This way we can understand how plants adapted during the expansion of agriculture across many latitudes with rapidity.
Keywords: ancient DNA, domestication, local adaptation, archaeogenomics
1. Introduction
During the closing phases of the last glacial stage that had predominated the climate system for previous 100 000 years or so, a number of plant species became adapted to an emergent human environment independently at different centres around the globe. This process led to the evolution of domesticated and commensal species. Initially, the evolution of domestication involved the selection of a characteristic group of traits collectively termed the domestication syndrome [1,2]. These traits, which included the loss of shattering, changes in seed size, loss of photoperiod sensitivity and changes in plant and floral architecture [3], enabled the better survival of plants in the human environment. That this was an adaptation to the human environment by plants is emphasized by the fact that a number of non-food plants such as small-seeded grasses and legumes also adapted to this environment under the same regime of cultivation and became commensals, and indeed also display traits of the domestication syndrome [4–6].
Evolution is an interminable process, and the story of the evolution of domestication did not end with the emergence of those first adaptors to the human environment. Some of the commensals later went on to become domesticated crops themselves, such as in the case of oats and rye (reviewed in reference [7]). The human environment to which the plants had adapted was dynamic and presented plants with new challenges, resulting in new adaptations and also new winners and losers among the domesticated species [8]. One of the greatest challenges was undoubtedly the spread of agriculture from various centres of origin to new latitudes, in almost all cases far away from the biogeographic distribution of the wild progenitor species. For plants, the environment changed in terms of temperature, rainfall and daylength on a grand scale, particularly for instance as crops were dispersed northwards into Europe. Further demands were placed on plants on a local and regional scale as they were moved to new soil types and specific environmental conditions, such as high altitude or arid environments. Alongside this, cultural innovation and changing agrarian practices altered the selection regime [9]. Evidence is emerging of the adaptation of different cultural complexes to specific ecological niches as agriculture spread into Europe [10], and stalls occurred in the spread of agriculture that are associated with a combination of the time required for the adaptation of crops to new environments as well as to the changing assemblage composition of the agrarian package itself [11,12]. In both Europe and Asia, the push northwards was associated with adaptations to changing daylength [13–15], and with commensals better adapted to the northern ecologies, such as European rye making the transition from commensal to domesticated species [16]. There is no definable endpoint to the evolution of domestication, and it is a process that should be considered as ongoing [17].
2. Crop adaptation to complex environments
(a). Ancient DNA as an approach to studying local adaptation
The challenges facing plants and humans were complex and dynamic. How and to what extent plants could adapt to complex environments and how much change could be embraced within their sphere of plasticity are questions of importance to understanding evolution in general, as well as the emergence and spread of agriculture. Ancient DNA (aDNA) provides an inroad to understanding that evolutionary process directly. Although the potential of aDNA in understanding the spread of agriculture was recognized in the 1990s [18], major obstacles became apparent. One major obstacle was that preservation of DNA was largely unsuitable for large-scale analyses with the technology of the time. There are two aspects to this obstacle. The first is that it is an inherent problem that the rise of agriculture took place in locales of low latitude where relatively warm temperatures limit to just a few thousand years the time depth from which ancient biomolecules can be retrieved (figure 1) [19]. The second is that the vast majority of archaeobotanical material are in the form of charred remains and a few species mainly found under waterlogged conditions. In the case of charring, evidence of DNA preservation was found to be at best sporadic [20]. Partly owing to these limitations, the number of studies of the evolution of domestication of plants that used aDNA was low relative to other research areas in aDNA [21–23]. Despite these limitations, some glimpses into the evolution of crops have been possible. An extinct expansion of a wheat crop type was detected using charred material that could have reflected an ecological limit and failure to adapt to the dynamic human environment [24]. However, insights using charred material were restricted to the sporadic establishment of the phylogeographic presence or absence of small markers from which little could be inferred about how evolution or local adaptation had occurred [25–27]. The potential to observe selection and adaptation to the human environment directly through aDNA was first achieved with desiccated remains of maize, in which three biologically significant genes were surveyed over time and space in a handful of samples [28]. While these incremental advances using aDNA offered tantalizing glimpses of the evolutionary process, the relatively small datasets generated meant that progress was prohibitively slow until the advent of next-generation sequencing a few years later.
Figure 1.
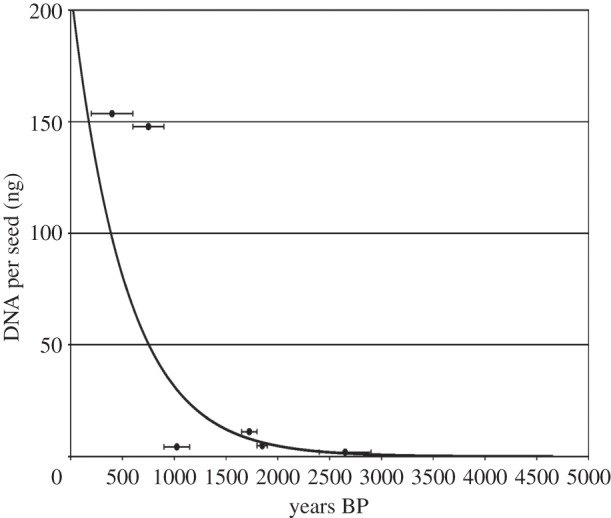
Graph shows DNA content of desiccated barley seeds over time in Qasr Ibrim, North Africa [19].
(b). Genetic expectations revealed through models
A second major obstacle to understanding how plants adapted to complex environments was a wider problem of accurate interpretation of genetic diversity that has been produced by complex processes [29–32]. This problem became apparent when interpretations of genetic data became increasingly divergent from the evidence unearthed in archaeology [30]. On the one hand, a long-held assumption of the high strength of artificial selection giving rise to a rapid and geographically definable origin of crop domestication was supported by many genome-wide-based analyses. On the other hand, archaeological evidence suggested a long protracted arrival of domesticated forms of cereal crops, with a hitherto unappreciated long period of pre-domestication cultivation that stretched thousands of years back into the Pleistocene [33,34], and a slow subsequent fixation of traits over a period of thousands of years [35].
Increasingly, computational models are being applied to phylogeographic data to assess alternative domestication history hypotheses. Modelling has revealed that the genetic inferences were based on analysis of data with low discriminatory power, and in fact, genetic data diversity is compatible with the notion of a protracted origin [30]. More precise estimates of the strength of domestication syndrome traits directly from the archaeological record have led to the further unexpected conclusion that the selection coefficients involved are low (in the order of 0.003) for traits as divergent as shattering, largely under monogenic control and increased seed size, under polygenic control [36]. This level of selection is more akin to natural selection than the popular perception of artificial selection. These results are surprising given that field experiments have shown that selection under cultivation can be strong [37]. Consequently, it has now become a central question to understand how plants became adapted to the human environment, and why it took as long as it did. This poses a second question: how much selection could have occurred? Haldane [38] first formally recognized that one could not have unbridled amounts of selection, because selection necessarily comes at a cost. In order for differential survival to occur, some individuals have to die (or fail to be born). For this reason, Haldane concluded that plant breeders are limited in the number of traits they can breed into varieties. A model of the number of genes, intensity of selection and probability of population survival shows that the limit to the number of genes that could be under selection is in the order of 50–100 for plant populations at the levels of intensity observed in the archaeological record [39] (electronic supplementary material, figure S1a). Interestingly, these values are similar to the number of genes showing signatures of selection from genome studies of crops such as maize, wheat and sunflower in which estimates vary from 27 to 70 genes under selection [40–43]. Another important insight from this model comes from the total amount of selection (the selection load) that occurs under different selection intensities per locus (electronic supplementary material, figure S1b). Here, we find that more selection can occur at lower selection intensities. An interpretation of this is that a greater amount of selection can occur under complexity than can occur under strong selection. Furthermore, a selective sweep may render a population vulnerable to further change, making it less able to cope with a dynamic human environment and restricted to effective adaptation within low complexity environments. To put it another way, we expect from these models for complex selection to be more robust than selective sweeps. This analysis begs the question about the nature of the human dynamic environment to which plants have adapted—can it be considered complex or simple? It may be tempting to speculate that cultivation should be considered a simple environment in which humans negate many of the issues plants would have to cope with in the wild, but the evidence from the archaeological record suggests otherwise [11].
The models outlined here suggest to us that we should expect a number of loci in the order of 50–100 under relatively weak selection. A corollary of this mode of change is that it seems likely that traits would be targeted at multiple loci weakly to effect strong selection rather than the more conventional perspective of a strong selective sweep at a single locus. It is therefore a prediction that we should see multiple changes in the interactions of genes and their products, such as regulatory and metabolic networks.
3. Complex adaptations viewed through ancient DNA and next-generation sequencing
(a). Large amounts of genome evolution over short time periods
The advent of next-generation sequencing (NGS) opened up the real possibility of using aDNA to track evolution directly and test the expectations of genetic diversity generated through models and the archaeological record. One approach to look at large-scale genome evolution is to monitor the change in the transposable element (TE) composition. Cotton provides a good example of a crop in which to study the evolution of genome architecture in this way, because evidence from interspecific comparisons suggests that there have been recent significant expansions and contractions of TEs [44–46]. We were astounded to observe the extent of change of retroelement composition in the diploid species Gossypium herbaceum (electronic supplementary material, figure S2). In this case, 454 Roche FLX shotgun metagenomic data were generated from four samples of desiccated archaeological cotton from Africa, Brazil and Peru, and compared with data from modern accessions [47]. G. herbaceum is thought to be a very young species, speculated to be little older than the Holocene [48]. This youth is supported by the observation that lineage sorting appears to be very incomplete between G. herbaceum and its sister taxon Gossypium arboreum in which we found out of 10 PCR systems none yielded alleles that were exclusive to one or other species in a sample of 91 accessions (SA Palmer, AJ Clapham, P Rose, F Freitas, BD Owen, D Beresford-Jones, JD Moore, JL Kitchen, RG Allaby 2012, unpublished data). We therefore find it surprising that such differentiation in TE proportions is observed within G. herbaceum. This finding contrasted with the tetraploids (G. hirsutum and G. barbadense) in which we found very little change within and between species. The tetraploid species are related to the diploid species through a genome donation of an ancestor of the diploids around 1.5 Ma [49]. The contrasting pattern between the diploids and tetraploids appears to be reminiscent of punctuated equilibrium, which has recently been linked to TE composition and turnover [50,51]. In this case, more work is needed to explore cotton genome evolution directly. For example, tracing older cotton genomes would enable us to see the development of expansions over time and establish whether we see a reduction of diversity closer to the origin of speciation, or whether there is standing variation that could better explain our results that lies in stark contrast to the invariant tetraploids which were sampled from a wider spatial and temporal range.
While the evidence of TE change over time would appear to support the expectation of large amounts of small change (assuming most transpositions had little effect on the genome functionality), they tell us little about the adaptive value of such change but hint at the potential pace of change and so capability of adaptation that could be possible. In this particular study, we considered fragments of retrieved DNA that fell in gene regions as a possible source of information about adaptive change. Gene variants from these types of data may represent allelic variants or sequencing errors (which occur at a rate of about 1% for the platform used). However, we would expect sequencing errors to be randomly distributed throughout the genome, but our expectation from the models outlined above is that variants are likely to be clustered non-randomly in gene networks. We identified 210 gene fragments from our cotton metagenomic dataset that differed from public database entries and of those we were able to map 20 to the KEGG (http://www.genome.jp/kegg/) metabolic pathways map (electronic supplementary material, figures S3 and S4). It is notable that of these 20 variants, 17 fall in close proximity to another variant in their respective part of the metabolic network, on average separated by three nodes from their nearest neighbour. In this analysis, six metabolic clusters are apparent, supporting the notion that they are not random and appear to fall in line with the model predictions of multiple changes within gene networks. The incorporation of such approaches in the study of local adaptation holds the promise of systems level insights through aDNA.
(b). Qasr Ibrim, Egypt: a site of local adaptation?
This early work with NGS in archaeobotanical remains established that complex genome level insights into the evolutionary process could be gained from samples of low latitude sites. The archaeological site at Qasr Ibrim, Egypt, provides an opportunity to expand on these approaches. Qasr Ibrim was a boundary settlement on the edge of the Nubian and Roman Empires located between the first and second cataracts of the Nile, and was occupied by five successive cultures: Napatan, Roman, Meroitic, Christian and Islamic [52]. The site is very dry, and the preservation of archaeobotanical remains is remarkably good [53]. There is a continuous record of occupation over 3000 years that provides an ideal opportunity to study crop evolution through time. Of particular interest is the barley found at the site that appears to be a two-row form that has evolved from a six-row form [19]. In modern barley, two-row architecture is the wild state, caused by a transcription factor Vrs1 that inhibits the development of the two lateral florets in a flower spike. The six-row architecture is caused by a loss of function of this transcription factor allowing lateral floret development [54]. The barley at Qasr Ibrim is curious because it has the non-functional version of the transcription factor, so should be six-row form. A simple model shows that the fecundity difference incurred by the architectural change between row types means that six-row barley is expected to quickly outcompete two-row, within the lifetime of a farmer [19]. Fitting with this expectation, the six-row type emerged very early in the domestication of barley [55]. The predomination of two-rows in the wild suggests that they must have a strong selective advantage over six-row in the face of the fecundity difference, and indeed, under conditions of water stress, two-row barleys fair better [56]. We hypothesized that the Qasr Ibrim barley may represent a local adaptation to the dry conditions of the Upper Nile. If so, then it may be the case that other genes show functional changes associated with adaptations to do with water usage and drought tolerance. We have explored several approaches to using NGS to study the ancient nucleic acids of the barley of Qasr Ibrim that illustrate what is possible, and which paint quite an unexpected picture of the history of barley in this region.
(c). Survival of and insights from ancient RNA
While DNA contains the evolutionary record of the genome, RNA has the potential to offer insights into the last activities of the organism through a record of gene expression. Although they were among the earliest in the field [57], few studies have been carried out on ancient RNA because it is expected to degrade about 50-fold faster than DNA, largely because it is highly prone to hydrolytic attack [58–60]. Consequently, under arid conditions, one might expect some preservation of RNA. Recently, NGS has been successfully applied to RNA from desiccated maize kernels [61]. We were surprised to find that the RNA content of the barley at Qasr Ibrim is actually higher than that of DNA [62]. At the point of death, RNA content is expected to be higher than DNA because of the multiple copies of RNA that exist relative to DNA gene copies. Assuming a ratio of between 5- and 100-fold more RNA than DNA at the point of deposition, we estimate that at Qasr Ibrim the rate of RNA decay is in the order of two- to fourfold greater than DNA, a much reduced rate than expected, most likely owing to the very arid conditions of this site and consequent reduced rate of hydrolytic attack. The diagenetic process of base modification appears to be similar in this RNA to that found in aDNA with frequent conversion of cytosine to uracil, most likely through deamination as with DNA (figure 2). There are, however, interesting differences also in the RNA degradation relative to DNA. The distribution of cytosine base modifications mapped through mapDamage v. 2.0 [63] is biased towards both ends of the molecule in RNA, rather than towards the 5′ end of reads in dsDNA where exposed overhangs are more prone to hydrolysis. We hypothesize that this may be due to secondary structures forming primarily over the central part of the molecule and sheltering it from chemical attack.
Figure 2.

Base modification frequencies of RNA observed from archaeological barley stripe mosaic virus (BSMV) [62].
We examined the RNA portion of the barley at Qasr Ibrim using Illumina sequencing technology to see if we could learn anything about the regulatory action of microRNAs [64]. Generally, we have succeeded in recovering miRNAs from archaeological barley, and we do view differences in the relative expression profiles of archaeological and modern barleys that seem to indicate that the barley of the Christian era was stressed. Briefly, our unpublished data (RG Allaby, R Gutaker, AC Clarke, N Pearson, R Ware, SA Palmer, JL Kitchen, O Smith 2013) show the presence of miRNAs in the Christian era associated with germination inhibition, suggesting the avoidance of growth under harsh environmental conditions. An unexpected result was the retrieval of the first RNA genome that belonged to the barley stripe mosaic virus (BSMV) [62]. Historical records attributed to this virus only go back for the past 100 years or so, and analysis of modern genomes suggests an age of origin no greater than 200 years. The inclusion of the ancient RNA genome indicates that the Qasr Ibrim virus is close to the base of the crown group, suggesting an expansion of this virus contemporaneous with the Crusades, with an origin some time before this. BSMV has no known vector and is spread by physical contact between grains or pollen [65], so its rise at this time may have been linked to the intensification of agriculture that occurred to support the medieval war machine.
The RNA results demonstrate that it is possible to identify activated points of gene networks directly from the past. Furthermore, viruses play an important role in the adaptation of organisms to new environments [66]. Viruses resident to an indigenous community may affect newcomers more severely than their indigenous hosts, and likewise the introduction of viruses by newcomers may affect the indigenous community severely. In this respect, viruses can be considered an important part of the adaptive arsenal carried by organisms rather than simply a burden. The movement of domesticated plants throughout history and particularly at a more global level in recent times is of concern regarding the emergence of new diseases that affect our food supply [67]. Therefore, viruses add an important dimension to the understanding of the local adaptation of crops that is visible through the archaeobotanical record.
(d). Methylation patterns
The global methylation state of a plant genome can be informative about the level of stress it is under. Methylation of cytosine bases causes the silencing of genes and is an effective genomic mechanism to control TEs. Typically, up to 90% of plant genomes can become methylated under conditions of stress [68]. Given the emergent picture of the barley at Qasr Ibrim, and our initial suspicions of water stress at the site, we were interested to know whether anything remained of the methylation signal under these preservation conditions [69]. Using the MethylMiner kit, the CpG methylation signal of archaeological barley through time was established (figure 3). The methylation signal falls exponentially over time, and extrapolation of the trend to modern times results in methylation levels which are in the normal range for barley. In ancient samples, the strength of the methylation signal is expected to be less than modern, because the shorter DNA fragments that are bead captured will contain fewer methylated cytosine sites. However, the decrease in signal, in this case, we believe is due to chemical modification of the methylation signal rather than DNA fragmentation, because the size distribution of the DNA fragments did not vary greatly between archaeobotanical samples of different ages. The barley that corresponds to the Christian strata is notable because its signal suggests 98% methylation of the living barley, indicating a high degree of stress. Therefore, the barley at Qasr Ibrim was not stressed as far as we can see, until the Christian era, and returned to normal methylation levels after that time in the Islamic era. We confirmed this pattern using bisulfite sequencing of a region of the eIF4 locus that again showed the high degree of methylation associated with the Christian era.
Figure 3.

A perspective of the history of barley in Nubia. After [62,69] and RG Allaby, R Gutaker, AC Clarke, R Ware, SA Palmer, O Smith, W Nicholson, L Kistler 2013, unpublished data.
(e). The possible utility of charred grains
A DNA capture approach was also applied to the barley of Qasr Ibrim through time. A chip of 183 genes selected for their possible roles in drought adaptation was used to capture DNA from multiple points in the strata, which was sequenced using Illumina technology (RG Allaby, R Gutaker, AC Clarke, R Ware, SA Palmer, O Smith, W Nicholson, L Kistler 2013, unpublished data). Some preliminary overviews of the results are presented here to help complete the picture of the use of NGS and aDNA to study local adaptation in barley at Qasr Ibrim. Of particular interest was a single 2000 year old (Roman) charred grain of barley from Kawa for which we obtained a few reads (amounting to 82 000 bp of reads in total). A frequency distribution of read lengths obtained from this sample, and compared with a desiccated sample of slightly greater age (Napatan) shows that the two are essentially identical in the size range and differ principally in absolute frequency (figure 4). While all interpretations should be cautious because of biases in size distribution introduced by the library preparation process, the similarity of these profiles is surprising and encouraging. Other research groups have managed to produce shotgun NGS data from charred wheat using alternative platforms [70], and sequencing technology has now reached a point at which the vast charred archaeobotanical record may be accessible to a useful degree.
Figure 4.

Size distribution of DNA reads from 2000 year old charred and 2500 year old desiccated barley (Napatan) obtained through DNA capture and Illumina sequencing.
(f). The crusades as an example of the introduction of poorly adapted crops
At the time of writing, we have recovered alleles of 86 loci from 52 single grains of barley from Napatan, Roman, Meroitic, Christian and Islamic levels at Qasr Ibrim (data not shown). Contrary to our expectations, the barley at Qasr Ibrim is not very distinct from the barley of Nubia and the Near East generally. However, we do see a distinct influx of different alleles during the Christian era.
In each of the ways we have examined the Qasr Ibrim barley, we have found that the Christian era is the distinct stratum. This era is contemporaneous with the arrival of the crusaders, during which time we see distinct barley, the arrival of a virus and a methylation signature of stress. An interpretation that we are now exploring is that the crusaders may have brought barley with them that was less able to cope with local conditions than the barley of the region. The original barley type appears to have generally been reasserted during the Late Christian and completely by the Islamic phases. If the barley that was resident at Qasr Ibrim before this time was truly locally adapted to the site, then the signature of that adaptation is more subtle than the level of resolution our analyses have currently reached. This would be in keeping with expectations from the models that demonstrate that the number of loci that can be under selection is limited, and the effect of any one locus likely small. In such a model, we might not expect fixation of differences, and that different combinations of alleles may achieve a selectively similar outcome. Qasr Ibrim still has a good deal to teach us about local adaptation, and further unexpected turns may come to light.
4. Concluding remarks: archaeogenomes to systems
(a). Getting behind introgressions
Technology has progressed to a level that allows the evolution of crops to be studied at a truly genomic level, and the next step is undoubtedly the retrieval of the first complete plant genomes from the archaeological record. This will enable us better to gauge the accuracy of the predictions of models for how evolution and selection have proceeded in domesticated crops. Models give us a framework in which we expect, for the most part, that selection has been weak. A corollary of this prediction is that if we sequence ancient plant genomes that come from a time closer to the onset of entering into the human environment at the beginning of the domestication trajectory then we should expect to see stronger signals of selection at loci in which we see no signal in modern genomes. Furthermore, as plants evolve along the domestication trajectory over long periods of time, and move to new environments where new wild populations are encountered, there is ample opportunity for introgressions to occur which have large functional consequences on the domesticated crop. For instance, the majority of the known domestication syndrome associated genes of Indian rice have been acquired, through introgression, from Japanese rice that arrived in the Indian subcontinent [71–76]. In this way, the domesticated crop was able to use adaptations of the different wild races to local environments. While modern genomes are the palimpsest of these complex histories and difficult to interpret, aDNA approaches will be able to unravel the sequence of acquisitions through introgression by sequencing genomes before and after such events. These approaches will help identify the origins of various parts of the genome and consequently the environments to which they had been adapted prior to introgression, as well as establishing the likely order of trait acquisition through introgression.
(b). Low complexity adaptations in crops to complex environments?
Our models lead us to expect a large number of genes under weak selection rather than under one or two genes under strong selection, which appears to be the emergent picture from studies of adaptive evolution in wild plants [77–79]. They also lead us to expect that gene networks and metabolic processes would be affected at multiple points. Interestingly, such a pattern has recently been observed in the adaptation of dogs to a starchy diet in the human environment [80]. This indicates that an important frontier to broach in genomic-level analyses is to account for how selection acts on genes that are interdependent in networks of interaction—to move to a systems level of analysis. At a systems level, the majority of adaptation is expected to be achieved through the up- and downregulation of members of gene networks, effecting rapid and complex responses to environmental stimuli [81]. The DNA binding sites for transcription factors involved in regulation are very simple and consequently frequently form and disappear spontaneously with mutation, answering to some extent Haldane's original assertion in his contribution to the modern synthesis that the majority of mutations are expected to result in the loss of function owing to the expectations of entropy [82]. This elegant description of evolution implies the retention of function in genes during most adaptive change. However, these expectations are not met when we consider the adaptation of domesticated plants, such as wheat, barley and rice, to higher latitudes. Instead of adjusting the expression of genes in the networks of the floral pathway to attune to these new latitudes as we might expect to have occurred naturally, we see irreversible loss-of-function mutations in the associated gene networks (reviewed in reference [13]). It was this that, in many cases, helped plants to move with the human environment into northerly latitudes in which survival of the winter season would not have been assured.
These adaptations of domesticated cereals to latitude appear crude, one-way and simplistic relative to what we would expect of natural systems. In many cases, a single mutation achieves the phenotype rather than a number of mutations with each contributing a small effect. Furthermore, these genotypes probably rose in frequency relatively rapidly given the rapid pace of the spread of agriculture [11]. Could these be examples of selective sweeps that are, by corollary, rapid adaptations of low complexity? Under this scenario, it could have been the pace of agricultural movement across latitude that drove the intensity of selection, demanding rapid adaptation by plants in the human environment. Our models suggest that populations under strong selection will be vulnerable to further selection pressures that could cause population collapse, because the overall cost of selection would be too high. Intriguingly, the archaeological record indicates that such processes may have occurred—we see repeatedly that agriculture arrived at certain latitudes and collapsed not long after [83,84].
Ancient DNA and models have an important role in facilitating understanding of local adaptation in the future at a systems level. In the case of adaptation to latitude, models need to be applied to determine what the expectations are for the evolution of gene networks moving over latitude [85]. Does rapid movement across a selective gradient lead to the expectation of the retention of loss-of-function mutations that effectively break gene network interactions? Would a slower pace of movement been more likely to have led to a more refined adjustment response in networks? Archaeogenomics provides a reasonable approach to track the timing and order of the occurrence and subsequent selection of the mutations involved. In the case of studying the adaptation to latitude, aDNA technology will have to make further inroads in to using DNA from charred material. The resulting insights into how evolution works will be of relevance to understanding how plants adapted to the complex and dynamic human environment in the past, and how they might do so in the future in an ever changing world.
Supplementary Material
Funding statement
This work was supported by NERC (NE/F000391/1, NE/G005974/1), BBSRC (BB/G0177941) and Leverhulme Trust (F/00 215/BC).
References
- 1.Harlan J, de Wet JMJ, Price EG. 1973. Comparative evolution of cereals. Evolution 27, 311–325. ( 10.2307/2406971) [DOI] [PubMed] [Google Scholar]
- 2.Hammer K. 1984. The domestication syndrome. Kulturpflanze 32, 11–34. ( 10.1007/BF02098682) [DOI] [Google Scholar]
- 3.Fuller DQ. 2007. Contrasting patterns in crop domestication and domestication rates: recent archaeobotanical insights from the Old World. Ann. Bot. 100, 903–924. ( 10.1093/aob/mcm048) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spahillari M, Hammer K, Gladis T, Diederichsen A. 1999. Weeds as part of agrobiodiversity. Outlook Agric. 28, 227–232. [Google Scholar]
- 5.Senda T, Hiraoka Y, Tominaga T. 2006. Inheritance of seed shattering in Lolium temulentum and L. persicum hybrids. Genet. Resour. Crop Evol. 53, 449–451. ( 10.1007/s10722-005-6096-6) [DOI] [Google Scholar]
- 6.Howard T, Archer JE, Turley RM. 2011. Evolution, physiology and phytochemistry of the psychotoxic arable mimic weed darnel (Lolium temulentum L.). Prog. Bot. 72, 73–104. [Google Scholar]
- 7.Fuller DQ, Willcox G, Allaby RG. 2011. Cultivation and domestication had multiple origins: arguments against the core area hypothesis for the origins of agriculture in the Near East. World Archaeol. 43, 628–652. ( 10.1080/00438243.2011.624747) [DOI] [Google Scholar]
- 8.Conolly J, Colledge S, Shennan S. 2008. Founder effect, drift, and adaptive change in domestic crop use in early Neolithic Europe. J. Archaeol. Sci. 35, 2797–2804. ( 10.1016/j.jas.2008.05.006) [DOI] [Google Scholar]
- 9.Fuller DQ, Allaby RG, Stevens C. 2010. Domestication as innovation: the entanglement of techniques, technology and chance in the domestication of cereal crops. World Archaeol. 42, 13–28. ( 10.1080/00438240903429680) [DOI] [Google Scholar]
- 10.Banks W, Antunes N, Rigaud S, d'Errico F. 2013. Ecological constraints on the first prehistoric farmers in Europe. J. Archaeol. Sci. 40, 2746–2753. ( 10.1016/j.jas.2013.02.013) [DOI] [Google Scholar]
- 11.Colledge S, Conolly J, Shennan S. 2005. The evolution of early Neolithic farming from SW Asian origins to NW European limits. Eur. J. Archaeol. 8, 137–156. ( 10.1177/1461957105066937) [DOI] [Google Scholar]
- 12.Coward F, Shennan S, Colledge S, Conolly J, Collard M. 2008. The spread of Neolithic plant economies from the Near East to northwest Europe: a phylogenetic analysis. J. Archaeol. Sci. 35, 42–56. ( 10.1016/j.jas.2007.02.022) [DOI] [Google Scholar]
- 13.Fuller DQ, Allaby RG. 2009. Seed dispersal and crop domestication: shattering, germination and seasonality in evolution under cultivation in fruit development and seed dispersal . Annu. Plant Rev. 38, 238–295. [Google Scholar]
- 14.Jones H, et al. 2008. Population-based resequencing reveals that the flowering time adaptation of cultivated barley originated east of the fertile crescent. Mol. Biol. Evol. 25, 2211–2219. ( 10.1093/molbev/msn167) [DOI] [PubMed] [Google Scholar]
- 15.Jones G, et al. 2012. Phylogeographic analysis of barley DNA as evidence for the spread of Neolithic agriculture through Europe. J. Archaeol. Sci. 39, 3230–3238. ( 10.1016/j.jas.2012.05.014) [DOI] [Google Scholar]
- 16.Küster H. 2000. Rye. In Cambridge world history of food (eds Kiple KF, Ornelas KC.), pp. 149–152. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 17.Brown TA, Jones MK, Powell W, Allaby RG. 2009. The complex origins of domesticated crops. Trends Ecol. Evol. 24, 103–109. ( 10.1016/j.tree.2008.09.008) [DOI] [PubMed] [Google Scholar]
- 18.Brown TA. 1999. How ancient DNA may help in understanding the origin and spread of agriculture. Phil. Trans. R. Soc. Lond. B 354, 89–98. ( 10.1098/rstb.1999.0362) [DOI] [Google Scholar]
- 19.Palmer SA, Moore JD, Clapham AJ, Rose P, Allaby RG. 2009. Archaeogenetic evidence of ancient Nubian barley evolution from six to two-row indicates local adaptation. PLoS ONE 4, e6301 ( 10.1371/journal.pone.0006301) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allaby RG, O'Donoghue K, Sallares R, Jones MK, Brown TA. 1997. Evidence for the survival of ancient DNA in charred wheat seeds from European archaeological sites. Anc. Biomol. 1, 119–129. [Google Scholar]
- 21.Gurgeli F, Parducci L, Petit R. 2005. Ancient plant DNA: review and prospects. New Phytol. 166, 409–418. ( 10.1111/j.1469-8137.2005.01360.x) [DOI] [PubMed] [Google Scholar]
- 22.Palmer S, Smith O, Allaby RG. 2012. The blossoming of plant archaeogenetics. Ann. Anat. 194, 146–156. ( 10.1016/j.aanat.2011.03.012) [DOI] [PubMed] [Google Scholar]
- 23.Schlumbaum A, Tensen M, Jaenicke-Despres V. 2008. Ancient plant DNA in archaeobotany. Veg. Hist. Archaeobot. 17, 233–244. ( 10.1007/s00334-007-0125-7) [DOI] [Google Scholar]
- 24.Brown TA, Allaby RG, Sallares R, Jones G. 1998. Ancient DNA in charred wheats: taxonomic identification of mixed and single grains. Anc. Biomol. 2, 185–193. [Google Scholar]
- 25.Allaby RG, Banerjee M, Brown TA. 1999. Evolution of the high molecular weight glutenin loci of the A, B, D and G genomes of wheat. Genome 42, 296–307. ( 10.1139/g98-114) [DOI] [PubMed] [Google Scholar]
- 26.Schlumbaum A, Jacomet S, Neuhaus JM. 1998. Coexistence of tetraploid and hexaploid naked wheat in a Neolithic lake dwelling of Central Europe: evidence from morphology and ancient DNA. J. Archaeol. Sci. 25, 1111–1118. ( 10.1006/jasc.1998.0338) [DOI] [Google Scholar]
- 27.Goloubinoff P, Pääbo S, Wilson AC. 1993. Evolution of maize inferred from sequence diversity of an Adh2 gene segment from archaeological specimens. Proc. Natl Acad. Sci. USA 90, 1997–2001. ( 10.1073/pnas.90.5.1997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaenicke-Despres V, Buckler ES, Smith BD, Gilbert MTP, Cooper A, Doebley J, Paabo S. 2003. Early allelic selection in maize as revealed by ancient DNA. Science 302, 1206–1208. ( 10.1126/science.1089056) [DOI] [PubMed] [Google Scholar]
- 29.Allaby RG, Brown TA. 2003. AFLP analysis and the origins of agriculture. Genome 46, 448–453. ( 10.1139/g03-025) [DOI] [PubMed] [Google Scholar]
- 30.Allaby RG, Fuller DQ, Brown TA. 2008. The genetic expectations of a protracted model for the origins of domesticated crops. Proc. Natl Acad. Sci. USA 105, 13 982–13 986. ( 10.1073/pnas.0803780105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allaby RG, Brown TA, Fuller DQ. 2010. A simulation of the effect of inbreeding on crop domestication genetics with comments on the integration of archaeobotany and genetics: a reply to Honne and Heun. Veg. Hist. Archaeobot. 19, 151–158. ( 10.1007/s00334-009-0232-8) [DOI] [Google Scholar]
- 32.Allaby RG. 2010. Integrating the processes in the evolutionary system of domestication. J. Exp. Bot. 61, 935–944. ( 10.1093/jxb/erp382) [DOI] [PubMed] [Google Scholar]
- 33.Weiss E, Kislev ME, Hartmann A. 2006. Autonomous cultivation before domestication. Science 312, 1608–1610. ( 10.1126/science.1127235) [DOI] [PubMed] [Google Scholar]
- 34.Willcox G, Stordeur D. 2012. Large-scale cereal processing before domestication during the tenth millennium cal BC in northern Syria. Antiquity 86, 99–114. [Google Scholar]
- 35.Tanno KI, Willcox G. 2006. How fast was wild wheat domesticated? Science 311, 1886 ( 10.1126/science.1124635) [DOI] [PubMed] [Google Scholar]
- 36.Purugganan M, Fuller DQ. 2011. Archaeological data reveal slow rates of evolution during plant domestication. Evolution 65, 171–183. ( 10.1111/j.1558-5646.2010.01093.x) [DOI] [PubMed] [Google Scholar]
- 37.Hillman GC, Davies MS. 1990. Domestication rates in wild-type wheats and barley under primitive cultivation. Biol. J. Linn. Soc. 39, 39–78. ( 10.1111/j.1095-8312.1990.tb01611.x) [DOI] [Google Scholar]
- 38.Haldane JBS. 1957. The cost of selection. J. Genet. 55, 511–524. ( 10.1007/BF02984069) [DOI] [Google Scholar]
- 39.Allaby RG, Fuller DQ, Kitchen JL. 2014. The limits of selection under plant domestication. (http://arxiv.org/abs/1403.1244) [DOI] [PMC free article] [PubMed]
- 40.Chapman MA, Pashley CH, Wenzler J, Hvala J, Tang S, Knapp SJ, Burke JM. 2008. A genomic scan for selection reveals candidates for genes involved in the evolution of cultivated sunflower (Helianthus annuus). Plant Cell 20, 2931–2945. ( 10.1105/tpc.108.059808) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peleg Z, Fahima T, Korol AB, Abbo S, Saranga Y. 2011. The genetic basis of wheat domestication and evolution under domestication. J. Exp. Bot. 62, 5051–5061. ( 10.1093/jxb/err206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peng J, Ronin Y, Fahima T, Röder MS, Li Y, Nevo E, Korol A. 2003. Domestication quantitative loci in Triticum dicoccoides, the progenitor of wheat. Proc. Natl Acad. Sci. USA 100, 2489–2494. ( 10.1073/pnas.252763199) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wright S, Bi IV, Schroeder SG, Yamasaki M, Doebley JF, McMullen MD, Gaut B. 2005. The effects of artificial selection on the maize genome. Science 308, 1310–1314. ( 10.1126/science.1107891) [DOI] [PubMed] [Google Scholar]
- 44.Hawkins JS, Kim HR, Nason JD, Wing RA, Wendel JF. 2006. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 16, 1252–1261. ( 10.1101/gr.5282906) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hawkins JS, Proulx SR, Rapp RA, Wendel JF. 2009. Rapid DNA loss as a counterbalance to genome expansion through retrotransposon proliferation in plants. Proc. Natl Acad. Sci. USA 106, 17 811–17 816. ( 10.1073/pnas.0904339106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu G, Hawkins JS, Grover CE, Wendel JF. 2010. The history and disposition of transposable elements in polyploid Gossypium. Genome 53, 599–607. ( 10.1139/G10-038) [DOI] [PubMed] [Google Scholar]
- 47.Palmer SA, et al. 2012. Archaeogenomic evidence of punctuated genome evolution in Gossypium. Mol. Biol. Evol. 29, 2031–2038. ( 10.1093/molbev/mss070) [DOI] [PubMed] [Google Scholar]
- 48.Fryxell PA. 1979. The natural history of the cotton tribe. Austin, TX: Texas A&M University Press. [Google Scholar]
- 49.Senchina DS, et al. 2003. Rate variation among genes and the age of polyploidy in Gossypium. Mol. Biol. Evol. 20, 633–643. ( 10.1093/molbev/msg065) [DOI] [PubMed] [Google Scholar]
- 50.Oliver KR, Greene WK. 2009. Transposable elements: powerful facilitators of evolution. Bioessays 31, 703–714. ( 10.1002/bies.200800219) [DOI] [PubMed] [Google Scholar]
- 51.Oliver KR, Greene WK. 2012. Transposable elements and viruses as factors in adaptation and evolution: an expansion and strengthening of the TE-thrust hypothesis. Ecol. Evol. 2, 2912–2933. ( 10.1002/ece3.400) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clapham AJ, Rowley-Conwy PA. 2007. New discoveries at Qasr Ibrim, Lower Nubia. In Fields of change–progress in African archaeobotany, vol. 5(ed. Cappers R.), pp. 157–164. Groningen, The Netherlands: Groningen Archaeological Studies. [Google Scholar]
- 53.O'Donoghue K, Clapham A, Evershed R, Brown TA. 1996. Remarkable preservation of biomolecules in ancient radish seeds. Proc. R. Soc. Lond. B 263, 541–547. ( 10.1098/rspb.1996.0082) [DOI] [PubMed] [Google Scholar]
- 54.Komatsuda T, et al. 2007. Six-rowed barley originated from a mutation in a homeodomainleucine zipper I-class homeobox gene. Proc. Natl Acad. Sci. USA 104, 1424–1429. ( 10.1073/pnas.0608580104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zohary D, Hopf M, Weiss E. 2012. Domestication of plants in the Old World, 4th edn Oxford, UK: Oxford University Press. [Google Scholar]
- 56.Forster BP, et al. 2004. Genotype and phenotype associations with drought tolerance in barley tested in North Africa. Ann. Appl. Biol. 144, 157–168. ( 10.1111/j.1744-7348.2004.tb00329.x) [DOI] [Google Scholar]
- 57.Rollo F. 1985. Characterisation by molecular hybridization of RNA fragments isolated from ancient (1400 BC) seeds. Theor. Appl. Genet. 71, 330–333. [DOI] [PubMed] [Google Scholar]
- 58.Eigner J, Boedtker H, Michaels G. 1961. The thermal degradation of nucleic acids. Biochim. Biophys. Acta 51, 165–168. ( 10.1016/0006-3002(61)91028-9) [DOI] [PubMed] [Google Scholar]
- 59.Fordyce SL, et al. 2013. Deep sequencing of RNA from ancient maize kernels. PLoS ONE 8, e50961 ( 10.1371/journal.pone.0050961) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thompson JE, Kutateladze TG, Schuster MC, Venegas FD, Messmore JM, Raines RT. 1995. Limits to catalysis by ribonuclease A. Bioorg. Chem. 23, 471–481. ( 10.1006/bioo.1995.1033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fordyce SL, Kampmann ML, van Doom NL, Gilbert MTP. 2013. Long-term RNA persistence in post mortem contexts. Invest. Genet. 4, 7 ( 10.1186/2041-2223-4-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith O, Clapham A, Rose P, Liu Y, Wang J, Allaby RG. 2014. A complete ancient RNA genome: identification, reconstruction and evolutionary history of archaeological barley stripe mosaic virus. Sci. Rep. 4, 4003 ( 10.1038/srep04003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jónsson H, Ginolhac A, Schubert M, Johnson P, Orlando L. 2013. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684. ( 10.1093/bioinformatics/btt193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith O. 2013. Small RNA-mediated regulation, adaptation and stress response in the barley archaeogenome. PhD thesis University of Warwick, Warwick, UK. [Google Scholar]
- 65.Jackson AO, Lim H-S, Bragg J, Lee MY. 2009. Hordeivirus replication, movement and pathogenesis. Annu. Rev. Phytopathol. 47, 385–422. ( 10.1146/annurev-phyto-080508-081733) [DOI] [PubMed] [Google Scholar]
- 66.Jones R. 2009. Plant virus emergence and evolution: origins, new encounter scenarios, factors driving emergence, effects of changing world conditions, and prospects for control. Virus Res. 141, 113–130. ( 10.1016/j.virusres.2008.07.028) [DOI] [PubMed] [Google Scholar]
- 67.Anderson PK, Cunningham AA, Patel NG, Morales FJ, Epstein PR, Daszak P. 2004. Emerging infectious diseases of plants: pathogen pollution, climate change and agrotechnology drivers. Trends Ecol. Evol. 19, 535–544. ( 10.1016/j.tree.2004.07.021) [DOI] [PubMed] [Google Scholar]
- 68.Dowen RH, Pelizzola M, Schmitz RJ, Lister R, Dowen JM, Nery JR, Dixon JE, Ecker JR. 2012. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl Acad. Sci. USA 109, E2183–E2191. ( 10.1073/pnas.1209329109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith O, Clapham A, Rose P, Liu Y, Wang J, Allaby RG. 2014. Genomic methylation patterns in archaeological barley show biotic stress-associated siRNA activity and time-dependent demethylation as a diagenetic process. Sci. Rep. 4, 5559 ( 10.1038/srep05559) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bunning SL, Jones G, Brown TA. 2012. Next generation sequencing of DNA in 3300-year-old charred cereal grains. J. Archaeol. Sci. 39, 2780–2784. ( 10.1016/j.jas.2012.04.012) [DOI] [Google Scholar]
- 71.Fuller DQ, Sato YI, Castillo C, Qin L, Weisskopf AR, Kingwell-Banham EJ, Song JX, Ahn SM, van Etten J. 2010. Consilience of genetics and archaeobotany in the entangled history of rice. Archaeol. Anthropol. Sci. 2, 115–131. ( 10.1007/s12520-010-0035-y) [DOI] [Google Scholar]
- 72.Ishii T, et al. 2013. OsLG1 regulates a closed panicle trait in domesticated rice. Nat. Genet. 45, 462–465. ( 10.1038/ng.2567) [DOI] [PubMed] [Google Scholar]
- 73.Kovach MJ, Calingacion MN, Fitzgerald MA, McCouch S. 2009. The origin and evolution of fragrance in rice (Oryza sativa L.). Proc. Natl Acad. Sci. USA 106, 14 444–14 449. ( 10.1073/pnas.0904077106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shomura A, Izawa Ebana K, Ebitani T, Kanegae H, Konishi S, Yano M. 2008. Deletion in a gene associated with grain size increased yields during rice domestication. Nat. Genet. 40, 1023–1028. ( 10.1038/ng.169) [DOI] [PubMed] [Google Scholar]
- 75.Sweeney MT, Thompson MJ, Cho YG, Park YJ, Williamson SH, Bustamante CD, McCouch S. 2007. Global dissemination of a single mutation conferring white pericarp in rice. PLoS Genet. 3, 1418–1424. ( 10.1371/journal.pgen.0030133) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yamanaka S, Nakamura I, Watanabe KN, Sato Y-I. 2004. Identification of SNPs in the waxy gene among glutinous rice cultivars and their evolutionary significance during the domestication process of rice. Theor. Appl. Genet. 108, 1200–1204. ( 10.1007/s00122-003-1564-x) [DOI] [PubMed] [Google Scholar]
- 77.Flowers JM, Hanzawa Y, Hall MC, Moore RC. 2009. Population genomics of the Arabidopsis thaliana flowering time gene network. Mol. Biol. Evol. 26, 2475–2486. ( 10.1093/molbev/msp161) [DOI] [PubMed] [Google Scholar]
- 78.Keller SR, Levsen N, Ingvarsson PK, Olson MS, Tiff P. 2011. Local selection across a latitudinal gradient shapes nucleotide diversity in balsam poplar, Populus balsamifera L. Genetics 188, 941–952. ( 10.1534/genetics.111.128041) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hall D, Ma X-F, Ingvarsson PK. 2011. Adaptive evolution of the Populus tremula photoperiod pathway. Mol. Ecol. 20, 1463–1474. ( 10.1111/j.1365-294X.2011.05014.x) [DOI] [PubMed] [Google Scholar]
- 80.Axelsson E, et al. 2013. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature 495, 360–365. ( 10.1038/nature11837) [DOI] [PubMed] [Google Scholar]
- 81.Alon U. 2007. Introduction to systems biology. London, UK: Chapman & Hall. [Google Scholar]
- 82.Haldane JBS. 1932. The causes of evolution. London, UK: Longmans, Green & Co. [Google Scholar]
- 83.Shennan S, Downey SS, Timpson A, Edinborough K, Kerig T, Manning K, Thomas MG. 2013. Regional population collapse followed initial agricultural booms in mid-Holocene Europe. Nat. Commun. 4, 2486 ( 10.1038/ncomms3486) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stephens CJ, Fuller DQ. 2012. Did Neolithic farming fail? The case for a Bronze Age agricultural revolution in the British Isles. Antiquity 86, 707–722. [Google Scholar]
- 85.Kitchen JL, Allaby RG. 2013. Systems modeling at multiple levels of regulation: linking systems and genetic networks to spatially explicit plant populations. Plants 2, 16–49. ( 10.3390/plants2010016) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.