

Abstract
The challenge of sequencing ancient DNA has led to the development of specialized laboratory protocols that have focused on reducing contamination and maximizing the number of molecules that are extracted from ancient remains. Despite the fact that success in ancient DNA studies is typically obtained by screening many samples to identify a promising subset, ancient DNA protocols have not, in general, focused on reducing the time required to screen samples. We present an adaptation of a popular ancient library preparation method that makes screening more efficient. First, the DNA extract is treated using a protocol that causes characteristic ancient DNA damage to be restricted to the terminal nucleotides, while nearly eliminating it in the interior of the DNA molecules, allowing a single library to be used both to test for ancient DNA authenticity and to carry out population genetic analysis. Second, the DNA molecules are ligated to a unique pair of barcodes, which eliminates undetected cross-contamination from this step onwards. Third, the barcoded library molecules include incomplete adapters of short length that can increase the specificity of hybridization-based genomic target enrichment. The adapters are completed just before sequencing, so the same DNA library can be used in multiple experiments, and the sequences distinguished. We demonstrate this protocol on 60 ancient human samples.
Keywords: ancient DNA, authenticity, library preparation, barcodes, flexibility, target capture
1. Introduction
Technical advances have made it possible to extract and sequence DNA from ancient samples in a way that obtains enough molecules to permit whole genome analysis while minimizing artefacts owing to contamination and damage [1,2]. Despite the technological breakthroughs, there are practical hurdles that need to be overcome in any ancient DNA study. In particular, many samples often need to be laboriously screened in order to obtain a subset that is promising for analysis.
One approach to screening is to perform PCR for target regions on ancient DNA extracts, and to use the results to prioritize samples for constructing next-generation sequencing (NGS) libraries. Alternatively, one can use NGS libraries directly for screening. While library construction requires more initial effort than PCR, a library has the advantage that it amplifies DNA into a renewable resource that can be used not only for screening, but also for larger-scale experiments. Once DNA molecules are ‘immortalized’ in a library, many experiments can be performed beyond screening, such as shotgun sequencing and target enrichment.
Most laboratories that use NGS libraries for sample screening construct an initial ‘test library’ for each sample, and then shotgun sequence or enrich it for a small region of interest: a few loci of the genome [3,4], whole mitochondrial DNA (mtDNA) [5,6] or the plasmids of a targeted pathogen [7]. The resulting data can be evaluated for features expected from authentic ancient DNA, such as short molecule sizes, and a high rate of C → U changes that are concentrated at the ends of molecules, which manifests as a high rate of C → T or G → A mismatches to the reference genome [8–11]. Additional evidence about authenticity comes from rates of mismatch to the consensus sequence at known polymorphisms, for example at mtDNA, where enough coverage can be obtained to build a consensus [12]. For samples that produce plausibly authentic DNA, additional ‘production’ libraries can be built. These are often built in the presence of the enzymes uracil–DNA–glycosylase (UDG), which cleaves deaminated cytosines (uracils), and Endo VIII [13], which cuts at the resulting abasic sites, driving down the rate of ancient DNA errors. While we focus here on UDG-treated libraries, non-UDG-treated libraries can also be of value for analyses that are tuned to handle ancient DNA damage [14,15]. Moreover, non-UDG-treated libraries have a particular advantage for analysis of contaminated samples, where restriction to damaged molecules can greatly reduce contamination [16,17].
Here, we describe a protocol for ancient DNA screening that requires producing only a single library per sample. The library is UDG-treated, but preserves a damage signal at the terminal bases of the molecules, so that the authenticity of the DNA in the library can be assessed. This single-library-based screening saves not only costs, but also time, because the same sample does not have to be subjected to multiple rounds of processing and authenticity checks. For successful samples, our screening procedure produces a complete mitochondrial genome as well as an assessment of the promise of the library for larger-scale analyses.
Our library preparation protocol also includes additional time- and cost-saving features. In particular, it is optimized to allow high-throughput target enrichment of samples in 96-well plates. Enrichment is important, as for many ancient samples, a large fraction of the DNA that enters the library is not endogenous to the remains being analysed, so large amounts of sequence data sometimes needs to be examined to generate adequate data for analysis. Moreover, only a small subset of the genome is often of interest (e.g. mtDNA or a set of single nucleotide polymorphisms). To increase the fraction of sequenced molecules that align to subsets of the genome of interest, a standard tool in ancient DNA analysis has become target enrichment via hybridization capture [6,7,18–22], which in its in-solution form is amenable to being carried out on 96-well plates and processed robotically [23,24]. Parallel handling of many samples, however, increases the opportunity for cross-contamination owing to spillover of liquid from neighbouring wells. Thus, we add molecular barcodes (tags) to each DNA molecule in the ligation step of the library construction. We adopt the idea of short (incomplete) adapters from modern DNA libraries [24], in order to increase the specificity of target enrichment via hybridization. Once target enrichment is finished, the adapters are completed for sequencing by an indexing PCR. Use of different indices has the additional advantage that the same sample can be assayed in multiple experiments (e.g. hybrid capture of the mtDNA and shotgun sequencing), and the products can then be sequenced together and distinguished by the index.
2. Material and methods
(a). Clean room
All DNA extractions and library preparations up to the set-up of the amplification step were performed in a dedicated ancient DNA laboratory at Harvard Medical School, according to established precautions for working with ancient human DNA [25,26].
(b). Samples
Sixty human bone samples from the Samara District in Russia dating to 3000–9000 years BP [27] were used for this study. Relative dating of the samples was performed based on archaeological context. For a subset of samples, 14C dates (University of Arizona, Tucson, USA) were available.
(c). DNA extraction
Between 50 and 75 mg of bone powder was used to extract DNA according to Dabney et al. [28]. Final elution was performed twice in 16–30 μl 1× Tris EDTA (TE) buffer (with 0.05% Tween-20).
(d). Barcoded adapter design and preparation
All oligonucleotides (adapter, primer, blockers) were ordered from IDT (Coralville, USA; no PTO-bonds, standard desalted and lyophilized). We designed 100 barcodes of 7 bp length, with at least a 3 bp difference to all other barcodes (the sequences are in the electronic supplementary material, table S1). Partial double-stranded adapters were prepared by hybridizing the long oligonucleotide (with truncated Illumina-specific universal adapter sites) to the reverse complementary short oligonucleotide, both with 7mer barcodes at the ends. The 100 barcoded P5-adapters and 100 barcoded P7-adapters were prepared independently as described in [29].
(e). Library preparation: no uracil–DNA–glycosylase treatment (I)
The protocol is based on [29,30]. Between 15 and 30 μl of DNA extract was used in a 50 μl blunting reaction with a final concentration of 1× buffer Tango, 100 μM each dNTP, 1 mM ATP, 25 U T4 polynucleotide kinase and 5 U T4 DNA polymerase (all reagents from Thermo Scientific Fermentas Molecular Biology Solutions Waltham, MA). After incubation at 25°C for 15 min and 12°C for 5 min, the reaction was cleaned up with the MinElute PCR purification kit (Qiagen, Hilden, Germany) by adding 250 μl of buffer PB to each reaction and applying the mixture to the MinElute column and centrifuging. Two washing steps with buffer PE were performed and after a dry-spin, DNA was eluted in 18 μl 10 mM Tris–HCl.
Each DNA extract was assigned a unique barcode combination and within each batch no single barcode was used more than once. For each library, 1 μl of a barcoded, partially double-stranded P5-adapter (10 μM) and 1 μl of a barcoded, partially double-stranded P7-adapter (10 μM) were added to the blunted DNA and mixed before adding the ligation mix to bring adapters and DNA into close proximity (final concentration 0.25 μM for each barcoded adapter). The concentrations in the 40 μl final ligation reactions were as follows: 1× T4 DNA ligase buffer, 5% PEG-4000, 5 U T4 DNA ligase (all reagents from Thermo Scientific Fermentas Molecular Biology Solutions, Waltham, MA). After mixing and a quick spin, the reaction was incubated for 30 min at room temperature. MinElute clean-up was performed by adding 200 μl buffer PB to each finished ligation reaction, washing with PE buffer twice, and eluting in 20 μl 10 mM Tris–HCl.
The fill-in reaction was performed in a final volume of 40 μl with 1× ThermoPol buffer (New England Biolabs (NEB), Ipswich, MA), 250 μM each dNTP (Thermo Scientific Fermentas Molecular Biology Solutions) and 16 U Bst polymerase, large fragment (NEB) and incubated at 37°C for 20 min followed by a heat-inactivation at 80°C for 20 min [29,30]. The entire 40 μl heat-inactivated fill-in reaction was used in the PCR that finished the library preparation. A total of 400 μl PCR mix (divided into four to eight reactions) per sample was prepared with the following final concentration: 1× Pfu Turbo Cx reaction buffer, 20U Pfu Turbo Cx Hotstart DNA Polymerase (both Agilent Technologies, Santa Clara, CA), 200 μM each dNTP (Thermo Scientific Fermentas Molecular Biology Solutions) and 400 nM of each of the two primers (PreHyb-F, PreHyb-R, sequences in the electronic supplementary material, table S1) that do not extend the adapter sites but keep them truncated (short). After an initial denaturation and activation of the polymerase at 95°C for 2 min, 30 cycles at 95°C for 30 s, 55°C for 30 s and 72°C for 1 min, we performed a final extension at 72°C for 5 min. Following PCR, the product was cleaned up with the MinElute PCR purification kit by adding 2 ml buffer PB to the 400 μl PCR and distributing the mix onto two MinElute columns. After washing the silica matrix twice with PE buffer and a dry-spin, each column was eluted in 25 μl 1× TE (with 0.05% Tween-20), resulting in 50 μl final library.
(f). Library preparation: partial uracil–DNA–glycosylase treatment (II)
Between 15 and 30 μl DNA extract was used in 60 μl blunting reactions with an initial USER enzyme treatment (NEB). The first part of the reaction (the partial UDG treatment, 52.2 μl total) consisted of 6 μl 10× buffer Tango, 0.24 μl 25 mM dNTP mix, 0.6 μl 100 mM ATP (all reagents from Thermo Scientific Fermentas Molecular Biology Solutions), and 3.6 μl USER enzyme mix (1 U μl−1, NEB). During the 30 min incubation period at 37°C, most deaminated cytosines were excised by UDG and abasic sites were cut by Endo VIII. Next, 3.6 μl of UGI (2 U μl, UDG inhibitor, NEB) was added and incubated for 30 min at 37°C, after which 3 μl T4 polynucleotide kinase and 1.2 μl T4 DNA polymerase were added, and the final 60 μl was incubated for 15 min at 25°C, followed by 5 min at 12°C. The clean-up of this blunting reaction with partial USER treatment was performed by adding 300 μl buffer PB, with all following steps performed as in §2e. A detailed working protocol is included in the electronic supplementary material.
(g). Library preparation: full uracil–DNA–glycosylase treatment (III)
Between 15 and 20 μl DNA extract was used in a 50 μl blunting reaction with simultaneous USER enzyme treatment. The final concentrations were as follows: 1× buffer Tango, 100 μM each dNTP, 1 mM ATP, 25 U T4 polynucleotide kinase (all reagents from Thermo Scientific Fermentas Molecular Biology Solutions) and 3U USER enzyme (NEB). An incubation was performed for 3 h at 37°C, followed by the addition of 1 μl T4 DNA polymerase (Thermo Scientific Fermentas Molecular Biology Solutions) and incubation at 25°C for 15 min and 12°C for 5 min. The subsequent steps were performed as described in §2e until after the heat-inactivation of the fill-in step. Then, the entire reaction from the fill-in step was amplified with AccuPrime Pfx DNA polymerase in a total of 400 μl (divided into four to eight reactions). The final concentration of the reaction consists of 1× AccuPrime Pfx reaction buffer, 10 U AccuPrime Pfx DNA polymerase and 400 nM of primer PreHyb-F and PreHyb-R. An initial denaturation and enzyme activation was performed at 95°C for 2 min, 30 cycles at 95°C for 15 s, 55°C for 30 s and 68°C for 1 min, followed by a final extension of 68°C for 5 min. Clean-up was performed the same way as for the other two protocols, resulting in 50 μl of library.
(h). Libraries for barcoded adapter tests
The influence of the barcodes on library characteristics was tested by ligating four different P5-barcoded adapters in all possible combinations with four P7-barcoded adapters to the same DNA extract. These 16 ligation reactions started from a large volume of cleaned-up and pooled blunting reaction products (partial UDG treatment). Barcoded adapter IDs 1–4 were chosen for the P5-site and IDs 97–100 for the P7-site (electronic supplementary material, table S1), so that all possible combinations of terminal nucleotides on both sides were used. Libraries were amplified with the regular PreHyb-primer set with Pfu Turbo Cx Hotstart DNA polymerase.
(i). Libraries for adapter length in hybridization test
Three different libraries were used: sample A was prepared without barcodes using universal Illumina adapter (IS1_adapter.P5 and IS3_adapter.P5 + P7; IS2_adapter.P7 and IS3_adapter.P5 + P7; PTO-bonds and high-performance liquid chromatography purified, as in [29], see the electronic supplementary material, table S1) and two barcoded libraries from the barcode adapter test were used that started from about 2.5 times less material (sample B: P5 no. 1, P7 no. 97; sample C: P5 no. 3, P7 no. 100). The amplification that finished the library construction was performed with the PreHyb-primer pair, and the adapter sites were left unfinished (‘short’). To test for the influence of different adapter lengths on hybridization specificity, the short libraries were further amplified with one indexing primer (i7 index primer and PreHyb-F) or two indexing primers (i5 and i7 indexing primers), resulting in ‘intermediate’ and ‘long’ adapter lengths. For library A, all three adapter lengths were prepared, whereas for libraries B and C, only ‘short’ and ‘long’ were prepared. Amplifications were performed with 1 μl Herculase II Fusion DNA Polymerase (Agilent Technologies) in 50 μl reactions consisting of 1× Herculase II reaction buffer, 400 nM each primer and 250 μM each dNTP using 1 μl of a 20-fold dilution of the ‘short’ library for 20 cycles (95°C for 2 min, 20 cycles at 95°C for 30 s, 58°C for 30 s, 72°C for 30 s, final extension 72°C for 10 min). MinElute clean-up was performed by adding 250 μl PB, followed by two PE-washes, and DNA was eluted in 15 μl 1× TE (with 0.05% Tween-20).
All libraries were evaluated on the BioAnalyzer and Nanodrop to assess library preparation success and to measure concentrations, allowing us to adjust the volume for the capture reaction later. Note that the real size distribution of the libraries is usually not retained after 30 PCR cycles, because the PCR was run into plateau and heteroduplices form that make insert sizes appear longer in gel electrophoresis.
(j). Target capture
Hybridization enrichment was performed as described elsewhere [19] with baits targeting the human mitochondrial genome (3 bp tiling based on NC_001807 as in [17]) using a semi-automated protocol in a 96-well plate set-up on an Evolution P3 (Perkin Elmer, Waltham, MA) for two consecutive rounds. This liquid handler has a 96-well tip head and is therefore suited for steps involving magnetic beads (i.e. from the streptavidin bead binding onwards and for the solid-phase reversible immobilization clean-ups of amplifications) in 96-well plates. This reduces hands-on time and is less expensive, because fewer pipette tips are used when compared with manual multi-channel pipetting. For a subset of the experiments, baits for 10–50 nuclear loci were spiked into the bait pool. The enrichment for mitochondrial sequences was not substantially affected by this addition (data not shown). For each hybridization reaction, we used 500 ng of single-stranded bait library together with 500 ng DNA library. The oligonucleotide blockers used for each of the respective adapter lengths are specified in the electronic supplementary material, table S1. All other parameters of the hybridization, capture and washing steps and amplifications can be found in the original paper [19]. Each sample was subsequently indexed prior to sequencing with a unique index (pair), using 7-mer index sequences as in Meyer & Kircher [29].
(k). Sequencing and analysis for the samples analysed in this study
We pooled indexed libraries (shotgun and mtDNA-captured with different index combinations) with several other libraries and sequenced them on a MiSeq instrument using v2 or v3 chemistry with the standard sequencing primers provided in the cartridges. Depending on the sequencing kit, we sequenced 2 × 75 or 2 × 150 cycles with the standard Nextera sequencing protocol (or TruSeq HT protocol) that reads both indices.
We used the automatic de-multiplexing provided by Illumina BaseSpace (allowing up to one mismatch per index). We then trimmed adapters and merged R1 and R2 sequences, requiring an overlap of at least 15 bp (allowing one mismatch) using SeqPrep [31] (modified to require more conservatively that quality scores in the merged regions use the best score rather than aggregating the two inferred reliabilities of the base call), prior to trimming the barcodes (if applicable) from both ends of the merged molecules. For each sample, we restricted to reads that had the expected indices. We then only analysed sequences that matched the expected 7mer barcodes, allowing up to two mismatches. Using bwa 0.6.1–r104 [32] we performed alignments twice: (i) to the human reference genome (hg19) which contains the rCRS mitochondrial genome, and to (ii) the reconstructed sapiens reference sequence mitochondrial sequence [33]. We computed target coverage with BEDtools v. 2.16.2 [34] and used MapDamage v. 2.0 [35] to compute damage rates and fragmentation patterns. We report the median length of all unique sequences aligned to the mitochondrial genome for samples for which we prepared multiple libraries with different protocols.
We estimated mtDNA contamination using a Markov chain Monte Carlo-based estimator (supplementary information 5 in [12]). More precisely, we built a consensus sequence using a minimum base quality of 30 and a minimum coverage of 5, stripping gaps and ignoring any spurious heterozygote positions. We realigned all reads to this consensus, and then used the resulting alignments for estimation of contamination (supplementary information in [12]), trimming the first and last five bases from every read to minimize errors owing to ancient DNA degradation.
3. Results
(a). Library construction
We built Illumina libraries following the protocol of Meyer & Kircher [29] with modifications as in Kircher et al. [30]. However, instead of ligating universal Illumina adapters to all samples and relying on indexing to differentiate samples, we ligated a unique combination of barcoded Illumina adapters directly to the DNA molecules [24,36,37] (for a schematic overview, see figure 1). The seven base barcodes were read as the beginning of sequence read1 and read2, respectively, and we removed them before alignment. Another option is to mask the barcodes during alignment, which can be done with bwa's alignment function B for paired end alignments [32]. On average, 98% of the reads per fastq file (demultiplexed) had the expected barcode combination for the 60 samples we examined on two different experiments each (mtDNA and shotgun, electronic supplementary material, table S3). These results show the value of the barcodes; they result in marginal loss of reads, while providing an extra layer of security in sample identification, even when several experiments of identical libraries are pooled for sequencing.
Figure 1.
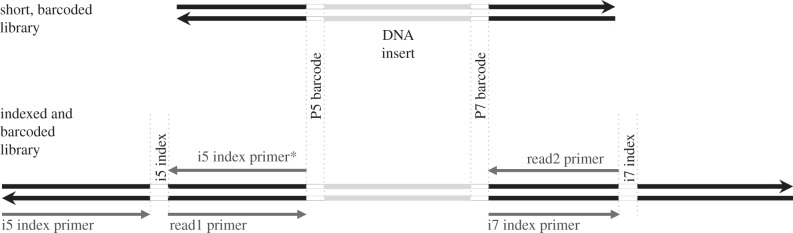
Schematic overview of our library architecture. After library preparation is finished with a PCR using the PreHyb primer pair, short, barcoded libraries can be used for, e.g. target enrichment via hybridization. Adapter sites of such short libraries need to be completed via indexing PCR before sequencing. If different experiments from the same libraries, such as mitochondrial target enrichment and shotgun sequencing, are pooled for sequencing, sequences from those experiments can be differentiated by the index sequences. The four regular Illumina sequencing primer sites for MiSeq, HiSeq and NextSeq instruments are shown, only the i5 index primer* is different for the NextSeq500.
A drawback of this strategy is that the Illumina sequencing software relies on the presence of a diverse mix of nucleotides in the first five cycles, as would be expected to occur in randomly fragmented DNA libraries. In the extreme case of a single barcoded library, all the nucleotides in the barcode positions are the same. This problem can be overcome by sequencing many screening libraries with diverse barcodes together. Another option is to spike in a PhiX control library (which wastes sequencing capacity), or to spike in a non-barcoded diverse library. A third option is to construct each library with a mixture of barcoded adapters that are balanced with respect to their representation of each nucleotide in the seven bases, which is the preferred option when a small subset of libraries needs to be sequenced to high coverage after the screening stage.
For two test samples, we constructed three different libraries: (I) without UDG treatment, (II) with our ‘new’ partial UDG treatment and (III) with regular UDG treatment. The protocols differ in that we use no USER enzyme for I, whereas we add USER (NEB) in the blunting step of both II and III. Protocol II differs from protocol III in that T4 DNA polymerase and T4 polynucleotide kinase are only added after USER treatment is completed and UDG is blocked by UDG inhibitor. This has the effect that some terminal deaminated cytosines of the ancient molecules are not efficiently removed, and means that ancient DNA damage is expected to persist at these positions [38,39]. The protocols also differ in the polymerases used for the final amplification of the library (for I and II, we use a polymerase that can read over uracil, whereas for III, several different polymerases can be used [40]).
For a set of six additional extracts, we prepared both non-UDG-treated (I) and partial UDG-treated (II) libraries to evaluate differences in the damage pattern between the protocols. On an even larger set of extracts (52), we prepared only partially UDG-treated libraries.
(b). Effectiveness of partial uracil–DNA–glycosylase treatment
Figure 2 shows the damage profile in the 5′-terminal 25 bases of two samples (we average the rates for all possible substitutions over all sequences and both ends, reverse complementing the 3′ end; electronic supplementary material, table S2). The untreated library (I) shows the familiar damage profile expected for ancient DNA: a high C → T rate of discrepancy compared with the reference at the 5′ ends of molecules that decreases towards the centre. The partial UDG-treatment (II) successfully removes uracils within the ancient molecules just like full UDG-treatment (III), but retains a substantial fraction of the terminal uracil substitutions. This experiment confirms that the partially UDG-treated libraries preserve a signal of damage. However, the terminal base damage rate is lower for the partial UDG-treatment (II) than for libraries without UDG-treatment (I). A possible explanation is that a large proportion of terminal uracils are phosphorylated and therefore removed by UDG [39], although we have not verified this.
Figure 2.

Damage profile of the terminal 25 nucleotides of two samples ((a) I–III) and ((b) I–III) treated in three different ways during library preparation: I, no UDG treatment; II, partial UDG treatment; III, full UDG treatment. The frequencies of all possible substitutions are plotted as a function of the distance from the 5′ end (average of the 5′ end and reverse complement of the 3′ end; between 1 and 25 bp on the x-axis) for all unique endogenous reads. Predominant substitution patterns (C → T and G → A) are highlighted. (Online version in colour.)
To test if the reduced damage in the terminal bases from partial UDG-treatment is useful as an assessment of authenticity, we compared the damage rates observed in extracts of the same samples that were made into both non-UDG-treated libraries (I) and partially UDG-treated libraries (II). On average, the reduction in both terminal bases is about threefold (from 27% in the non-UDG-treated library to 9% in the partially UDG-treated library), whereas the reduction is about 26-fold for the second bases of the molecules (table 1 and figure 3a). For non-UDG-treated libraries, Sawyer et al. [41] found that samples older than 500 years have a damage rate of at least 10% and suggested this as a threshold to call a library plausibly authentic. Taking into account the approximately threefold reduction of substitutions seen in partially UDG-treated libraries, we propose using a damage rate of 3% or higher. Although uracils are excised and DNA strands cleaved at abasic sites (and therefore miscoding substitutions mostly removed from the DNA molecules), the UDG and Endo VIII treatment of ancient DNA leaves a trace in the alignment one nucleotide upstream and downstream of the analysed DNA molecules when compared with the average base composition in the genome. Specifically, we find an elevated rate of cytosines in the site preceding the 5′ end of the alignment and guanines following the 3′ end (electronic supplementary material, figure S1, II). This is consistent with the expectation for UDG-treated libraries, in which uracils (deaminated cytosines) are removed and the strands are cut by the USER enzyme mixture [13] (electronic supplementary material, figure S1, III). Finally, we observe that the median fragment lengths are reduced for libraries prepared with partially UDG-treatment compared with non-UDG-treated libraries. The difference is 5–17 bp over eight samples (table 1).
Table 1.
Damage in the terminal and penultimate bases (5′ C → T, and the reverse complement 3′ G → A) and median fragment length of libraries from eight extracts (a–h) prepared with non-UDG-treatment (I) and partial UDG-treatment (II). (Damage rates are assessed in all reads as frequencies of C to T substitutions at 5′ ends and G to A substitutions at 3′ ends using MapDamage.)
| sample ID | UDG treatment | reads aligning to mt genome | damage rate in terminal position (%) | damage rate in penultimate position (%) | ratio: terminal : penultimate | median length (bp) |
|---|---|---|---|---|---|---|
| a | I | 15 010 | 32.74 | 24.19 | 1.4 | 75 |
| II | 61 630 | 10.92 | 0.46 | 19.2 | 59 | |
| b | I | 8054 | 27.71 | 20.22 | 1.4 | 74 |
| II | 81 543 | 9.30 | 0.23 | 40.7 | 67 | |
| c | I | 57 550 | 38.50 | 33.80 | 1.1 | 69 |
| II | 86 461 | 6.49 | 0.32 | 20.6 | 52 | |
| d | I | 20 042 | 26.52 | 18.90 | 1.4 | 70 |
| II | 68 761 | 12.03 | 0.96 | 12.5 | 65 | |
| e | I | 41 624 | 29.28 | 22.11 | 1.3 | 78 |
| II | 5238 | 10.07 | 0.66 | 15.3 | 61 | |
| f | I | 30 569 | 27.37 | 20.46 | 1.3 | 74 |
| II | 123 598 | 11.39 | 0.44 | 25.7 | 69 | |
| g | I | 45 264 | 28.05 | 20.34 | 1.4 | 76 |
| II | 104 215 | 9.21 | 0.47 | 19.6 | 66 | |
| h | I | 27 549 | 32.44 | 25.43 | 1.3 | 59 |
| II | 125 596 | 7.39 | 0.85 | 8.7 | 53 |
Figure 3.

Partial UDG treatment (a) damage pattern in terminal bases (circles) and penultimate bases (crosses) for eight samples when libraries from the same extract were prepared without UDG-treatment (x-axis) and with partial UDG treatment (y-axis); average is shown in filled markers with 1 s.e. bars. The median reduction is three times for terminal bases and 26× for penultimate bases. (b) Histogram of terminal damage rate of 51 libraries prepared with the partial UDG treatment with average mitochondrial coverage >2.0×. (c) Terminal damage rate as a function of contamination estimate using reads aligning to the mitochondrial reference for all libraries prepared with the partial UDG-treatment protocol with coverage of >10× on the mitochondrial genome. (d) Terminal damage rates as a function of sample age for all partially UDG-treated libraries. Samples of similar age were grouped together and bars show 1 s.e.; triangles show results for individual samples with direct 14C dates.
We examined the damage pattern from a larger number of samples with partially UDG-treated libraries. We restricted to 51 samples that yielded at least a two times covered mitochondrial genome after the initial mtDNA screening. All samples show on average at least 4% of terminal cytosines being damaged (electronic supplementary material, table S3 and figure 3b) and most samples show around 8%. The minimum of 4% supports our suggested threshold of 3%. We caution, however, that all the samples in this study are from one temperate region (the Samara Valley region of Russia) and come from a threefold range of ages (3000–9000 years ago). Further studies may reveal that a modified threshold for declaring a sample to be authentic will be preferable to greater than or equal to 3%.
We correlated the damage rate with the estimated contamination rate for samples with mtDNA coverage greater than or equal to 10× after duplication removal (figure 3c). We observe no significant correlation, which is unsurprising, because the two tests are interrogating different contamination scenarios. The ancient DNA damage signal is sensitive to large fractions of contamination; if there is only a modest amount, we still expect to detect substantial ancient damage. By contrast, the assessment of contamination requires a consensus, and this works well only if the great majority of molecules are from one individual.
We also tested for a correlation between the damage rate and the age of the samples. All samples are from a threefold date range, which limits the value of this test. Figure 3d shows no correlation, either for direct or relative dates. For non-UDG-treated libraries, Sawyer et al. [41] and Allentoft et al. [42] explored a wider range of dates and found a correlation.
(c). Barcode combination test
A potential concern with our barcoded library construction is that different pairs of barcodes may be differently efficient at ligating to ancient molecules. To test this, we prepared 16 libraries from one large volume of blunt-end repaired partial UDG-treated extract by using four different barcoded adapters with the P5-sequence and four different barcoded adapters with the P7-sequence in all possible combinations. Every barcode in each set of four had a different nucleotide at the terminal base of the barcode, which is the base that gets attached to the DNA molecule and that we hypothesize is the most influential base of the barcode during ligation. We enriched the libraries for the mitochondrial genome with two rounds of hybridization, and tested for differences in the characteristics of these 16 libraries.
Table 2 shows that the percentage of reads mapping to the human reference genome (hg19) in the unenriched shotgun data varies between 2.6% and 3.2% with an average of 2.9%. After two rounds of enrichment for the mitochondrial genome and randomly downsampling to the number of reads for the library with the lowest number of reads, we obtain very similar on-target rates (67%–72%) for all 16 experiments. After we remove duplicates, the variation becomes larger (81× versus 44×). We have not been able to determine whether the differences in complexity of these 16 libraries originates from different ligation efficiencies of the different barcoded adapters, or simply reflects variability of the multi-step process, which includes a reaction clean-up and therefore potential loss of molecules. The mean insert sizes vary to some degree (62 ± 3 bp), and the damage rate of the terminal cytosine also varies (between 7% and 10%). The results for one P7 barcode (CTAGGTG) are the largest outlier, as libraries with this barcode have the lowest damage rate and the lowest uniqueness rate and therefore lowest mitochondrial coverage. This barcode has a terminal G, and we speculate that the double-stranded G–C terminal base pair of the barcode may ligate with reduced efficiency to the damaged cytosines at the 5′-ends that T4 DNA polymerase has filled in with the complementary A (U–A base pair). However, other mechanism(s) must also be influencing these results, because the P5-barcode with a terminal G does not show the pattern to the same extent.
Table 2.
Barcode combination experiment. (Basic results before and after enrichment for mtDNA of 4 × 4 libraries with all possible barcode combinations. For the assessment after enrichment, we downsampled all reads to 125 406, to facilitate comparability across samples. Italicized numbers represent the number of reads that aligned to the mitochondrial (mt) genome.)
| before enrichment |
after enrichment |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P5 barcode | P7 barcode | all reads | reads aligning to hg19 (mt) | % endogenous (%) | all reads | reads aliging to mt genome | % of reads aligning to mt genome (%) | unique reads aligning to mt genome | median length in bp | damage rate in terminal bases (%) | % of unique reads aligning to mt genome (%) | duplication rate (%) | average mt coverage |
| ATCGATT | GACTTAT | 44 884 | 1256 (3) | 2.80 | 125 406 | 90 118 | 71.9 | 17 455 | 62 | 8.72 | 13.9 | 80.6 | 71.6 |
| ATCGATT | TCGAACA | 45 866 | 1287 (8) | 2.81 | 125 406 | 88 429 | 70.5 | 17 395 | 65 | 9.37 | 13.9 | 80.3 | 73.2 |
| ATCGATT | AGTCCGC | 35 813 | 935 (2) | 2.61 | 125 406 | 88 335 | 70.4 | 18 253 | 64 | 9.70 | 14.6 | 79.3 | 76.6 |
| ATCGATT | CTAGGTG | 29 091 | 870 (4) | 2.99 | 125 406 | 86 795 | 69.2 | 13 970 | 59 | 8.05 | 11.1 | 83.9 | 55.3 |
| CAGTCAA | GACTTAT | 34 104 | 888 (1) | 2.60 | 125 406 | 90 026 | 71.8 | 19 122 | 65 | 9.46 | 15.2 | 78.8 | 81.2 |
| CAGTCAA | TCGAACA | 38 693 | 1060 (3) | 2.74 | 125 406 | 89 294 | 71.2 | 15 242 | 64 | 8.14 | 12.2 | 82.9 | 62.8 |
| CAGTCAA | AGTCCGC | 37 276 | 996 (5) | 2.67 | 125 406 | 88 251 | 70.4 | 18 240 | 64 | 9.17 | 14.5 | 79.3 | 77.0 |
| CAGTCAA | CTAGGTG | 29 494 | 952 (2) | 3.23 | 125 406 | 85 767 | 68.4 | 11 060 | 60 | 6.98 | 8.8 | 87.1 | 43.5 |
| GCTAGCC | GACTTAT | 24 227 | 707 (2) | 2.92 | 125 406 | 86 112 | 68.7 | 16 948 | 63 | 8.90 | 13.5 | 80.3 | 71.3 |
| GCTAGCC | TCGAACA | 40 675 | 1157 (1) | 2.84 | 125 406 | 83 702 | 66.7 | 14 922 | 59 | 9.63 | 11.9 | 82.2 | 59.4 |
| GCTAGCC | AGTCCGC | 38 108 | 1191 (3) | 3.13 | 125 406 | 85 471 | 68.2 | 16 518 | 61 | 8.39 | 13.2 | 80.7 | 67.6 |
| GCTAGCC | CTAGGTG | 33 780 | 1092 (3) | 3.23 | 125 406 | 89 368 | 71.3 | 13 421 | 63 | 7.72 | 10.7 | 85.0 | 54.0 |
| TGACTGG | GACTTAT | 54 050 | 1555 (12) | 2.88 | 125 406 | 89 679 | 71.5 | 18 298 | 65 | 9.56 | 14.6 | 79.6 | 77.9 |
| TGACTGG | TCGAACA | 27 733 | 790 (2) | 2.85 | 125 406 | 87 788 | 70.0 | 16 149 | 62 | 8.52 | 12.9 | 81.6 | 66.1 |
| TGACTGG | AGTCCGC | 35 675 | 1097 (4) | 3.07 | 125 406 | 86 835 | 69.2 | 18 303 | 63 | 9.23 | 14.6 | 78.9 | 76.9 |
| TGACTGG | CTAGGTG | 36 744 | 1102 (2) | 3.00 | 125 406 | 88 364 | 70.5 | 13 550 | 59 | 7.15 | 10.8 | 84.7 | 53.4 |
In conclusion, the results for the 16 barcode combination experiment show some variability that might, in part, result from different ligation efficiencies of the barcodes, especially to damaged molecules. Nevertheless, the observed maximal twofold difference in complexity and differences in damage rates and insert size may be acceptable given the other advantages of this library preparation procedure.
(d). Effect of adapter length on specificity of hybridization enrichment
To explore the influence of adapter length on target enrichment efficiency, we prepared three independent partial UDG-treated libraries from the same extract using different input amounts. We prepared one library (A) using universal adapters (without barcodes) and two (B and C) with barcodes. We finished library preparation with an initial amplification with primers not extending the adapter sites, resulting in ‘short’ libraries (A.short, B.short, C.short). After cleaning with the MinElute PCR purification kit, we amplified these three short libraries with indexing primers that encode an index sequence in the oligonucleotide sequence of the primer and extend the short adapters to full-length adapter sites. We performed three different PCR's for library A.short: A.intermediate with a i7-index primer and the universal PreHyb-F primer resulting in a completed P7-site and short P5-site, and A.before and A.long, each with a unique i5 and i7 index combination, resulting in both sites completed for sequencing (long). The A.before and A.long libraries are technically identical libraries with different index combination in order to be able to sequence the unenriched library (A.before) together with the enriched library (A.long, after two rounds of enrichment) in the same sequencing run. For the barcoded libraries B and C, we performed two indexing PCRs each, with unique combinations of index primers resulting in B.before, B.long, C.before and C.long.
For all three libraries (A–C), the ‘short’ category is associated with a higher endogenous percentage (as measured by reads mapping to the reference genome) for all sequenced reads and also for the unique reads (table 3). For the experiment with intermediate adapter length (A.intermediate), the proportion of reads mapping to the reference lies between that for the short and long adapters, indicating that libraries with the longest adapters are captured least efficiently. The average coverage after duplication removal is highest for the ‘short’ adapters and lowest for the ‘long’ adapters, and does not differ much within each library group, probably reflecting the fact that the complexity of these three libraries is limited and all these libraries were sequenced to saturation.
Table 3.
Influence of adapter length on mitochondrial target enrichment specificity. (Three different libraries (A–C) were prepared and subjected to target enrichment for mtDNA with different adapter lengths. Basic results before and after enrichment (reads were downsampled to have the identical number of reads examined) show higher target enrichment specificity of libraries with short adapters with the settings used. Italics indicate that no significance can be given for these reports of fractions of the reads.)
| library | barcodes (P5–P7) | adapter length during capture | indices during capture | all reads | aligned reads to mt | % endogenous | unique aligned reads to mt | unique % endogenous | average mt coverage | coefficient of variation |
|---|---|---|---|---|---|---|---|---|---|---|
| A.before | none | — | — | 37 344 | 3 | 0.008 | 3 | 0.008 | — | — |
| A.short | none | short | none | 331 224 | 296 075 | 89.4 | 28 344 | 8.6 | 139 | 0.12 |
| A.intermediate | none | intermediate | i7 | 331 224 | 240 086 | 72.5 | 27 256 | 8.2 | 130 | 0.15 |
| A.long | none | long | i5 + i7 | 331 224 | 198 777 | 60.0 | 26 946 | 8.1 | 129 | 0.15 |
| B.before | ATCGATT-GACTTAT | — | — | 34 104 | 1 | 0.002 | 1 | 0.002 | — | — |
| B.short | ATCGATT-GACTTAT | short | none | 189 334 | 136 140 | 71.9 | 20 540 | 10.8 | 87 | 0.16 |
| B.long | ATCGATT-GACTTAT | long | i5 + i7 | 189 334 | 112 560 | 59.5 | 20 313 | 10.7 | 84 | 0.19 |
| C.before | GCTAGCC-CTAGGTG | — | — | 33 780 | 2 | 0.006 | 2 | 0.006 | — | — |
| C.short | GCTAGCC-CTAGGTG | short | none | 125 406 | 89 368 | 71.3 | 13 421 | 10.7 | 54 | 0.18 |
| C.long | GCTAGCC-CTAGGTG | long | i5 + i7 | 125 406 | 70 764 | 56.4 | 13 302 | 10.6 | 52 | 0.22 |
It is notable that barcoded libraries with short adapters perform similarly to non-barcoded libraries with intermediate adapter length, with both showing approximately 72% of the reads mapping to the target. Thus, the barcoding and single indexing procedures are compromising capture efficiency, although not as much as for dual indexing (long adapters). The best capture specificity is for non-barcoded, non-indexed libraries, but we do not recommend this library architecture for screening as sample mix-ups and cross-contamination cannot be traced.
In conclusion, this experiment shows that the capture specificity can be increased and therefore sequencing costs reduced when barcoded libraries with short (incomplete) adapter sites are used, or alternatively, when libraries with one incomplete adapter site and an index (complete adapter site) on the other site, as opposed to fully indexed libraries are used. While it is possible that alternative hybridization conditions could make target capture more efficient in the presence of long adapters, these results show that the use of short adapters can be of value.
4. Summary
We have presented a modified double-stranded library preparation protocol involving partial UDG treatment. This protocol speeds up the screening of ancient DNA samples. Unique barcodes that we attach to all the DNA molecules in the library minimize the risk of wrongly assigned sequences owing to sample mix-ups or spillover. Finally, our use of short adapter libraries during oligonucleotide enrichment via hybridization makes enrichment more specific and allows efficient analysis in conjunction with robotics. While our new protocol increases the efficiency of ancient DNA screening, it is important to recognize that analysis of ancient DNA will always be somewhat slow, as each sample needs to be mechanically pulverized in clean conditions and complexity is usually limited. In addition, various elements of our protocol that we introduced to increase robustness and efficiency also results in some limitations at the sequencing stage, particularly related to our use of barcodes. For samples that emerge from screening as particularly useful, and for which it seems of interest to generate additional data and libraries, it may be of value to prepare new libraries that are non-barcoded, but indexed, to allow sequencing to become independent of barcode balancing. This is especially relevant for cases in which deep coverage of a small number of promising samples is the goal.
Acknowledgements
We are grateful to David Anthony and to Dorcas Brown for sharing bone samples. We thank Matthias Meyer and Philip L. Johnson and the ancient DNA group in the Reich Laboratory for helpful discussions, Steven McCarroll for sharing of laboratory equipment, and two anonymous reviewers for helpful comments.
Data accessibility
A manuscript about the data including population genetics findings is in preparation. The raw sequences are available on request from the corresponding author.
Funding statement
This work was supported by HOMINID grant no. 1032255. D.R. is an Investigator at the Howard Hughes Medical Institute.
References
- 1.Paabo S. 1989. Ancient DNA: extraction, characterization, molecular cloning, and enzymatic amplification. Proc. Natl Acad. Sci. USA 86, 1939–1943. ( 10.1073/pnas.86.6.1939) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shapiro B, Hofreiter M. 2014. A paleogenomic perspective on evolution and gene function: new insights from ancient DNA. Science 343, 1236573 ( 10.1126/science.1236573) [DOI] [PubMed] [Google Scholar]
- 3.Briggs AW, Good JM, Green RE, Krause J, Maricic T, Stenzel U, Pääbo S. 2009. Primer extension capture: targeted sequence retrieval from heavily degraded DNA sources. J. Vis. Exp. 31, e1573 ( 10.3791/1573) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Enk J, Rouillard JM, Poinar H. 2013. Quantitative PCR as a predictor of aligned ancient DNA read counts following targeted enrichment. Biotechniques 55, 300–309. [DOI] [PubMed] [Google Scholar]
- 5.Horn S. 2012. Case study: enrichment of ancient mitochondrial DNA by hybridization capture. Methods Mol. Biol. 840, 189–195. ( 10.1007/978-1-61779-516-9_22) [DOI] [PubMed] [Google Scholar]
- 6.Maricic T, Whitten M, Paabo S. 2010. Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS ONE 5, e14004 ( 10.1371/journal.pone.0014004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuenemann VJ, et al. 2011. Targeted enrichment of ancient pathogens yielding the pPCP1 plasmid of Yersinia pestis from victims of the black death. Proc. Natl Acad. Sci. USA 108, E746–E752. ( 10.1073/pnas.1105107108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Briggs AW, et al. 2007. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl Acad. Sci. USA 104, 14 616–14 621. ( 10.1073/pnas.0704665104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofreiter M, et al. 2001. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 29, 4793–4799. ( 10.1093/nar/29.23.4793) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brotherton P, Endicott P, Sanchez JJ, Beaumont M, Barnett R, Austin J, Cooper A. 2007. Novel high-resolution characterization of ancient DNA reveals C > U-type base modification events as the sole cause of post mortem miscoding lesions. Nucleic Acids Res. 35, 5717–5728. ( 10.1093/nar/gkm588) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stiller M, et al. 2006. Patterns of nucleotide misincorporations during enzymatic amplification and direct large-scale sequencing of ancient DNA. Proc. Natl Acad. Sci. USA 103, 13 578–13 584. ( 10.1073/pnas.0605327103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu Q, et al. 2013. A revised timescale for human evolution based on ancient mitochondrial genomes. Curr. Biol. 23, 553–559. ( 10.1016/j.cub.2013.02.044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Briggs AW, Stenzel U, Meyer M, Krause J, Kircher M, Paabo S. 2010. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res. 38, e87 ( 10.1093/nar/gkp1163) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raghavan M, et al. 2014. Upper Palaeolithic Siberian genome reveals dual ancestry of Native Americans. Nature 505, 87–91. ( 10.1038/nature12736) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rasmussen M, et al. 2014. The genome of a Late Pleistocene human from a Clovis burial site in western Montana. Nature 506, 225–229. ( 10.1038/nature13025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skoglund P, Northoff BH, Shunkov MV, Derevianko AP, Paabo S, Krause J, Jakobsson M. 2014. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proc. Natl Acad. Sci. USA 111, 2229–2234. ( 10.1073/pnas.1318934111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyer M, et al. 2014. A mitochondrial genome sequence of a hominin from Sima de los Huesos. Nature 505, 403–406. ( 10.1038/nature12788) [DOI] [PubMed] [Google Scholar]
- 18.Carpenter ML, et al. 2013. Pulling out the 1%: whole-genome capture for the targeted enrichment of ancient DNA sequencing libraries. Am. J. Hum. Genet. 93, 852–864. ( 10.1016/j.ajhg.2013.10.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu Q, Meyer M, Gao X, Stenzel U, Burbano HA, Kelso J, Paabo S. 2013. DNA analysis of an early modern human from Tianyuan Cave, China. Proc. Natl Acad. Sci. USA 110, 2223–2227. ( 10.1073/pnas.1221359110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gnirke A, et al. 2009. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 27, 182–189. ( 10.1038/nbt.1523) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hodges E, et al. 2007. Genome-wide in situ exon capture for selective resequencing. Nat. Genet. 39, 1522–1527. ( 10.1038/ng.2007.42) [DOI] [PubMed] [Google Scholar]
- 22.Enk JM, et al. 2014. Ancient whole genome enrichment using baits built from modern DNA. Mol. Biol. Evol. 31, 1292–1294. ( 10.1093/molbev/msu074) [DOI] [PubMed] [Google Scholar]
- 23.Fisher S, et al. 2011. A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol. 12, R1 ( 10.1186/gb-2011-12-1-r1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rohland N, Reich D. 2012. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 22, 939–946. ( 10.1101/gr.128124.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cooper A, Poinar HN. 2000. Ancient DNA: do it right or not at all. Science 289, 1139 ( 10.1126/science.289.5482.1139b) [DOI] [PubMed] [Google Scholar]
- 26.Knapp M, Clarke AC, Horsburgh KA, Matisoo-Smith EA. 2012. Setting the stage: building and working in an ancient DNA laboratory. Ann. Anat. 194, 3–6. ( 10.1016/j.aanat.2011.03.008) [DOI] [PubMed] [Google Scholar]
- 27.Anthony DW, et al. 2005. The Samara valley project: late Bronze Age economy and ritual in the Russian steppes. Eurasia Antiqua 11, 395–417. [Google Scholar]
- 28.Dabney J, et al. 2013. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl Acad. Sci. USA 110, 15 758–15 763. ( 10.1073/pnas.1314445110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyer M, Kircher M. 2010. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, pdb prot5448 ( 10.1101/pdb.prot5448) [DOI] [PubMed] [Google Scholar]
- 30.Kircher M, Sawyer S, Meyer M. 2012. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 40, e3 ( 10.1093/nar/gkr771) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.John JS. 2011. See https://github.com/jstjohn/SeqPrep.
- 32.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. ( 10.1093/bioinformatics/btp324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Behar DM, et al. 2012. A ‘Copernican’ reassessment of the human mitochondrial DNA tree from its root. Am. J. Hum. Genet. 90, 675–684. ( 10.1016/j.ajhg.2012.03.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. ( 10.1093/bioinformatics/btq033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jonsson H, Ginolhac A, Schubert M, Johnson PLF, Orlando L. 2013. MapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684. ( 10.1093/bioinformatics/btt193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Craig DW, et al. 2008. Identification of genetic variants using bar-coded multiplexed sequencing. Nat. Methods 5, 887–893. ( 10.1038/nmeth.1251) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stiller M, Knapp M, Stenzel U, Hofreiter M, Meyer M. 2009. Direct multiplex sequencing (DMPS): a novel method for targeted high-throughput sequencing of ancient and highly degraded DNA. Genome Res. 19, 1843–1848. ( 10.1101/gr.095760.109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meyer M, et al. 2012. A high-coverage genome sequence from an archaic Denisovan individual. Science 338, 222–226. ( 10.1126/science.1224344) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varshney U, van de Sande JH. 1991. Specificities and kinetics of uracil excision from uracil-containing DNA oligomers by Escherichia coli uracil DNA glycosylase. Biochemistry 30, 4055–4061. ( 10.1021/bi00230a033) [DOI] [PubMed] [Google Scholar]
- 40.Dabney J, Meyer M. 2012. Length and GC-biases during sequencing library amplification: a comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries. Biotechniques 52, 87–94. ( 10.2144/000113809) [DOI] [PubMed] [Google Scholar]
- 41.Sawyer S, Krause J, Guschanski K, Savolainen V. 2012. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS ONE 7, e34131 ( 10.1371/journal.pone.0034131) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allentoft ME, et al. 2012. The half-life of DNA in bone: measuring decay kinetics in 158 dated fossils. Proc. R. Soc. B 279, 4724–4733. ( 10.1098/rspb.2012.1745) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
A manuscript about the data including population genetics findings is in preparation. The raw sequences are available on request from the corresponding author.