

Abstract
As obligate intracellular parasites, viruses must traverse the host-cell plasma membrane to initiate infection. This presents a formidable barrier, which they have evolved diverse strategies to overcome. Common to all entry pathways, however, is a mechanism of specific attachment to cell-surface macromolecules or ‘receptors’. Receptor usage frequently defines viral tropism, and consequently, the evolutionary changes in receptor specificity can lead to emergence of new strains exhibiting altered pathogenicity or host range. Several classes of molecules are exploited as receptors by diverse groups of viruses, including, for example, sialic acid moieties and integrins. In particular, many cell-adhesion molecules that belong to the immunoglobulin-like superfamily of proteins (IgSF CAMs) have been identified as viral receptors. Structural analysis of the interactions between viruses and IgSF CAM receptors has not shown binding to specific features, implying that the Ig-like fold may not be key. Both proteinaceous and enveloped viruses exploit these proteins, however, suggesting convergent evolution of this trait. Their use is surprising given the usually occluded position of CAMs on the cell surface, such as at tight junctions. Nonetheless, the reason for their widespread involvement in virus entry most probably originates in their functional rather than structural characteristics.
Keywords: virus, receptor, cell entry, cryo-electron microscopy, cell adhesion
1. Introduction
Fundamentally, viruses are infectious nucleic acids that have evolved efficient mechanisms for shuttling their genomes between the host cells that they depend upon for replication. A key stage in the viral replication cycle is cell entry. To initiate infection, all viruses must traverse the host-cell's plasma membrane and in many cases a cell wall. For those viruses that infect animals, the first stage of this process is attachment to a cell-surface macromolecule, the viral receptor. There is considerable interest in understanding the virus–receptor interaction at the structural level. As the first step in the infection process, viral attachment represents an attractive target for intervention. The process of receptor engagement leads to initiation of the internalization pathway. Furthermore, receptor binding is frequently the trigger for conformational changes in the virion itself. These structural rearrangements are thought to initiate the uncoating process—the controlled, targeted release of the genome to the site of replication.
2. Viral entry pathways
Viruses employ diverse entry pathways following attachment. The host-cell plasma membrane presents a significant barrier, penetration of which may be facilitated by the presence of a viral membrane or envelope. Enveloped viruses acquire their membrane from the host either by budding from the plasma membrane of an infected cell, or by budding into cellular compartments. In either case, the membrane will bear viral glycoproteins that mediate attachment and entry. These functions may be performed by a single glycoprotein or may be divided between two or more. Following attachment, the glycoprotein responsible for mediating cell entry is activated to become fusogenic, undergoing conformational changes resulting in insertion of a hydrophobic ‘fusion-peptide’ into the host-cell plasma membrane. Further structural rearrangements then bring the viral and cellular membranes together leading to the formation of a fusion pore. The contents of the virion, including the encapsidated viral genome (nucleocapsid) are then delivered into the cytosol.
Not all enveloped viruses initiate fusion at the plasma membrane. Influenza viruses, for example, enter through the clathrin-mediated endocytic pathway [1]. Acidification of the late endosome triggers the activity of the viral fusion protein haemagglutinin, leading to release of the virion contents into the cytosol [2]. Endocytosis is also the most common entry mechanism for non-enveloped (proteinaceous) viruses; however, the manner in which these viruses leave the endosome is, in general, poorly understood. It is thought that this is accomplished either by the formation of a pore in the endosomal membrane through which the genome is ejected, or destruction of the endosomal membrane by viral-encoded gene products [3,4].
3. Receptor usage and viral tropism
Receptor usage is a key factor in defining tropism in many viruses; for example, influenza viruses bind to sialic acid moieties on the apical surfaces of epithelial cells in the respiratory tract of mammals or the gut of avian species. These are the primary sites of viral replication in the respective hosts. Those viruses that infect humans have evolved to bind α2,6 sialic acid which is found primarily in the upper respiratory tract. The haemagglutinin protein of avian viruses, on the other hand, binds to α2,3 sialic acid, which is the predominant form in the avian gut epithelium [5]. Thus, evolving to bind differently linked sialic acids is thought to be one important step required for avian viruses to transmit readily between human hosts. Evolution of receptor usage is therefore a key event that may lead to emergence of new pathogens with altered pathogenicity or host ranges.
4. Viral entry via multiple receptor molecules
Binding to sialic acid is a widely used strategy for attachment to the cell surface in diverse groups of viruses. Indeed, several classes of receptor molecule have been identified that are repeatedly found to be used by apparently unrelated viral species. These include integrins and cell-adhesion molecules that are members of the immunoglobulin-like superfamily (IgSF CAMs), the latter being the main focus of this review. Interestingly, two quite different viruses, feline calicivirus (FCV) and reovirus, have been found to employ both sialic acid and the IgSF CAM junctional adhesion molecule A (JAM-A) [6–9], while the picornavirus encephalomyocarditis virus binds sialic acid and a different IgSF CAM, vascular cell-adhesion molecule 1 (VCAM-1) [10,11]. A requirement for more than one receptor molecule is not uncommon and many viruses have evolved multi-step attachment processes. One extreme example is hepatitis C virus (HCV), which has been shown to require several molecules for cell entry including heparan sulfate [12], liver specific intercellular adhesion molecule-3-grabbing non-integrin (L-SIGN) or dendritic cell intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) [13–15], low-density lipoprotein receptor (LDL-R) [16,17], transferrin receptor 1 (TfR1) [18], Niemann-Pick C1-like protein 1 (NPC1L1) [19], scavenger receptor class B type I (SR-B1) [20], the tetraspanin CD81 [21] and the tight-junction components claudin-1 (CLDN-1) [22] and occludin (OCLN) [23]. SR-B1, CD81, CLDN-1 and OCLN are considered the minimal requirements for cell entry, while attachment to L-SIGN is postulated to confer tissue tropism in vivo. Lying at the centre of the HCV entry pathway is the interaction between the viral envelope glycoprotein E2 and CD81, which triggers actin-dependent trafficking of the virus to tight junctions where it comes into contact with CLDN-1 and OCLN, leading to viral entry by endocytosis.
Trafficking of virus to tight junctions from the apical cell surface was first demonstrated for group B coxsackie viruses (CVBs). Many CVBs bind to coxsackievirus and adenovirus receptor (CAR—an IgSF CAM) as well as the complement control protein decay-accelerating factor (DAF, also known as CD55). Clustering of DAF molecules by virus attachment to the apical cell surface stimulates remodelling of the actin cytoskeleton by Abl kinase. This, in turn, leads to delivery of the virus to the tight junction where entry occurs by caveolin-mediated endocytosis [24].
5. The structure and functions of Ig-like cell-adhesion molecules
An intriguing aspect of viral receptor usage is the widespread exploitation of cell-surface glycoproteins that are found predominantly in intercellular junctions of polarized cells. Perhaps the most widely used class of adhesion molecule is the IgSF CAMs (table 1). The immunoglobulin-like superfamily of proteins is characterized as consisting of seven to nine anti-parallel beta-strands that form two beta-sheets in a Greek-key motif, having a barrel shape. The superfamily is subdivided according to the number of beta-strands and topological similarities to the constant (c) or variable (v) chains of antibodies (V, C1, C2, I). Figure 1a shows the topology of the V-set Ig-like fold (reviewed in [37]).
Table 1.
Diverse groups of viruses have been shown to bind to immunoglobulin-like superfamily cell-adhesion molecules to gain entry to the host cell. Both DNA and RNA viruses exploit this class of molecules, as do enveloped and proteinaceous viruses.
| virus | receptor name | abbreviation | references |
|---|---|---|---|
| proteinaceous viruses | |||
| Adenoviridae | |||
| human adenovirus C | coxsackievirus–adenovirus receptor | CAR | [25] |
| Caliciviridae | |||
| feline calicivirus | junctional adhesion molecule A | JAM-A | [8] |
| Picornaviridae | |||
| coxsackie A virus type 21 | intercellular adhesion molecule 1 | ICAM-1/CD54 | [26] |
| coxsackie B virus | coxsackievirus–adenovirus receptor | CAR | [25] |
| encephalomyocarditis virus | vascular cell-adhesion molecule 1 | VCAM-1/CD106 | [10] |
| major receptor group rhinovirus | intercellular adhesion molecule 1 | ICAM-1/CD54 | [27] |
| poliovirus | poliovirus receptor | PVR/CD155 | [28] |
| Reoviridae | |||
| reovirus | junctional adhesion molecule A | JAM-A | [6] |
| enveloped viruses | |||
| Coronaviridae | |||
| mouse hepatitis virus | carcinoembryonic antigen-related cell-adhesion molecule 1 | CEACAM-1/CD66a | [29] |
| Herpesviridae | |||
| herpes simplex virus | nectin-1 nectin-2 |
HvecC/CD111 HvecB/CD112 |
[30,31] |
| Paramyxoviridae | |||
| measles virus | nectin-4 | [32] | |
| Rhabdoviridae | |||
| rabiesvirus | neuronal cell-adhesion molecule 1 | NCAM-1/CD56 | [33] |
Figure 1.

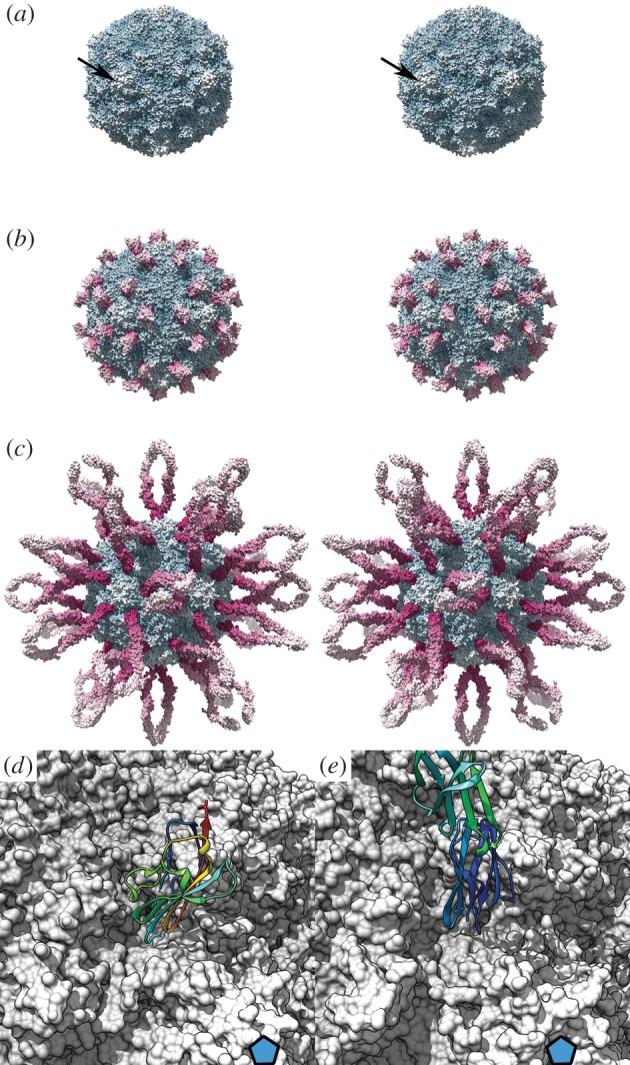
The immunoglobulin superfamily of proteins are characterized as having domains of between seven and nine beta-strands arranged in two antiparallel sheets that form a sandwich structure, stabilized by a conserved disulfide bridge (a; rainbow coloured from the N-terminus (blue) to the C-terminus (red)). The crystal structure of a typical IgSF CAM—human JAM-A (b; PDB 1NBQ [34]) reveals that the two-domain molecule exists as a dimer in solution that is thought to represent the native structure at tight junctions (c). The ribbon diagrams are rainbow coloured across two domains, hence the colours of individual strands in (b) and (c) do not correspond to those in (a). Caliciviruses such as feline calicivirus (FCV) are RNA containing viruses that have a T = 3 icosahedral capsid (d; PDB 3M8L) [35]. This is composed of 180 capsid proteins (VP1) arranged as two classes of dimer: AB dimers (light and mid-blue) and CC dimers (dark blue). Cryo-electron microscopy of FCV (e) decorated with a soluble fragment of feline JAM-A (f) reveals that the receptor binds to the tip of the protruding domain of VP1. Receptor engagement induces conformational changes in the viral capsid such that the AB dimer rotates 15° anticlockwise and the CC dimer tilts away from the twofold symmetry axis (g; arrows). Docking high-resolution coordinates to the three-dimensional reconstruction led to the calculation of a quasi-atomic resolution map of the virus—receptor complex (h; FCV coloured blue, fJAM-A coloured magenta) that allowed the identification of putative contact residues (i). The VP1 AB dimer is viewed from the virus exterior, fJAM molecules viewed as if peeled away from the capsid surface and rotated 180o [36]. Panels (d–i) are presented as wall-eyed stereo pairs. (e–i) adapted from [36].
JAM-A is a prototypic tight junction associated IgSF CAM expressed on epithelial and endothelial cells as well as on leucocytes and platelets [38]. It comprises two extracellular Ig-like domains, a single transmembrane region and a short cytoplasmic tail. X-ray crystallography of an ectodomain soluble fragment of human JAM-A reveals that the N-terminal, membrane-distal D1 domain has nine beta-strands and is therefore classified as similar to the antibody variable domain (V-set). The membrane proximal D2 domain, on the other hand, is classed as I-set, having only eight strands (figure 1b).
Analytical ultracentrifugation analysis of recombinant soluble murine JAM-A revealed that it forms homodimers. Both murine and human JAM-A crystal structures show a similar non-covalent interaction between the membrane-distal D1 domains at the GFCC’ face, and this is thought to represent the dimeric state of JAM-A at the cell surface (figure 1c). Homotypic and heterotypic interactions with JAM-A and other adhesion molecules, respectively, on adjacent cells, are then thought to regulate tight-junction formation and facilitate leucocyte transmigration [34,39].
6. Feline calicivirus binding to JAM-A
FCV is one of only a handful of tractable models for the Caliciviridae, a family of positive-sense RNA containing icosahedral viruses, which includes norovirus, the cause of winter vomiting disease. These small proteinaceous viruses assemble a T = 3 icosahedral capsid from 90 dimers of a single major capsid protein VP1 (figure 1d) [35]. The virion is characterized by the presence of protruding (P) domains that give rise to the appearance of cups on the surface of the particles when viewed by negative stain electron microscopy; hence their name, which derives from the Latin calyx.
FCV has been shown to bind to both N-linked α2,6 sialic acid and feline JAM-A (fJAM-A) [8,9]. Both molecules are important for cell entry, which is by clathrin-mediated endocytosis and requires acidification of the endosome [40].
fJAM-A is the only protein receptor to be identified for any member of the Caliciviridae. Experiments demonstrated that transfection of the fJAM-A gene into non-permissive cells rendered them susceptible to FCV infection, while antibodies raised against fJAM-A blocked infection [8]. To investigate the structural basis for fJAM-A receptor engagement, we used cryogenic electron microscopy (cryoEM) combined with computational three-dimensional image reconstruction to determine the structure of purified FCV particles decorated with a soluble fragment of fJAM-A (figure 1e–g) [36,41]. This revealed that the fragment binds to the outer face of the capsid P domain. Two fJAM-A molecules lie in a head-to-tail arrangement about the twofold symmetry axis of each VP1 dimer. Docking high-resolution coordinates for FCV and fJAM-A—derived from crystallographic analysis and homology modelling respectively—led to the synthesis of a quasi-atomic resolution model of the receptor decorated virion (figure 1h). This showed that the membrane-distal D1 domain was primarily responsible for the interaction, and key residues in both virus and receptor that are involved in viral attachment were identified (figure 1i). Interestingly, the soluble fJAM-A fragment did not bind to the capsid in the dimeric state seen in the structures of human and murine forms solved by X-ray crystallography. The oligomeric state of fJAM-A used in our study is not known; however, the site of virus attachment to JAM-A does not suggest that viral binding would directly compete with the homodimer interaction. The FCV-binding site on JAM-A resides in the beta-sheet comprising strands ABE while the homodimer interface is at the opposite GFCC’ face. It may, however, be the case that binding to FCV disrupts the JAM-A homodimer by inducing a structural change in the receptor.
In our study, substantial conformational changes in the viral capsid protein were seen upon receptor binding. The P-domain of the AB dimer was seen to rotate 15° counterclockwise, while at the CC dimer the P-domain tilted away from the icosahedral twofold symmetry axis. We hypothesize that these changes in virion conformation may reflect the early stages of uncoating, priming the capsid for subsequent genome release.
7. Picornavirus attachment
The Picornaviridae, like the Caliciviridae, are small icosahedral, non-enveloped, positive-sense RNA-containing viruses. Many viruses in this family have been found to have IgSF CAM receptors. Interestingly, these receptors also induce profound conformational changes in the virion upon binding, destabilizing the capsid and leading to genome release. However, those viruses that bind to non-IgSF receptors do not appear to undergo such receptor-induced rearrangements.
Picornaviruses have generally well-conserved virion morphology. The icosahedral capsid assembles from four structural proteins designated VP1–4. VP1–3 occupy positions conventionally taken by multiple copies of a single protein species in a T = 3 icosahedral lattice; thus picornaviruses are described as pseudo T = 3 or P3. Most picornaviruses have pronounced star-shaped mesas at their icosahedral fivefold symmetry axes that are surrounded by deep canyons (figure 2) [46]. CryoEM studies of picornavirus–CAM complexes show that the tip of the membrane-distal Ig-like domain inserts into the canyon such that the receptor is oriented more or less perpendicular to the capsid surface [42,45]. This manner of receptor engagement is rather different to that seen in FCV, which binds to one side of the D1 domain. Surprisingly, however, detailed comparison of the interactions between picornaviruses and their respective IgSF CAM receptors revealed that they bind to different faces of the D1 domain [47].
Figure 2.
Picornaviruses are small proteinaceous icosahedral viruses, many of which have pronounced star-shaped mesas at their fivefold symmetry axes (a; arrow) that are surrounded by canyons which contains the receptor-binding sites for IgSF CAMs. Coxsackie B virus type 3 binds to the coxsackievirus and adenovirus receptor (b; CAR domain 1 only is shown, figure generated using PDB 1COV, 1KAC based on 1JEW [42–44]). Coxsackie A virus type 21 binds to intercellular adhesion molecule 1 (ICAM-1) (c; PDB 1Z7Z [45]). Both molecules are oriented perpendicular to the capsid surface. A close up view of the interaction (receptors shown as ribbon diagrams) shows that CAR (d) and ICAM-1 (e) bind to the canyon in a similar but not identical orientation (fivefold symmetry axis indicated by a blue pentagon). (a–c) Stereoscopic views in which virus is radially coloured blue-white and receptor is radially coloured magenta-white. Panels (d) and (e) are monoscopic and the virus surface is shown in grey; the receptor is shown as a rainbow-coloured ribbon diagram.
8. Evolution of virus–receptor interactions
Structural analysis of viral attachment proteins complexed to Ig-like SF CAMs provides us with detailed descriptions of the first step of the infectious process. In all cases studied so far, the virus binds to the V-set membrane-distal D1 domain. It has been suggested that viruses engage Ig-like SF molecules in a manner that parallels immunoglobulin pathogen recognition [48]. The above-mentioned comparison of interactions in the Picornaviridae and our analysis of FCV JAM-A binding are at odds with this, however, suggesting that viruses do not exploit a common binding site. The FCV-binding site of fJAM-A comprises residues in strands A, B and E, while entry of reoviruses is mediated by an attachment protein σ-1, which engages the dimer interface of monomeric JAM-A at residues in the C and C’ beta-strands (figure 3) [49]. Thus, opposite faces of the D1 domain are required for entry of these two viruses. In the case of reovirus attachment it is possible that virus binding may directly disrupt JAM-A dimers.
Figure 3.
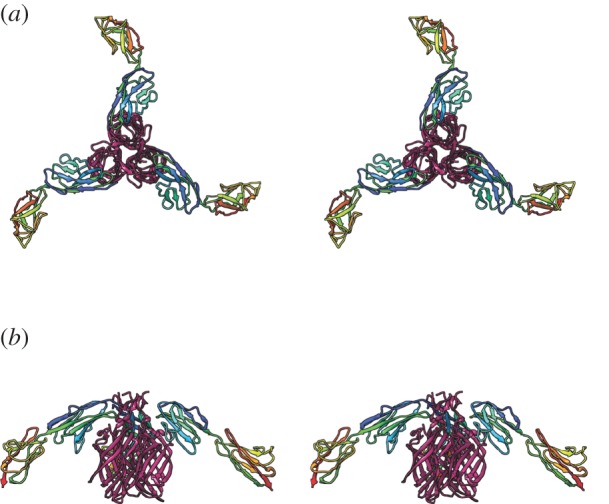
(a,b) Views of the interaction between JAM-A (rainbow coloured) and the reovirus attachment protein σ-1 (magenta; wall-eyed stereo pairs—PDB 3EOY, 1NBQ [34,49]).
Reo- and caliciviruses are quite distinct classes of viruses. The Orthoreovirinae are large proteinaceous double-stranded RNA containing viruses with a complex multi-layered capsid. Protruding from each fivefold vertex are trimeric fibres of the attachment protein σ-1 that terminate in a globular knob domain comprising three beta-barrels. Evolution of JAM-A and sialic acid binding in reo- and caliciviruses is therefore most probably a product of convergent evolution. The fact that IgSF CAM binding is widely observed in both proteinaceous and enveloped viruses also argues that the exploitation of such molecules is a highly advantageous capability that has emerged repeatedly and independently.
Virus–receptor interactions in animal viruses are subject to continual selective pressure in the face of immune surveillance. A consequence of this is that the outer surfaces of virus particles are characterized by the presence of elaborate loop structures and hypervariable regions that disrupt antibody-mediated neutralization and serve to camouflage the more highly conserved receptor-binding site. Under these circumstances it is likely that the binding site itself will nonetheless undergo constant modification. It iseasy to imagine how, through mutation of the capsid or attachment proteins, the receptor may ‘walk’ over the surface of the virus. Likewise, the virus may, over time, evolve to bind a different face of the receptor molecule or even to bind a different structurally related molecule. Strong evidence for this can be seen in the diverse interactions displayed by distantly related picornaviruses that bind to the complement control protein DAF. DAF (also known as CD55) is not a member of the immunoglobulin-like superfamily of proteins but displays a striking divergence of binding modes by viruses that exploit it as a receptor. In research published by ourselves and others, cryoEM analysis of echovirus type 12 (EV12) and coxsackie virus type B3 (CVB3) decorated with DAF revealed a marked difference in the orientation of the receptor molecule on the virion surface (figure 4) [50,51]. DAF comprises four short-consensus repeat domains. CVB3 binding occurs primarily at SCR2 while EV12 binds to SCR3. Moreover, there is a rotation of almost 90o in the position of the DAF molecule relative to the capsid surface. It has been suggested that the differences in DAF binding in these two viruses is the product of convergent evolution to bind a common receptor. Given the high mutation rates in RNA containing viruses and the above-mentioned selective pressure, however, divergent evolution from a common DAF binding ancestor would seem equally or more plausible.
Figure 4.
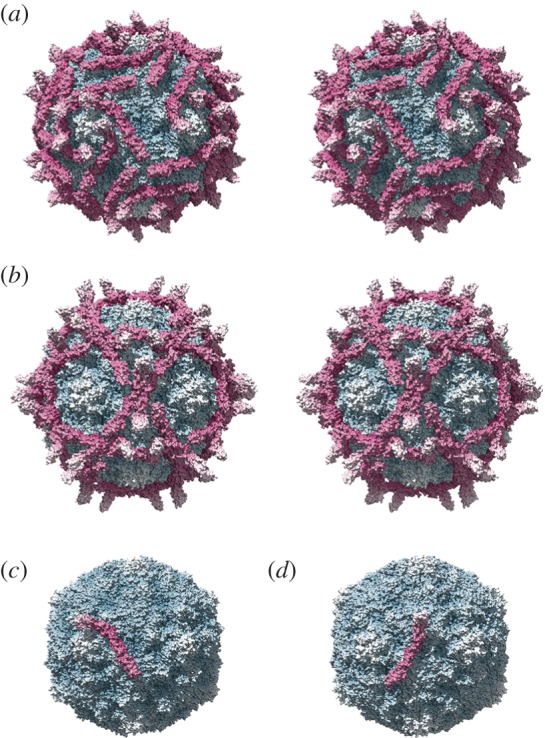
Evolution of virus–receptor interactions. Coxsackie B virus type 3 (CVB3) binds to the complement control protein decay-accelerating factor (DAF or CD55) at the apical cell surface prior to trafficking to tight junctions where entry is mediated by the coxsackievirus and adenovirus receptor (CAR). Comparison of the CVB3–DAF interaction (a; PDB 1COV, 3J24 [50]) with that of the distantly related picornavirus echovirus type 12 (EV12) (b; PDB 2C8I [51]) shows a markedly different receptor orientation. The structure is more easily interpreted when only a single DAF molecule is shown. CVB3 (c) binds primarily to domain 2 of DAF. EV12 binds predominantly domain 3 (d); moreover, there is approximately 90° rotation in the orientation of the two molecules on the capsid surface. These two viruses have quite different receptor interactions but have probably evolved from a common DAF-binding picornavirus ancestor. In each panel, the virus is radially coloured blue-white; DAF-CD55 is radially coloured magenta-white.
As we have seen, IgSF CAM binding occurs widely among diverse classes of viruses, suggesting convergent evolution. On the other hand, we can see how a process of continual evolution in the face of immune surveillance may have led to diversification of canyon-binding IgSF CAM interactions within the Picornaviridae. Both convergent and divergent evolution of this trait among different viruses argues that there is a strong selective pressure to gain and retain a capability to bind these molecules. As noted above, however, evidence for a conserved binding motif is not compelling. Thus, the requirement to bind to cell-adhesion molecules may originate in their functional properties despite the apparent inconvenience of requiring the virus to traffic across the cell surface to intercellular junctions to effect cell entry. Recent research in this area is beginning to provide insights into the possible reasons for the persistence of this phenomenon.
9. Disruption of CAR homotypic interactions leads to endocytosis
Group C adenoviruses bind to CAR in a manner that closely parallels the reovirus JAM-A interaction. A trimeric fibre knob protein engages the normally dimeric CAR molecule at an interface that overlaps its dimerization site. The recent finding by Salinas et al. [52] that disruption of the CAR homodimer by adenovirus fibre knob protein stimulates endocytosis provides one possible explanation for the conservation of IgSF CAM binding among diverse groups of viruses. Endocytosis is an important aspect of IgSF CAM function, serving as a means of intracellular signal transduction. Moreover, depletion of CAMs from the cell surface is critical in regulation of cell migration. Thus exploitation of this important functional quality of IgSF CAMs could allow viruses to gain entry to the cell interior. Several viruses that engage IgSF CAMs are, however, enveloped and postulated to enter by fusion at the plasma membrane. Thus triggering endocytosis may not be the sole reason for IgSF CAM usage, and further work is necessary to establish whether those enveloped viruses undergo endocytosis at intercellular junctions.
10. JAM-A facilitates dissemination of reovirus infection
Much of our understanding of virus entry derives from studies of viruses grown in cell culture. Our comprehension of the complexities of virus behaviour within tissue or the whole organism is therefore limited. To investigate the role of JAM-A in reovirus infectious processes, Antar et al. [53] investigated pathogenesis in JAM-A null mice. Animals were perorally inoculated with a neurotropic strain of the virus. The primary site of infection, the intestine, was infected normally; however, JAM-A−/− mice showed no sign of neurological disease. In these studies, virus replication at sites of secondary infection was significantly reduced as a consequence of a failure of the virus to enter the blood-stream. Neural spread was, however, unaffected. The authors suggest that JAM-A plays a crucial role in establishment of viraemia, either by facilitating infection of endothelial cells leading to release of virus from apical cell surfaces into the bloodstream or receptor-specific transcytosis of virus across endothelial cells. An alternative hypothesis is that dissemination of virus through the bloodstream might be mediated by infection of blood leucocytes.
11. Viral exploitation of IgSF proteins expressed on cells of the immune system
In addition to the above-mentioned exploitation of IgSF CAMs as viral receptors, there are several viruses that have evolved specifically to infect cells of the immune system through engagement of other IgSF molecules. These include T-cell membrane protein 1 (TIM-1) which is bound by Ebola, dengue and hepatitis A viruses [54–56], the HIV receptor CD4 [57], and signalling lymphocyte-activation molecule (SLAM) [58], which is exploited by measles virus. The ability to infect migratory cells of the immune system is a strategy that allows viral dissemination within the host without danger of exposure to the adaptive immune response. It seems plausible, then, that the viral strategy of infecting via IgSF CAM mediated entry may also confer this advantage. Immune cells display IgSF CAMs to mediate transmigration through tissues. In the case of FCV and reovirus infection of the respiratory and intestinal epithelium, respectively, leucocytes responding to the viral attack may themselves become infected and facilitate dissemination to secondary sites of infection.
12. Summary
Many diverse groups of viruses bind to IgSF CAMs at the cell surface to mediate cell entry. Structural analyses of virus receptor complexes do not, however, indicate a common mode of binding, suggesting that this phenomenon is not a simple exploitation of the adhesive properties of these molecules. Cell-adhesion molecules play a critical role in maintaining tissue integrity and also mediating migration of immune cells. This is achieved through control of surface expression levels. Endocytosis of CAMs is the primary means of modulating surface expression, and there is evidence that this is mediated by disruption of the CAM homodimer interface. Exploitation of this phenomenon by viruses to gain access to the cell interior is a satisfying explanation for the IgSF CAM binding phenotype. However, the exploitation of these receptors by enveloped viruses that are thought to enter by fusion at the plasma membrane argues against this being the sole reason. Studies of reovirus infection in animal models show that JAM-A is required for viral dissemination through the bloodstream. It is postulated that this is related to the expression of JAM-A on endothelial cells, which may allow for transcytosis of virus into blood, or perhaps infection of the endothelium may lead to apical release of virus into the bloodstream. An alternative hypothesis is that IgSF CAM expression allows for viral dissemination from sites of primary infection in epithelial cells by transmission to responding migratory cells of the immune system.
It is hoped that a detailed understanding of processes of virus attachment and entry will provide researchers with new targets for anti-viral development. In particular, mechanisms found to be widespread among diverse groups of viruses, such as entry via IgSF CAM binding, are appealing targets as interventions have the potential for broad-spectrum activity. As targeting host systems, such as regulation of cell adhesion and immune cell transmigration, would seem to be a strategy liable to significant toxicity, detailed dissection of the manner in which viruses exploit these processes is critical.
References
- 1.Matlin KS, Reggio H, Helenius A, Simons K. 1981. Infectious entry pathway of influenza virus in a canine kidney cell line. J. Cell Biol. 91, 601–613. ( 10.1083/jcb.91.3.601) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yoshimura A, Ohnishi S. 1984. Uncoating of influenza virus in endosomes. J. Virol. 51, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prchla E, Plank C, Wagner E, Blaas D, Fuchs R. 1995. Virus-mediated release of endosomal content in vitro: different behavior of adenovirus and rhinovirus serotype 2. J. Cell Biol. 131, 111–123. ( 10.1083/jcb.131.1.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiethoff CM, Wodrich H, Gerace L, Nemerow GR. 2005. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 79, 1992–2000. ( 10.1128/JVI.79.4.1992-2000.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rogers GN, Paulson JC. 1983. Receptor determinants of human and animal influenza virus isolates: differences in receptor specificity of the H3 hemagglutinin based on species of origin. Virology 127, 361–373. ( 10.1016/0042-6822(83)90150-2) [DOI] [PubMed] [Google Scholar]
- 6.Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104, 441–451. ( 10.1016/S0092-8674(01)00231-8) [DOI] [PubMed] [Google Scholar]
- 7.Gentsch JR, Pacitti AF. 1985. Effect of neuraminidase treatment of cells and effect of soluble glycoproteins on type 3 reovirus attachment to murine L cells. J. Virol. 56, 356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makino A, Shimojima M, Miyazawa T, Kato K, Tohya Y, Akashi H. 2006. Junctional adhesion molecule 1 is a functional receptor for feline calicivirus. J. Virol. 80, 4482–4490. ( 10.1128/JVI.80.9.4482-4490.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stuart AD, Brown TD. 2007. α2,6-linked sialic acid acts as a receptor for feline calicivirus. J. Gen. Virol. 88, 177–186. ( 10.1099/vir.0.82158-0) [DOI] [PubMed] [Google Scholar]
- 10.Huber SA. 1994. VCAM-1 is a receptor for encephalomyocarditis virus on murine vascular endothelial cells. J. Virol. 68, 3453–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tavakkol A, Burness AT. 1990. Evidence for a direct role for sialic acid in the attachment of encephalomyocarditis virus to human erythrocytes. Biochemistry 29, 10 684–10 690. ( 10.1021/bi00499a016) [DOI] [PubMed] [Google Scholar]
- 12.Barth H, et al. 2003. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 278, 41 003–41 012. ( 10.1074/jbc.M302267200) [DOI] [PubMed] [Google Scholar]
- 13.Gardner JP, Durso RJ, Arrigale RR, Donovan GP, Maddon PJ, Dragic T, Olson WC. 2003. L-SIGN (CD 209L) is a liver-specific capture receptor for hepatitis C virus. Proc. Natl Acad. Sci. USA 100, 4498–4503. ( 10.1073/pnas.0831128100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lozach PY, et al. 2003. DC-SIGN and L-SIGN are high affinity binding receptors for hepatitis C virus glycoprotein E2. J. Biol. Chem. 278, 20 358–20 366. ( 10.1074/jbc.M301284200) [DOI] [PubMed] [Google Scholar]
- 15.Pohlmann S, et al. 2003. Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J. Virol. 77, 4070–4080. ( 10.1128/JVI.77.7.4070-4080.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX. 1999. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl Acad. Sci. USA 96, 12 766–12 771. ( 10.1073/pnas.96.22.12766) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Monazahian M, Bohme I, Bonk S, Koch A, Scholz C, Grethe S, Thomssen R. 1999. Low density lipoprotein receptor as a candidate receptor for hepatitis C virus. J. Med. Virol. 57, 223–229. () [DOI] [PubMed] [Google Scholar]
- 18.Martin DN, Uprichard SL. 2013. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc. Natl Acad. Sci. USA 110, 10 777–10 782. ( 10.1073/pnas.1301764110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sainz B, Jr, et al. 2012. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 18, 281–285. ( 10.1038/nm.2581) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scarselli E, et al. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21, 5017–5025. ( 10.1093/emboj/cdf529) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pileri P, et al. 1998. Binding of hepatitis C virus to CD81. Science 282, 938–941. ( 10.1126/science.282.5390.938) [DOI] [PubMed] [Google Scholar]
- 22.Evans MJ, et al. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446, 801–805. ( 10.1038/nature05654) [DOI] [PubMed] [Google Scholar]
- 23.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457, 882–886. ( 10.1038/nature07684) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coyne CB, Bergelson JM. 2006. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124, 119–131. ( 10.1016/j.cell.2005.10.035) [DOI] [PubMed] [Google Scholar]
- 25.Carson SD. 2001. Receptor for the group B coxsackieviruses and adenoviruses: CAR. Rev. Med. Virol. 11, 219–226. ( 10.1002/rmv.318) [DOI] [PubMed] [Google Scholar]
- 26.Shafren DR, Dorahy DJ, Greive SJ, Burns GF, Barry RD. 1997. Mouse cells expressing human intercellular adhesion molecule-1 are susceptible to infection by coxsackievirus A21. J. Virol. 71, 785–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Staunton DE, Merluzzi VJ, Rothlein R, Barton R, Marlin SD, Springer TA. 1989. A cell adhesion molecule, ICAM-1, is the major surface receptor for rhinoviruses. Cell 56, 849–853. ( 10.1016/0092-8674(89)90689-2) [DOI] [PubMed] [Google Scholar]
- 28.Mendelsohn CL, Wimmer E, Racaniello VR. 1989. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 56, 855–865. ( 10.1016/0092-8674(89)90690-9) [DOI] [PubMed] [Google Scholar]
- 29.Dveksler GS, Pensiero MN, Cardellichio CB, Williams RK, Jiang GS, Holmes KV, Dieffenbach CW. 1991. Cloning of the mouse hepatitis virus (MHV) receptor: expression in human and hamster cell lines confers susceptibility to MHV. J. Virol. 65, 6881–6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280, 1618–1620. ( 10.1126/science.280.5369.1618) [DOI] [PubMed] [Google Scholar]
- 31.Warner MS, Geraghty RJ, Martinez WM, Montgomery RI, Whitbeck JC, Xu R, Eisenberg RJ, Cohen GH, Spear PG. 1998. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 246, 179–189. ( 10.1006/viro.1998.9218) [DOI] [PubMed] [Google Scholar]
- 32.Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, Richardson CD. 2011. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 7, e1002240 ( 10.1371/journal.ppat.1002240) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thoulouze MI, Lafage M, Schachner M, Hartmann U, Cremer H, Lafon M. 1998. The neural cell adhesion molecule is a receptor for rabies virus. J. Virol. 72, 7181–7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prota AE, et al. 2003. Crystal structure of human junctional adhesion molecule 1: implications for reovirus binding. Proc. Natl Acad. Sci. USA 100, 5366–5371. ( 10.1073/pnas.0937718100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ossiboff RJ, Parker JS. 2007. Identification of regions and residues in feline junctional adhesion molecule required for feline calicivirus binding and infection. J. Virol. 81, 13 608–13 621. ( 10.1128/JVI.01509-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhella D, Goodfellow IG. 2011. The cryo-electron microscopy structure of feline calicivirus bound to junctional adhesion molecule A at 9-angstrom resolution reveals receptor-induced flexibility and two distinct conformational changes in the capsid protein VP1. J. Virol. 85, 11 381–11 390. ( 10.1128/JVI.05621-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barclay AN. 2003. Membrane proteins with immunoglobulin-like domains—a master superfamily of interaction molecules. Semin. Immunol. 15, 215–223. ( 10.1016/S1044-5323(03)00047-2) [DOI] [PubMed] [Google Scholar]
- 38.Bazzoni G. 2003. The JAM family of junctional adhesion molecules. Curr. Opin. Cell Biol. 15, 525–530. ( 10.1016/S0955-0674(03)00104-2) [DOI] [PubMed] [Google Scholar]
- 39.Kostrewa D, et al. 2001. X-ray structure of junctional adhesion molecule: structural basis for homophilic adhesion via a novel dimerization motif. EMBO J. 20, 4391–4398. ( 10.1093/emboj/20.16.4391) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stuart AD, Brown TD. 2006. Entry of feline calicivirus is dependent on clathrin-mediated endocytosis and acidification in endosomes. J. Virol. 80, 7500–7509. ( 10.1128/JVI.02452-05) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhella D, Gatherer D, Chaudhry Y, Pink R, Goodfellow IG. 2008. Structural insights into calicivirus attachment and uncoating. J. Virol. 82, 8051–8058. ( 10.1128/JVI.00550-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He Y, et al. 2001. Interaction of coxsackievirus B3 with the full length coxsackievirus-adenovirus receptor. Nat. Struct. Biol. 8, 874–878. ( 10.1038/nsb1001-874) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bewley MC, Springer K, Zhang YB, Freimuth P, Flanagan JM. 1999. Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, CAR. Science 286, 1579–1583. ( 10.1126/science.286.5444.1579) [DOI] [PubMed] [Google Scholar]
- 44.Muckelbauer JK, Kremer M, Minor I, Tong L, Zlotnick A, Johnson JE, Rossmann MG. 1995. Structure determination of coxsackievirus B3 to 3.5 A resolution. Acta Crystallogr. D Biol. Crystallogr. 51, 871–887. ( 10.1107/S0907444995002253) [DOI] [PubMed] [Google Scholar]
- 45.Xiao C, Bator-Kelly CM, Rieder E, Chipman PR, Craig A, Kuhn RJ, Wimmer E, Rossmann MG. 2005. The crystal structure of coxsackievirus A21 and its interaction with ICAM-1. Structure 13, 1019–1033. ( 10.1016/j.str.2005.04.011) [DOI] [PubMed] [Google Scholar]
- 46.Rossmann MG, et al. 1985. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature 317, 145–153. ( 10.1038/317145a0) [DOI] [PubMed] [Google Scholar]
- 47.Organtini LJ, Makhov AM, Conway JF, Hafenstein S, Carson SD. 2014. Kinetic and structural analysis of coxsackievirus B3 receptor interactions and formation of the A-particle. J. Virol. 88, 5755–5765. ( 10.1128/JVI.00299-14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dermody TS, Kirchner E, Guglielmi KM, Stehle T. 2009. Immunoglobulin superfamily virus receptors and the evolution of adaptive immunity. PLoS Pathog. 5, e1000481 ( 10.1371/journal.ppat.1000481) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kirchner E, Guglielmi KM, Strauss HM, Dermody TS, Stehle T. 2008. Structure of reovirus sigma1 in complex with its receptor junctional adhesion molecule-A. PLoS Pathog. 4, e1000235 ( 10.1371/journal.ppat.1000235) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoder JD, Cifuente JO, Pan J, Bergelson JM, Hafenstein S. 2012. The crystal structure of a coxsackievirus B3-RD variant and a refined 9-angstrom cryo-electron microscopy reconstruction of the virus complexed with decay-accelerating factor (DAF) provide a new footprint of DAF on the virus surface. J. Virol. 86, 12 571–12 581. ( 10.1128/JVI.01592-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pettigrew DM, Williams DT, Kerrigan D, Evans DJ, Lea SM, Bhella D. 2006. Structural and functional insights into the interaction of echoviruses and decay-accelerating factor. J. Biol. Chem. 281, 5169–5177. ( 10.1074/jbc.M510362200) [DOI] [PubMed] [Google Scholar]
- 52.Salinas S, Zussy C, Loustalot F, Henaff D, Menendez G, Morton PE, Parsons M, Schiavo G, Kremer EJ. 2014. Disruption of the coxsackievirus and adenovirus receptor-homodimeric interaction triggers lipid microdomain- and dynamin-dependent endocytosis and lysosomal targeting. J. Biol. Chem. 289, 680–695. ( 10.1074/jbc.M113.518365) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Antar AA, Konopka JL, Campbell JA, Henry RA, Perdigoto AL, Carter BD, Pozzi A, Abel TW, Dermody TS. 2009. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 5, 59–71. ( 10.1016/j.chom.2008.12.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaplan G, Totsuka A, Thompson P, Akatsuka T, Moritsugu Y, Feinstone SM. 1996. Identification of a surface glycoprotein on African green monkey kidney cells as a receptor for hepatitis A virus. EMBO J. 15, 4282–4296. [PMC free article] [PubMed] [Google Scholar]
- 55.Meertens L, Carnec X, Lecoin MP, Ramdasi R, Guivel-Benhassine F, Lew E, Lemke G, Schwartz O, Amara A. 2012. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 12, 544–557. ( 10.1016/j.chom.2012.08.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kondratowicz AS, et al. 2011. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl Acad. Sci. USA 108, 8426–8431. ( 10.1073/pnas.1019030108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T, Gluckman JC, Montagnier L. 1984. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature 312, 767–768. ( 10.1038/312767a0) [DOI] [PubMed] [Google Scholar]
- 58.Tatsuo H, Ono N, Tanaka K, Yanagi Y. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406, 893–897. ( 10.1038/35022579) [DOI] [PubMed] [Google Scholar]