

Abstract
Anchorage-dependent cells are of great interest for various biotechnological applications. (i) They represent a formidable production means of viruses for vaccination purposes at very large scales (in 1000–6000 l reactors) using microcarriers, and in the last decade many more novel viral vaccines have been developed using this production technology. (ii) With the advent of stem cells and their use/potential use in clinics for cell therapy and regenerative medicine purposes, the development of novel culture devices and technologies for adherent cells has accelerated greatly with a view to the large-scale expansion of these cells. Presently, the really scalable systems—microcarrier/microcarrier-clump cultures using stirred-tank reactors—for the expansion of stem cells are still in their infancy. Only laboratory scale reactors of maximally 2.5 l working volume have been evaluated because thorough knowledge and basic understanding of critical issues with respect to cell expansion while retaining pluripotency and differentiation potential, and the impact of the culture environment on stem cell fate, etc., are still lacking and require further studies. This article gives an overview on critical issues common to all cell culture systems for adherent cells as well as specifics for different types of stem cells in view of small- and large-scale cell expansion and production processes.
Keywords: adherent cells, virus production, cell therapy, stem cells, engineering issues, large scale expansion
1. Introduction
The development of animal cell technology began with the initiation of the production of viruses for vaccine purposes, starting with the cultivation of primary cells (monkey kidney cells and chicken embryo fibroblasts), followed by the development of diploid cell cultures (from humans, such as human fetal lung fibroblasts (WI-38, MRC-5) or from monkeys, such as DBC-FRhL-2 (lung fibroblasts from a male rhesus fetus)) and finally evolving towards the use of continuous cell lines. This included, in the first instance, anchorage-dependent cells (Vero and MDCK) and later suspension cells (BHK21, EB66, insect cells and designer cells (HEK293, Per.C6, CAP, AGE1.CR and AGE1.CR.pIX), etc.). However, with the development of these cells and the much simpler large-scale cell culture technology available for suspension cells, the tendency is towards the use of suspension cells for the production of biologicals (viruses, viral vectors, recombinant proteins). Despite this general trend many production processes, in particular, for viruses and viral vectors, are still based on the use of surface adherent cells because of the advantages of these cell substrates for virus production. In addition, the recent arrival of stem cell technology for stem cell therapy and regenerative medicine, and the requirement for growth on surfaces of many stem cell types, has given a kick to the further evolution of culture systems of anchorage-dependent cells. This has led to a reappraisal of microcarrier technology and new developments in cell culture devices for these novel purposes.
Furthermore, it should be mentioned here that the culture of anchorage-dependent cells is not only limited to biomass or recombinant protein/viral particle production, but it serves also as an in vitro model for drug screening or disease modelling. However, this application is not further discussed in this review. In each case and as learned for stem cell/primary cell culture, the modulation/retention of a particular phenotype (i.e. in terms of productivity for cell lines, the differentiation stage for stem cells or the functional phenotype of primary cells, such as chondrocytes, osteocytes, hepatocytes or neurons) may be an issue as important as cell growth.
This article provides an overview on critical issues in cell culture of anchorage-dependent cells and provides perspectives for future developments, in particular, with respect to the large-scale amplification of anchorage-dependent stem cells for vaccine and cell therapy purposes.
2. Anchorage-dependent cells and their cultivation
(a). Biological properties of anchorage-dependent cells
All normal tissue-derived cells (except those derived from the haematopoietic system) are anchorage-dependent cells and need a surface/cell culture support for normal proliferation. By contrast, cells derived from the haematopoietic system as well as transformed cells (tumour cells) are substantially different and are able to proliferate in suspension and do not need any surface for cell growth. Tumoural cell transformation is accompanied by a modification of the phenotype (large nucleus to cytoplasma ratio, less attached and extended when adherent, tendency to round up, easier to adapt to serum-free culture condition) [1]. This modification also includes an increased resistance to apoptotic stress, partial or complete independence of growth factors and shift of the metabolism to abnormal glycolysis (anaerobic). All normal non-transformed anchorage-dependent cells require a culture surface for proliferation and its absence leads to growth arrest and induction of anoikis (a form of programmed cell death which is induced by anchorage-dependent cells detaching from the surrounding extracellular matrix (ECM)) [2].
As mentioned, normal primary tissue-derived cells (including stem cells, with the exception of cells from the haematopoietic system) absolutely require a culture support for self-renewal and differentiation. In contrast to transformed cells, stem cells need an environment comparable to the naturally existing stem cell niche consisting of soluble (such as growth factors and cytokines) and surface-bound signalling factors, cell–cell contacts, the presence of ECM and a local biomechanical microenvironment. Cell expansion and/or phenotype retention/modulation depends on the interaction of the cellular integrins with the integrin-binding molecules and other molecules of the ECM as well as a favourable biomechanical microenvironment. Both the adhesion substrate itself, the soluble and insoluble factors, as well as the mechanical microenvironment (including stress) are involved in modifications in cell expansion, morphology and differentiation (stem cell fate).
All adherent cells, but in particular, primary as well as stem cells are sensitive to shear stress. Shear stress generated by large-scale cell culture devices using microcarriers essentially results in growth reduction, cell detachment or cell death (see §3b(ii)). Moreover, in recent years it was established that the biomechanical microenvironment has an important impact on stem cells and, together with growth factor-mediated signalling pathways, regulates stem cell fate (see §5b). A careful engineering of the cell culture device and the operation conditions are thus an indispensable premise for a successful implementation of this technology for large-scale use.
(b). Strategies for the culture of anchorage-dependent cells
Since all cells initially developed and used for the production of viral vaccines for human use and for many veterinary vaccines were primary and later diploid and continuous (but anchorage-dependent) cells, initially only cell culture systems for surface adherent cells were developed and scaled-up. Since the way to scale-up cell cultures of anchorage-dependent cells is based on the increase in the cell culture surface, different solutions have been proposed and developed for the generation of large amounts of cell biomass for production purposes. This search for large cell culture surfaces passed by the evaluation of the simplest culture systems (different types of bottles), via more evolved culture systems, like stack-plate propagators or fixed-bed reactors, and led to the development of microcarrier-based culture systems which are, from a technological point of view, the most advanced culture systems for adherently growing cells.
Though traditional culture plate (max. surface: 175 cm2), roller bottle (max. surface: 1750 cm2; figure 1) or multitray (CellFactory (CF-10–10 stack unit: 6320 cm2, figure 2; CF-40–40 stack unit: 25 280 cm2, figure 3), CellStack) systems are most appropriate for small-scale cell culture, and more recent developments of these systems such as the HYPERStack cell culture vessel (Corning) have led to a certain scale-up, these culture systems are characterized by two major drawbacks: (i) their scalability is limited (largest cell culture unit: 60 000 cm2; HYPERStack 120) and can only be alleviated by the addition of parallel culture units (scale-out approach), and (ii) the culture conditions are not controlled with respect to pH or pO2 leading to a continuous change of the physico-chemical parameters over the culture duration (in parallel to the changing conditions during a batch culture). Thus, at least to alleviate the drawbacks with respect to control of the physico-chemical parameters, ATMI has introduced the Integrity Xpansion system consisting of multiple parallel plates and providing pH and pO2 control/regulation (via gas diffusion and medium circulation) in addition to a maximal surface area per reactor unit of 122 400 cm2 (Xpansion-200) [3].
Figure 1.
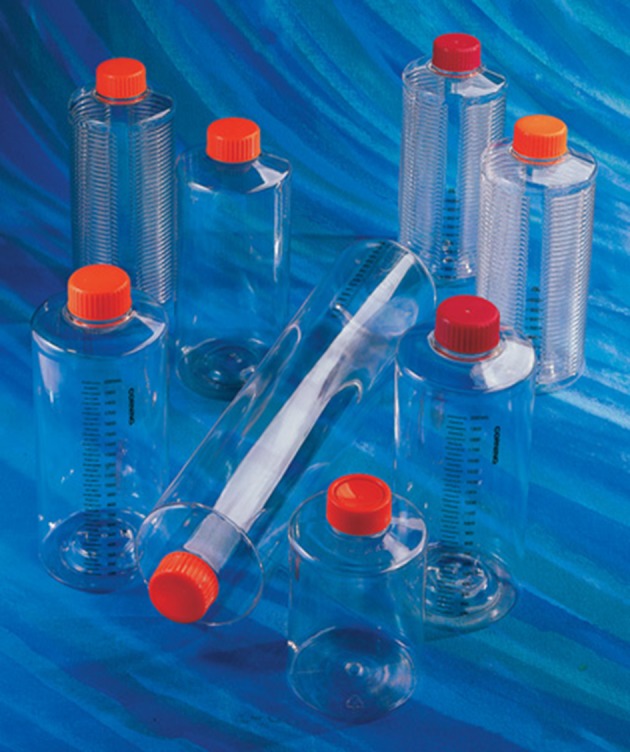
Roller bottles. Three different sizes are shown: 490, 850 and 1750 cm2 (Corning). (Online version in colour.)
Figure 2.

CellFactory (10 stack version) (Nunc). (Online version in colour.)
Figure 3.

CellFactory (40 stack version) (Nunc).
However, in view of real scalability (scale-up approach), a suspension process based on the use of microcarrier technology (see §3b) is the choice performed by the vaccine industry using mostly Vero or MDCK cells (figure 4 shows the growth of MDCK cells on Cytodex 1 carriers) for the routine production of viral vaccines at a maximum scale of 6000 l (estimated surface available: approx. 2430 m2, table 4) [5]. A further advantage of the use of such a homogeneous culture system (using stirred-tank reactors) is that it is controlled for pH and pO2 levels. This technology is also of potential interest for the expansion of stem cells for allogeneic stem cell therapy because for this type of cell therapy huge amounts of stem cells will be necessary for the treatment of large patient groups (→ off-the-shelf products).
Figure 4.
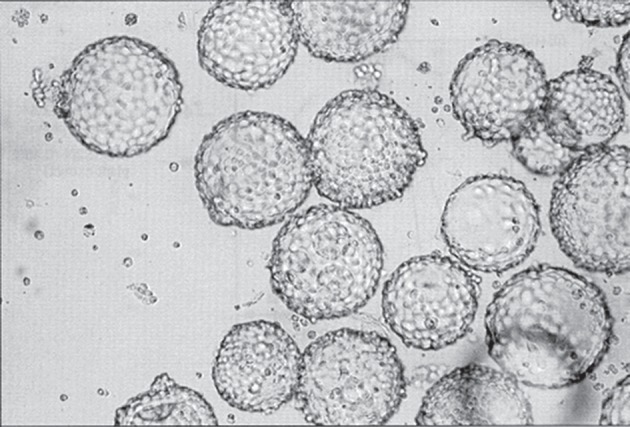
MDCK cells grown in serum-free medium on Cytodex I microcarriers. MDCK cells were grown on Cytodex I microcarriers (5 g l−1) to a cell density of 5.8 × 106 c ml−1 in MDSS2N medium containing 0.45% plant extract. The diameter of the microcarrier beads is ≈180 µm. (Adapted from Merten et al. [4], reproduced with permission from IABS, Geneva.)
Table 4.
Large-scale manufacturing facilities for the production of viral vaccines and proteins using anchorage-dependent cells.
| production technology | principle | largest scale available | cell line | product(s) | references |
|---|---|---|---|---|---|
| microcarrier cultures in stirred-tank reactors | Cytodex 3 (unknown concentration) | 6000 l—2430 m2a | Vero | human vaccine (influenza) | [5,21] |
| Cytodex 1 (1.5 g l−1) | 1000 l—660 m2 | Vero | human vaccines (polio, rabies) | [17–19] | |
| roller bottles | 7200 glass roller bottles/ production unit | 446.4 l—446.4 m2 | BHK, calf kidney cells | veterinary vaccines (FMD, IBR, PI3, Aujesky virus) | [8] |
| fully automated disposable roller bottle system: 96 racks with 45–90 roller bottles/rackb | 1st: 45 rollers/rack: 432–2100 l—367.2 m2, or 2nd: 90 rollers/rack: 864–4320 l—734.4 m2 |
rCHO | EPO | [9] | |
| fixed-bed reactor system | CellCube (unit 340 000 cm2) | unknown volumec—34m2 | MRC-5 | human vaccine (hepatitis A) | [57] |
| CellFactory | 4 CF-40 units | 9.6 l–10.1 m2 | human fibroblasts | β-interferon | [10] |
aSupposition of use of 1.5 g l−1 of carriers.
bThe exact number of roller bottles/rack is not known, and it is supposed that a rack can carry either 45 or 90 roller bottles (850 cm2); since a roller bottle can be filled with 125–500 ml of medium, the minimal and maximal volume per production batch is given.
cSince hepatitis A virus is not released from the cells, only the cell lysate is processed.
Although microcarrier technology is well adapted for large-scale production of cell biomass as well as of various biological products, other culture systems for the expansion of adherent cells should not be neglected, in particular for stem cell technology. In the case of autologous stem cell therapy (patient-specific therapy), estimated culture volumes of up to 70 l, range 2–70 l depending on the therapeutic approach [6], will be required per patient signifying that culture systems of a small or intermediate scale (see §§3c and 4b), such as fixed-bed reactors, can be envisaged. In most cases, they are characterized by the same advantages as classical stirred-tank reactors with respect to the control of physico-chemical parameters, with the advantage of reduced shear stress to cells but increased technical difficulties to harvest the cells for further use if the carriers/scaffolds are not used for direct cell delivery, as for bone or cartilage repair, for instance [7].
3. Culture systems for large-scale culture of anchorage-dependent cells—purpose: production of viral vaccines and recombinant proteins
As mentioned, the expansion/amplification of anchorage-dependent cells is only possible on culture surfaces and, according to the production needs, the production scale depends mainly on the available culture surface. In dependence on the purpose, different scales have been developed using various technologies (see above). In the following, the largest developed production scales established for different purpose are reviewed.
(a). Parallelized monolayer culture devices
The simplest way to expand surface adherent cells is the use of large surface areas based initially on glass (at the beginning of the large-scale vaccine production using anchorage-dependent cells) and later on tissue culture polystyrene-based culture devices. These devices include Povitsky bottles, roller bottles, stack plate propagators and multitrays (CellFacory, CellStack). The productions are performed using multiple processing systems consisting of the multiplication and parallel processing of single culture units. Such a production system, for instance, was established for the production of veterinary viral vaccines. The Instituto Zooprofilatico Sperimentale in Brescia/I has installed four individual units (incubators) each with 7200 glass roller bottles for the production of different viral vaccines using baby hamster kidney (BHK) and calf kidney cell cultures. One production unit of 7200 rollers of a surface area of 620 cm2 per roller provided in such a production facility a culture surface of 446.4 m2 [8]. The main drawbacks were that it was mainly a manual system requiring many operators, and that such a system is generally characterized by potential biohazard problems and higher losses due to contamination than when using reactor processes.
Although, from a technological point of view, this is an archaic production system, probably for reducing the time from development to market Kirin had opted to install an automated large-scale production system based on the use of an estimated number of 4320–8640 rollers (available culture surface: 367.2–734.4 m2) allowing the production of erythropoietin (EPO) using adherently grown recombinant Chinese hamster ovary (CHO) cells [9]. This automation reduced the number of operators required for running such a production plant and led also to an improvement of biohazard and contamination issues.
Another, in principle, similar culture system is the multitray system (today CellFactory—CF) which had been scaled to a 40-layer stack (figure 3) providing on its own a culture surface of 25 280 cm2. As for the roller bottle system, the use of several units in parallel allows the scale-up of the production. In this context, a robot for the parallel handling of 4 CF-40 stacks had been developed providing a surface of 10.1 m2 per handling unit. The development for this type of production system had been essentially driven by the need for the production of large quantities of β-interferon using human fibroblast cultures [10].
The main disadvantages of all these systems are the absence of pH and pO2 control—thus absence of possibility for real process optimization—and the limited scalability—only a linear scalability is possible which is based on the addition of further culture units for increasing the surface area for cellular biomass propagation. These drawbacks could finally be alleviated by the introduction of microcarrier technology [11], the identification of the optimal range of exchange capacity of positive charge carrying microcarriers at about 2 meq g–1 microcarrier [12,13] and general optimization of culture conditions (see §3b).
(b). Use of microcarrier technology in stirred-tank reactors for large-scale culture of adherent cells
(i). Microcarrier technology: basics
The first description of the possibility of growing anchorage-dependent cells on microcarriers in suspension was published by van Wezel in 1967 [11]. This was a breakthrough allowing the alleviation of the limited scalability of the classical cell culture systems, in particular because of their high surface/volume ratio, leading to a considerably reduced footprint for the culture device. Further advantages of the microcarrier technology were the use of a homogeneously stirred suspension allowing monitoring and controlling of various environmental parameters, including pH, pO2 and concentration of medium components, leading to a more optimal and reproducible cell culture process. In addition, representative cell samples could be taken and analysed.
However, before implementing this technology for routine manufacturing, many improvements and engineering studies had to be performed. The improvements directly related to the microcarriers are presented in the following, whereas details on the hydrodynamic studies for better understanding the strengths and weaknesses of microcarrier technology are presented in §3b(ii).
Surface charge. Levine et al. [12] identified the optimal exchange capacity (positive charge–DEAE-dextran) of microcarriers to be between 1 and 2 meq g−1 carrier for a range of cell lines. Charge density is a critical parameter. If charge density is too low, cell attachment will be insufficient and below 1 meq g−1 no growth is observed [12], whereas too high a charge density will have a toxic effect leading to limited or no cell growth.
Carrier diameter. Maroudas [14] could show that there was a considerable growth reduction on spherical carriers with a diameter of less than 50–70 µm, whereas beyond this limit no differences in comparison to cell growth on planar culture surfaces were seen. As a general rule, microcarriers should have a diameter of 100–200 µm; however, in order to ensure a homogeneous culture as much as possible with most carriers becoming confluent at approximately the same moment, the variations of the diameter of the carriers should be as small as possible. This is, in particular, valid for the expansion of stem cells because a better defined size distribution (with a reduced s.d.) leads in general to a more homogeneous cell culture and time point of achieving confluence/desired harvesting cell density.
Density. The density of the microcarriers should be slightly greater than that of the culture medium thus facilitating an easy separation of cells and medium. However, the density should also be sufficiently low to allow complete suspension of the carriers at a minimum stirring rate in order to avoid hydrodynamic damage to the culture (see below). Moreover, it should be mentioned that the density of the carriers is gradually increasing with cell attachment and growth. Microcarrier densities of 1.03–1.05 g ml−1 are considered optimal.
Microcarrier types. Since the development of the concept of microcarriers by van Wezel [11] many different types of carriers have been developed and they can be divided into positively charged carriers, such as Cytodex 1 (dextran-based, GE Healthcare); collagen or ECM-coated carriers, such as Cytodex 3 (dextran-based, GE Healthcare) or HyQspheres Pro-F 102–4 (polystyrene-based, Thermo Scientific); non-charged carriers, like HyQspheres P 102–4; or macroporous carriers based on gelatin (Cultisphere, Percell Biolytica) or cellulose (Cytopore, GE Healthcare) (table 1).
Table 1.
Different types of microcarriers commercially available (from Chen et al. [15], reproduced with permission from Elsevier).
| type | microcarrier | matrix | dimension (pore size) | surface feature |
|---|---|---|---|---|
| positive charged | Cytodex 1 | dextran | spherical ∅︀ 190 ± 58 µm | diethylaminoethyl (1.2–1.6 meq g−1 dry) |
| DE-52 | cellulose | cylindrical L 130 ± 60 µm × ∅︀ 35 ± 7 µm | diethylaminoethyl (0.88–1.08 meq g−1 dry) | |
| DE-53 | diethylaminoethyl (1.8–2.2 meq g−1 dry) | |||
| QA-52 | quaternary ammonium (1.09 meq g−1 dry) | |||
| HLX II-170 | polystyrene | spherical ∅︀ 170 ± 10 µm | triethylamine | |
| P Plus 102-L | spherical ∅︀ 169 ± 44 µm | cationic charged | ||
| FACT 102-L | cationic charged and type 1 porcine collagen | |||
| collagen coated | CGEN 102-L | type 1 porcine collagen | ||
| Cytodex 3 | dextran | spherical ∅︀ 175 ± 36 µm | denatured pig skin type 1 collagen | |
| ECM coated | Pro-F 102-L | polystyrene | spherical ∅︀ 169 ± 44 µm | recombinant fibronectin |
| non/negative charged | P 102-L | uncoated | ||
| 2D Microhex | hexagon L 125 µm × W 25 µm | tissue culture treated | ||
| macro-porous | Cultisphere G | gelatin | spherical ∅︀ 255 ± 125 µm (10–20 µm) | gelatin |
| Cultisphere S | ||||
| Cultisphere GL | spherical ∅︀ 255 ± 125 µm (50–70 µm) | |||
| Cytopore 1 | cellulose | spherical ∅︀ 240 ± 40 µm (30 µm) | diethylaminoethyl (0.9–1.2 meq g−1 dry) | |
| Cytopore 2 | diethylaminoethyl (1.65–1.95 meq g−1 dry) | |||
| weighted | Cytoline | polyethylene and silica | lens-shape L 2.1 ± 0.4 mm × W 0.75 ± 0.35 mm (10–400 µm) | a slight negative charge |
However, at the end of the day, the choice of the carrier to be taken for cell mass production depends essentially on the cell line to be cultivated. ‘Normal’ (diploid and continuous) cell lines can easily be expanded on positively charged carriers, such as Cytodex 1. Such carriers had been initially used for the production of foot and mouth disease (FMD) virus vaccine using pig kidney cells [16], and today are extensively used for the manufacturing of polio and rabies virus vaccines using Vero cells [17–19].
Coated carriers (collagen, ECM, other coatings) are particularly useful for establishing primary cell cultures, for cells that are difficult to grow [20] and more recently for the cultivation of different types of stem cells [15]. In addition, collagen-coated carriers are also used for the production of influenza virus vaccine using Vero cells [21]. Collagen-coated or gelatin-based carriers have the advantage that cell detachment can easily be performed with various proteases with minor damage to the cells. Moreover, carriers consisting entirely of gelatin can be completely disintegrated leaving the cells in suspension.
The microcarrier type chosen has a direct impact on the amplifiable cell number per carrier. Whereas solid microcarriers, such as Cytodex 1 or 3, provide a surface area of 4400 and 2700 cm2 g−1 dry weight, respectively, porous carriers (such as Cultisphere) provide higher (but unspecified) surface area for cell growth because the cells can also colonize the pores of these carriers allowing the generation of more than four times the cell biomass normally obtained with dextran-based carriers (http://www.percell.se/principles40.gif). A further advantage of porous carriers is that cells are shielded from shear-induced damage [22] and shear stress [23]. A detailed description of different carriers available today can be found in Chen et al. [15].
In view of their use for the expansion of stem cells, in particular, solid microcarriers have some critical issues requiring further study. First, stem cells need specialized coating for expansion as well as differentiation. However, microcarrier surface pattern and distribution of the ECM are poorly controlled. And second, the mechanical properties of microcarriers have to be characterized and controlled for improved cell functions because the stiffness of the support has an impact on stem cell fate (see §5a,b for more details).
Seeding density. Hu et al. [24] performed a mechanistic analysis on inoculum requirements for the cultivation of mammalian cells on microcarriers and established a minimum critical cell number per carrier for colonizing a maximum number of carrers (Poisson distribution). Using a human fibroblast cell line, the critical cell number was identified to be five to six cells per carrier (type: positive-charged dextran-based carrier—Cytodex 1-like); however, this number can be reduced by employing an improved culture medium. An equivalent optimal cell to bead ratio, 7, was determined for MDCK cells [25].
In the case of macroporous carriers (Cutlisphere-G), Ng et al. [23] established 30 cells (Vero) per carrier as minimum cell number to ensure an even distribution of cells on the available microcarriers with a low proportion of unoccupied beads.
(ii). Use of stirred-tank reactors for large-scale culture of adherent cells on microcarriers: engineering issues
The development of the microcarrier technology by van Wezel [11] made it possible to use agitated suspension culture systems for the propagation of anchorage-dependent cells. At laboratory scale, the simplest system consists of an agitated spinner flask of several tens to hundreds of millilitres with either a stirred magnetic bar, a derivative of it (figure 5a) or a ball-shaped eccentrically rotating agitator (figure 5b), whereas at a large scale, stirred-tank reactors of 1000 l [19] to 6000 l [5] have been implemented for routine manufacturing.
Figure 5.
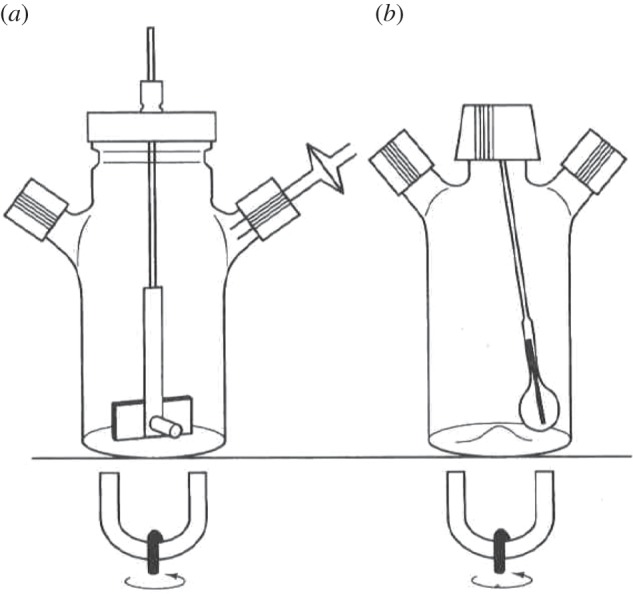
Two different types of spinner flasks: (a) spinner with a magnetic bar stirrer and (b) spinner with a ball-shaped eccentrically rotating agitator.
In addition to the availability of a controlled environment, the advantage of a stirred-tank reactor is the fact that under ideal conditions a homogeneous culture situation is achieved, meaning that anywhere in the culture vessel the culture environment is identical. Though the agitation should result in a homogeneous culture situation, in reality this is not the case because any agitation will lead to the generation of turbulences which are required for mixing purposes but which are also characterized by several drawbacks. Thus, mixing of microcarrier-based cultures at a large scale is a critical issue and needs careful optimization; however, details on mixing conditions and choice of the agitator at a large scale are proprietary and are not available. For a better comprehension of critical issues of agitation at a large scale, basic engineering issues are described in the following. For more details, the reader is referred to the articles by Cherry & Papoutsakis [26] and Croughan & Wang [27].
Reactor hydrodynamics and shear sensitivity. Cells grown on microcarriers in an agitated culture system are exposed to shear stress and turbulences with potentially adverse effects on cell growth and/or production. Although in the 1990s considered as particular shear sensitive, cells are much more robust than previously thought [28].
Turbulences (table 2). In order to obtain efficient mixing and thus a homogeneous culture it is necessary that the suspension culture be performed under a turbulent regime, signifying that Reynold's number (Re, a dimensionless quantity that is used to help predict similar flow patterns in different fluid flow situations; http://en.wikipedia.org) exceeds approximately 10 000 [29,30]. Reynold's number is defined as
where N is the impeller rotation rate, Di is the impeller diameter and ν is the kinematic viscosity. Turbulence distribution throughout the reactor depends on impeller and vessel geometry.
Table 2.
Formulae for modelling hydrodynamic effects on animal cells grown on microcarriers and cell aggregates in suspension cultures.
| parameter | formula | explanations | significance, remark | reference |
|---|---|---|---|---|
| Reynolds number | Re = (NDi2)/ν |
N = impeller rotation rate Di = impeller diameter ν = kinematic viscosity Re = dimensionless quantity that is used to help predict similar flow patterns in different fluid flow situations |
Re > 10 000—turbulent regime turbulence distribution throughout the reactor depends on impeller and vessel geometry |
[29,30] |
| Kolmogorov eddy length | L = (ν3/ɛ)¼ |
ɛ = power dissipation per unit mass (or specific energy dissipation rate) ɛ = NpDi5N3/Vd, where Np = dimensionless power number Vd = dissipation volume |
the eddy size decreases as the agitation speed increases. Kolmogorov eddies with sizes similar to approximately 70% of the carrier/aggregate diameter are detrimental for the adherent cells | [31] |
| maximum mean aggregate size | Dmax = Cɛανβ | parameters C, α and β can be estimated from experiments (e.g. [32]) | the equation permits the estimation of the maximal aggregate/clump size dependent on kinematic viscosity and viscous energy dissipation | [33] |
| maximum shear stress | τmax = 5.33ρ(ɛν)½ | ρ = density | suspended single cells are damaged at high agitation rates; the formula provides estimation of the maximum shear stress which a cell experiences in suspension | [34] |
The effects of turbulent eddies on cells growing on microcarriers have been examined using the Kolmogorov eddy length model. Under conditions of isotropic equilibrium in the viscous dissipation regime, the size of the smallest eddies can be roughly given by the Kolmogorov eddy length (L):
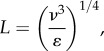 |
where ɛ is the power dissipation per unit mass (or specific energy dissipation rate).
Croughan et al. [31] have shown that the Kolmogorov eddy length can be correlated with relative growth extent. Eddy lengths comparable to the microcarrier size and greater have little effect on cell growth, while cell death becomes apparent when the average eddy length falls below two-thirds of the microcarrier diameter. In this situation, eddies directly interact with the adherent cells and thus damage them whereas they are too small to efficiently move the carriers. The smaller the eddy sizes are the more the relative specific growth rate is reduced.
It is important to note that turbulent energy dissipation rate in a stirred-tank reactor culture is always given as an average value; the maximal energy dissipation rate is reported near the stirrer, whereas the lowest values are found near the top surface. The ratio between the real energy dissipation rate at a defined locus and the average value can vary between 30 (near the stirrer) and 0.2 (near the top surface) [35]. This signifies that the cells growing on the microcarriers suffer considerable variations in turbulences when moving through the culture medium with potential impact on expansion and stem cell fate in the case that stem cells are expanded using such a culture system. Further details can be found in Nienow [28].
Further damage to cells may result from bead-to-bead collision or collision between microcarrier-bound cells and impeller, vessel wall and baffles and other reactor inserts (in particular when higher microcarrier concentrations are used) [36,37]. Work on these issues suggests that the potential damage increases rapidly with the average energy dissipation rate, with increasing microcarrier size and also with increasing solids concentration [37]. Thus, it is interesting to note that the use of smaller microcarriers has been reported to reduce cell death and increase growth rates [26], by respecting the required minimum diameter [14]. For more details on cell damage due to bead-to-bead collision or collision between carrier-bound cells and reactor inserts, see Cherry & Papoutsakis [38] and Croughan & Wang [27].
In addition to the modification of the agitation regime and the size of the microcarriers, there are further approaches to reduce potential shear damage to cells grown on carriers. On the one hand, in case of very fragile cells, in particular cells infected by viruses for virus production, it was shown that the replacement of solid carriers such as Cytodex 1 by Fibra-cel discs, in which the cells are immobilized, led to higher (rabies) virus production probably because of reduced cell loss caused by bead collisions [39]. On the other hand, cells can also be protected from turbulences at high agitation by addition of thickening agents (polymers to increase viscosity) [40].
Shear stress (table 2). Each agitation induces some shear stress in microcarrier cultures, in particular, when using solid microcarriers. Shear at a low level is required to prevent sedimentation and aggregation of cells and microcarriers at high cell concentrations. However, the maximal acceptable level depends on the culture support and the cell line. The following equation provides an estimate of the maximum shear stress which a cell experiences [34]:
It was established that shear stress of 0.26 N m−2 had no effect on cell viability, whereas higher shear levels of 0.65–1.30 N m−2 led to morphological changes, loss of viability and cell removal. At even higher levels (more than 2.6 N m−2) more than 75% cell detachment was observed [41]. Moreover, it has also to be considered that increased shear stress has a modulating effect on the cellular metabolism.
However, since its calculation is based on Kolmogorov's theory and gives the stress as the Kolmogorov length a much better estimation for keeping suspension cells viable and able to produce the product with the required quality is the comparison of the Kolmogorov length and the cell size.
Scale-up. Scale-up is the predictable—engineered—increase in the production capacity. It is the base for the establishment of large-scale/industrial-scale production methods using stirred-tank reactors. The critical issue is that cell damage can occur due to the elevated energy dissipation at large scale signifying that scale-up is performed by keeping constant energy input or Reynold's number across the reactor scales. More details can be found in the electronic supplementary material (SM1).
Other issues. Aeration of large-scale cell culture can only be efficiently performed via injection of gas into the medium (sparging) in order to get an efficient gas transfer. The main risk is the potential foam formation, because microcarriers accumulate at the liquid–foam interface and thus are lost for the culture. The use of microspargers (reduction of bubbles) and/or the addition of tensioactive substances to the medium can prevent foam formation. In addition, hydrodynamic stress induced due to rising bubbles (probably due to power dissipation in the fluid adjacent to rising bubbles) can be alleviated by the addition of protective polymers [42,43].
(iii). Subcultivation of anchorage-dependent cells at large scale
For passaging adherently growing cells, in general, they have to be detached from their culture surface, diluted and plated into another culture device. At a large scale, this is essentially performed by the use proteases, of which porcine trypsine ± ethylenediaminetetraacetic acid was the most often used detachment agent. Today recombinant and non-animal-derived proteases and trypsine-like proteolytic enzymes are available for replacing porcine trypsine for industrial use (for details, see Merten [44]). At large scale, trypsinizers allow washing of the cells still bound to the carriers after short trypsine incubation as well as the separation of the detached cells from carriers. Such systems had been developed as glass/stainless steel devices [45] or more recently as disposable devices (HyQ Harvestainer, Thermo Scientific).
The efficient replacement of proteolytic cell detachment by a non-proteolytic method would be interesting, because no agent would be added and have to be removed after passaging. However, this has not yet been achieved for routine use at a large scale.
One possibility is the direct bead-to-bead transfer which was shown to work for several cells, in particular, for those which show less adhesion strength to the support than those which are very well attached. This depends obviously on the cell line as well as on the nature of the microsupport. It has been shown that continuous cell lines, such as BHK-21 [46], different clones of CHO [47–49], Vero [46,50,51] and MDCK cells [46] could colonize fresh carriers. Colonization capacity was described for Cytodex 1 [46,48], Cytodex 3 [46,48,50,51], Cytopore 1 [48,49], Cytopore 2 [48], Cultisphere G [47] and for Biosilon [52], although others [48] have seen a less efficient cell transfer for another CHO clone using different culture conditions. The evaluation of other microcarriers with respect to the efficiency of bead-to-bead transfer of CHO cells showed that collagen, fast attachment collagen-treated and different Cultisphere carriers were less efficient than the different Cytodex and Cytopore carriers [48]. On another side, diploid cells, like human fibroblasts, are completely unable to perform bead-to-bead transfer [24]. More details are presented in table 3.
Table 3.
Bead-to-bead transfer of adherent cells.
| cell line | microcarrier type | colonization efficiency of newly added beads | scale (working volume) | references |
|---|---|---|---|---|
| Vero | Cytodex 3 | addition of new beads (1 : 1) at confluence; intermittent agitation for 2 days after addition of new beads; 2 days later most of the carriers were confluent | 200 ml spinner scale | [50] |
| efficiency of bridge formation of importance for bead-to-bead transfer at confluence; 8 h intermittent agitation after addition of new beads; 2% of residual empty beads 6 days post-addition | 30 ml spinner scale, addition of 60 ml of a carrier suspension | [51] | ||
| BHK21 | 1/7 split evaluated; 1 day post-addition of fresh carriers about 80% of carriers contained more than or equal to 2 cells per carrier; 2 days post-addition, more than 90% of the carriers were populated | 30 l reactor | [46] | |
| CHO clones | Cytodex 3 | at more than 5 × 105 c ml−1, split 1/5, addition of new carriers: 1 day post-addition, 100% of carriers contain cells | spinner scale | [48] |
| Cytodex 1 | 1 day post-addition, 100% of carriers contain cells | |||
| Cytopore 2 | 1 day post-addition, 84% of carriers contain cells, at 4 days: 100% | |||
| Cytopore 1 | 1 day post-addition, 75% of carriers contain cells, at 4 days: 100% | |||
| 3- to 20-fold scale-up possible via addition of new carriers, complete confluence within some days after addition of fresh carriers | 200 ml spinner flasks—1.5–20 l reactor | [49] | ||
| Biosilon | threefold scale-up via addition of fresh carriers, all fresh carriers were covered 3 days later | 200 ml spinner flasks—1.5 l reactor | [52] | |
| CHO-K1 | Cultisphere G | 5–50 cells/bead are sufficient for effective seeding; within 1 day of bead addition, most of added beads show evenly attached cells | 3.1 l reactor | [47] |
| human fibroblasts (FS-4) | positive-charged dextran carriers | diploid fibroblasts are unable to colonize newly added beads | spinner scale | [24] |
Concerning implementation at a large scale (beyond a reactor scale of 30 l [46]), the open literature does not report on any study on or large scale use of bead-to-bead transfer of cells. In view of high cell density culture processes, bead-to-bead transfer is of high interest, because it allows the augmentation of the carrier concentration and thus available culture surface for surface adherent cells. That this can be a feasible approach was shown for CHO cells by Ohlson et al. [47] and by Xiao et al. [49,52].
A general problem with this type of cell transfer is the fact that a high percentage of microcarrier aggregation and multilayering of cells is observed, meaning a certain degree of heterogeneity within the aggregates. This is probably not optimal for routine processes [46], although when producing recombinant proteins using rCHO cells this might be a lesser problem than when producing viruses, for which a more homogeneous culture is of higher importance for quality reasons.
(c). Bioreactor systems for the culture of adherent cells at intermediate scale
For intermediate (medium)-scale cell culture expansion other reactor systems including WAVE and packed-bed reactors can be used, which are well characterized by reduced shear-fields and are thus better placed for the expansion of shear-sensitive cells or the production of shear-sensitive products. Whereas the WAVE reactor can be used at a maximum scale of 500 l (batch mode) a packed-bed reactor of a bed volume of 21 l can generate up to 1500 l d−1 (perfusion mode, for the production of secreted products). In addition, WAVE reactor systems are often used for the generation of biomass for the inoculation of large-scale reactor cultures, whereas packed-bed reactors are also of interest for the expansion of stem cells. Details on both systems can be found in the electronic supplementary material, section SM2.
(d). Use of microcarrier and packed-bed technology for the production of biologicals at an intermediate/large scale
Today the general tendency is towards the use of culture processes using suspension cells because such processes are, from a technological point of view, less complex than cell culture processes for anchorage-dependent cells. However, most of the viral vaccines (those developed in the 1970s, but also those recently developed) are produced using anchorage-dependent cells using mainly a microcarrier-based manufacturing platform.
Thus, based on the initial development done for the establishment of a large-scale routine production process of FMD virus vaccine using microcarriers [16], other large-scale production processes of human vaccines (polio and rabies virus vaccine) have been developed at a 1000 l scale using Vero cells [19]. Over the last 10 years, many novel viral vaccines for human use have been developed using the Vero cell platform technology (for details, see Barrett et al. [5]), and more recently this technology had been scaled-up to 6000 l for the production of influenza virus vaccine [5]. The only other surface-adherent cell line which had been scaled-up for large-scale production of a viral vaccine is the MDCK. Several different clones of MDCK (anchorage-dependent and suspension-adapted clones) have been developed as cell substrate for influenza virus vaccine with a maximum production scale of 2500 l (microcarrier-based process) and of 10 000 l (suspension cell-based process) [53,54].
All these production processes are low-density processes (less than 5 g l−1 Cytodex) and only relatively recently have high-density processes with a Cytodex concentration of up to 25 g l−1 in a perfusion mode been set up (e.g. [55]). The availability of porous microcarriers has greatly increased the scope of microcarrier technology by increasing unit yield and specific productivity due to enhanced perfusion efficiency and protecting fragile cells from culture turbulences.
For medium-scale manufacturing, for instance, based on the use of human diploid fibroblasts, systems other than microcarriers are used, such as fixed-bed reactors of the CellCube style [56,57], because fibroblast cultures tend to heterogeneous cell distribution and to aggregation when the cultures get confluent as well as during virus production (hepatitis A) [56,58]. For the same reason, the ‘large-scale’ CellFactory system had been initially implemented for the production of fibroblast interferon [10] (see §3a). Table 4 presents a short update of the largest manufacturing scales using anchorage-dependent cells implemented in the past for the production of viral vaccines and proteins, and clearly puts into perspective the advantages of a microcarrier-based production system with respect to available cell culture surface and produced supernatant volume.
4. Anchorage dependence: evolution to novel applications
Large-scale standard production platforms have been established for the production of biologicals, including viruses and recombinant proteins. However, initially these platforms had not been designed for the generation of biomass for further use, in particular, for regenerative medicine or cell therapy purposes using stem cells.
(a). Stem cells for tissue engineering
In contrast to traditional continuous cell lines, including Vero or MDCK cells, stem cells have the following basic properties: on the one hand, they are characterized by self-renewal meaning by unlimited or at least long-term survival with self-renewal capacity, and on the other hand by their differentiation potential meaning that they can be induced to differentiate into different lineages. The maintenance of self-renewal and the induction of differentiation are tightly linked with regulation of a specific microenvironment, including biochemical and biomechanical components.
In contrast to haematopoietic stem cells which can be cultured as single cell suspension in culture, mesenchymal stem cells (MSCs) and pluripotent stem cells (PSCs) require culture devices for anchorage-dependent cells or when cultured as aggregates culture devices for suspension cultures. If cultured as adherent cells, the attachment needs are different for both stem cell types.
MSCs display in vitro features of mesenchymal progenitor cells. They are human fibroblast-like, plastic-adherent cells and are characterized by expression of fibroblastic markers and an in vitro differentiation potential into adipocytes, chondrocytes and osteoblasts [59]. Although they have a limited lifespan in vitro [60] their large spectrum of trophic activities enables their clinical use. Today, it is believed that the secretion of a broad spectrum of bioactive molecules (growth factors, cytokines, chemokines) is the main mechanism by which MSCs achieve their therapeutic effects. These activities can be divided into six categories: immunomodulation, anti-apoptosis, angiogenesis, support of growth and differentiation of local stem progenitor cells, anti-scarring and chemoattraction [61].
PSCs include embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). The first human ESCs (hESCs) were derived from frozen or fresh blastocysts left over from infertility treatments whereby the inner cell masses were isolated from cultured blastocysts [62]. Human iPSCs were first established by Takahashi et al. [63] via transduction of human fibroblasts with four genes (Oct3/4, Sox2, Klf4 and c-Myc) leading to their reprogramming. Both types of PSCs have unlimited self-renewal ability and the capacity to differentiate into lineages of the three germ layers [62,63].
The culture requirements are different for MSCs and PSCs. Despite their requirements differing from those of continuous cell lines used for the production of viral vaccines and recombinant protein, the existing technology can be used after adaptation for the generation of cell mass for clinical use and screening purposes. However, before any development or scale-up of a cell manufacturing platform it is necessary to get an idea of what quantity of cells will be needed per patient or per clinical trial phase, as well as which kind of stem cells will be required. Today, most of the clinical trials (more than 320; www.clinicaltrials.com) make use of MSCs and only very few trials (in total: three) deal with the assessment of hESCs.
Depending on the indication, the number of cells (cell dose) can vary profoundly, from 50 000 (treatment of macular degeneration using hESC) to 6 × 109 cells (treatment of osteogenesis imperfecta using MSCs) [15]. Moreover, depending on the stem cells to be produced—either for autologous (use of expanded patient-derived cells) or for allogeneic (use of cells from universal donors) cell therapy—the scale of the cell culture devices will vary considerably. In the case of an autologous treatment mode, usually many small-scale productions have to be done, whereas for allogeneic cell therapy, large cell stocks for many patients have to be generated, meaning that in this case, very large culture systems are required.
Another very important difference between expanded MSCs and PSCs is the fact that MSCs do not require separation from undifferentiated cells because they can be directly used after expansion. However, this is not the case for PSCs because these cells have to be differentiated at least to the intermediate progenitor cell state. Since non-differentiated PSCs have the capacity to continue to grow indefinitively residual non-differentiated PSCs in a preparation of differentiated cells can form teratomas in the recipient—they have a tumorigenic potential. The use of PSCs for clinical purposes requires therefore a 100% efficient method to remove non-differentiated cells in order to preclude this potential safety problem (see §6b for further details). Basically due to this safety concern, up to now only a very limited number of clinical trials have been performed using PSCs.
(b). Culture configurations to expand stem cells
As mentioned earlier, the culture device as well as its scale has to be adapted to the cell mass to be produced. For large-scale cell expansion for the production of huge cell biomass as required for allogeneic cell therapy, either microcarrier-based or parallel-plate systems such as multitrays or HYPERstack vessels can be used (see §4(c) and §5), whereas small-scale expansion systems are used for the generation of small amounts of biomass as required for autologous cell therapy. Although the same culture principles as for large-scale biomass production can be used (microcarrier or parallel-plate systems), other culture devices might be better applied for this application.
(c). Stem cell expansion at small scale
For the production/expansion of smaller quantities of stem cells for autologous but also for allogeneic cell therapeutic purposes, when only small amounts of cells are required per patient, the following culture systems can be used, including hollow fibre cartridges in which the cells grow at tissue-like densities (up to 2 × 108 c ml−1; more as adherently growing cells) and packed-bed reactors, in particular of interest for the expansion of cells in scaffolds which are then directly implanted (perfusion reactor culture). Where stem cells are grown as aggregates, attached to microcarriers or immobilized in scaffolds, suspension culture systems are employed. A reactor system (stirred-tank reactor) is the culture system of choice when an unlimited scale-up under controlled conditions is required. However, stirred-tank reactor systems are characterized by potential hydrodynamic stress-related problems when not carefully optimized (see §4c). In this specific context, the WAVE reactor system, of interest for medium-scale cell expansion, is characterized by reduced hydrodynamic shear stress (see the electronic supplementary material, section SM2). Finally, the culture devices generating the lowest shear stress are the rotating wall perfused bioreactors. They are limited in scalability as well as in controlling aggregate size due to very low shear stress. More details can be found in the electronic supplementary material, section SM3.
As mentioned, stem cells can be expanded in the form of aggregates, clumps with or without microcarriers or microencapsulated (as single cells, aggregates or after attachment to microcarriers) [64]. This is a more natural form for expansion of stem cells because it reiterates more accurately cell adhesion and signalling found in an in vivo situation [65,66]. It is particularly prevalent for ESCs expressing E-cadherin in their pluripotent state enabling the cells to aggregate spontaneously [67]. These aggregates (embryoid bodies, EBs) have a tendency to differentiate into the three germ layers, and even when using microcarriers for expansion of PSCs this natural tendency to aggregation is preserved.
For the expansion of PSCs as aggregate cultures, stirred-tank reactor systems (spinner flasks, bioreactors) at low [64,68,69] or high cell densities using perfusion devices [70], WAVE reactors [68] and the rotating wall perfused bioreactors such as the slow turning lateral vessel (STLV) [71] have been used. Improved homogeneity of the size and morphology of EBs was demonstrated for stirred [72] and rotary [73] orbital culture systems. Moreover, several studies suggested that suspension culture devices were superior to a static culture system for hESC proliferation [72,74] as well as the generation of homogeneous-sized EBs [73]. However, there are also considerable differences among the various agitation systems: spinner flasks equipped with bulb-shaped impellers and STLVs were capable of forming EBs from single ESCs, whereas spinners equipped with paddle-type impellers and HARVs (high aspect ratio vessel) led to massive cell and EB aggregation [75,76]. In the context of the expansion of stem cell aggregate cultures in agitated reactor systems, Kinney et al. [77] have published a study on the impact of hydrodynamic culture environments on the relationships between stem cell aggregation, phenotype and metabolism. For a review on the use of these expansion systems for PSCs, see Placzek et al. [78].
Aggregate or clump cultures are less common for MSCs, because they have been traditionally cultured on two-dimensional plastic culture plates. However, they also have the tendency to form aggregates creating thus an ‘in vivo-like’ microenvironment which seems to better preserve their phenotype and properties [79,80]. A comparison of MSCs expanded on two-dimensional monolayers with those propagated as aggregates clearly indicated that aggregation enhances the differentiation potential, the secretion of trophic factors and of ECM molecules. The advantages for in vivo application of MSC aggregates have been demonstrated in animal models for bone and cartilage regeneration, wound healing, angiogenesis and cardiac transplantation. As for PSCs, aggregates of MSCs have been expanded in spinner flasks, rotating wall perfused and cyclic compression reactors (for more details, see Sart et al. [81]).
In addition to the expansion of MSCs as aggregates, they can also be expanded using scaffolds in a sort of derivative of an aggregate culture. Scaffold cultures are either performed as suspension (micro-)carrier cultures or as ‘fixed support’ cultures [82].
Zhang et al. [82] compared four different reactors for the expansion of hfMSCs (human fetal MSCs) on polycaprolactone/tri-calcium phosphate-based scaffold and established that a specifically developed bioreactor (BXR or biaxial rotating bioreactor) was superior for cell expansion as well as homogeneous distribution of the cells across the scaffold to the other three tested reactor types. These were a perfusion reactor (which is essentially a packed-bed reactor consisting of the scaffold as packed bed), a spinner flask reactor (in which the scaffolds were fixed to the cap of the spinner flask by pins) and the rotating wall perfused reactor STLV (in which the scaffolds were free floating; see electronic supplementary material, SM3); the order of the reactor systems corresponds to the performance of neo-tissue volume distribution in the scaffolds. The advantages of the BXR reactor could be explained by multiaxial flow across the scaffold preventing the loss of cells seen when a unidirectional flow is used (washout phenomenon, as in the case of the perfusion reactor) or due to random collision occurring between scaffolds themselves and culture chamber (in the case of the rotating wall perfused reactor). In addition, the microgravity condition of the STLV is known to have adverse effects on proliferation and maturation of osteogenic cells [83]. The disadvantage of the spinner flask reactor could be explained by the strong turbulent flow generated by the stirrer shown to be detrimental to the cells and newly formed ECM [82,84]. Since the assessed culture systems are not scalable, they can only be used for the expansion of stem cells for a patient destined therapy.
In view of treatment of cartilage defects, it could be shown that microcarriers/scaffolds consisting of PLGA (poly (lactic-co-glycolic acid)) and PLLA (poly-(l)-lactic acid) are suitable as an injectable delivery system for chondrocytes [85–87]. Moreover, the same type of scaffolds can be used for the expansion of osteoblast-like cells [88] and of MSCs. More details on different scaffolds can be found in a review by Placzek et al. [78].
(d). Stem cell expansion at large scale: theoretical considerations
From a cell culture device point of view, classically planar (two-dimensional) culture systems have been used for the generation of cell mass; however, for large-scale applications, such culture systems are often insufficient (i.e. same problems as for the large-scale production of viral vaccines, e.g. see §3a and table 4). In addition, it has to be taken into account that different types of stem cells are able to generate largely differing maximal harvest densities of cells per cm2; the harvest densities vary from 25 000 cells (MSCs) to 80 000 cells (human diploid fibroblasts) and to 160 000 cells per cm2 (human PSCs) [89]. Thus, for instance, using the largest planar cell culture system (robotic version of a 120 layers HYPERstack vessel (60 000 cm2 unit−1), consisting of 64 subunits providing a culture surface area of 3.84 × 106 cm2) an expansion run can generate between 120 × 109 cells (MSCs) and 768 × 109 cells (human PSCs).
Based on culture surface area, a similar cell quantity can be produced using a 350 l reactor culture with a microcarrier density of 2.5 g l−1 (calculation base: Cytodex 1 as example). With respect to minimal medium requirements (no medium change), the HYPERstack device will need about 830 l, whereas a microcarrier culture requires only 350 l. Goh et al. [90] reported on the advantages of the use of Cytodex 3 microcarriers showing their improved expansion capacity of hfMSCs with respect to planar culture systems. The expansion capacities are 12- to 16-fold and 4- to 6-fold, respectively [90]. A similar improvement was also reported for human iPSCs [91].
For providing a decision base for the use of different culture systems (normal planar versus automated planar versus microcarrier-based stirred-tank reactor system), Simaria et al. [92] have published a study on bioprocess economics and optimization in the context of allogeneic cell therapy using MSCs and confirmed the analysis provided by Rowley et al. [89]. Using technology S-curves by plotting the performance of each cell expansion technology in terms of 109 cells produced per lot against R&D effort/investment, they demonstrated that the use of microcarrier technology was only of interest when very large cell biomasses ranging from 47 × 109 to 4700 × 109 cells per lot (use of 8 × 20 l to 8 × 2000 l single use stirred-tank reactors) have to be produced. Automated planar cell culture systems are preferable for the production of smaller lot sizes with a maximum production limit of about 400 × 109 cells/lot (use of 80 units of the automated versions of 120 layers HYPERstack vessels). The production of larger lot sizes of 1013 cells/lot would require the use of more parallel 2000 l microcarrier cultures or the use of microcarriers with a larger surface area (e.g. use of macroporous carriers with the advantage of protecting/shielding the cells from stress) [93]. Figure 6 shows some details of the optimal production ranges of the different cell culture production systems but indicates also the R&D efforts required to move from a T-flask-based process to a large-scale expansion process.
Figure 6.
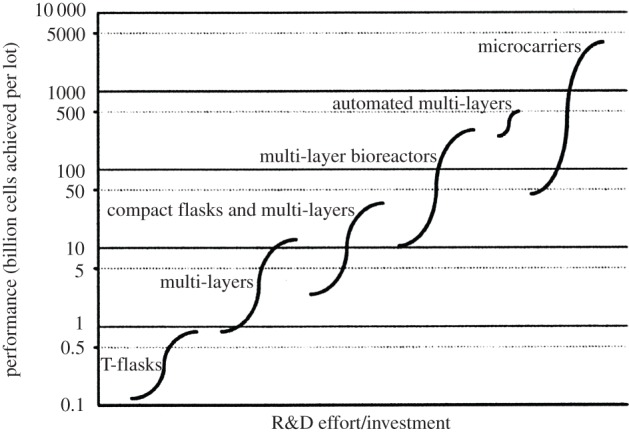
Conceptual illustration of a technology S-curve showing the evolution of expansion technologies used in cell therapy manufacture. The limits of each S-curve correspond to the amount of cells achieved by the smallest and largest size of each technology type when using the maximum number of units (80 for planar culture systems and eight for microcarriers). The culture systems are the following: T-flasks (75–500 cm2); multi-layers (CF-1–CF-10 CellFactories, 636–6360 cm2); compact flasks and multi-layers (HYPERflask–HYPERstack-36: 1720–18 000 cm2); multi-layer bioreactors (Integrity Xpansion-10–180: 6360–114 480 cm2); automated multi-layers (automated CF-40 CellFactories, 25 440 cm2—HYPERstack-120, 60 000 cm2); microcarriers (20–2000 l disposable stirred-tank reactor; chosen mid-point values: 2930 cm2 g−1 carrier, 6.3 g carrier l−1). The x-axis represents qualitatively the research and development effort required for a company currently using T-flasks to change to other cell expansion technologies. (Adapted from Simaria et al. [92], with kind permission from Wiley & Sons.)
It should be recalled here that in addition to the advantage of a bioreactor related to the fact of being very closely controlled for keeping the culture conditions optimal (such as control of pH or pO2, homogeneous culture system) which is not possible for the planar culture systems, cost (including cost of goods) differences and the much simpler handling of stirred tank reactors (including scalability; cf. §3) are also in favour of a microcarrier/reactor-based cell expansion system. Thus, as for the production of viruses using surface adherent cells, the development and use of microcarrier-based culture processes for the generation of large quantities of stem cells is the most straightforward and scalable manufacturing mode.
5. Specific issues regarding stem cell expansion and differentiation in microcarrier-based bioreactors
The most straightforward way to generate large quantities of stem cells for clinical application is the use of microcarrier culture systems. In principle, the acquired know-how obtained during the development of large-scale viral vaccine or recombinant protein manufacturing processes using microcarriers [26,27] will also serve for the development of large-scale cell expansion processes for stem cells, although engineering aspects have only partially been assessed [94]. Since the cells will ultimately be used in vivo, it is required that cell expansion is performed under serum-free/xeno-free culture conditions rendering large-scale cell expansion slightly more difficult because of the absence of the cell protective properties of serum. In the case of autologous cell therapy applications, autologous serum or platelet extracts are also applicable for clinical uses. Whereas, for MSCs, microcarriers without supplementary coating can be used for cell expansion in the presence of serum this is not the case when serum-free media are used. In the case of iPSCs, a coating is indispensable for cell attachment and proliferation.
(a). Choice of carrier and attachment factors
Since MSCs and PSCs behave differently, the choice of the microcarrier as well as their eventual coating have to be adapted to the needs of the specific stem cell type. A list of available carriers is presented in table 1.
(i). Requirements for the microcarrier expansion and differentiation of mesenchymal stem cells
Using serum-containing media, it was shown that MSCs could be expanded on gelatin (Cultisphere, Cytodex 3) as well as on positive-charged carriers (Cytodex 1) [15]. Kehoe et al. [95] compared different types of microcarriers and established that Cytodex 1 and 3 were most optimal for cell expansion but rather inefficient for cell recovery, whereas solid collagen-coated microcarriers (from Solohill) were 20–30% less efficient for cell expansion but most optimal for viable cell recovery. However, in all cases, the ECM proteins required for the interaction of the cells with the carriers were delivered by the serum-containing medium. When serum-free media are used for the expansion of MCSs using microcarrier cultures, these media have to provide the ECM proteins (such as fibronectin or vitronectin) normally delivered by the serum-containing medium for ensuring a satisfying interaction of the cellular integrins with the carriers via these factors [15,96]. Other possibilities would be the use of microcarriers coated with xeno-free proprietary cell adhesive-culture substrate (CELLstart from Life Technologies) as, for instance, evaluated by dos Santos et al. [97] for propagation of bone marrow and adipose-derived MSCs. Though CELLstart is xeno-free it is as ill-defined as serum [98].
It was observed that the expansion of MSCs was different among different cell sources, tissues of origin and age of donor [96] which can be explained by the large heterogeneity in the integrin expression profile among MSCs of different sources (e.g. [99,100]). Fetal MSCs, for instance, have a significantly higher proliferation and differentiation potential and, in addition, a lower immunogenicity than MSCs isolated from perinatal or postnatal origins [101].
The final aim of the expansion of MSCs is to differentiate them to the lineage of choice, and it should be recalled that a microcarrier which is optimal for expansion of the MSCs is not necessarily optimal for a certain lineage-specific differentiation. Recently, a relationship between actin organization of the cells on microcarriers and the ability of MSCs to differentiate to chondrocytes was shown. It was established that microcarriers favouring round shape and disorganized actin, such as Cytopore 2, promote chondrogenesis while microcarriers favouring cell spreading and formation of stress fibres inhibit chondrogenesis [102] and promote osteogenesis. Furthermore, gelatin- and collagen-based carriers were found to be suitable for promoting osteogenesis independent of tissue origin and species [90,103,104]. For more details on the use of microcarriers and their impact on stem cell expansion and differentiation, the reader is referred to the very comprehensive review recently published by Sart et al. [96].
(ii). Requirements for microcarrier expansion and differentiation of pluripotent stem cells
PSCs can also grow on microcarriers, but in contrast to MSCs they grow mainly as aggregates between cells and carriers or as pure aggregates. Despite the advances in the development of this specific technology, today there is not yet a clear view on which would be the most suitable microcarrier(s). Chen et al. [105] did a systematic study for establishing the impact of different carrier characteristics on propagation of PSCs: large spherical (e.g. Cytodex 1 and 3) and cylindrical carriers (e.g. DE-53) promoted higher growth due to a more open structure compared with small carriers (e.g. Tosoh 10 PR). The latter carriers lead to more compact aggregates whose degree of compactness increased with the decrease in the carrier size. Cell growth is probably reduced due to lack of nutrients caused by the compactness (diffusion limitation). Nonetheless, carriers have to be coated with ECM in order to allow cell attachment and propagation. For more details, the reader is referred to the review by Chen et al. [15].
Traditionally, PSCs are cultured using either inactivated feeder cells (mouse embryonic fibroblasts) or Matrigel (basement material prepared from mouse Engelbreth-Holm-Swarm sarcoma, containing laminin-111, collagen IV, perlecan, nidogen, fibronectin, entactin and other factors) -coated cell culture support. Obviously, both coatings are of animal origin, variable in their composition and characteristics (undefined nature) and have to be replaced by more defined coatings ideally of synthetic origin.
In principle, microcarriers can also be seeded with feeder cells, such as irradiated mouse embryonic fibroblasts, and hESCs can be expanded as on culture plates seeded with feeder cells although at a reduced growth rate. The main drawback is that the microcarriers have to be seeded twice, initially with feeder cells at day 0 and then the next day with the hESCs to be expanded, which might be impractical at a larger scale [106]. What is interesting about this approach is the possibility to freeze directly expanded hESCs adherent to microcarriers because of improved thawing recovery.
Classically, Matrigel, but also vitronectin and laminin have been used with success as coatings for microcarrier cultures of hiPSCs. For instance, the use of vitronectin and laminin as coating for microcarriers was evaluated for the expansion of hESCs by Heng et al. [107] and cell expansion (over 20 passages) similar to that on planar culture surfaces coated with the same attachment factors was obtained. The coating of carriers with ECM proteins, in particular, with laminin, via a cationic charge (poly-l-lysine) in between the carrier and the ECM protein further improved cell attachment and spreading efficiency and allowed direct seeding in serum-free medium under agitation [108].
However, the final aim is the use of either recombinant attachment factor-based coatings or better fully synthetic coatings. In this context, with respect to the expansion of hESCs in planar culture systems, different coatings based on the use of recombinant attachment factors, such as laminin 511, laminin 332 or laminin 111 (in order of efficiency of supporting adhesion and proliferation of hESCs) [109,110], or (semi-)synthetic, such as carboxylic acid-containing acrylate grafted with peptide sequences of vitronectin and sialoprotein via EDC/NHS conjugation—Synthemax (Corning) [111], tissue culture plates grafted with high-affinity cyclic RGD peptide (GACRGDCLGA) via NHS-(PEG)12-maleimide [112], or use of a synthetic polymer (poly[2-(methacryloyloxy)ethyl dimethyl-(3-sulfopropyl)ammonium hydroxide]) [113] have been evaluated. For all coatings, the cells could be subcultured for at least 10 passages, maintained their pluripotency and showed cell doubling times similar or shorter than those observed for the control cultures done on Matrigel coatings. Future design of synthetic microcarriers without ECM coating or using synthetic polymers as coatings is expected to provide a completely defined system for iPSC expansion in bioreactors. Further details can be found in the review by Villa-Diaz et al. [114].
It should be added here that in view of a large-scale reactor use the choice of the carrier coating has a potential impact on the sensibility of the attached cells to shear stress (vide infra).
(b). Influence of mechanical stress on the fate of mesenchymal stem cells and pluripotent stem cells
Stem cells are not only impacted by soluble factors, including growth factors and cytokines, but also by physical and mechanical cues. In the case of MSCs, it was shown that a stiff culture surface (increased matrix stiffness (11–30 kPa)) together with an adapted biochemical matrix such as polyallylamine promotes spindle-like shape and osteogenic differentiation [115], that a matrix with E = 2.5–5 kPa leads predominantly to adipogenic differentiation, whereas soft matrices (E < 1 kPa) lead to neural differentiation [116–118]. A similar situation exists for PSCs. Zoldan et al. [119] demonstrated that hESCs grown on hard surfaces promoted mesodermal commitment, surfaces with intermediate elastic moduli endodermal and soft surfaces ectodermal differentiation. In addition, the self-renewal of hESCs was promoted by hard surfaces (greater than 6 MPa). More information on mechanical cues can be found in the electronic supplementary material, section SM4.
The mechanosensitivity of stem cells as well as mechanical stimuli are of high practical interest for stem cell expansion for later in vivo use because the choice of the microcarriers or, in general, of the cell culture supports and thus their stiffness has a direct impact on the final stem cell fate.
(c). Shear stress in carrier/aggregate cultures
As described at the beginning of this article any agitation in spinner bottles or stirred tank reactors leads to the generation of shear stress, which might have an impact on the cells attached to the carrier. For continuous cell lines, dependent on the shear stress level the impact is relatively limited leading to reduced growth, cell detachment and cell death with negative effects on production; however, in the case of stem cells, physical signals in the local cellular environment can strongly influence stem cell fate in addition to the commonly observed cell damage due to shear stress in bioreactors (see §3b(ii)) [120].
In addition to growth on microcarriers, stem cells can grow as aggregates/clumps in suspension. They are influenced by shear stress as when cultured on microcarriers, thus a careful optimization of agitation is important to avoid negative impact on the cells.
(i). Hydrodynamic effects
Aggregates of mouse ESCs could be successfully expanded in spinner flasks at an agitation of 80–100 r.p.m. corresponding to a maximum shear stress of 0.45–0.61 N m−2, whereas extensive damage and no further proliferation were observed at a maximum shear stress of 0.78 N m−2 (approx. 120 r.p.m.) [121]. An optimization study using factorial experimental design established an agitation of 100 r.p.m. optimal for expanding hESCs as aggregates in spinner flasks (NDS Technologies) [122]. The maximum shear stress at this agitation rate was 0.845 N m−2 and thus somewhat higher than that established by Cormier et al. [122]. However, as for Cormier et al. [122], an agitation rate of 120 r.p.m. (max. shear stress of 1.081 N m−2) was too high and led to reduced cell growth/increased cell death. In this context, Wolfe & Ahsan [123] showed that shear stress in the range from 0.15 to 1.5 N m−2 was a potent inducer of mesodermal commitment of ESCs through the modulation of FLK1 membrane protein. These results infer that after a better insight into the effect of the interrelationship between shear stress and cell signalling on differentiation/cell fate of hiPSCs it will be possible to use mechanical cues that can be readily incorporated into scalable bioreactors to substitute for exogenously added reagents in the clinical production of cells [123]. For getting more insights, microfluidic systems have been implemented allowing very precise studies on the effects of shear fields/shear stress in conjunction with ECM and medium composition (study of the effects of cytokines, growth factors, etc.) on cell renewal and stem cell fate in view to the development of an optimal culture system for expansion and differentiation of stem cells [124–126].
(ii). Maintenance of optimal aggregate size using hydrodynamics
When using aggregate cultures, it is obvious that a critical parameter is the aggregate size (table 2). It has an impact on the level of nutrients and metabolic waste products within the aggregates (in particular in the centre), with the possibility of nutrient limitations when the aggregate size exceeds a certain maximum. The first encountered limitation is related to the oxygen levels becoming limited in the centre of the aggregates. Bliem & Katinger [127] reported that for cell aggregates with a diameter below 300 µm there was no oxygen diffusion limitation and as oxygen is the nutrient factor in cell culture with the lowest bulk concentration there are no other limitations, such as of glucose or glutamine, to be expected. Sen et al. [128] and Wu et al. [129] confirmed that no hypoxic conditions were observed in the centre of aggregates with diameters of less than 300 µm, neither for neural stem cells [128] nor for hESC or mESCs (mESCs show a higher oxygen consumption than hESCs; spinner cultures, r.p.m. = 60) [129], whereas at larger aggregate diameters, hypoxia became apparent. In contrast to continuous cell lines used for the production of biologicals, the residual oxygen level is of much higher importance in the case of stem cells because the oxygen level has an impact on the stem cell fate; for example, in the case of hESCs lower pO2 (e.g. at 3%) promotes differentiation into lineages including endothelial cells [130] and chondrocytes [131] comparable to in vivo processes as well as growth/expansion of iPSCs [70]. Moreover, lower oxygen tension is preferable for expansion of PSCs because the frequency of spontaneous differentiation and the occurrence of chromosomal aberrations are reduced [132,133]. The maximal aggregate size might be limited due to transport limitations hindering the growth of aggregates. This was confirmed by Cameron et al. [72], who observed a stabilization of the size of hESC aggregates at a diameter of 400–500 µm beyond 10 days of culture. In the context of mass transfer limitations and in particular of oxygen transfer limitations at high cell densities including cell aggregates and scaffolds, Martin & Vermette [134] have presented a theoretical evaluation of reactor optimization.
Thus, it is important to maintain a certain predefined aggregate size and thus a stable environment for the cells of the aggregates for maintaining cell expansion and differentiation capacity. Culture systems with insufficient agitation, including the rotating wall perfused reactors (see electronic supplementary material, SM3) which are characterized by very low turbulence/shear stress levels, lead to the generation of aggregate diameters greater than 500 µm which, according to Bliem & Katinger [127], are characterized by oxygen limitation resulting in low expansion rates and partial cell differentiation in spinner flasks [135]. This signifies that an agitation generating higher turbulence/shear stress levels is required in order to maintain a certain maximal aggregate size [33]. There is an inverse relationship between agitation speed and aggregate size. Based on the assumption that hydrodynamics within a suspension reactor can control aggregate diameter [33] the maximal aggregate size in a stirred-tank reactor can be calculate as follows:
where ɛ is the power dissipation per unit mass or specific energy dissipation rate and ν is the kinematic viscosity. The relationship with the Kolmogorov eddy length model is obvious (see §3b(ii)). Parameters C, α and β can be estimated from experiments (e.g. [32]). This relationship was initially developed by Moreira et al. [33] for BHK-21 clump cultures and proven valid for aggregate cultures of neural stem cells [32].
The overall culture conditions, including agitation and medium, have a direct impact on the maximal aggregate size. In this context, Lam et al. [108] established that the optimal size of carrier-iPSC aggregates was about greater than or equal to 300 µm with greater than 50 aggregates per ml. In the case of pure stem cell aggregates (without carriers), different authors have optimized the conditions to achieve relatively homogeneous aggregate sizes. Whereas Olmer et al. [136] obtained aggregates ranging from 93 to 125 µm, not exceeding 200 µm, mainly based on the choice of the agitator design (axial eight-blade pitched impellers with a diameter of 40 mm, vessel diameter 70 mm, with blade angles differing at the centre and the edge of the impeller) and the agitation rate (60 r.p.m.), Abbasalizadeh et al. [69] used a Cellspin spinner flask with two pendula (i.e. very soft agitation) and influenced the aggregate size with the agitation rate (50 r.p.m.) and an increase in the medium viscosity (addition of 0.1% carboxymethylcellulose) to 40 cp. Thus, by precise control of the agitation rates and the addition of shear protectants the aggregate size could be controlled. Abbasalizadeh et al. [69] could maintain the aggregate size very homogeneously, ranging from 190 to 215 µm. This size range was also achieved by Hunt et al. [121] with an agitation of 100 r.p.m.
(iii). Protection against shear stress in stirred-tank reactors
Under serum-free conditions, when using Matrigel and laminin-111 coatings of microcarriers it was observed that different hESCs showed different sensitivities towards fluid shear stress. Whereas HES-2 cells could grow without significant differences on laminin- and Matrigel-coated DE-53 carriers in agitated spinner flasks, HES-3 cells exhibited a decrease in cell yield, viability and pluripotent markers on laminin- as compared to Matrigel-coated carriers [105]. The reason seems to be that the Matrigel coating offers shear protection arising from the gelatinous polymeric nature of the thick coating, which is not the case for the laminin coating. Cell detachment of HES-3 cells under agitation conditions was probably enhanced due to a lower cell contact area when compared with Matrigel [110]. Only two different coatings were evaluated in this study [105] and other potential coatings providing a more intense attachment and eventually a better cell protection from fluid shear stress should be evaluated in the future. In this context, it was shown that the combination of poly-l-lysine with a coating of ECM proteins (in particular of laminin) improved cell attachment and expansion under serum-free agitated conditions. Using such a modified coating, the hESC HES-3, which was particularly sensitive to shear effects [105], could be expanded without losing pluripotency and maintaining differentiation potential [108]. Another complementary measure would be the use of smaller carriers (as was the case in the report by Lam et al. [108]) because their use led to the generation of more compact aggregates, which might show a higher tolerance to shear stress in stirred-tank reactor cultures.
In principle, adverse fluid effects might be alleviated by the addition of ‘protective’ agents, including Pluronic F68, methylcellulose or dextran. However, Leung et al. [137] could not identify any protective agent, classically known to protect cell cultures from shear effects, being able to protect shear-sensitive hESCs from loss of pluripotent markers or from decrease in cell growth. These results are in agreement with the observation that shear stress can impact stem cell fate probably due to indirect cellular signalling effects (see above). Further research will be necessary to identify the differences between shear-sensitive and shear-non-sensitive (or less sensitive) iPSCs in view to the establishment of robust cell culture expansion processes based on the use shear-non-sensitive iPSCs.
The addition of antifoam at 0.0125% necessary for alleviating foam formation and the stress due to aeration was shown to have no effect on mESC viability and EB formation [76].
For the moment, the carrier coating has to be chosen depending on the cell line to be expanded as well as with respect to the culture system and the expected fluid stress. Although there is no general comprehensive view of the effects of shear on stem cells to be amplified under reactor conditions using microcarriers, the reader is referred to a recent review by Kinney et al. [77] covering the interdependence between stem cell aggregates, metabolism and phenotype in relation to hydrodynamic culture environments.
6. Large-scale retrieval of stem cells for clinical applications
(a). Cell detachment
Cell detachment is a critical step for subculturing surface adherent cells. At a small scale, mechanical detachment methods are well established for different types of cells and are traditionally used for PSCs. However, for large-scale cell expansion these methods cannot be used. In principle, the efficiency of cell detachment depends on the cells to be detached, the microcarrier and carrier surface, and thus the interaction of the cells with the microcarrier and, of course, also the detachment agent. Traditionally, proteolytic enzymes, in particular, trypsine or its recombinant and/or animal-free substitutes, are used for the efficient generation of single cell suspensions.
In addition to genomic instability seen to be readily generated in systems where enzymatic dissociation is used [138], there are further disadvantages when using proteases for cell detachment. Goh et al. [90] showed that trypsin was the most efficient detachment agent with respect to speed and efficiency for detaching human fetal MSCs from Cytodex 3 carriers, followed by collagenase I and TrypLE Express with an overall viability of the detached cells of more than 95% for all agents. However, all of the cells detached with protease showed (osteogenic) differentiation efficiency significantly lower than that observed for the non-treated/non-harvested cells; the differentiation efficiencies were 26–56 µg Ca++-deposition/106 cells (i.e. indicator for osteogenic differentiation) and 71 ± 4 µg Ca++-deposition/106 cells, respectively.
This indicates very clearly the negative impact of protease treatment for cell detachment on cell fate due to the degradation of ECM proteins, destruction of cytoskeletal structures and cell-to-cell contacts. Similar effects caused by proteolytic cell detachment, for instance, have been reported by Canavan et al. [139] for the detachment of bovine aortic endothelial cell monolayers. Specifically for hMSCs, Potapova et al. [140] reported a decrease of CD105 expression with time of exposure to trypsin over a range of 5–90 min. These results clearly indicate that the exposure to trypsin or any other proteolytic agent should be as short as possible. Based on this observation, Nienow et al. [141] evaluated a novel two-step protocol for trypsinization of hMSCs comprised of the addition of and incubation with trypsin-ETDA for 7 min at a fivefold increased agitation rate (30 → 150 r.p.m., followed by 200 r.p.m. for the last 5 s) leading to an increase in the maximal specific energy dissipation rate (ɛ) from approximately 9 × 10−4 to 0.11 W kg−1. This equates to a reduction of the Kolmogorov length from about 180 µm (well above the eddy size at which damaging occurs, as suggested by Croughan et al. [31,36]) to 55 µm, which is well above the size critical for isolated cells, however sufficient to detach cells. The second step consisted of the inactivation of trypsin and the removal of the carriers via filtration. Nienow et al. [141] reported a recovery of more than 95% of the cells which maintained their specific characteristics.
With respect to the detachment of hiPSCs, there is only limited literature available. In principle, when aiming to get to a single cell suspension, trypsin or equivalent (recombinant) proteases, including Accutase, Accumax or TrypLE, are preferable because the protein bridges between the cells are also degraded. However, the generation of single cell suspension of hiPSCs is characterized by high cell loss due to dissociation-induced apoptosis after passaging as single cells, which could be solved by the use of Rho kinase inhibitors, such as Y-27632 or ROCKi [142]. Another way to circumvent this problem is dissociation into aggregates because it has been shown that microcarrier cultures of PSCs can also be seeded with cell aggregates/clumps and that single cell suspensions are not required [143]. The only issue of importance is the fact that the size of aggregates has to be uniform during the culture, which is achieved by agitation generating the required turbulences (see above).
Dissociation of cultures into aggregates can be achieved by using other proteases and agents, including collagenase IV, dispase or hypertonic Na-citrate solution. In this context, Nie et al. [106] showed that the use of hypertonic Na-citrate solution led to superior results with respect to viability and total viable cell number obtained over several passages when compared with detachment using collagenase, dispase or mechanical methods (order of decreasing efficiency). The cells detached using the hypertonic citrate solution kept their ability to express pluripotency markers and could differentiate to all three germ layers after 30 passages.
Thus, in general, the use of non-proteolytic cell detachment means (absence of proteases) has an undeniable positive effect on subcultivation and differentiation of stem cells. These few references show very clearly that any proteolytic cell detachment should be avoided in order to maintain the organization of the cellular cytoskeleton as well as the cell-to-cell interaction as much as possible and to reduce cell damage. For regenerative medicine purposes, protocols for non-proteolytic cell sheet detachments have been developed (e.g. [144]), mainly based on the use of a temperature-responsive poly(N-isopropylacrylamide (pNIPAAm) modified polystyrene surface allowing the detachment of cells by a reduction of temperature (below the lower critical solution temperature) leading to hydrophilic surface properties and cell detachment [145]. Since, no proteases are used, the cells detach as cell sheets because the intercellular proteinous bridges are preserved. In order to improve cell adhesion and proliferation such thermo-responsive surfaces can be functionalized, including grafting on heparine for binding bFGF [146], the cell-binding domain RGDS [147] or the cyclic RGD peptide CRGDC [148] because of its high affinity binding to the αvβ3, αvβ5 or α5β1 integrins of cells, to mention just a few possibilities.
Only recently, microcarriers grafted with poly(N-isopropylacrylamide allowing the non-invasive harvesting of anchorage-dependent cells had been evaluated. That this principle can be implemented was shown by Kenda-Ropson et al. [149] and Tamura et al. [150] for the detachment of CHO-K1 from modified microhex carriers (pNIPAAm-grafted 2D-polystyrene carriers) and from chloro-methylated polystyrene beads (pNIPAAm-grafted), respectively. Furthermore, Yang et al. [151] and Gümüşderelioğlu et al. [152] showed the efficient detachment of human bone marrow-derived MSCs from Cytodex 3 grafted with pNIPAAm and of mouse fibroblasts (L929) and human keratinocytes (HS2) from cross-linked poly(2-hydroxyethyl methacrylate) (PHEMA) beads grafted with pNIPAAm. In all cases, the reduction of the temperature from 37°C to less than 30°C led to cell detachment, though more in the form of cell clumps than isolated cells. Although this is a very promising development, few advances have been performed in this specific domain with respect to thermo-responsive microcarriers and further efforts are necessary in order to solve many still existing technological hurdles before large-scale implementation.
With respect to the use of such temperature-responsive culture supports for the expansion of iPSCs, it is highly probable that they can be used for this type of cells without any drawbacks, since their differentiation potential as well as genetic stability is not affected by temperature reduction and exposure to 25°C and 4°C for up to 2 days [153]. More details on the possibilities and use of thermo-responsive and, in general, of stimuli-responsive cell culture supports can be found in the review by Brun-Graeppi et al. [154].
(b). Separation of non-differentiated cells and of carriers: problem of carrier breakage
Removal of undifferentiated cells is a particular concern in clinical applications of differentiated progenies derived from iPSCs because undifferentiated PSCs are teratogenic [78]. On a small scale, fluorescence activated cell sorting for the isolation of differentiated cells, such as cardiomyocytes [155], neural [156] or endothelial cells [157], can be used. A similar technique is immunomagnetic cell sorting [158], allowing separation at a larger scale. The selective removal of undifferentiated cells using cytotoxic antibodies, such as the cytotoxic mAb 84 [159], is also of high interest and can be coupled to immunomagnetic cell sorting [158] reducing further the risk of teratoma formation. In certain cases, such as the separation of PSC-derived cardiomyocytes from non-cardiomyocytes and undifferentiated cells, the separation is enabled by distinct metabolic flow. Tohyama et al. [160] reported that in a glucose-depleted and lactate-enriched medium only cardiomyocytes survived and that no tumours were formed after transplantation. More extended information can be found in recent reviews by Chen et al. [15], Abbasalizadeh & Bahavand [161] and Diogo et al. [162]. Nevertheless, it has to be mentioned that to date no systematic studies on harvesting PSCs and separation their differentiated progenies have been performed.
Another important issue should be touched here very briefly: Gupta et al. [163] found that ‘excessive’ stirring of microcarrier (Cytodex 3) cultures (r.p.m. > 25) in Bellco spinner flasks can lead to bead breakage. This is a known problem for soft carriers like Cytodex 1 or 3, but is also of concern when the expanded and differentiated cells are used in vivo, because these preparations have to be free of any residual microcarrier or sub-particles derived from these carriers. Sieving is a principle way to get rid of entire carriers; however, breakage products are removed with difficulty or not at all. There are two main potential solutions to this problem: either using rigid carriers which are resistant to breakage, like beads made from polystyrene or glass, or using soft carriers which can be dissolved using proteases, such as gelatin- or collagen-based carriers. However, it should be recalled that the choice is critical because the rigidity/stiffness of the support may have an influence on the stem cell fate (see above). On the other hand, it is evident that aggregate cultures without using carriers as an initial support would also present a solution for this specific problem.
7. ‘Large-scale’ expansion of stem cells using microcarrier cultures
‘Large-scale’ manufacturing of stem cells has mainly been done using classical two-dimensional planar cell culture devices, such as CellFactory, Xpansion or HYPERStack, allowing a linear scale-up. However, as mentioned earlier, the real volumetric scale-up happens via an increase in the culture volume, going up to 1000 l per vessel and beyond. Nevertheless, the implementation of stirred-tank bioreactor systems for the expansion/large-scale expansion of stem cells is still in its infancy. Effectively no large scale and only small laboratory scale reactor cultures have been performed up to now.
Concerning MSCs, the largest reactor scale ever used was a 5 l scale with a working volume of 2.5 l using non-porous P-102L plastic carriers [164]. The fold cell number increase was more than sixfold over 12 days of culture, which was equivalent to cell expansion in tissue culture flasks (i.e. 65× T-175 flasks) and can be explained by the improved control of the culture conditions (pH, pO2). This is also the reason why bioreactor cultures outperformed the cultures done in spinner flasks. Cell harvesting was performed via trypsinization, increased agitator speed in a separate vessel (reduced volume) followed by sieving through a Steriflip 60 µm filtration unit, comparable to passaging cells at a large scale [45]. The use of this optimized detachment protocol [141] allowed a cell recovery of almost 100%. Very importantly, the cells after expansion in the bioreactor and harvesting showed the same phenotype and differentiation capability as before expansion.
The use of bioreactors for the expansion of hESCs was performed at a much smaller scale (0.5 l stirred-tank reactor, working volume 300 ml, use of Matrigel-coated Cytodex 3 carriers in a perfusion culture) [165]. Aggregates were formed between the carriers and the hESCs. Since the culture conditions were controlled, a considerably improved cell expansion was obtained in comparison to the planar two-dimensional cultures or carrier-based three-dimensional cultures performed in spinner flasks: 15-fold for the reactor cultures and sixfold in the spinner flasks (culture duration: 10–12 days). The expanded cells retained their pluripotency and differentiation capabilities. The protocol could be used for the expansion of other PSCs. When employing aggregate cultures, stirred bioreactor cultures (50–200 ml) allowed an eightfold increase for each passage. Thus within 30 days, an overall amplification of 128- to 192-fold is possible providing at the end of the expansion about 2 × 109 cells [69]. The use of environment-controlled bioreactors will probably allow a better cell expansion efficiency approaching that communicated by Serra et al. [165].
Although no large-scale cell expansion has been performed for MSCs or PSCs up to now, the reactor results clearly show the potential of reactor amplification of stem cells. However, before implementation for large-scale cell expansion, culture parameters including physico-chemical parameters, growth medium, differentiation media, etc., have to be systematically studied and optimized using either step-wise approaches [69] or preferentially, factorial experimental designs for also modelling the interactions between different parameters, for instance as done by Hunt et al. [121]. The use of microfluidic devices will be of considerable help to deepen and accelerate the optimization of the cell expansion process at large scale [124–126]. In addition, it is very important to keep in mind that stem cells are not identical and that each stem cell has its proper characteristics and needs, signifying that a very flexible platform process has to be set up which can easily be adapted to the specific needs of the stem cell to be expanded.
8. Conclusion and outlook
Surface adherent cells can be used and expanded for different purposes. Interest in the large-scale amplification of these cells started with the development of manufacturing processes for viral vaccines for which reactor scales of several thousand litres have been established using microcarrier processes. In the case that novel suspension cells are not used for virus vaccine production purposes, the originally developed, established and largely validated process technologies using Vero and MDCK cells can be used for the development of novel viral vaccines, signifying that there is only limited novel development necessary for the setting up of new viral vaccine production processes.
The situation for the expansion of different types of stem cells is rather different. This is a very new domain and the requirements for the expansion of stem cells are rather different from those of continuous cell lines traditionally used for virus vaccine production. Dependent on the stem cell type and their specific needs, expansion processes are under development, ideally using adapted microcarriers or carrier-cell clump-based protocols to preserve the cells' pluripotency and differentiation potential. They can be impacted by the culture environment, including fluid shear stress, culture (reactor) conditions, microcarrier surface coating and means of cell detachment, these already studied many years ago in the context of the production of cell biomass for virus vaccine production purposes. Despite the fact that different stem cells behave differentially, in particular with respect to the impact of the culture environment, recent optimization studies show the overall direction of development with large-scale reactor cultures at the end. The presently evaluated reactor scales of 5 and 0.5 l (working volumes: 2.5 l [164] and 0.3 l [165], respectively) for the amplification of MSCs and PSCs, respectively, using microcarriers or clump cultures are the first steps along the way to the large-scale expansion of stem cells which will see the light within the years to come.
Supplementary Material
Conflict of interests
I have no competing interests.
References
- 1.Griffiths JB, Riley PA. 1985. Cell biology: basic concepts. In Animal cell biotechnology, vol. 1 (eds Spier RE, Griffiths JB.), pp. 17–48. London, UK: Academic Press. [Google Scholar]
- 2.Frisch SM, Screaton RA. 2001. Anoikis mechanisms. Curr. Opin. Cell Biol. 13, 555–562. ( 10.1016/S0955-0674(00)00251-9) [DOI] [PubMed] [Google Scholar]
- 3.Castillo J, Egloff M, Collignon F, Goffinet J, Drugmand J. 2013. Transfer of hepatic progenitor stem cell culture process from multiple tray stacks to the Xpansion multiplate bioreactor. Cytotherapy 15, Suppl. S20. ( 10.1016/j.jcyt.2013.01.074) [DOI] [Google Scholar]
- 4.Merten O-W, Manuguerra JC, Hannoun C, van der Werf S. 1999. Production of influenza virus in serum-free mammalian cell cultures. Dev. Biol. Stand. 98, 23–37; discussion 73–74. [PubMed] [Google Scholar]
- 5.Barrett PN, Mundt W, Kistner O, Howard MK. 2009. Vero cell platform in vaccine production: moving towards cell culture-based viral vaccine. Expert Rev. Vaccines 8, 607–618. ( 10.1586/erv.09.19) [DOI] [PubMed] [Google Scholar]
- 6.Eaker S, et al. 2013. Concise review: guidance in developing commercializable autologous/patient-specific cell therapy manufacturing. Stem Cells Transl. Med. 2, 871–883. ( 10.5966/sctm.2013-0050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malda J, Frondoza CG. 2006. Microcarriers in the engineering of cartilage and bone. Trends Biotechnol. 24, 299–304. ( 10.1016/j.tibtech.2006.04.009) [DOI] [PubMed] [Google Scholar]
- 8.Panina GF. 1985. Monolayer growth systems: multiple processes. In Animal cell biotechnology, vol. 1 (eds Spier RE, Griffiths JB.), pp. 211–242. London, UK: Academic Press. [Google Scholar]
- 9.Kunitake R, Suzuki A, Ichihashi H, Matsuda S, Hirai O, Morimoto K. 1997. Fully-automated roller bottle handling system for large scale culture of mammalian cells. J. Biotechnol. 52, 289–294. ( 10.1016/S0168-1656(96)01654-9) [DOI] [PubMed] [Google Scholar]
- 10.Pakos V, Johansson A. 1985. Large scale production of human fibroblast interferon in multitray battery systems. Dev. Biol. Stand. 60, 317–320. [PubMed] [Google Scholar]
- 11.van Wezel AL. 1967. Growth of cell-strains and primary cells on micro-carriers in homogeneous culture. Nature 216, 64–65. ( 10.1038/216064a0) [DOI] [PubMed] [Google Scholar]
- 12.Levine DW, Wong JS, Wang DI, Thilly WG. 1977. Microcarrier cell culture: new methods for research-scale application. Somatic Cell Genet. 3, 149–155. ( 10.1007/BF01551811) [DOI] [PubMed] [Google Scholar]
- 13.Levine DW, Wong JS, Wang DIC, Thilly WG. 1979. Optimization of growth surface parameters in microcarrier cell cultures. Biotechnol. Bioeng. 21, 821–846. ( 10.1002/bit.260210507) [DOI] [Google Scholar]
- 14.Maroudas NG. 1972. Anchorage dependence correlation between amount of growth and diameter of bead, for single cells grown on individual glass beads. Exp. Cell Res. 74, 337–342. ( 10.1016/0014-4827(72)90385-0) [DOI] [PubMed] [Google Scholar]
- 15.Chen AK-L, Reuveny S, Oh SKW. 2013. Application of human mesenchymal and pluripotent stem cell microcarrier cultures in cellular therapy: achievements and future direction. Biotechnol. Adv. 31, 1032–1046. ( 10.1016/j.biotechadv.2013.03.006) [DOI] [PubMed] [Google Scholar]
- 16.Meignier B, Mougeot H, Favre H. 1980. Foot and mouth disease virus production on microcarrier-grown cells. Dev. Biol. Stand. 46, 249–256. [PubMed] [Google Scholar]
- 17.Montagnon BJ, Fanget B, Vincent-Falquet JC. 1984. Industrial scale production of inactivated poliovirus vaccine prepared by culture of Vero cells on microcarrier. Rev. Infect. Dis. 6, S341–S344. ( 10.1093/clinids/6.Supplement_2.S341) [DOI] [PubMed] [Google Scholar]
- 18.Montagnon B, Vincent-Falquet JC, Fanget B. 1984. Thousand litre scale microcarrier culture of Vero cells for killed polio virus vaccine. Promising Results Dev. Biol. Stand. 55, 37–42. [PubMed] [Google Scholar]
- 19.Montagnon BJ, Vincent-Falquet JC, Saluzzo JF. 1999. Experience with vero cells at Pasteur Mérieux Connaught. Dev. Biol. Stand. 98, 137–140; discussion 167. [PubMed] [Google Scholar]
- 20.Elsdale T, Bard J. 1972. Cellular interactions in mass cultures of human diploid fibroblasts. Nature 236, 152–155. ( 10.1038/236152a0) [DOI] [PubMed] [Google Scholar]
- 21.Kistner O, Barrett PN, Mundt W, Reiter M, Schober-Bendixen S, Eder G, Dorner F. 1999. Development of a Vero cell-derived influenza whole virus vaccine. Dev. Biol. Stand. 98, 101–110; discussion 111. [PubMed] [Google Scholar]
- 22.Moran E. 1999. A microcarrier-based cell culture process for the production of a bovine respiratory syncytial virus vaccine. Cytotechnology 29, 135–148. ( 10.1023/A:1008022828736) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng YC, Berry JM, Butler M. 1996. Optimization of physical parameters for cell attachment and growth on macroporous microcarriers. Biotechnol. Bioeng. 50, 627–635. () [DOI] [PubMed] [Google Scholar]
- 24.Hu WS, Meier J, Wang DI. 1985. A mechanistic analysis of the inoculum requirement for the cultivation of mammalian cells on microcarriers. Biotechnol. Bioeng. 27, 585–595. ( 10.1002/bit.260270507) [DOI] [PubMed] [Google Scholar]
- 25.Butler M, Thilly WG. 1982. MDCK microcarrier cultured: seeding density effects and amino acid utilization. In Vitro 18, 213–219. ( 10.1007/BF02618573) [DOI] [PubMed] [Google Scholar]
- 26.Cherry RS, Papoutsakis ET. 1990. Understanding and controlling fluid-mechanical injury of animal cells in bioreactors. In Animal cell biotechnology, vol. 4 (eds Spier RE, Griffiths JB.), pp. 71–121. London, UK: Academic Press. [Google Scholar]
- 27.Croughan MS, Wang DIC. 1991. Hydrodynamic effects on animal cells in microcarrier bioreactors. In Animal cell bioreactors (eds Ho CS, Wang DIC.), pp. 213–249. Stoneham, MA: Butterworth-Heinemann. [DOI] [PubMed] [Google Scholar]
- 28.Nienow AW. 2014. Re ‘Development of a scale-down model of hydrodynamic stress to study the performance of an industrial CHO cell line under simulated production scale bioreactor conditions’ [Sieck JB, Cordes T, Budach WE, Rhiel MH, Suemeghy Z, Leist C, Villiger TK, Morbidelli M, Soos M. 2013 J. Biotechnol. 164, 41–49] J. Biotechnol. 171, 82–84. ( 10.1016/j.jbiotec.2013.12.002) [DOI] [PubMed] [Google Scholar]
- 29.Rushton JH, Costich EW, Everett HJ. 1950. Power characteristics of mixing impellers. Chem. Eng. Proc. 46, 467–471. [Google Scholar]
- 30.Paul EL, Atiemo-Obeng VA, Kresta SM. 2004. Handbook of industrial mixing. Hoboken, NJ: Wiley-Interscience. [Google Scholar]
- 31.Croughan MS, Hamel JF, Wang DIC. 1987. Hydrodynamic effects on animal cells grown in microcarrier cultures. Biotechnol. Bioeng. 29, 130–141. ( 10.1002/bit.260290117) [DOI] [PubMed] [Google Scholar]
- 32.Sen A, Kallos MS, Behie LA. 2002. Expansion of mammalian neural stem cells in bioreactors: effect of power input and medium viscosity. Brain Res. Dev. Brain Res. 134, 103–113. ( 10.1016/S0165-3806(01)00328-5) [DOI] [PubMed] [Google Scholar]
- 33.Moreira JL, Santana PC, Feliciano AS, Cruz PE, Racher AJ, Griffiths JB, Carrondo MJ. 1995. Effect of viscosity upon hydrodynamically controlled natural aggregates of animal cells grown in stirred vessels. Biotechnol. Prog. 11, 575–583. ( 10.1021/bp00035a012) [DOI] [PubMed] [Google Scholar]
- 34.Cherry RS, Kwon KY. 1990. Transient shear stresses on a suspension cell in turbulence. Biotechnol. Bioeng. 36, 563–571. ( 10.1002/bit.260360603) [DOI] [PubMed] [Google Scholar]
- 35.Geisler R, Krebs R, Forschner P. 1994. Local turbulent shear stress in stirred vessels and its significance for different mixing tasks. In Proc. 8th Eur. Conf. on Mixing, IChemE Symposium Series no. 136, pp. 241–251. London, UK: IChemE. [Google Scholar]
- 36.Croughan MS, Hamel JF, Wang DI. 1988. Effects of microcarrier concentration in animal cell culture. Biotechnol. Bioeng. 32, 975–982. ( 10.1002/bit.260320805) [DOI] [PubMed] [Google Scholar]
- 37.Nienow AW. 1997. The mixer as a reactor—liquid/solid systems. In Mixing in the process industries (eds Harnby N, Edwards MF, Nienow AW.), pp. 394–411. London, UK: Butterworth-Heinemann. [Google Scholar]
- 38.Cherry RS, Papoutsakis ET. 1989. Growth and death rates of bovine embryonic kidney cells in turbulent microcarrier bioreactors. Bioproc. Eng. 4, 81–89. ( 10.1007/BF00373735) [DOI] [Google Scholar]
- 39.Hassanzadeh SM, Zavareh A, Shokrgozar MA, Ramezani A, Fayaz A. 2011. High vero cell density and rabies virus proliferation on fibracel disks versus cytodex-1 in spinner flask. Pak. J. Biol. Sci. 14, 441–448. ( 10.3923/pjbs.2011.441.448) [DOI] [PubMed] [Google Scholar]
- 40.Croughan MS, Sayre ES, Wang DIC. 1989. Viscous reduction of turbulent damage in animal cell culture. Biotechnol. Bioeng. 33, 862–872. ( 10.1002/bit.260330710) [DOI] [PubMed] [Google Scholar]
- 41.Stathopoulos NA, Hellums JD. 1985. Shear stress effects on human embryonic kidney cells in vitro. Biotechnol. Bioeng. 27, 1021–1026. ( 10.1002/bit.260270713) [DOI] [PubMed] [Google Scholar]
- 42.Ma N, Chalmers JJ, Auniņs JG, Zhou W, Xie L. 2004. Quantitative studies of cell-bubble interactions and cell damage at different pluronic F-68 and cell concentrations. Biotechnol. Prog. 20, 1183–1191. ( 10.1021/bp0342405) [DOI] [PubMed] [Google Scholar]
- 43.Nienow AW. 2006. Reactor engineering in large scale animal cell culture. Cytotechnology 50, 9–33. ( 10.1007/s10616-006-9005-8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Merten O-W. 2010. Cell detachment. In Encyclopedia of industrial biotechnology: bioprocess, bioseparation, and cell technology (ed. Flickinger MC.), pp. 1–22. New York, NY: John Wiley & Sons Ltd. [Google Scholar]
- 45.Van Wezel AL, van der Velden-de-Groot CAM, van Herwaarden JAM. 1980. The production of inactivated poliovaccine on serially cultivated kidney cells from captive-bred monkeys. Dev. Biol. Stand. 46, 151–158. [PubMed] [Google Scholar]
- 46.Shevitz J, LaPorte TL, Stinnett TE. 1990. Production of viral vaccines in stirred bioeactors. In Viral vaccines in the series of advances in biotechnological processes, vol. 14 (ed. Mizrahi A.), pp. 1–35. New York, NY: Wiley-Liss. [PubMed] [Google Scholar]
- 47.Ohlson S, Branscomb J, Nilsson K. 1994. Bead-to-bead transfer of Chinese hamster ovary cells using macroporous microcarriers. Cytotechnology 14, 67–80. ( 10.1007/BF00772197) [DOI] [PubMed] [Google Scholar]
- 48.Kong D, Chen M, Gentz R, Zhang J. 1999. Cell growth and protein formation on various microcarriers. Cytotechnology 29, 149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiao C, Huang Z, Li W, Hu X, Qu W, Gao L, Liu G. 1999. High density and scale-up cultivation of recombinant CHO cell line and hybridomas with porous Cytopore. Cytotechnology 30, 143–147. ( 10.1023/A:1008038609967) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y, Ouyang F. 1999. Bead-to-bead transfer of Vero cells in microcarrier culture. Cytotechnology 31, 221–224. ( 10.1023/A:1008079013375) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luo F, Sun H, Gang T, Qi N. 2008. Application of Taguchi's method in the optimization of bridging efficiency between confluent and fresh microcarriers in bead-to-bead transfer of Vero cells. Biotechnol. Lett. 30, 645–649. ( 10.1007/s10529-007-9604-2) [DOI] [PubMed] [Google Scholar]
- 52.Xiao C, Huang Z, Zhang Z, Ye L, Gao L, Guo Z, Cheng D, Zhou H, Kong W. 1994. High density cultivation of a recombinant CD-1 cell line producing prourokinase using a Biosilon microcarrier culture system. Chin. Med. Sci. J. 9, 203–208. [PubMed] [Google Scholar]
- 53.Genzel Y, Reichl U. 2009. Continuous cell lines as a production system for influenza vaccines. Expert. Rev. Vaccines 8, 1681–1692. ( 10.1586/erv.09.128) [DOI] [PubMed] [Google Scholar]
- 54.Merten O-W, Bakker WAM, Vorlop J, Reiter M, Visnovsky G, Jäger V, Merabishvili M, Reichl U. 2014. Virus production under suspension conditions. In Industrial scale suspension culture of living cells (eds Meyer H-P, Schmidhalter D.), pp. 503–554. Weinheim, Germany: Wiley-VCH. [Google Scholar]
- 55.Yu P, Huang Y, Zhang Y, Tang Q, Liang G. 2012. Production and evaluation of a chromatographically purified Vero cell rabies vaccine (PVRV) in China using microcarrier technology. Hum. Vaccine Immunother. 8, 1230–1235. ( 10.4161/hv.20985) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aunins JG, et al. 1997. Reactor development for the hepatitis A vaccine VAQTA. In Animal cell technology: from vaccines to genetic medicins (eds Carrondo MJT, Griffiths B, Moreira JLP.), pp. 175–183. Dordrecht, The Netherlands: Kluwer Academic Publishers. [Google Scholar]
- 57.Hagen A, et al. 2000. Development, preparation, and testing of VAQTA, a highly purified hepatits A vaccine. Bioproc. Eng. 23, 439–449. ( 10.1007/s004499900157) [DOI] [Google Scholar]
- 58.Junker BH, et al. 1992. Evaluation of a microcarrier process for large-scale cultivation of attenuated hepatitis A. Cytotechnology 9, 173–187. ( 10.1007/BF02521745) [DOI] [PubMed] [Google Scholar]
- 59.Domenici M, et al. 2006. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement . Cytotherapy 8, 315–317. ( 10.1080/14653240600855905) [DOI] [PubMed] [Google Scholar]
- 60.Baxter MA, Wynn RF, Jowitt SN, Wraith JE, Fairbairn LJ, Bellantuono I. 2004. Study of telomere length reveals rapid aging of human marrow stromal cells following in vitro expansion. Stem Cells 22, 675–682. ( 10.1634/stemcells.22-5-675) [DOI] [PubMed] [Google Scholar]
- 61.da Silva Meirelles L, Fontes AM, Covas DT, Caplan AI. 2009. Mechanisms involved in the therapeutic properties of mesenchymal stem cells. Cytokine Growth Factor Rev. 20, 419–427. ( 10.1016/j.cytogfr.2009.10.002) [DOI] [PubMed] [Google Scholar]
- 62.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. 1998. Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. ( 10.1126/science.282.5391.1145) [DOI] [PubMed] [Google Scholar]
- 63.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. ( 10.1016/j.cell.2007.11.019) [DOI] [PubMed] [Google Scholar]
- 64.Serra M, Correia C, Malpique R, Brito C, Jensen J, Bjorquist P, Carrondo MJ, Alves PM. 2011. Microencapsulation technology: a powerful tool for integrating expansion and cryopreservation of human embryonic stem cells. PLoS ONE 6, e23212 ( 10.1371/journal.pone.0023212) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Akins RE, Rockwood D, Robinson KG, Sandusky D, Rabolt J, Pizarro C. 2010. Three-dimensional culture alters primary cardiac cell phenotype. Tissue Eng. Part A 16, 629–641. ( 10.1089/ten.tea.2009.0458) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chang TT, Hughes-Fulford M. 2009. Monolayer and spheroid culture of human liver hepatocellular carcinoma cell line cells demonstrate distinct global gene expression patterns and functional phenotypes. Tissue Eng. Part A 15, 559–567. ( 10.1089/ten.tea.2007.0434) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dasgupta A, Hughey R, Lancin P, Larue L, Moghe PV. 2005. E-cadherin synergistically induces hepatospecific phenotype and maturation of embryonic stem cells in conjunction with hepatotrophic factors. Biotechnol. Bioeng. 92, 257–266. ( 10.1002/bit.20676) [DOI] [PubMed] [Google Scholar]
- 68.Correia C, et al. 2014. Combining hypoxia and bioreactor hydrodynamics boosts induced pluripotent stem cell differentiation towards cardiomyocytes. Stem Cell Rev. Rep. 10, 786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abbasalizadeh S, Larijani MR, Samadian A, Baharvand H. 2012. Bioprocess development for mass production of size-controlled human pluripotent stem cell aggregates in stirred suspension bioreactor. Tissue Eng. Part C 18, 831–851. ( 10.1089/ten.tec.2012.0161) [DOI] [PubMed] [Google Scholar]
- 70.Baptista RP, Fluri DA, Zandstra PW. 2012. High density continuous production of murine pluripotent cells in an acoustic perfused bioreactor at different oxygen concentrations. Biotechnol. Bioeng. 112, 648–655. (doi:10.1002/bit.24717) [DOI] [PubMed] [Google Scholar]
- 71.Côme J, Nissan X, Aubry L, Tournois J, Girard M, Perrier AL, Peschanski M, Cailleret M. 2008. Improvement of culture conditions of human embryoid bodies using a controlled perfused and dialyzed bioreactor system. Tissue Eng. Part C 14, 289–298. ( 10.1089/ten.tec.2008.0029) [DOI] [PubMed] [Google Scholar]
- 72.Cameron CM, Hu WS, Kaufman DS. 2006. Improved development of human embryonic stem cell-derived embryoid bodies by stirred vessel cultivation. Biotechnol. Bioeng. 94, 938–948. ( 10.1002/bit.20919) [DOI] [PubMed] [Google Scholar]
- 73.Carpenedo RL, Sargent CY, McDevitt TC. 2007. Rotary suspension culture enhances the efficiency, yield, and homogeneity of embryoid body differentiation. Stem Cells 25, 2224–2234. ( 10.1634/stemcells.2006-0523) [DOI] [PubMed] [Google Scholar]
- 74.Yirme G, Amit M, Laevsky I, Osenberg S, Itskovitz-Eldor J. 2008. Establishing a dynamic process for the formation, propagation, and differentiation of human embryoid bodies. Stem Cells Dev. 17, 1227–1241. ( 10.1089/scd.2007.0272) [DOI] [PubMed] [Google Scholar]
- 75.Gerecht-Nir S, Cohen S, Itskovitz-Eldor J. 2004. Bioreactor cultivation enhances the efficiency of human embryoid body (hEB) formation and differentiation. Biotechnol. Bioeng. 86, 493–502. ( 10.1002/bit.20045) [DOI] [PubMed] [Google Scholar]
- 76.Schroeder M, Niebruegge S, Werner A, Willbold E, Burg M, Ruediger M, Field LJ, Lehmann J, Zweigerdt R. 2005. Differentiation and lineage selection of mouse embryonic stem cells in a stirred bench scale bioreactor with automated process control. Biotechnol. Bioeng. 92, 920–933. ( 10.1002/bit.20668) [DOI] [PubMed] [Google Scholar]
- 77.Kinney MA, Sargent CY, McDevitt TC. 2011. The multiparametric effects of hydrodynamic environments on stem cell culture. Tissue Eng. Part B 17, 249–262. ( 10.1089/ten.teb.2011.0040) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Placzek MR, et al. 2009. Stem cell bioprocessing: fundamentals and principles. J. R. Soc. Interface 6, 209–232. ( 10.1098/rsif.2008.0442) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frith JE, Thomson B, Genever PG. 2010. Dynamic three-dimensional culture methods enhance mesenchymal stem cell properties and increase therapeutic potential. Tissue Eng. Part C 16, 735–749. ( 10.1089/ten.tec.2009.0432) [DOI] [PubMed] [Google Scholar]
- 80.Bartosh TJ, Ylöstalo JH, Mohammadipoor A, Bazhanov N, Coble K, Claypool K, Lee RH, Choi H, Prockop DJ. 2010. Aggregation of human mesenchymal stromal cells (MSCs) into 3D spheroids enhances their antiinflammatory properties. Proc. Natl Acad. Sci. USA 107, 13 724–13 729. ( 10.1073/pnas.1008117107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sart S, Tsai AC, Li Y, Ma T. 2013. Three-dimensional aggregates of mesenchymal stem cells: cellular mechanisms, biological properties, and applications. Tissue Eng. Part B 20, 365–380. ( 10.1089/ten.teb.2013.0537) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang ZY, Teoh SH, Teo EY, Chong MSK, Shin CW, Tien FT, Choolani MA, Chan JK. 2010. A comparison of bioreactors for culture of fetal mesenchymal stem cells for bone tissue engineering. Biomaterials 31, 8684–8695. ( 10.1016/j.biomaterials.2010.07.097) [DOI] [PubMed] [Google Scholar]
- 83.Mukundakrishnan K, Ayyaswamy PS, Risbud M, Hu HH, Shapiro IM. 2004. Modeling of phosphate ion transfer to the surface of osteoblasts under normal gravity and simulated microgravity conditions. Ann. N Y Acad. Sci. 1027, 85–98. ( 10.1196/annals.1324.009) [DOI] [PubMed] [Google Scholar]
- 84.Sikavitsas VI, Bancroft GN, Mikos AG. 2004. Formation of three-dimensional cell/polymer constructs for bone tissue engineering in a spinner flask and a rotating wall vessel bioreactor. J. Biomed. Mater. Res. 62, 136–148. ( 10.1002/jbm.10150) [DOI] [PubMed] [Google Scholar]
- 85.Chun KW, Yoo HS, Yoon JJ, Park TG. 2004. Biodegradable PLGA microcarriers for injectable delivery of chondrocytes: effect of surface modification on cell attachment and function. Biotechnol. Prog. 20, 1797–1801. ( 10.1021/bp0496981) [DOI] [PubMed] [Google Scholar]
- 86.Mercier NR, Costantino HR, Tracy MA, Bonassar LJ. 2005. Poly(lactide-co-glycolide) microspheres as a moldable scaffold for cartilage tissue engineering. Biomaterials 26, 1945–1952. ( 10.1016/j.biomaterials.2004.06.030) [DOI] [PubMed] [Google Scholar]
- 87.Hong Y, Gao C, Xie Y, Gong Y, Shen J. 2005. Collagen-coated polylactide microspheres as chondrocyte microcarriers. Biomaterials 26, 6305–6313. ( 10.1016/j.biomaterials.2005.03.038) [DOI] [PubMed] [Google Scholar]
- 88.Botchwey EA, Pollack SR, Levine EM, Laurencin CT. 2001. Bone tissue engineering in a rotating bioreactor using a microcarrier matrix system. J. Biomed. Mater. Res. 55, 242–253. () [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rowley J, Abraham E, Campbell A, Brandwein H, Oh S. 2012. Meeting lot-size challenges of manufacturing adherent cells for therapy. BioProcess. Int. 103, 16–20. [Google Scholar]
- 90.Goh TW-P, Zhang Z-Y, Chen AK-L, Reuveny S, Choolani M, Chan JKY, Oh SK-W. 2013. Microcarrier culture for efficient expansion and osteogenic differentiation of human fetal mesenchymal stem cells. BioResearch Open Access 2, 84–97. ( 10.1089/biores.2013.0001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bardy J, Chen AK, Lim YM, Wu S, Wei S, Weiping H, Chan K, Reuveny S, Oh SK. 2013. Microcarrier suspension cultures for high-density expansion and differentiation of human pluripotent stem cells to neural progenitor cells. Tissue Eng. Part C 19, 166–180. ( 10.1089/ten.tec.2012.0146) [DOI] [PubMed] [Google Scholar]
- 92.Simaria AS, Hassan S, Varadaraju H, Rowley J, Warren K, Vanek P, Farid SS. 2013. Allogeneic cell therapy bioprocess economics and optimization: single-use cell expansion technologies. Biotechnol. Bioeng. 111, 69–83. ( 10.1002/bit.25008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sart S, Schneider YJ, Agathos SN. 2010. Influence of culture parameters on ear mesenchymal stem cells expanded on microcarriers. J. Biotechnol. 150, 149–160. ( 10.1016/j.jbiotec.2010.08.003) [DOI] [PubMed] [Google Scholar]
- 94.Want AJ, Nienow AW, Hewitt CJ, Coopman K. 2012. Large-scale expansion and exploitation of pluripotent stem cells for regenerative medicine purposes: beyond the T flask. Regen. Med. 7, 71–84. ( 10.2217/rme.11.101) [DOI] [PubMed] [Google Scholar]
- 95.Kehoe D, Schnitzler A, Simler J, DiLeo A, Ball A. 2012. Scale-up of human mesenchymal stem cells on microcarriers in suspension in a single-use bioreactor. BioPharm. Int. 25, 28–38. [Google Scholar]
- 96.Sart S, Agathos SN, Li Y. 2013. Engineering stem cell fate with biochemical and biomechanical properties of microcarriers. Biotechnol. Prog. 29, 1354–1366. ( 10.1002/btpr.1825) [DOI] [PubMed] [Google Scholar]
- 97.dos Santos F, et al. 2011. Toward a clinical-grade expansion of mesenchymal stem cells from human sources: a microcarrier-based culture system under xeno-free conditions. Tissue Eng. Part C 17, 1201–1210. ( 10.1089/ten.tec.2011.0255) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hughes CS, Radan L, Betts D, Postovit LM, Lajoie GA. 2011. Proteomic analysis of extracellular matrices used in stem cell culture. Proteomics 11, 3983–3991. ( 10.1002/pmic.201100030) [DOI] [PubMed] [Google Scholar]
- 99.Roubelakis MG, Pappa KI, Bitsika V, Zagoura D, Vlahou A, Papadaki HA, Antsaklis A, Anagnou NP. 2007. Molecular and proteomic characterization of human mesenchymal stem cells derived from amniotic fluid: comparison to bone marrow mesenchymal stem cells. Stem Cells Dev. 16, 931–952. ( 10.1089/scd.2007.0036) [DOI] [PubMed] [Google Scholar]
- 100.Maijenburg MW, Noort WA, Kleijer M, Kompier CJ, Weijer K, van Buul JD, van der Schoot CE, Voermans C. 2010. Cell cycle and tissue of origin contribute to the migratory behaviour of human fetal and adult mesenchymal stromal cells. Br. J. Haematol. 148, 428–440. ( 10.1111/j.1365-2141.2009.07960.x) [DOI] [PubMed] [Google Scholar]
- 101.Zhang ZY, Teoh SH, Chong MS, Schantz JT, Fisk NM, Choolani MA, Chan J. 2009. Superior osteogenic capacity for bone tissue engineering of fetal compared with perinatal and adult mesenchymal stem cells. Stem Cells 27, 126–137. ( 10.1634/stemcells.2008-0456) [DOI] [PubMed] [Google Scholar]
- 102.Sart S, Errachid A, Schneider YJ, Agathos SN. 2013. Modulation of mesenchymal stem cell actin organization on conventional microcarriers for proliferation and differentiation in stirred bioreactors. J. Tissue Eng. Regen. Med. 7, 537–551. ( 10.1002/term.545) [DOI] [PubMed] [Google Scholar]
- 103.Weber C, Pohl S, Pörtner R, Wallrapp C, Kassem M, Geigle P, Czermak P. 2007. Expansion and harvesting of hMSC-TERT. Open Biomed. Eng. J. 1, 38–46. ( 10.2174/1874120700701010038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang Y, Rossi FM, Putnins EE. 2007. Ex vivo expansion of rat bone marrow mesenchymal stromal cells on microcarrier beads in spin culture. Biomaterials 28, 3110–3120. ( 10.1016/j.biomaterials.2007.03.015) [DOI] [PubMed] [Google Scholar]
- 105.Chen AK-L, Chen X, Choo AB, Reuveny S, Oh SK. 2011. Critical microcarrier properties affecting the expansion of undifferentiated human embryonic stem cells. Stem Cell Res. 7, 97–111. ( 10.1016/j.scr.2011.04.007) [DOI] [PubMed] [Google Scholar]
- 106.Nie Y, Walsh P, Clarke DL, Rowley JA, Fellner T. 2014. Scalable passaging of adherent human pluripotent stem cells. PLoS ONE 9, e88012 ( 10.1371/journal.pone.0088012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Heng BC, Li J, Chen AK, Reuveny S, Cool SM, Birch WR, Oh SK. 2012. Translating human embryonic stem cells from 2-dimensional to 3-dimensional cultures in a defined medium on laminin- and vitronectin-coated surfaces. Stem Cells Dev. 21, 1701–1715. ( 10.1089/scd.2011.0509) [DOI] [PubMed] [Google Scholar]
- 108.Lam AT-L, Li J, Chen AK, Reuveny S, Oh SK, Birch WR. 2014. Cationic surface charge combined with either vitronectin or laminin dictates the evolution of hESC/microcarrier aggregates and cell growth in agitated cultures. Stem Cells Dev. 23, 1688–1703. ( 10.1089/scd.2013.0645) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Miyazaki T, et al. 2008. Recombinant human laminin isoforms can support the undifferentiated growth of human embryonic stem cells. Biochem. Biophys. Res. Commun. 375, 27–32. ( 10.1016/j.bbrc.2008.07.111) [DOI] [PubMed] [Google Scholar]
- 110.Rodin S, Domogatskaya A, Ström S, Hansson EM, Chien KR, Inzunza J, Hovatta O, Tryggvason K. 2010. Long-term self-renewal of human pluripotent stem cells on human recombinant laminin-511. Nat. Biotechnol. 28, 611–615. ( 10.1038/nbt.1620) [DOI] [PubMed] [Google Scholar]
- 111.Melkoumian Z, et al. 2010. Synthetic peptide-acrylate surfaces for long-term self-renewal and cardiomyocyte differentiation of human embryonic stem cells. Nat. Biotechnol. 28, 606–610. ( 10.1038/nbt.1629) [DOI] [PubMed] [Google Scholar]
- 112.Kolhar P, Kotamraju VR, Hikita ST, Clegg DO, Ruoslahti E. 2010. Synthetic surfaces for human embryonic stem cell culture. J. Biotechnol. 146, 143–146. ( 10.1016/j.jbiotec.2010.01.016) [DOI] [PubMed] [Google Scholar]
- 113.Villa-Diaz LG, Nandivada H, Ding J, Nogueira-de-Souza NC, Krebsbach PH, O'Shea KS, Lahann J, Smith GD. 2010. Synthetic polymer coatings for long-term growth of human embryonic stem cells. Nat. Biotechnol. 28, 581–583. ( 10.1038/nbt.1631) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Villa-Diaz LG, Ross AM, Lahann J, Krebsbach PH. 2013. Concise review: The evolution of human pluripotent stem cell culture: from feeder cells to synthetic coatings. Stem Cells 31, 1–7. ( 10.1002/stem.1260) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Huebsch N, Arany PR, Mao AS, Shvartsman D, Ali OA, Bencherif SA, Rivera-Feliciano J, Mooney DJ. 2010. Harnessing traction-mediated manipulation of the cell/matrix interface to control stem-cell fate. Nat. Mater. 9, 518–526. ( 10.1038/nmat2732) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Engler AJ, Sen S, Sweeney HL, Discher DE. 2006. Matrix elasticity directs stem cell lineage specification. Cell 126, 677–689. ( 10.1016/j.cell.2006.06.044) [DOI] [PubMed] [Google Scholar]
- 117.Lanniel M, Huq E, Allen S, Buttery L, Williams PM, Alexander MR. 2011. Substrate induced differentiation of human mesenchymal stem cells on hydrogels with modified surface chemistry and controlled modulus. Soft Matter 7, 6501–6514. ( 10.1039/c1sm05167a) [DOI] [Google Scholar]
- 118.Shih YR, Tseng KF, Lai HY, Lin CH, Lee OK. 2011. Matrix stiffness regulation of integrin-mediated mechanotransduction during osteogenic differentiation of human mesenchymal stem cells. J. Bone Miner. Res. 26, 730–738. ( 10.1002/jbmr.278) [DOI] [PubMed] [Google Scholar]
- 119.Zoldan J, Karagiannis ED, Lee CY, Anderson DG, Langer R, Levenberg S. 2011. The influence of scaffold elasticity on germ layer specification of human embryonic stem cells. Biomaterials 32, 9612–9621. ( 10.1016/j.biomaterials.2011.09.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kehoe DE, Jing D, Lock LT, Tzanakakis ES. 2010. Scalable stirred-suspension bioreactor culture of human pluripotent stem cells. Tissue Eng. Part A 16, 405–421. ( 10.1089/ten.tea.2009.0454) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hunt MM, Meng G, Rancourt DE, Gates ID, Kallos MS. 2014. Factorial experimental design for the culture of human embryonic stem cells as aggregates in stirred suspension bioreactors reveals the potential for interaction effects between bioprocess parameters. Tissue Eng. Part C 20, 76–89. ( 10.1089/ten.tec.2013.0040) [DOI] [PubMed] [Google Scholar]
- 122.Cormier JT, zur Nieden NI, Rancourt DE, Kallos MS. 2006. Expansion of undifferentiated murine embryonic stem cells as aggregates in suspension culture bioreactors. Tissue Eng. 12, 3233–3245. ( 10.1089/ten.2006.12.3233) [DOI] [PubMed] [Google Scholar]
- 123.Wolfe RP, Ahsan T. 2013. Shear stress during early embryonic stem cell differentiation promotes hematopoietic and endothelial phenotypes. Biotechnol. Bioeng. 110, 1231–1242. ( 10.1002/bit.24782) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Przybyla LM, Voldman J. 2012. Probing embryonic stem cell autocrine and paracrine signaling using microfluidics. Ann. Rev. Anal. Chem. 5, 293–315. ( 10.1146/annurev-anchem-062011-143122) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Przybyla LM, Voldman J. 2012. Attenuation of extrinsic signaling reveals the importance of matrix remodeling on maintenance of embryonic stem cell self-renewal. Proc. Natl Acad. Sci. USA 109, 835–840. ( 10.1073/pnas.1103100109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Toh Y-C, Voldman J. 2011. Fluid shear stress primes mouse embryonic stem cells for differentiation in a self-renewing environment via heparan sulfate proteoglycans transduction. FASEB J. 25, 1208–1217. ( 10.1096/fj.10-168971) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bliem R, Katinger H. 1988. Scale-up engineering in animal-cell technology. Parts I and II. Trends Biotechnol. 6, 190–195, 224–230 ( 10.1016/0167-7799(88)90045-5) [DOI] [Google Scholar]
- 128.Sen A, Kallos MS, Behie LA. 2001. Effects of hydrodynamics on cultures of mammalian neural stem cell aggregates in suspension bioreactors. Ind. Eng. Chem. Res. 40, 5350–5357. ( 10.1021/ie001107y) [DOI] [Google Scholar]
- 129.Wu J, Rostami MR, Cadavid Olaya DP, Tzanakakis ES. 2014. Oxygen transport and stem cell aggregation in stirred-suspension bioreactor cultures. PLoS ONE 9, e102486 ( 10.1371/journal.pone.0102486) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Prado-Lopez S, et al. 2010. Hypoxia promotes efficient differentiation of human embryonic stem cells to functional endothelium. Stem Cells 28, 407–418. (doi:10.1002/stem.295) [DOI] [PubMed] [Google Scholar]
- 131.Koay EJ, Athanasiou KA. 2008. Hypoxic chondrogenic differentiation of human embryonic stem cells enhances cartilage protein synthesis and biomechanical functionality. Osteoarthrit. Cartilage 16, 1450–1456. ( 10.1016/j.joca.2008.04.007) [DOI] [PubMed] [Google Scholar]
- 132.Ezashi T, Das P, Roberts RM. 2005. Low O2 tensions and the prevention of differentiation of hES cells. Proc. Natl Acad. Sci. USA 102, 4783–4788. ( 10.1073/pnas.0501283102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Forsyth NR, Musio A, Vezzoni P, Simpson AH, Noble BS, McWhir J. 2006. Physiologic oxygen enhances human embryonic stem cell clonal recovery and reduces chromosomal abnormalities. Cloning Stem Cells 8, 16–23. ( 10.1089/clo.2006.8.16) [DOI] [PubMed] [Google Scholar]
- 134.Martin Y, Vermette P. 2005. Bioreactors for tissue mass culture: design, characterization, and recent advances. Biomaterials 26, 7481–7503. ( 10.1016/j.biomaterials.2005.05.057) [DOI] [PubMed] [Google Scholar]
- 135.Singh H, Mok P, Balakrishnan T, Rahmat SN, Zweigerdt R. 2010. Up-scaling single cell-inoculated suspension culture of human embryonic stem cells. Stem Cell Res. 4, 165–179. ( 10.1016/j.scr.2010.03.001) [DOI] [PubMed] [Google Scholar]
- 136.Olmer R, Lange A, Selzer S, Kasper C, Haverich A, Martin U, Zweigerdt R. 2012. Suspension culture of human pluripotent stem cells in controlled, stirred bioreactors. Tissue Eng. Part C 18, 772–784. ( 10.1089/ten.tec.2011.0717) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Leung HW, Chen A, Choo AB, Reuveny S, Oh SK. 2011. Agitation can induce differentiation of human pluripotent stem cells in microcarrier cultures. Tissue Eng. Part C 17, 165–172. ( 10.1089/ten.tec.2010.0320) [DOI] [PubMed] [Google Scholar]
- 138.Mitalipova MM, Rao RR, Hoyer DM, Johnson JA, Meisner LF, Jones KL, Dalton S, Stice SL. 2005. Preserving the genetic integrity of human embryonic stem cells. Nat. Biotechnol. 23, 19–20. ( 10.1038/nbt0105-19) [DOI] [PubMed] [Google Scholar]
- 139.Canavan HE, Cheng X, Graham DJ, Ratner BD, Castner DG. 2005. Cell sheet detachment affects the extracellular matrix: a surface science study comparing thermal liftoff, enzymatic, and mechanical methods. J. Biomed. Mater. Res. Part A 75, 1–13. ( 10.1002/jbm.a.30297) [DOI] [PubMed] [Google Scholar]
- 140.Potapova IA, Brink PR, Cohen IS, Doronin SV. 2008. Culturing of human mesenchymal stem cells as three-dimensional aggregates induces functional expression of CXCR4 that regulates adhesion to endothelial cells. J. Biol. Chem. 283, 13 100–13 107. ( 10.1074/jbc.M800184200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nienow AW, Rafiq QA, Coopman K, Hewitt CJ. 2014. A potentially scalable method for the harvesting of hMSCs from microcarriers. Biochem. Eng. J. 85, 79–88. ( 10.1016/j.bej.2014.02.005) [DOI] [Google Scholar]
- 142.Watanabe K, et al. 2007. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 25, 681–686. ( 10.1038/nbt1310) [DOI] [PubMed] [Google Scholar]
- 143.Oh SKW, Chen AK, Mok Y, Chen X, Lim UM, Chin A, Choo AB, Reuveny S. 2009. Long-term microcarrier suspension cultures of human embryonic stem cells. Stem Cell Res. 2, 219–230. ( 10.1016/j.scr.2009.02.005) [DOI] [PubMed] [Google Scholar]
- 144.Yang J, Yamato M, Nishida K, Ohki T, Kanzaki M, Sekine H, Shimizu T, Okano T. 2006. Cell delivery in regenerative medicine: the cell sheet engineering approach. J. Control. Release 116, 193–203. ( 10.1016/j.jconrel.2006.06.022) [DOI] [PubMed] [Google Scholar]
- 145.Okano T, Yamada N, Okuhara M, Sakai H, Sakurai Y. 1995. Mechanism of cell detachment from temperature-modulated, hydrophilic-hydrophobic polymer surfaces. Biomaterials 16, 297–303. ( 10.1016/0142-9612(95)93257-E) [DOI] [PubMed] [Google Scholar]
- 146.Arisaka Y, Kobayashi J, Yamato M, Akiyama Y, Okano T. 2013. Switching of cell growth/detachment on heparin-functionalized thermoresponsive surface for rapid cell sheet fabrication and manipulation. Biomaterials 34, 4214–4222. ( 10.1016/j.biomaterials.2013.02.056) [DOI] [PubMed] [Google Scholar]
- 147.Higuchi A, et al. 2006. Temperature-induced cell detachment on immobilized pluronic surface. J. Biomed Mater. Res. A 79, 380–392. ( 10.1002/jbm.a.30773) [DOI] [PubMed] [Google Scholar]
- 148.Ebara M, Yamato M, Aoyagi T, Kikuchi A, Sakai K, Okano T. 2004. Immobilization of cell-adhesive peptides to temperature-responsive surfaces facilitates both serum-free cell adhesion and noninvasive cell harvest. Tissue Eng. 10, 1125–1135. ( 10.1089/1076327041887691) [DOI] [PubMed] [Google Scholar]
- 149.Kenda-Ropson N, Lenglois S, Miller AO. 2002. Microsupport with two-dimensional geometry 2D-MS. 4. Temperature-induced detachment of anchorage-dependent CHO-K1 cells from cryoresponsive MicroHex((R)) (CryoHex). Cytotechnology 39, 163–170. ( 10.1023/A:1023949716842) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tamura A, Kobayashi J, Yamato M, Okano T. 2012. Temperature-responsive poly(N-isopropylacrylamide)-grafted microcarriers for large-scale non-invasive harvest of anchorage-dependent cells. Biomaterials 33, 3803–3812. ( 10.1016/j.biomaterials.2012.01.060) [DOI] [PubMed] [Google Scholar]
- 151.Yang HS, Jeon O, Bhang SH, Lee SH, Kim BS. 2010. Suspension culture of mammalian cells using thermosensitive microcarrier that allows cell detachment without proteolytic enzyme treatment. Cell Transplant. 19, 1123–1132. ( 10.3727/096368910X516664) [DOI] [PubMed] [Google Scholar]
- 152.Gümüşderelioğlu M, Çakmak S, Timuçin HÖ, Çakmak AS. 2013. Thermosensitive PHEMA microcarriers: ATRP synthesis, characterization, and usabilities in cell cultures. J. Biomater. Sci. Polym. Ed. 24, 2110–2125. ( 10.1080/09205063.2013.827104) [DOI] [PubMed] [Google Scholar]
- 153.Heng BC, Vinoth KJ, Liu H, Hande MP, Cao T. 2006. Low temperature tolerance of human embryonic stem cells. Int. J. Med. Sci. 3, 124–129. ( 10.7150/ijms.3.124) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Brun-Graeppi AK, Richard C, Bessodes M, Scherman D, Merten OW. 2011. Cell microcarriers and microcapsules of stimuli-responsive polymers. J. Control. Release 149, 209–224. ( 10.1016/j.jconrel.2010.09.023) [DOI] [PubMed] [Google Scholar]
- 155.Dubois NC, Craft AM, Sharma P, Elliott DA, Stanley EG, Elefanty AG, Gramolini A, Keller G. 2011. SIRPA is a specific cell-surface marker for isolating cardiomyocytes derived from human pluripotent stem cells. Nat. Biotechnol. 29, 1011–1018. ( 10.1038/nbt.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pruszak J, Sonntag KC, Aung MH, Sanchez-Pernaute R, Isacson O. 2007. Markers and methods for cell sorting of human embryonic stem cell-derived neural cell populations. Stem Cells 25, 2257–2268. ( 10.1634/stemcells.2006-0744) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Levenberg S, Golub JS, Amit M, Itskovitz-Eldor J, Langer R. 2002. Endothelial cells derived from human embryonic stem cells. Proc. Natl Acad. Sci. USA 99, 4391–4396. ( 10.1073/pnas.032074999) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Schriebl K, Satianegara G, Hwang A, Tan HL, Fong WJ, Yang HH, Jungbauer A, Choo A. 2012. Selective removal of undifferentiated human embryonic stem cells using magnetic activated cell sorting followed by a cytotoxic antibody. Tissue Eng. Part A 18, 899–909. ( 10.1089/ten.tea.2011.0311) [DOI] [PubMed] [Google Scholar]
- 159.Tan HL, Fong WJ, Lee EH, Yap M, Choo A. 2009. mAb 84, a cytotoxic antibody that kills undifferentiated human embryonic stem cells via oncosis. Stem Cells 27, 1792–1801. ( 10.1002/stem.109) [DOI] [PubMed] [Google Scholar]
- 160.Tohyama S, et al. 2013. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 12, 127–137. ( 10.1016/j.stem.2012.09.013) [DOI] [PubMed] [Google Scholar]
- 161.Abbasalizadeh S, Baharvand H. 2013. Technological progress and challenges towards cGMP manufacturing of human pluripotent stem cells based therapeutic products for allogeneic and autologous cell therapies. Biotechnol. Adv. 31, 1600–1623. ( 10.1016/j.biotechadv.2013.08.009) [DOI] [PubMed] [Google Scholar]
- 162.Diogo MM, da Silva CL, Cabral JM. 2012. Separation technologies for stem cell bioprocessing. Biotechnol. Bioeng. 109, 2699–2709. ( 10.1002/bit.24706) [DOI] [PubMed] [Google Scholar]
- 163.Gupta P, Ismadi M-Z, Verma PJ, Fouras A, Jadhav S, Bellare J, Hourigan K. In press. Optimization of agitation speed in spinner flask for microcarrier structural integrity and expansion of induced plutipotent stem cells. Cytotechnology. (doi:10.1007/s10616-014-9750-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Rafiq QA, Brosnan KM, Coopman K, Nienow AW, Hewitt CJ. 2013. Culture of human mesenchymal stem cells on microcarriers in a 5 l stirred-tank bioreactor. Biotechnol. Lett. 35, 1233–1245. ( 10.1007/s10529-013-1211-9) [DOI] [PubMed] [Google Scholar]
- 165.Serra M, et al. 2010. Improving expansion of pluripotent human embryonic stem cells in perfused bioreactors through oxygen control. J. Biotechnol. 148, 208–215. ( 10.1016/j.jbiotec.2010.06.015) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.