

Abstract
Investigations of interictal epileptiform spikes and seizures have played a central role in the study of epilepsy. The background EEG activity, however, has received less attention. In this chapter we discuss the characteristic features of the background activity of the brain when individuals are at rest and awake (resting wake) and during sleep. The characteristic rhythms of the background EEG are presented, and the presence of 1/f β behavior of the EEG power spectral density is discussed and its possible origin and functional significance. The interictal EEG findings of focal epilepsy and the impact of interictal epileptiform spikes on cognition are also discussed.
Keywords: Electroencephalogram, Epilepsy, Local Field Potential Oscillations, High Frequency Oscillations, Sleep
Introduction
The electrical activity of mammalian brain, defined by the electroencephalogram (EEG), has long been a focus of scientific and clinical brain research (Brazier 1961). The mechanisms underlying various EEG changes associated with brain maturation, behavioral states, cognition, motor function, and neurological disease represent fundamental discoveries of neuroscience. Epilepsy in particular has benefited from EEG investigations (Brazier 1960). A disorder characterized by unprovoked recurrent seizures, epilepsy has many underlying pathological causes but is unified by the common clinical expression of seizures and the associated pathological brain electrical activity. Not long after the discovery of the human EEG (Berger 1929), Berger also reported that epileptic seizures had an abnormal EEG signature, and that between the seizures (interictal) there were also transient epileptiform abnormalities not seen in controls (translated in (Berger et al. 1969)). Thereafter, the significance of interictal epileptiform spikes (IIS) and abnormal transient oscillatory network activity in the development of epilepsy (epileptogenesis), seizure generation (ictogenesis), and associated functional impairments (e.g., cognition, memory, and reaction times) have been active areas of research.
Physiological Electrical Activity in the Normal Mammalian Brain
Since the first observation of the occipital alpha rhythm (Berger 1929) (translated in (Berger et al. 1969)) the interest in brain oscillations and their physiological and pathological correlates has occupied a central position in human neuroscience. Historically clinical and basic research focused on specific oscillations that are prominent in the EEG intermittently, for example the occipital alpha rhythm (α; 8-12 Hz) recorded at rest with eyes closed, beta (β; 12-30 Hz) and gamma frequency activity (γ; 30-50 Hz) during mental and motor tasks, theta frequency oscillations (θ; 4-8 Hz) during memory tasks or sleep, and delta frequency activity (δ; 0.5-4 Hz) that characterizes slow wave sleep. Similarly, the EEG activity in traditional frequency bands (δ, θ, α, β, γ) became the focus of EEG research and intensively studied in brain maturation (Neidermeyer et al. 2005) , normal function, and disease states. However, it is widely recognized that the brain generates activities well outside these classic EEG bands. In fact, the high amplitude EEG activity below δ (< 0.5 Hz) including direct current (DC) changes were some of the earliest electrical activities recorded (Aladjalova 1957)..
While EEG research has largely focused on narrow band EEG oscillations (δ, θ, α, β, γ) it is well recognized that there is a broad spectrum of on-going, arrhythmic, background activity that does not contain a dominant characteristic oscillation, but rather is composed of intermixed spectral frequencies (Buzsaki 2009, He et al. 2010). It is out of this arrhythmic background that the traditional EEG oscillations discussed above, e.g. the posterior dominant alpha rhythm, may be evoked or spontaneously emerge (Figure 1). More recently, this broad spectrum of on-going background activity has been a focus of attention (Buzsaki 2009, He et al. 2010, Linkenkaer-Hansen et al. 2001).
Figure 1.

Interictal Background Activity from human hippocampus recorded with intracranial depth electrode. There is an ongoing background activity followed by a paroxysmal gamma frequency oscillation (bold arrows), and an interictal epileptiform spike (IIS, arrow). The wavelet transform (1 - 600 Hz, Morlet basis) spectrogram of raw intracranial EEG shows the background theta activity, and the emergent low amplitude gamma oscillation preceding the IIS. The scale bar is normalized units standard deviations from background. Time base is 100 msec per division. (Courtesy of Liankun Ren, M.D. unpublished)
This composition of background electrical activity in mammalian brain generally follows power-law behavior, i.e. the spectral power scales with frequency as 1/f β where β is called a scaling exponent (Figure 2). This 1/f β (“one-over-f”) spectrum with lower frequency activities exhibiting higher amplitudes than faster frequencies is characterized by the scaling exponent β that can be obtained by plotting log power vs. log frequency (log(Power) v.s. -β log(f )) and ranges over 0 < β < 4 (He et al. 2010, Freeman et al. 2003, Parish et al. 2004, Worrell et al. 2002). The spectral peaks embedded in the 1/f β represent ongoing persistent oscillations or organized emergent oscillations that arise out of the ongoing EEG background, such as the traditional EEG rhythms of the human EEG (Figure 2).
Figure 2.

Power spectral density (PSD) from 5 minutes of human hippocampus during sleep recorded with intracranial EEG (0.05 - 10,000 Hz, sampled at 32 kHz) using micro- and clinical macro- electrodes. The wide bandwidth recording exhibits 1/f β behavior with different scaling regions characterized by different scaling exponents β = 1 and 2. The inset shows an expansion of the PSD in the 0.05 - 40 Hz range and spectral peaks from a low delta frequency oscillation (~ 0.75 Hz) and a theta-alpha frequency oscillation (7.45 Hz). The characteristic 0.75 Hz oscillation is persistent throughout the 5 minutes and modulates the 7.45 Hz intermittent oscillation. (Unpublished Data)
The arrhythmic background activity has more recently received attention within the context of the advancing understanding of complex systems. It is recognized that 1/f β behavior in complex systems can be a signature of a self-organized system with scale-free dynamics (He et al. 2010, Freeman et al. 2003, Plenz et al. 2007). It turns out that 1/f β patterns are ubiquitous in nature, from the statistics of earthquakes to stock market dynamics (Bak 1996). The origin of 1/f β behavior in EEG and local field potential (LFP) recordings remains unclear (He et al. 2010, Bédard et al. 2006, Bédard et al. 2010), but perhaps one of the most intriguing ideas is that it results from hierarchal nesting of brain activity (He et al. 2010, Tort et al. 2010, Canolty et al. 2010) whereby lower frequency activity modulates higher frequency activity (He et al. 2010). The modulation of gamma oscillations by theta oscillations is a classic example (Canolty et al. 2006, Bragin et al. 1995, Belluscio et al. 2012). At the cellular level, multiunit activity is correlated with EEG gamma power and phase-locked to the negative-going phase of the delta frequency activity (Whittingstall et al. 2009). Synchronization between neuronal assemblies also occurs within arrhythmic brain activity (Eckhorn 1994, Thivierge et al. 2008, Manning et al. 2009).
Maturation of EEG
The continuous maturation of EEG activity through young adulthood reflects brain development, e.g. myelination, and organization (Neidermeyer et al. 2005). In premature infants (24 - 27 weeks), the EEG is discontinuous and may alternate between periods containing bursts of high amplitude slow (0.1 - 1 Hz) activity and intermixed faster rhythms (8 - 14 Hz). From these earliest electrical rhythms in the infant brain there are long periods of continuous development through late childhood (~12 y.o) when the posterior dominant alpha rhythm reaches ~10 Hz (Neidermeyer et al. 2005).
Electrical activity of the sleeping brain
There exists substantial evidence for the physiological importance of sleep and in particular the requirement of sleep for normal memory (Diekelmann et al. 2010). To better understand how memory benefits from sleep, it would be helpful to first describe briefly the EEG during the two main types of sleep – rapid eye movement (REM) and non-REM sleep – and then how the neurophysiology of sleep might support aspects of memory formation. Since patients with epilepsy often report deficits in sleep and impairment in memory, subsequent sections describe electrophysiological disturbances in the epileptic brain and their likely functional implications for cognition.
EEG of REM sleep
During REM or desynchronized sleep, arising from a background of low-voltage, mixed frequency EEG, are spontaneous synchronous bursts of neuronal activity generated by the pontine tegmentum that spread to the lateral geniculate nucleus and visual cortices in the occipital lobe that are termed “PGO waves”. Conspicuous in the EEG of rats and cats and less in humans, PGO waves coincide with rapid eye movements and can become phase-locked with theta oscillations. In rodents, theta oscillations occur with largest amplitude in the hippocampal CA1 area driven by inputs from septum, entorhinal cortex, and CA3. In addition to REM sleep, hippocampal theta can also be observed during awake behaviors in rodents. Theta also occurs in humans during wakefulness, but is more apparent in neocortical areas and less coherent in hippocampal areas.
EEG of non-REM sleep
Non-REM sleep is characterized by high-voltage slow wave activity that includes slow oscillations < 1 Hz and delta activity. The slow oscillation persists in isolated neocortical tissue and is abolished if thalamocortical cells are deafferented from cortical inputs, suggesting slow oscillations are generated largely within neocortex (Timofeev et al. 2000, Sanchez-Vives et al. 2000). In scalp EEG, the alternating sequence of surface positive (depth negative) and negative (depth positive) waves correspond with periods of neuronal membrane depolarization and hyperpolarization respectively. Periods of membrane depolarization occur within excitatory and inhibitory cells that produces sustained neuronal firing commonly referred to as “UP-states”, whereas periods of membrane hyperpolarization are accompanied by neuronal silence denoted as “DOWN-states”. The mechanisms generating slow oscillations are not yet clear, although evidence to date suggests UP-states could arise from widespread summation of calcium- and persistent sodium inward current-mediated excitatory postsynaptic potentials in cortical cells, while neuronal disfacilitation associated with DOWN-states could be due to calcium- and sodium-dependent potassium currents, inactivation of persistent sodium currents, and possibly GABA-mediated inhibition.
Slow oscillations strongly modulate two other transient oscillations that occur during non-REM sleep – spindles and sharp wave-ripple complexes – and is another classic example of frequency nesting. Spindle waves are beta frequency oscillations that wax and wane between 10 and 16 Hz and last 0.5 to 2 seconds that characterize stage 2 of NREM sleep. Spindles arise from interactions between GABA-containing neurons in the thalamic reticular nucleus as well as thalamocortical cells that facilitate the synchrony and spread of spindles throughout neocortex. Human studies have identified two types of spindles designated slow and fast; however, whether these two types of spindles arise from different neuronal mechanisms or reflect the modulation of a common spindle generator is not known. Slow (10-12 Hz) spindles occur primarily over frontal cortical areas and more frequently during slow wave sleep than stage 2 sleep, and fast (13-15 Hz) spindles appear broadly over central and parietal cortices and are often coincident with increased hippocampal activity.
In hippocampus during non-REM sleep, spontaneous extra-hippocampal impulses drive neuronal firing in CA3 that projects forward via Schaffer collaterals onto dendritic processes of CA1 pyramidal cells and some types of interneurons. This briefly irregular (30-120 milliseconds in duration), increase in neuronal firing registers in the depth EEG as a large amplitude sharp wave with maximum negativity in stratum lucidum, stratum radiatum, and inner third of stratum moleculare corresponding to input layers of CA3, CA1 and dentate gyrus respectively. In CA1, a similarly brief high-frequency oscillation (HFO; 80-200 Hz) termed “ripple” arises from synchronous firing between pyramidal cells and basket cells that is largest in amplitude in stratum pyramidale and superimposed on the sharp wave. During widespread neuronal depolarization associated with the slow oscillation UP-state, hippocampal ripples can co-occur with neocortical fast spindles to form spindle-ripple events with ripples that temporally coincide with the troughs of spindle waves (Siapas et al. 1998).
Concept of memory function and putative neuronal mechanisms
Memory function generally involves processes of encoding, consolidation, and retrieval. In the awake brain, encoding occurs when perception of the stimuli produces a new, yet unstable, memory trace. During subsequent sleep, the labile memory trace becomes more stable and eventually integrated into brain networks supporting long-term storage of knowledge in a process termed “consolidation”. During retrieval, the stored memory is accessed and recalled. A number of theories have been proposed on how sleep supports memory consolidation with some more than others supported by compelling data from animal and human studies (for extensive review, see (Rasch et al. 2013). Central to current theories (e.g., “active system consolidation”) is the concept of reactivation that involves a sleep-related replay of neuronal firing patterns corresponding to the neuronal firing patterns that occurred while encoding, i.e., during prior wakefulness, as well as specific roles for different types of sleep in memory consolidation.
Considerable research has focused on identifying the neuronal mechanisms that provide sleep-related benefits on memory formation. Current models emphasize precisely coordinated neuronal activity between neocortex and hippocampus for hippocampal-dependent memories (Ekstrom et al. 2003). During non-REM sleep, neocortical slow oscillation UP-states provide a temporal window for increased ripple activity and associated increase in neuronal firing that could reflect reactivation of hippocampal memories. The coincidence of hippocampal ripples with neocortical fast spindles (spindle-ripple events) is thought to promote the transfer and ultimately storage of the hippocampal memory to neocortex (Siapas et al. 1998), which is reflected presumably by long-term functional and structural changes that strengthen synaptic transmission (e.g., long-term potentiation). In addition, evidence suggests REM sleep PGO- and theta-related neuronal activity could also be involved with synaptic modifications with theta possibly playing a role in synaptic downscaling, which extends the “synaptic homeostasis” hypothesis that links the regulation of sleep with mechanisms of synaptic plasticity (Tononi et al. 2003).
Human single neuron correlates of sleep and memory
Microelectrode unit recordings during natural sleep in humans are few, but available data indicate hippocampal neuronal firing increases during non-REM sleep and declines during REM sleep (Ravagnati et al. 1979, Staba et al. 2002). Furthermore, the propensity for burst discharge is highest during non-REM sleep compared to awake and REM sleep episodes. These results are similar to the rates and pattern of hippocampal pyramidal cell firing during non-REM and REM sleep in rodents (Siapas et al. 1998), and are generally consistent with levels of hippocampal activity that could be involved with reactivation described in the preceding paragraphs. Work using the same microelectrode recordings from single neurons in humans has primarily focused on memory and navigation. These studies have led to the discovery of place cells in the human mesial temporal lobe underlying spatial navigation (Ekstrom et al. 2003), which resembles the location-specific firing patterns of some pyramidal cells in non-primate hippocampus described in the sections that follow. In addition, studies in humans have found evidence for neurons that encode category specific images (Kreiman et al. 2000, Kreiman et al. 2000).
Abnormal Electrical Activity in the Epileptic Brain
In addition to sleep and wake behavioral states of normal brain, epileptic brain is characterized by interictal state (between seizures), ictal state (seizures), and post-ictal states (after the seizure). It should be noted that while seizures are generally limited to a minute or so, the post-ictal state as determined by subtle EEG or cognitive and physical changes can be prolonged (Fisher et al. 2010). In addition to the interictal and post-ictal state, there is emerging evidence for a pre-ictal state that is associated with increase in probability of seizure occurrence (Cook et al. 2013, Elger et al. 2013, Mormann et al. 2007).
Interictal Epileptiform Discharges
Interictal EEG spikes (IIS) are brief, sharply contoured voltage fluctuations of less than 200 msec that are a signature of epileptic brain. The intracellular correlate of IIS is the paroxysmal depolarizing shift (Ayala et al. 1973) seen in the neuronal membrane potential and is associated paroxysmal burst of neuronal population firing, but also involves a more complex interaction of inhibitory and excitatory neurons (Ayala et al. 1973). Depth electrode recordings during overnight polysomnographic sleep studies show that in patients with temporal lobe epilepsy (TLE), the highest rates of IIS regularly occur during non-REM stage 3, and in some cases stages 1 and 2, sleep compared to waking and REM sleep (Lieb et al. 1980, Sammaritano et al. 1991). In addition, the spatial distribution of IIS is often broader during non-REM sleep than waking or REM sleep, i.e., IIS appear at electrode recording sites within and remote from where seizures begin (Sammaritano et al. 1991).
At the level of single neurons, patient studies have not consistently found differences in interictal firing rates and bursting inside versus outside the seizure onset zone (SOZ) during awake episodes (Colder et al. 1996, Colder et al. 1996). However, during non-REM and REM sleep compared to wakefulness, interictal firing rates, bursting, and synchrony of discharges are significantly higher in mesial temporal lobe (MTL) ipsilateral to the SOZ than contralateral MTL (Staba et al. 2002, Staba et al. 2002). These results provide evidence for sleep-related facilitation of interictal neuronal hyperexcitability within the SOZ of patients with temporal lobe epilepsy.
Pathological HFO
In the epileptic brain, transient abnormally synchronous discharges of principal cells can summate in the extracellular space that give rise to a burst of population spikes commonly termed pathological HFO or pHFO (Bragin et al. 1999, Bragin et al. 1999). Chronic animal models of epilepsy and studies in patients with epilepsy indicate hippocampal and neocortical pHFOs are strongly associated with brain areas capable of generating spontaneous seizures (Engel 2011). With respect to the wake-sleep cycle, recordings in epileptic rats and patients show the highest rates of hippocampal pHFOs occur during non-REM sleep compared to awake and REM sleep, while equivalent rates can be found during the latter two desynchronized EEG states (Staba et al. 2002). By contrast, ripples are highest during non-REM sleep, while rates are lower during wakefulness and lowest in REM sleep, which is consistent with their occurrence in the normal non-primate hippocampus (Staba et al. 2002).
Pathological Synchrony
Synchronization of neuronal assemblies is thought to underlie normal brain functions such as perception, learning, and cognition. Alterations in neuronal synchrony are thought to underlie the clinical manifestations of many neurological diseases. Hypersynchrony of pathological neuronal assemblies as the generator of epileptiform activity has been a central theme of epileptic brain electrophysiology (Penfield et al. 1954). Jasper and Penfield speculated that the local high amplitude interictal epileptiform activity recorded directly from human cortex during surgery was generated by a burst of hypersynchronous neuronal activity (Penfield et al. 1954). Interestingly, however, many seizures appear to begin with an apparent “asynchronous state” - low amplitude LFP activity that evolves into a hypersynchronous state with high amplitude rhythmic activity (Penfield et al. 1954). Multiple studies have reported increased local synchrony, i.e. hypersynchrony, within epileptic brain using a range of quantitative measures of synchrony, including spectral coherence (Towle et al. 1999), magnitude squared coherence (Zaveri et al. 2009), and mean phase coherence (Schevon et al. 2007). In addition, investigations of LFP synchrony during spontaneous human seizures have consistently demonstrated a decrease in local LFP synchrony at seizure onset compared to baseline (Mormann et al. 2000, Schindler et al. 2007, Wendling et al. 2003) .
Analysis of long records of interictal iEEG from patients with focal epilepsy and control subjects with intractable facial pain found that the spatial distribution of LFP synchronization fell rapidly with the distance between electrodes (Warren et al. 2010). Consistent with the hypothesis that the generators of normal and pathological HFOs are more spatially localized than lower frequency oscillations, the synchrony fall off is frequency dependent (Logothetis et al. 2007). Synchrony in the epileptic brain, however, was shown to be markedly reduced in electrodes bridging connections between the SOZ and surrounding brain (Figure 3). In effect, the SOZ is functionally disconnected and isolated from surrounding brain regions (Warren et al. 2010).
Figure 3.
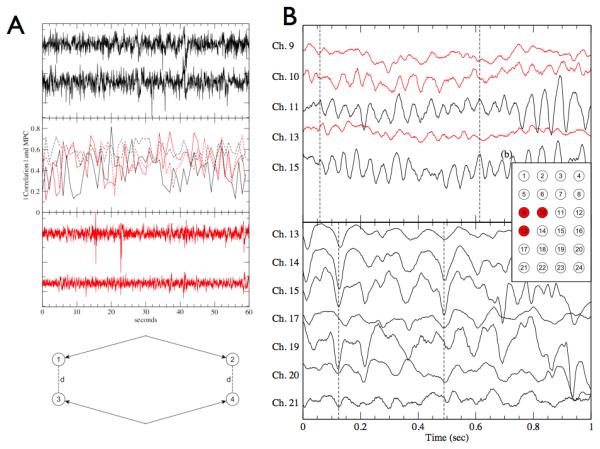
Data and from 2 patients, one with intractable facial pain (black) and other with focal epilepsy (gray). Data from the patient with intractable facial pain and no history of seizures serves as a control recording for quantitative comparison. A) (a) Sample signals from two electrodes of the control brain recording. (b) The correlation magnitudes (solid lines) and mean phase coherence (dashed lines) of the signal pairs in (a) and (c). Both the correlation and mean phase coherence (MPC) show significant temporal variability over the course of 60 seconds, with values primarily ranging from (0.2 - 0.7) (c) Sample signals from two electrodes in epileptic cortex outside the seizure onset zone. (d) A sample layout of the bipolar reference pair measurement. The distance between one corresponding pair of electrodes 1 and 3 is equal to the distance between the other pair, electrodes 2 and 3, and this is the distance referenced in our bipolar measurements (Fig. 3). B) Sample interictal iEEG signals from Patient 1 with epilepsy from both inside the seizure onset zone (SOZ), shown in gray, and near signals outside the SOZ (black). Dashed line marks significant phase lag between inside and outside the SOZ. (b) Spatial layout of the intracranial electrodes for Patients 1 and A with the SOZ electrodes (9, 10 and 13) of Patient 1 shown in red. (c) Sample signals from the control Patient A. The spatial numbering is as shown in (b). For clarification, signals are offset vertically. (Reproduced from Warren CP et al. J Neurophysiol 2010, Dec;104(6):3530-9)
Focal EEG Slowing
In addition to the IIS and pHFO that have been widely investigated, focal slowing on the EEG is common in the region of epileptic brain. Focal delta frequency slowing was initially described in patients with focal structural abnormalities, such as tumors and strokes (Gloor et al. 1977, Walter 1936), but is also common in TLE (Blume et al. 1993). When the slowing occurs as intermittent oscillations of monomorphic delta activity in the temporal lobe region it is termed, temporal intermittent rhythmic delta activity and is a signature of focal epilepsy (Reiher et al. 1989, Tao et al. 2011). Focal delta frequency slowing has also recently been shown to be more common than IIS following febrile status epilepticus (Nordli et al. 2012).
Hypsarrhythmia
Hypsarrhythmia is the an EEG pattern characterized by disorganized high-amplitude spikes and spike- and/or sharp-slow wave discharges and commonly observed in infants with West syndrome (Infantile spasms, Hypsarrhthymia, and Developmental regression) (Figure 4). Hypsarrhythmia is modulated by the sleep-wake cycle. During non-REM sleep, there is a tendency for runs of these high-amplitude discharges to become grouped or clustered with a period consistent with non-REM slow wave activity that typically is then followed by episodes of EEG attenuation (Gibbs et al. 1941). By contrast, during REM sleep, there is a significant reduction and in some cases disappearance of hypsarrhythmia that reappears toward the end of the REM sleep episode, and if awakening then hypsarrhythmia continues often with clinical manifestations.
Figure 4.
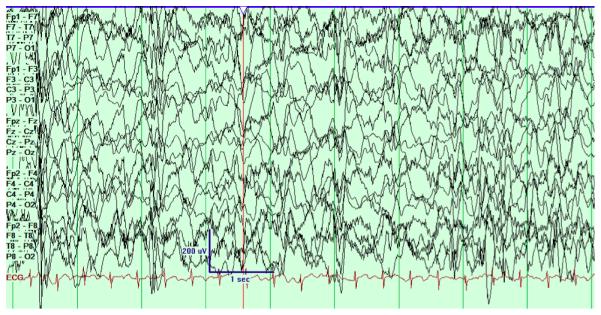
The pattern of hypsarrhythmia is a specific EEG pattern associated with West Syndrome. First described in detail by Gibbs (66) the pattern consists of high voltage, disorganized EEG with multifocal and generalized epileptiform spikes and sharp waves. The characteristic pattern, often described as disorganized or chaotic, is unique in that the normal pattern of spatial synchrony over multiple brain regions is absent. The EEG tracing from each channel (e.g. Fp1 and F7) appear independent of each other despite the anatomical proximity. This is distinct from normal brain activity where there is widespread synchronous activity over multiple brain regions. In the hypsarryhthmia pattern the periods of generalized synchrony are due to generalized epileptiform discharges. The epileptiform spikes fluctuate in time and space, and are various focal, multifocal, and generalized.
IIS impact on cognition
While epilepsy in general could have detrimental effects on sleep, sleep-related epileptiform activity in particular could contribute to cognitive impairment (Elger et al. 2004, Noebels et al. 2012). Epileptic encephalopathies refer to conditions in some patients with epilepsy who have neurological deterioration associated with frequency or severity of seizures and/or interictal epileptiform discharges and not due to the original cause or etiology (Nabbout et al. 2003). Hypsarrhthymia in West syndrome and Electrical Status Epilepticus During Sleep associated with continuous spike-wave of sleep and Landau-Kleffner syndrome reflect interictal EEG patterns that predominate during non-REM sleep that could support abnormal activity-dependent synaptic plasticity which in turn produce cognitive impairments. Improved control of seizures and interictal EEG in these patients is often associated with improved cognitive performance that suggests ictal and interictal discharges contribute to these deficits (Holmes et al. 2006). Indeed, studies in adults and children show IIS can be associated with brief episodes of impaired cognitive functioning referred to as “transient cognitive impairment” (Aarts et al. 1984). The functional disruption coincides with the location were the IIS occur, e.g., verbal memory task impairment with left-side IIS, spatial task impairment with right-sided interictal epileptiform discharges (IEDs). Others studies observed IIS in occipital cortices disrupted visual stimuli presented in the contralateral visual field (Shewmon et al. 1988). Work using pilocarpine-treated epileptic rats showed that in a hippocampal-dependent memory task, hippocampal IIS occurring during memory retrieval, but not encoding or maintenance, reduced performance (Kleen et al. 2010, Kleen et al. 2013). Similar results were observed in presurgical patients during a short-term memory task (Kleen et al. 2013). In this study, when depth electrode-recorded hippocampal IIS contralateral to SOZ or bilateral occurred during memory retrieval or maintenance, performance was lower. Recent evidence implicates brain network activity underlying memory processing in suppression of the IEDs, however, which raises an important confound of the interplay between cognitive processing on IIS (Matsumoto et al. 2013).
How IIS disrupt memory processes is not known, but studies in epileptic rats show hippocampal IIS are associated with a decrease in CA1 cell firing (Zhou et al. 2007). Moreover, a significant reduction in firing was found after compared to before the IIS in interneurons, but not CA1 pyramidal cells that preferentially discharge when the rat is a specific location in the environment (“place cell”). A separate study of CA3 cell firing found significantly lower firing rates in interneurons and pyramidal cells before and after IIS compare to rates during random episodes (Zhou et al. 2007). These data indicate that neuronal firing surrounding and particularly after IIS could reflect episodes associated with reduced activity-dependent synaptic plasticity. By contrast, spontaneous pHFOs reflect brief episodes of increased neuronal discharges that are two-fold greater or more than the level of activity that occurs during spontaneous hippocampal ripples. Unlike ripples that involves interneuron-mediated regulation of pyramidal cell firing, it appears that the effects of interneurons are diminished during pHFOs. The abnormally high spatial and temporal coincidence of discharges could contribute to pathological synaptic plasticity that is functionally disruptive to memory consolidation. One study of resective sclerotic hippocampal tissue, which is often associated with pHFOs and hyperexcitability in patients with drug-resistant temporal lobe epilepsy, found significantly lower levels of long-term potentiation and its synaptic counterpart long-term depression in sclerotic compared to non-sclerotic tissue (Beck et al. 2000). These data suggest morphological alterations associated with hippocampal sclerosis and persistent abnormal interictal activity contribute to diminished capacity for physiological activity-dependent synaptic plasticity.
Disruptions in sleep and sleep-related oscillatory activity could interfere with aspects of memory formation. Patients with epilepsy are two times more likely to complain of sleep disturbances, chiefly excessive daytime sleepiness and insomnia that can have a negative influence on quality of life measures (de Weerd et al. 2004, Piperidou et al. 2008). Studies indicate seizures, comorbidity (e.g., sleep apnea), and anti-seizure drugs can cause disruptions in the amount and architecture of sleep. During nights with nocturnal seizures, patients often have a greater number of nighttime stage-shifts or awakenings, increased amounts of non-REM stage 1 sleep, and reduced amounts of non-REM stage 3 sleep and REM sleep (Tononi et al. 2003). Daytime seizures, which themselves could prevent or interfere with learning, also reduce the amount of REM sleep during the subsequent night (Tononi et al. 2003). Furthermore, older types of anti-seizure drugs, such as barbiturates and benzodiazepines, can reduce the amount of REM sleep and in some cases stage 3 non-REM sleep. Newer drugs have no effect or can even increase the amount of REM sleep, although some reduce amounts of non-REM sleep (e.g., Gabapentin, Lamotrigine). Loss of slow wave-rich non-REM sleep or REM sleep in terms of absolute amounts and relative to when daytime learning occurs could negate the time-dependent benefits of sleep on memory formation.
Conclusions
Is the “background” activity important?
The answer to this question is clearly yes. There is good evidence that the background EEG activity contains important information about brain function and dysfunction in human epilepsy. While the diagnostic importance of IIS and seizures is clear, there is also evidence that the background EEG during sleep and wake is important prognostic tool. In addition, investigations of LFP synchrony and neuronal assemblies are providing mechanistic insights about brain function, cognition, and epilepsy related comorbidities.
Is interictal background really “normal”?
In some epilepsy cases there are clear EEG background abnormalities. In West syndrome, Electrical Status Epilepticus in Slow-wave Sleep and other epileptic encephalopathies the EEG background is markedly abnormal. Whether there are more subtle abnormalities in the background in primary generalized epilepsy is an area of active study, but often the EEG on visual review appears normal. In focal epilepsy there may be focal slowing in the region of epileptic brain. In drug resistant epilepsy there are abnormalities in LFP synchrony that are present even in the absence of IIS.
Our cognition and behavior rely upon precisely-timed interactions among neurons forming brain networks by coordinated activity of anatomically distributed neuronal networks mediated via rhythmic brain oscillations (Uhlhaas et al. 2010). Research into the common cognitive (Elger et al. 2004) and behavioral (Kanner 2011, Téllez-Zenteno et al. 2007) comorbidities of epilepsy is only beginning to emerge. As our understanding of the cellular mechanisms of cognition and behavior advance the impact on understanding the impact on epilepsy comorbidities should be significant.
Acknowledgements
We are honored to have the opportunity to participate in this book celebrating Philip Schwartzkroin’s many contributions to epilepsy research.
Other Acknowledgements: This research was supported by NIH R01-NS071048 (RS) and NIH R01-NS63039 (GW)
References:
- Aarts JH, Binnie CD, Smit AM, Wilkins AJ. Brain. 1984;107:293–308. doi: 10.1093/brain/107.1.293. Pt 1. [DOI] [PubMed] [Google Scholar]
- Aladjalova NA. Nature. 1957;4567:957–9. doi: 10.1038/179957a0. [DOI] [PubMed] [Google Scholar]
- Ayala GF, Dichter M, Gumnit RJ, Matsumoto H, Spencer WA. Brain Res. 1973;52:1–17. doi: 10.1016/0006-8993(73)90647-1. [DOI] [PubMed] [Google Scholar]
- Bak P. Nature. 1996;383:772–3. [Google Scholar]
- Beck H, Goussakov IV, Lie A, Helmstaedter C, Elger CE. The Journal of Neuroscience. 2000;20:7080–6. doi: 10.1523/JNEUROSCI.20-18-07080.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belluscio MA, Mizuseki K, Schmidt R, Kempter R, Buzsáki G. The Journal of neuroscience. 2012;32:423–35. doi: 10.1523/JNEUROSCI.4122-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger H. I Mitteilung Arch Psychiatr Nervenkr. 1929;87:527–70. [Google Scholar]
- Berger H, Gloor P. Hans Berger on the electroencephalogram of man: The fourteen original reports on the human electroencephalogram. Elsevier Publishing Company; Amsterdam: 1969. [Google Scholar]
- Bédard C, Kröger H, Destexhe A. Phys Rev Lett. 2006;97:118102. doi: 10.1103/PhysRevLett.97.118102. [DOI] [PubMed] [Google Scholar]
- Bédard C, Rodrigues S, Roy N, Contreras D, Destexhe A. J Comput Neurosci. 2010;29:389–403. doi: 10.1007/s10827-010-0250-7. [DOI] [PubMed] [Google Scholar]
- Blume WT, Borghesi JL, Lemieux JF. Ann Neurol. 1993;34:703–9. doi: 10.1002/ana.410340513. [DOI] [PubMed] [Google Scholar]
- Bragin A, Engel J, Wilson CL, Fried I, Buzsaki G. Hippocampus. 1999;9:137–42. doi: 10.1002/(SICI)1098-1063(1999)9:2<137::AID-HIPO5>3.0.CO;2-0. J. [DOI] [PubMed] [Google Scholar]
- Bragin A, Engel J, Wilson CL, Fried I, Mathern GW. Epilepsia. 1999;40:127–37. doi: 10.1111/j.1528-1157.1999.tb02065.x. J. [DOI] [PubMed] [Google Scholar]
- Bragin A, Jando G, Nadasdy Z, Hetke J, et al. J Neurosci. 1995;15:47–60. doi: 10.1523/JNEUROSCI.15-01-00047.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazier MA. Epilepsia. 1960;1:328–36. doi: 10.1111/j.1528-1157.1959.tb04270.x. [DOI] [PubMed] [Google Scholar]
- Brazier MA. A history of the electrical activity of the brain: The first half-century. Macmillan; 1961. [Google Scholar]
- Buzsaki G. Rhythms of the Brain. Oxford University Press; 2009. [Google Scholar]
- Canolty RT, Edwards E, Dalal SS, Soltani M, et al. Science. 2006;313:1626–8. doi: 10.1126/science.1128115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canolty RT, Knight RT. Trends Cogn Sci. 2010;14:506–15. doi: 10.1016/j.tics.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colder BW, Frysinger RC, Wilson CL, Harper RM, Engel J. Epilepsia. 1996;37:113–21. doi: 10.1111/j.1528-1157.1996.tb00001.x. J. [DOI] [PubMed] [Google Scholar]
- Colder BW, Wilson CL, Frysinger RC, Harper RM, Engel J. Brain Res. 1996;719:96–103. doi: 10.1016/0006-8993(96)00107-2. J. [DOI] [PubMed] [Google Scholar]
- Cook MJ, O'Brien TJ, Berkovic SF, Murphy M, et al. Lancet Neurol. 2013;12:563–71. doi: 10.1016/S1474-4422(13)70075-9. [DOI] [PubMed] [Google Scholar]
- Diekelmann S, Born J. Nature Reviews Neuroscience. 2010;11:114–26. doi: 10.1038/nrn2762. [DOI] [PubMed] [Google Scholar]
- Eckhorn R. Prog Brain Res. 1994;102:405–26. doi: 10.1016/S0079-6123(08)60556-7. [DOI] [PubMed] [Google Scholar]
- Ekstrom AD, Kahana MJ, Caplan JB, Fields TA, et al. Nature. 2003;425:184–8. doi: 10.1038/nature01964. [DOI] [PubMed] [Google Scholar]
- Elger CE, Helmstaedter C, Kurthen M. The Lancet Neurology. 2004;3:663–72. doi: 10.1016/S1474-4422(04)00906-8. [DOI] [PubMed] [Google Scholar]
- Elger CE, Mormann F. Lancet Neurol. 2013;12:531–2. doi: 10.1016/S1474-4422(13)70092-9. [DOI] [PubMed] [Google Scholar]
- Engel J. Biomark Med. 2011;5:537–44. doi: 10.2217/bmm.11.62. [DOI] [PubMed] [Google Scholar]
- Fisher RS, Engel JJ. Epilepsy Behav. 2010;19:100–4. doi: 10.1016/j.yebeh.2010.06.038. [DOI] [PubMed] [Google Scholar]
- Freeman WJ, Holmes MD, Burke BC, Vanhatalo S. Clin Neurophysiol. 2003;114:1053–68. doi: 10.1016/s1388-2457(03)00045-2. [DOI] [PubMed] [Google Scholar]
- Gibbs FA, Gibbs EL. Atlas of electroencephalography. FA Gibbs, Boston City Hospital; 1941. [Google Scholar]
- Gloor P, Ball G, Schaul N. Neurology. 1977;27:326. doi: 10.1212/wnl.27.4.326. [DOI] [PubMed] [Google Scholar]
- He BJ, Zempel JM, Snyder AZ, Raichle ME. Neuron. 2010;66:353–69. doi: 10.1016/j.neuron.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes GL, Lenck-Santini PP. Epilepsy Behav. 2006;8:504–15. doi: 10.1016/j.yebeh.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Kanner AM. Epilepsy Currents. 2011;11:90–1. doi: 10.5698/1535-7511-11.3.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleen JK, Scott RC, Holmes GL, Lenck-Santini PP. Ann Neurol. 2010;67:250–7. doi: 10.1002/ana.21896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleen JK, Scott RC, Holmes GL, Roberts DW, et al. Neurology. 2013;81:18–24. doi: 10.1212/WNL.0b013e318297ee50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiman G, Koch C, Fried I. Nat Neurosci. 2000;3:946–53. doi: 10.1038/78868. [DOI] [PubMed] [Google Scholar]
- Kreiman G, Koch C, Fried I. Nature. 2000;408:357–61. doi: 10.1038/35042575. [DOI] [PubMed] [Google Scholar]
- Lieb JP, Joseph JP, Engel J, Walker J, Crandall PH. Electroencephalogr Clin Neurophysiol. 1980;49:538–57. doi: 10.1016/0013-4694(80)90396-x. J. [DOI] [PubMed] [Google Scholar]
- Linkenkaer-Hansen K, Nikouline VV, Palva JM, Ilmoniemi RJ. J Neurosci. 2001;21:1370–7. doi: 10.1523/JNEUROSCI.21-04-01370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis NK, Kayser C, Oeltermann A. Neuron. 2007;55:809–23. doi: 10.1016/j.neuron.2007.07.027. [DOI] [PubMed] [Google Scholar]
- Manning JR, Jacobs J, Fried I, Kahana MJ. J Neurosci. 2009;29:13613–20. doi: 10.1523/JNEUROSCI.2041-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto JY, Stead M, Kucewicz MT, Matsumoto AJ, et al. Brain. 2013;136:2444–56. doi: 10.1093/brain/awt159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormann F, Andrzejak RG, Elger CE, Lehnertz K. Brain. 2007;130:314–33. doi: 10.1093/brain/awl241. [DOI] [PubMed] [Google Scholar]
- Mormann F, Lehnertz K, David P, Elger CE. Physica D: Nonlinear Phenomena. 2000;144:358–69. [Google Scholar]
- Nabbout R, Dulac O. J Clin Neurophysiol. 2003;20:393–7. doi: 10.1097/00004691-200311000-00002. [DOI] [PubMed] [Google Scholar]
- Neidermeyer E, Da Silva FL. Electroencephalography: basic Principals, Clinical Applications, and Related Fields. Lippincott and Wilkins; Philadelphia: 2005. [Google Scholar]
- Noebels JL, Avoli M, Rogawski MA, Olsen RW, et al. 2012.
- Nordli DR, Moshé SL, Shinnar S, Hesdorffer DC, et al. Neurology. 2012;79:2180–6. doi: 10.1212/WNL.0b013e3182759766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish LM, Worrell GA, Cranstoun SD, Stead SM, et al. Neuroscience. 2004;125:1069–76. doi: 10.1016/j.neuroscience.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Penfield Jasper. Epilepsy and the functional anatomy of the human brain. Little Brown; Boston: 1954. [Google Scholar]
- Piperidou C, Karlovasitou A, Triantafyllou N, Terzoudi A, et al. Seizure. 2008;17:588–94. doi: 10.1016/j.seizure.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Plenz D, Thiagarajan TC. Trends Neurosci. 2007;30:101–10. doi: 10.1016/j.tins.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Rasch B, Born J. Physiol Rev. 2013;93:681–766. doi: 10.1152/physrev.00032.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravagnati L, Halgren E, Babb TL, Crandall PH. Sleep. 1979;2:161–73. doi: 10.1093/sleep/2.2.161. [DOI] [PubMed] [Google Scholar]
- Reiher J, Beaudry M, Leduc CP. Can J Neurol Sci. 1989;16:398–401. doi: 10.1017/s0317167100029450. [DOI] [PubMed] [Google Scholar]
- Sammaritano M, Gigli GL, Gotman J. Neurology. 1991;41:290–7. doi: 10.1212/wnl.41.2_part_1.290. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vives MV, McCormick DA. Nat Neurosci. 2000;3:1027–34. doi: 10.1038/79848. [DOI] [PubMed] [Google Scholar]
- Schevon CA, Cappell J, Emerson R, Isler J, et al. Neuroimage. 2007;35:140–8. doi: 10.1016/j.neuroimage.2006.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler K, Leung H, Elger CE, Lehnertz K. Brain. 2007;130:65–77. doi: 10.1093/brain/awl304. [DOI] [PubMed] [Google Scholar]
- Shewmon DA, Erwin RJ. Ann Neurol. 1988;23:131–7. doi: 10.1002/ana.410230205. [DOI] [PubMed] [Google Scholar]
- Siapas AG, Wilson MA. Neuron. 1998;21:1123–8. doi: 10.1016/s0896-6273(00)80629-7. [DOI] [PubMed] [Google Scholar]
- Staba RJ, Wilson CL, Bragin A, Fried I, Engel J. J Neurophysiol. 2002;88:1743–52. doi: 10.1152/jn.2002.88.4.1743. [DOI] [PubMed] [Google Scholar]
- Staba RJ, Wilson CL, Bragin A, Fried I, Engel J. J Neurosci. 2002;22:5694–704. doi: 10.1523/JNEUROSCI.22-13-05694.2002. J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staba RJ, Wilson CL, Fried I, Engel J. Hippocampus. 2002;12:724–34. doi: 10.1002/hipo.10026. J. [DOI] [PubMed] [Google Scholar]
- Tao JX, Chen XJ, Baldwin M, Yung I, et al. Epilepsia. 2011;52:467–76. doi: 10.1111/j.1528-1167.2010.02918.x. [DOI] [PubMed] [Google Scholar]
- Téllez-Zenteno JF, Dhar R, Hernandez-Ronquillo L, Wiebe S. Brain. 2007;130:334–45. doi: 10.1093/brain/awl316. [DOI] [PubMed] [Google Scholar]
- Thivierge JP, Cisek P. J Neurosci. 2008;28:7968–78. doi: 10.1523/JNEUROSCI.0870-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeev I, Grenier F, Bazhenov M, Sejnowski TJ, Steriade M. Cereb Cortex. 2000;10:1185–99. doi: 10.1093/cercor/10.12.1185. [DOI] [PubMed] [Google Scholar]
- Tononi G, Cirelli C. Brain Res Bull. 2003;62:143–50. doi: 10.1016/j.brainresbull.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Tort AB, Komorowski R, Eichenbaum H, Kopell N. J Neurophysiol. 2010;104:1195–210. doi: 10.1152/jn.00106.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towle VL, Carder RK, Khorasani L, Lindberg D. J Clin Neurophysiol. 1999;16:528–47. doi: 10.1097/00004691-199911000-00005. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. Nature Reviews Neuroscience. 2010;11:100–13. doi: 10.1038/nrn2774. [DOI] [PubMed] [Google Scholar]
- Walter G. Lancet. 1936;2:305–8. [Google Scholar]
- Warren CP, Hu S, Stead M, Brinkmann BH, et al. J Neurophysiol. 2010;104:3530–9. doi: 10.1152/jn.00368.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Weerd A, de Haas S, Otte A, Trenité DK, et al. Epilepsia. 2004;45:1397–404. doi: 10.1111/j.0013-9580.2004.46703.x. [DOI] [PubMed] [Google Scholar]
- Wendling F, Bartolomei F, Bellanger JJ, Bourien J, Chauvel P. Brain. 2003;126:1449–59. doi: 10.1093/brain/awg144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittingstall K, Logothetis NK. Neuron. 2009;64:281–9. doi: 10.1016/j.neuron.2009.08.016. [DOI] [PubMed] [Google Scholar]
- Worrell GA, Cranstoun SD, Echauz J, Litt B. Neuroreport. 2002;13:2017–21. doi: 10.1097/00001756-200211150-00005. [DOI] [PubMed] [Google Scholar]
- Zaveri HP, Pincus SM, Goncharova II, Duckrow RB, et al. Neuroreport. 2009;20:891–5. doi: 10.1097/WNR.0b013e32832c78e0. [DOI] [PubMed] [Google Scholar]
- Zhou JL, Lenck-Santini PP, Zhao Q, Holmes GL. Epilepsia. 2007;48:720–31. doi: 10.1111/j.1528-1167.2006.00972.x. [DOI] [PubMed] [Google Scholar]