

Abstract
Data along several lines of evidence have suggested that a systemic autoimmune response may be provoked in glaucoma and could contribute to retinal ganglion cell (RGC) loss. If such an autoimmune response exists one could predict that in cases of unilateral glaucoma, autoantibodies generated would affect both eyes, leading to damage in the unaffected, contralateral, eye in an intraocular pressure (IOP) independent fashion. However, such an effect has not yet been reported. There are currently no data to reconcile these contrasting observations but a review of the literature suggests a possible explanation.
The Contralateral Eye is frequently not Normal In Experimental Glaucoma Models
Elevated IOP reliably leads to the progressive RGC loss and optic nerve axonal damage in mice, rats and monkeys. A number of experimental approaches are available to induce elevated IOP in one eye of these animals including laser coagulation of the episcleral veins, injection of microbeads or hyaluronan into the anterior chamber, or episcleral vein injection of hypertonic saline.1–5
One of the perceived benefits of inducible models was that glaucoma could be induced in one eye, the contralateral eye serving as an internal control. However, observations suggest that the contralateral eye is not normal in these animals and exhibits clear differences from eyes obtained from naïve animals. For example Gallego et al6 found elevated levels of glial fibrillary acid protein (GFAP), major histocompatibility complex class II molecule (MHC-II), and neurofilament of 200 kD (NF200) positive RGC in the control eyes of mice with unilaterally elevated IOP, indicating macro- and microglial activation and RGC damage. There was a mild progressive RGC loss in the uninduced eyes in a model of ischemia/reperfusion damage.7 As a consequence many investigators have now moved away from using the contralateral eye as a normal control, relying on eyes from naïve animals instead.
How, then, could a neurodegenerative stimulus be transmitted to the unaffected eye in induced animal models? One mechanism might be through cytokines secreted into the circulation by the affected eye, but to date little data exist to support the notion of elevated serum levels of pro-inflammatory cytokines and it is difficult to imagine that the retina would synthesize sufficiently large quantities of such compounds to raise steady-state levels systemically. Alternatively, it is also possible that degenerative impulses are transmitted to the contralateral eye via the visual centers of the brain. There is good evidence of degenerative changes in the lateral geniculate nucleus in primates with elevated IOP and in human glaucoma patients.8–10 It is conceivable that this process also affects the synaptic terminals of RGC in the unaffected eye that extend ipsilateral projections to the same lateral geniculate nucleus. However, there is currently no data to either support or discount this possibility.
Serum-Antibodies Against Retinal Antigens are Frequently Observed
In contrast, there is considerable evidence to suggest that glaucomatous degeneration is frequently accompanied by the presence of serum autoantibodies directed against retinal antigens.11–13 These have been observed in both primary and secondary glaucomas, including exfoliation glaucoma, suggesting that their appearance is not the primary cause of RGC death, but is most likely a consequence thereof. It appears that antibodies appear to be capable to exit the retinal vasculature and binding to targets within the retinal ganglion cell layer.14
The presence of anti-RGC antibodies are potentially pathologic and indeed injection of antibodies directed against heat shock proteins or preparations of optic nerve proteins into the tail veins of mice or rats have been reported to result in RGC loss15,16. While these data demonstrate that it is in principle possible for serum antibodies to cause RGC death, it must be cautioned that in these experiments antibodies were administered with Freud’s incomplete adjuvant or pertussis toxin, which might create an unphysiological degree of retinal vessel leakage or an excessively pro-inflammatory environment. Nevertheless, these experiments indicate that under the right circumstances, IgG accumulation in the retina can lead to RGC death. Binding of IgG to RGC can also be observed in the retinas of human eye donors.14 Immunohistochemical detection of human IgG in retinas of donors with or without glaucoma reveals that approximately 1% of all ganglion cells are bound by autoantibodies (Figure 1). The fraction of antibody-bound RGC appears to be slightly higher in glaucomatous retina, but eyes from older donors without glaucoma also contain an appreciable number of such cells. The presence of IgG-bound RGC and the fact that the serum of older non-glaucomatous patients also contains anti-retinal IgG raises the question: If autoantibodies are capable of inducing RGC damage why does this not occur in non-glaucomatous individuals or in the second eye of a unilateral glaucoma case?
Figure 1.
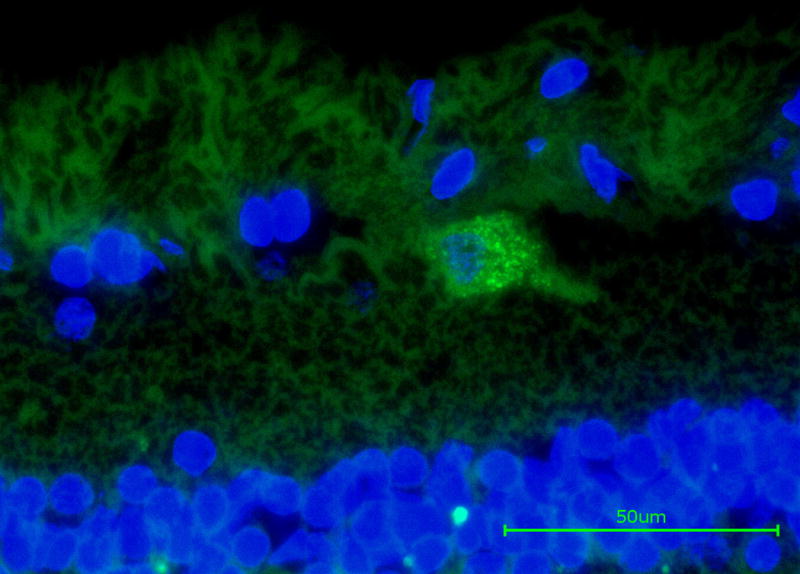
immunohistochemical detection of endogenous IgG (green label) bound to retinal ganglion cells in the retina of a human eye donor with glaucoma. In the sagittal section IgG was detected following incubation with an anti-human IgG antibody. Nuclei were counterstained with DAPI (blue) to facilitate orientation. Image courtesy of Dr. O. Gramlich (University of Mainz)
The Role of the Complement Cascade in Neuroinflammation
One explanation might be that effective mechanisms exist to avoid destruction of RGC through a retinal immune response. Cells bound by antibody are not necessarily condemned to cell death, particularly in an environment such as the retina without cytotoxic T cells, macrophages, or natural killer cells. However, one process that can quickly result in the degeneration of an antibody bound cell in the retina is the activation of the classical complement cascade. This process, which is frequently initialized by immunoglobulins binding to the surface of a pathogen or a degenerating cell, can result in the formation of the Membrane Attack Complex (MAC) and lead to cell lysis if left uninhibited. The degeneration of RGC in the retina secondary to, for example IOP elevation, is accompanied by the marked accumulation of components of the classical complement cascade, including complement component 1Q (C1Q) and complement component 3 (C3), in association with RGC and the formation of MAC.17,18 In human donor eyes with advanced glaucoma, MAC labeling can be regionally observed on the majority of RGCs, presumably associated with areas of active RGC degeneration (Figure 2).
Figure 2.
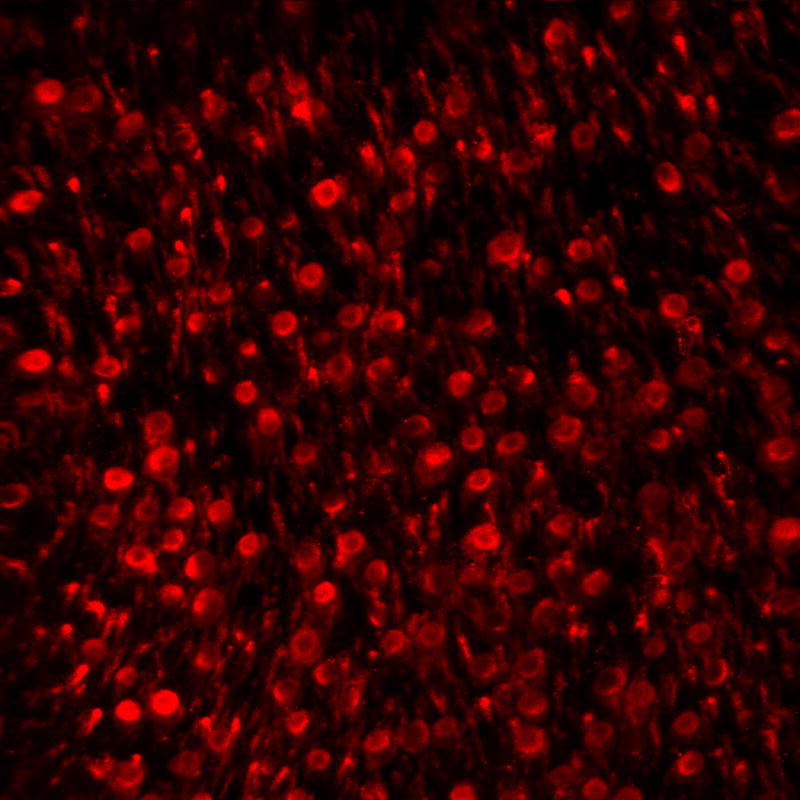
Immunohistochemical detection of MAC associated with retinal ganglion cells. In this image of a flat mounted retina of a human donor with advanced glaucoma profound labeling is observed in distinct regions. Other retinal regions of the same eye exhibit far fewer MAC positive cells or none at all.
Studies using complement-deficient mice demonstrate that in animals lacking a functional complement cascade, RGC death occurs more slowly, although ultimately a similar number of RGC are lost.7,19,20 These findings suggest that the role of complement activation is to actively promote the rapid demise and clearance of damaged RGC cells. Such a mechanism might be desirable in order to avoid the development of an autoimmune response (reviewed in21). Patients with C1 and C2 deficiencies frequently develop autoimmune disease, and it has been hypothesized that this is the result of inefficient clearance of debris following cell death thus allowing an opportunity for the immune system to mount a response.22,23 One could hypothesize that an autoimmune response in the retina might result either from the prolonged presence of degenerating RGCs or perhaps from a brief, but catastrophic, disruption of the blood-retina barrier as in the case of splinter hemorrhage.
Lack of a Pro-inflammatory Environment Could Protect the Healthy Eye from Autoimmunity
Glaucoma-associated activation of complement in the retina is accompanied by synthesis of C1q, C3, and perhaps, C4 by retinal cells.17 Local synthesis of these initiating components not only avoids a systemic response of the innate immune system, but also allows a response that is attuned to the severity of the RGC damage. Importantly C1q synthesis is readily detectable in glaucomatous eyes but is very low or absent in healthy eyes. This local control over the magnitude of the complement response may explain why the presence of autoantibodies in patients without glaucoma does not lead to the development of RGC loss, or why damage to the contralateral eye in cases of unilateral glaucoma might be comparatively mild or even absent: If activation of complement and the formation of MAC serves to eliminate RGC bound by IgG, then the absence of C1Q synthesis in otherwise healthy eyes prevents initiation of this process. Consequently the presence of anti-RGC antibodies in the serum might result in IgG bound RGC in the second eye of unilateral glaucoma cases, but not in the destruction of these cells.
There are currently few data to support or refute this hypothesis. However some predictions could be made:
Depending on the type of autoantibody created and the predisposition of the patient, significant differences may exist between individuals.
Effects on the contralateral eye will generally be mild, but progressive.
The establishment of a pro-inflammatory environment, even due to non-ocular conditions, could significantly influence the extent to which autoimmune processes exert damage. Analogous to the events observed in the brain primed micro- and macroglia in the contralateral eye may become damaging in response to systemic inflammation24
Finally, studies testing the notion that the second eye in patients with unilateral glaucoma remains unaffected would contribute much to this question. Such studies might involve nerve fiber layer thickness measurements in the second eye over several years, using consistent instrumentation and parameters. Patients with moderate to advanced glaucoma may develop damage through mechanisms that are unaffected by modulating IOP. Consequently, an autoimmune component to glaucoma, if it indeed contributes to pathology in humans, would require treatment paradigms that are far different from current medical practice.
References
- 1.Morrison JC, Moore CG, Deppmeier LM, et al. A rat model of chronic pressure-induced optic nerve damage. Experimental Eye Res. 1997;64:85–96. doi: 10.1006/exer.1996.0184. [DOI] [PubMed] [Google Scholar]
- 2.Moreno MC, Marcos HJ, Oscar Croxatto J, et al. A new experimental model of glaucoma in rats through intracameral injections of hyaluronic acid. Experimental Eye Res. 2005;81:71–80. doi: 10.1016/j.exer.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 3.Grozdanic SD, Kwon YH, Sakaguchi DS, et al. Functional evaluation of retina and optic nerve in the rat model of chronic ocular hypertension. Experimental Eye Res. 2004 Jul;79:75–83. doi: 10.1016/j.exer.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 4.Chen H, Wei X, Cho KS, et al. Optic neuropathy due to microbead-induced elevated intraocular pressure in the mouse. Investigative ophthalmology & visual science. 2011 Jan;52:36–44. doi: 10.1167/iovs.09-5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rasmussen CA, Kaufman PL. Primate glaucoma models. J Glaucoma. 2005;14:311–314. doi: 10.1097/01.ijg.0000169409.01635.bc. [DOI] [PubMed] [Google Scholar]
- 6.Gallego BI, Salazar JJ, de Hoz R, et al. IOP induces upregulation of GFAP and MHC-II and microglia reactivity in mice retina contralateral to experimental glaucoma. J Neuroinflammation. 2012;9:92. doi: 10.1186/1742-2094-9-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuehn MH, Kim CY, Jiang B, et al. Disruption of the complement cascade delays retinal ganglion cell death following retinal ischemia-reperfusion. Experimental Eye Res. 2008;87:89–95. doi: 10.1016/j.exer.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 8.Yucel YH, Zhang Q, Weinreb RN, et al. Effects of retinal ganglion cell loss on magno-, parvo-, koniocellular pathways in the lateral geniculate nucleus and visual cortex in glaucoma. Progress in retinal and eye Res. 2003;22:465–481. doi: 10.1016/s1350-9462(03)00026-0. [DOI] [PubMed] [Google Scholar]
- 9.Yucel YH, Zhang Q, Weinreb RN, et al. Atrophy of relay neurons in magno- and parvocellular layers in the lateral geniculate nucleus in experimental glaucoma. Investigative ophthalmology & visual science. 2001 Dec;42:3216–3222. [PubMed] [Google Scholar]
- 10.Gupta N, Greenberg G, de Tilly LN, et al. Atrophy of the lateral geniculate nucleus in human glaucoma detected by magnetic resonance imaging. The British journal of ophthalmology. 2009;93:56–60. doi: 10.1136/bjo.2008.138172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altintas O, Yuksel N, Sonmez GT, et al. Serum antiphospholipid antibody levels in pseudoexfoliation. J Glaucoma. 2012 Jun-Jul;21:326–330. doi: 10.1097/IJG.0b013e31821206cd. [DOI] [PubMed] [Google Scholar]
- 12.Dervan EW, Chen H, Ho SL, et al. Protein macroarray profiling of serum autoantibodies in pseudoexfoliation glaucoma. Investigative ophthalmology & visual science. 2010 Jun;51:2968–2975. doi: 10.1167/iovs.09-4898. [DOI] [PubMed] [Google Scholar]
- 13.Joachim SC, Bruns K, Lackner KJ, et al. Analysis of IgG antibody patterns against retinal antigens and antibodies to alpha-crystallin, GFAP, and alpha-enolase in sera of patients with “wet” age-related macular degeneration. Graefe’s archive for clinical and experimental ophthalmology. 2007;245:619–626. doi: 10.1007/s00417-006-0429-9. [DOI] [PubMed] [Google Scholar]
- 14.Gramlich OW, Beck S, von Thun Und Hohenstein-Blaul N, et al. Enhanced insight into the autoimmune component of glaucoma: IgG autoantibody accumulation and pro-inflammatory conditions in human glaucomatous retina. PloS one. 2013;8:e57557. doi: 10.1371/journal.pone.0057557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wax MB, Tezel G, Yang J, et al. Induced autoimmunity to heat shock proteins elicits glaucomatous loss of retinal ganglion cell neurons via activated T-cell-derived fas-ligand. J Neuroscience. 2008;28:12085–12096. doi: 10.1523/JNEUROSCI.3200-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laspas P, Gramlich OW, Muller HD, et al. Autoreactive antibodies and loss of retinal ganglion cells in rats induced by immunization with ocular antigens. Investigative ophthalmology & visual science. 2011;52:8835–8848. doi: 10.1167/iovs.10-6889. [DOI] [PubMed] [Google Scholar]
- 17.Kuehn MH, Kim CY, Ostojic J, et al. Retinal synthesis and deposition of complement components induced by ocular hypertension. Experimental Eye Res. 2006;83:620–628. doi: 10.1016/j.exer.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Stasi K, Nagel D, Yang X, et al. Complement component 1Q (C1Q) upregulation in retina of murine, primate, and human glaucomatous eyes. Investigative ophthalmology & visual science. 2006;47:1024–1029. doi: 10.1167/iovs.05-0830. [DOI] [PubMed] [Google Scholar]
- 19.Howell GR, Macalinao DG, Sousa GL, et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. The Journal of clinical investigation. 2011;121:1429–1444. doi: 10.1172/JCI44646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howell GR, Soto I, Ryan M, et al. Deficiency of complement component 5 ameliorates glaucoma in DBA/2J mice. J Neuroinflammation. 2013;10:76. doi: 10.1186/1742-2094-10-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexander JJ, Anderson AJ, Barnum SR, et al. The complement cascade: Yin-Yang in neuroinflammation–neuro-protection and -degeneration. J Neurochemistry. 2008;107:1169–1187. doi: 10.1111/j.1471-4159.2008.05668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Truedsson L, Bengtsson AA, Sturfelt G. Complement deficiencies and systemic lupus erythematosus. Autoimmunity. 2007;40:560–566. doi: 10.1080/08916930701510673. [DOI] [PubMed] [Google Scholar]
- 23.Fraser DA, Tenner AJ. Directing an appropriate immune response: the role of defense collagens and other soluble pattern recognition molecules. Current drug targets. 2008;9:113–122. doi: 10.2174/138945008783502476. [DOI] [PubMed] [Google Scholar]
- 24.Perry VH. Contribution of systemic inflammation to chronic neurodegeneration. Acta Neuropathologica. 2010;120:277–286. doi: 10.1007/s00401-010-0722-x. [DOI] [PubMed] [Google Scholar]