

Abstract
Previously, we observed that mir-155 is induced during dendritic cell (DC) differentiation. We now demon-strated convincing evidence indicating that mir-155 promotes DC maturation and regulates its capacity for antigen presentation and induction of alloreactive T cell activation. Interestingly, the induction of miR-155 expression in DCs is dependent on the TLR4/Myd88/NF-κB signaling. Our mechanistic studies further revealed that SOCS1 is a direct target for mir-155, and by binding to its 3’UTR, mir-155 is likely to affect SOCS1 translation. Suppression of mir-155 expression in DCs significantly attenuated LPS-induced DC maturation along with reduced capability to stimulate allogeneic T cell proliferation. As a result, administration of antagomiR-155 provided protection for cardiac allografts from rejection. Together, our data support that suppression of miR-155 in DCs could be a viable therapeutic strategy for prevention and treatment of allograft rejection in clinical setting of transplantation.
Keywords: miR-155, dendritic cells, immune response, allograft
Introduction
Dendritic cells (DCs) are the most potent antigen-presenting cells in immune system, whose maturation and functional state determine the outcome of immune response [1]. DCs initiate adaptive immune responses by presenting antigen peptide with MHC molecules, as well as providing co-stimulatory signals and supporting cytokines. Over the last decade, studies have shown that immature DCs with low levels of expression of MHC and costimulatory molecules possess the capability to induce antigen-specific T-cell hyporesponsiveness and tolerance [2,3]. Therefore, manipulation of DC maturation state appeared to be a promising approach for tolerance induction in organ transplantation [4].
MicroRNAs (miRNA) are a class of highly conserved noncoding RNA that regulate the expression of target genes involved in numerous physiologic processes [5]. miRNAs exert profound effects on the development, differentiation and function of immune cells and play important roles in the regulation of innate and acquired immune responses [6-8]. Previously, we demonstrated that during the course of RNA virus infection, miR-155 can be induced to promote host anti-virus response by targeting negative regulator of IFN-γ downstream singling [9]. Furthermore, our MPSS data mining analysis revealed a marked donw-regulation for miR-155 after DC maturation [10], but its potential biological relevant is yet to be clarified. We thus in the present study investigated the functional relevance for miR-155 in the regulation of immune response and allograft survival in the setting of organ transplantation. Our studies revealed that miR-155 regulates DC phenotype and its allostimulatory capability. Knockdown of miR-155 inhibited DC secretion of IL-12 and expression of CD40/80/86 along with suppressed T cell activation and proliferation. As a result, administration of antagomiR-155 could significantly prolong cardiac allograft survival in mice. Our data support that miR-155 could be a potential therapeutic target for treatment of allograft rejection.
Materials and methods
Mice and reagents
C57BL/6 (H-2b), BALB/C (H-2d) male mice (6 weeks) were purchased from Joint Ventures Sipper BK Experimental Animal Company (Shanghai, China). TLR3, TLR4, TLR9, RIG-I and MyD88 knockout mice were obtained as described previously [9,11]. All animal experiments were approved by the Institutional Animal Care and Use Committee of Second Military Medical University. The mice were maintained in IVC systems with temperature at 24°C on a 12-h light/dark cycle. Mice were anesthetized by pentobarbital sodium and decapitated before collecting bone marrow cells. Pyrrolidinecarbodithoic acid (PDTC, an inhibitor of NF-κB), SB203580, an inhibitor for p38), PD98059, a MEK/ERK inhibitor), and SP600125, a JNK inhibitor) were purchased from Calbiochem. Lipopolysaccharides (LPS) were obtained from Sigma. Ovalbumin (OVA, ISQAVHAAHAEINEAGR) was purchased from InvivoGen (French) with peptide sequence. Antibodies specific for GAPDH and SOCS1 were from Cell Signaling Technology (Danvers, MA). Murine CD4 magnetic beads for MACS were bought from Miltenyi Biotec (Germany). Mus-CD11c-FITC Ab was purchased from BD Biosciences (Franklin Lakes, NJ).
Cell culture and transfection
Murine dendritic cell line DC2.4 was obtained from the American Type Culture Collection (ATCC) (Manassas, VA) and cultured in DM/F12 with 10% FBS. THP-1 cell line was obtained from ATCC and cultured in RPMI-1640 with 10% FBS. Thioglycolate-elicited murine peritoneal macrophages were collected from the abdominal cavity of mice by washing with RPMI-1640 medium and purified by differential time attachment. Murine DCs were generated from bone marrow (BM) cells, cultured and induced as previously reported [12]. The cells (5 × 105 cells in 0.75 mL) were seeded into each well of a 24-well plate, or 2 × 106 cells in 3 mL were seeded into each well of a 6-well plate. HEK293 cell line was obtained from ATCC and cultured in RPMI-1640 with 10% FBS, and 2 × 104 cells were seeded into each well of a 96-well plate. Lipofectamine RNAiMax was used for RNA transfection and JetSI-ENDO was used for co-transfecting plasmids with RNAs. To generate dendritic cells, monocytes were cultured at 1 × 106/mL in a 2 mL volume of RPMI-1640 medium containing 50 ng/mL GM-CSF and 10ng/mL IL-4 for approximately 48 hours at 37°C.
Bioinformatic analysis
The online software TargetScan (http://www.targetscan.org) was used to predict miR-155 target sites in cellular RNA sequences using the following settings: maximum number of allowed unpaired bases = 0 in 6 nt seed/nucleus sequence, minimum number of paired-up bases in heteroduplex = 12, and maximum folding energy for heteroduplex (Kcal/mol) = 225. The KEGG enrichment analysis was carried by genecodis (http://genecodis2.dacya.ucm.es/).
miR-155 mimics, inhibitor and antgomir
The miR-155 mimics and miR-155 inhibitor were from GenePharma (Shanghai, China) and used to manipulate miR-155 by overexpression or inhibition, respectively, in murine dendritic cells or cell line. Dendritic cells or cell line described above were transfected as described previously [9]. Negative control mimics or inhibitor (GenePharma) were transfected into cells as the respective controls for miR-15 mimics or inhibitor. The micrOFFTM mmu-miR-155 antagomir (Antago miR-155; Ribobio) or negative control antagomir (AntagomiR-NC, Ribobio) were used for miR-155 in vivo research. Antagomir (100 nM) was dissolved in 50 µL PBS and then administrated via intravenous instillation.
Heart transplantation model
The mouse cervical heart transplantation was performed by cuff technique as described perversely using BALB/c mice as donors and C57BL/6 mice as recipients [13,14]. The time of cold ischemia was within 30 minutes and operation can be completed within 1 hour. Usually, the success rate is over 90%, and the mean survival time of allograft is 7 ± 1 days.
Mixed lymphocyte reaction (MLR) and assay for allogeneic T-cell responsiveness
CD4+ T cells were purified by MACS. For MLR assay, CD4+ T-cells (2 × 105/well) were from BALB/c mice, and cocultured with DC2.4 cells at various ratios. T cell proliferation was assessed by pulsing with [3H] thymidine (1Ci/well; Amersham Pharmacia Biotech) for the final 18 h of culture, and thymidine incorporation was determined using a liquid scintillation counter (Wallac, Turku, Finland).
RNA interference
The sequences for SOCS1-specific small interfering RNA (siRNA) used in the experiments were designed by GenePharma (Shanghai, China). Four siRNAs, si-1 (5’-CAG CCA GUU UAG GUA AUA AUU-3’ and 5’-UUG UCG GUC AAA UCC AUU AUU-3’), si-2 (5’-GCG CCU UAU UAU UU CUU AUU U-3’ and 5’-UUC GCG GAA UAA UAA AGA AUA-3’), si-3 (5’-GAA UUC CAC UCC UAC UCU UU-3’ and 5’-UUC UUA AGG UGA GGA UGG AGA-3’), and si-4 (5’-CCC AGU AUC UUU GCA CAA AUU-3’ and 5’-UUG GGU CAU AGA AAC GUG UUU-3’) were employed for the study. A scrambled siRNA provided by GenePharma was used as a control. siRNA duplexes were transfected into cells indicated with a final concentration of 20 nM.
RNA quantification
Total RNA was extracted by Trizol regent and then reverse transcribed for real-time PCR as described previously [9,15]. For miRNA analysis, the reverse transcription primer for miR-155 was 5’-GTC GTA TCC AGT GCA GGG TCC GAG GTA TTC GCA CTG GAT ACG ACC CCC TA-3’. Quantitative PCR primers were 5’-CTC GTG GTT AAT GCT AAT TGT GA-3’ and 5’-GTG CAG GGT CCG AGG T-3’. The relative expression levels for miRNA were assessed by GAPDH normalization as described previously [16]. Primers for each target gene was obtained from PrimerBank (pga.mgh.harvard.edu/primerbank/).
3’-UTR luciferase reporter assays
The luciferase reporter plasmid for SOCS1 3’-UTR was constructed by PCR amplifying the mouse SOCS1 mRNA 3’-UTR sequence containing miR-155 binding site, and then cloned into the pMIR-REPORTTM vector (Invitrogen) using the SpeI site. HEK293 cells were transfected with 100ng of luciferase reporter and 50ng of pRL-TK-Renilla-luciferase plasmid along with an indicated miRNA mimic (e.g., si-4) or inhibitor (final concentration, 20 nM). After 24 hours, the cells were lysed and firefly luciferase activities were detected in a 96-well plate reader as reported [17].
Western blot analysis
The cells were washed twice with cold PBS and lysed with cell lysis buffer (Cell Signaling Technology) supplemented with protease inhibitor mixture (Calbiochem). The total protein concentration was measured by BCA assays (Pierce, Rockford, IL). Equal amount of proteins were loaded onto 10% Polyacrylamide gels for Western blot analysis as described previously [18].
Statistical analysis
Data from multiple groups were analyzed using one-way ANOVA with post-hoc Bonferroni’s correction (GraphPad Prism 5.0, GraphPad Software, San Diego, CA). Data derived from two groups were analyzed using an unpaired Student’s t-test. All data are expressed as mean ± SD. In all cases, differences achieving values of p < 0.05 were considered to be statistically significant.
Results
miR-155 upregulation is associated with DC maturation
By massively parallel signature sequencing (MPSS) analysis we noted that the counting number of miR-155 was significantly higher in matured DCs as compared with that of immature DCs (Figure 1A). qRT-PCR further confirmed that miR-155 was significantly upregulated in matured DCs as compared that in immature DCs and monocytes (Figure 1B). Indeed, LPS induced DCs expression of marked higher levels of miR-155 12 h after stimulation, and peaked at 24 h (Figure 1C). A similar result could be obtained when DCs were challenged with 100 ng/mL TNF-α (Figure 1D), and dose-dependent effect was also noted (Figure 1E). Consistently, LPS stimulation induced murine peritoneal macrophages or human mononuclear cell THP-1 expression of higher levels of miR-155 (Figure 1F).
Figure 1.
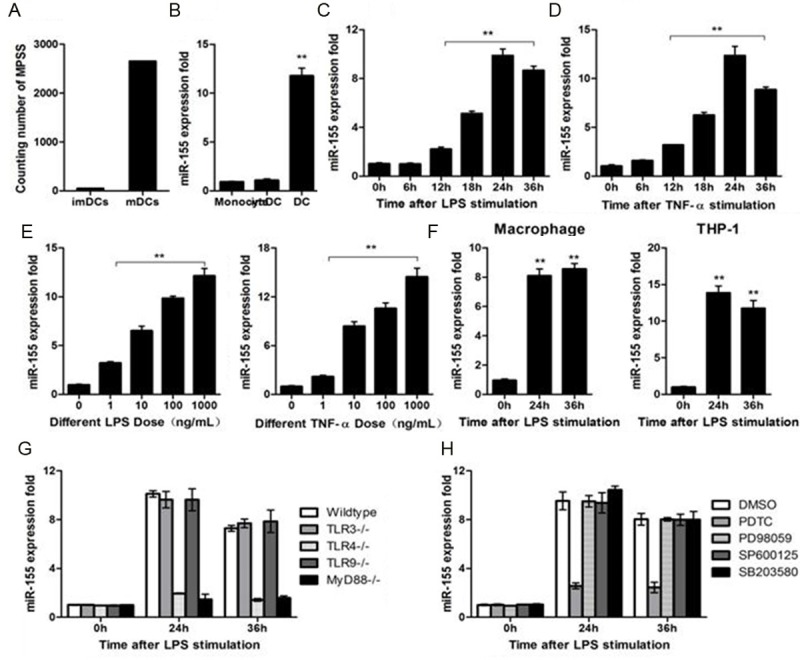
miR-155 expression in dendritic cells depend on TLR4/MyD88/NF-κB pathway. (A) Counting number of miR-155 in immature DCs and mature DCs by MPSS. (B) miR-155 expression level was measured by qRT-PCR in monocytes, immature DCs and mature DCs, U6 expression were used as internal control. (C) miR-155 expression in DC2.4 cell, challenged with 100 ng/mL LPS. (D) miR-155 expression in DC2.4 cell, challenged with TNF-α (100 ng/mL). (E) miR-155 expression in DC2.4 cell challenged with LPS or TNF-α at different doses for 24 h. (F) Murine macrophages or THP1-cells were stimulated by LPS (100 ng/mL) and miR-155 was measured by qRT-PCR. (G) Murine dendritic cells from wild-type-, TLR3-, TLR4-, TLR9-, or MyD88-deficient mice were stimulated with LPS at 100 ng/mL for indicated time. Expression of miR-155 was measured as in (A). (H) Murine dendritic cells were pretreated with DMSO, SB203580 (10 mM), PD98059 (10 mM), PDTC (100 mM), or SP600125 (10 mM) as indicated for 30 min and then stimulated with LPS at 100 ng/mL for indicated time. MiR-155 expression was measured. Data are shown as the mean ± s.d. (n = 3) from one representative experiment. Similar results were obtained in three independent experiments. **p < 0.01.
Given that TLR signaling has been proved to modulate miRNAs expression [9,15,16], we next examined the impact of TLR signaling on LPS induced upregulation of mir-155 in DCs. As expected, LPS failed to induce TLR4 or MyD88- deficient DCs expression of higher levels of mir-155 (Figure 1G). To further define the down-stream signaling, we pretreated DCs before LPS challenge with inhibitors for NF-κB (PDTC), ERK (PD98059), JNK (SP600125), and P38 (SB203590), respectively. Interesting, only PDTC attenuated LPS-induced upregulation of miR-155 (Figure 1H), indicating that LPS induction of mir-155 expression in DCs is dependent on NF-κB signaling.
miR-155 regulates DC phenotype and cytokine secretion
To determine the functional relevance of miR-155 in DC phenotype, DCs were transfected with a miR-155 mimics or an inhibitor, respectively. Indeed, miR-155 mimics significantly upregulated miR-155 expression in DCs (Figure 2A), and the inhibitor effectively knocked down miR-155 expression (Figure 2B). Importantly, Upregulation of miR-155 significantly promoted DCs to express IL-12p40 (Figure 2C), and in contrast, knockdown of mir-155 significantly suppressed IL-12p40 expression (Figure 2D), and similar results for the production of IL-12 in the culture supernatants were noted (Figure 2E). Next, flow cytometry analysis was conducted to examine surface marker expressions. In line with IL-12 results, upregulation of miR-155 significantly promoted DC maturation manifested by the significantly higher levels of CD40, CD80 and CD86 expressions (Figure 3F-H). Together, our data support that mir-155 could be implicated in the regulation of DC maturation and cytokine production.
Figure 2.
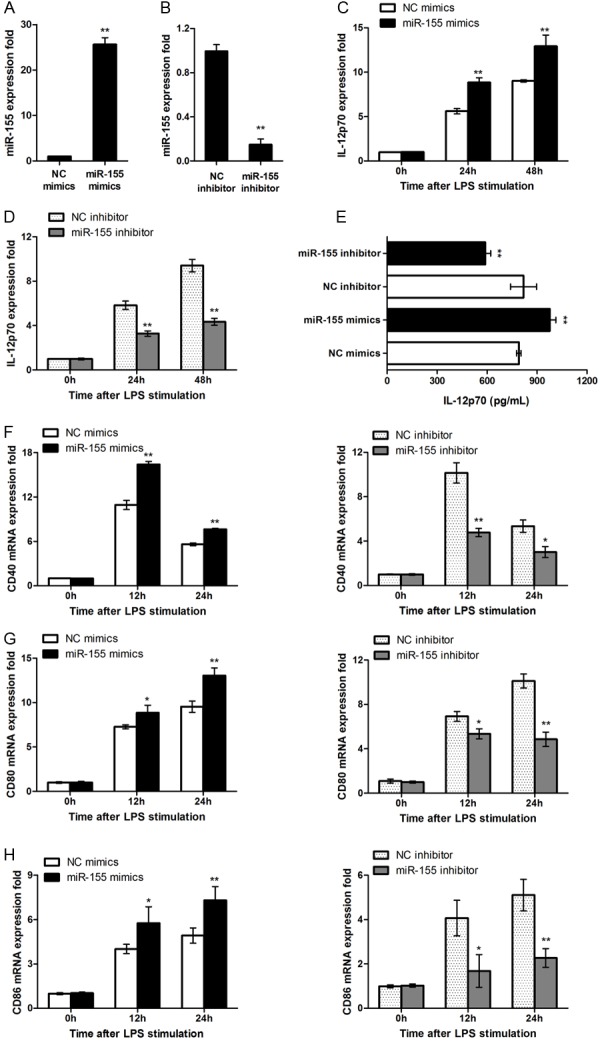
miR-155 regulate DCs phenotype and cytokine production. (A) A total of 0.5 ml 5 × 105 DCs were transfected with control mimics or miR-155 mimics. After 48 h, miR-155 level was measured by qRT-PCR and normalized to that of U6 in each sample. (B) DCs were transfected with control inhibitor or miR-155 inhibitor, miR-155 level was measured by qRT-PCR 72 hrs after transfection. (C, D) DCs were transfected with miR-155 mimics, inhibitor or ctrl, IL-12p40 mRNA expression were measured by qRT-PCR. (E) IL-12p70 level was measured by ELISA. (F-H) DCs were transfected as in (C, D), after stimulating by LPS for indicating time, CD40, CD80 and CD86 mRNA expression was measured by qRT-PCR. Data are shown as mean ± SD (n = 3) of one representative experiment. Similar results were obtained in three independent experiments. **p < 0.01; *p < 0.05.
Figure 3.
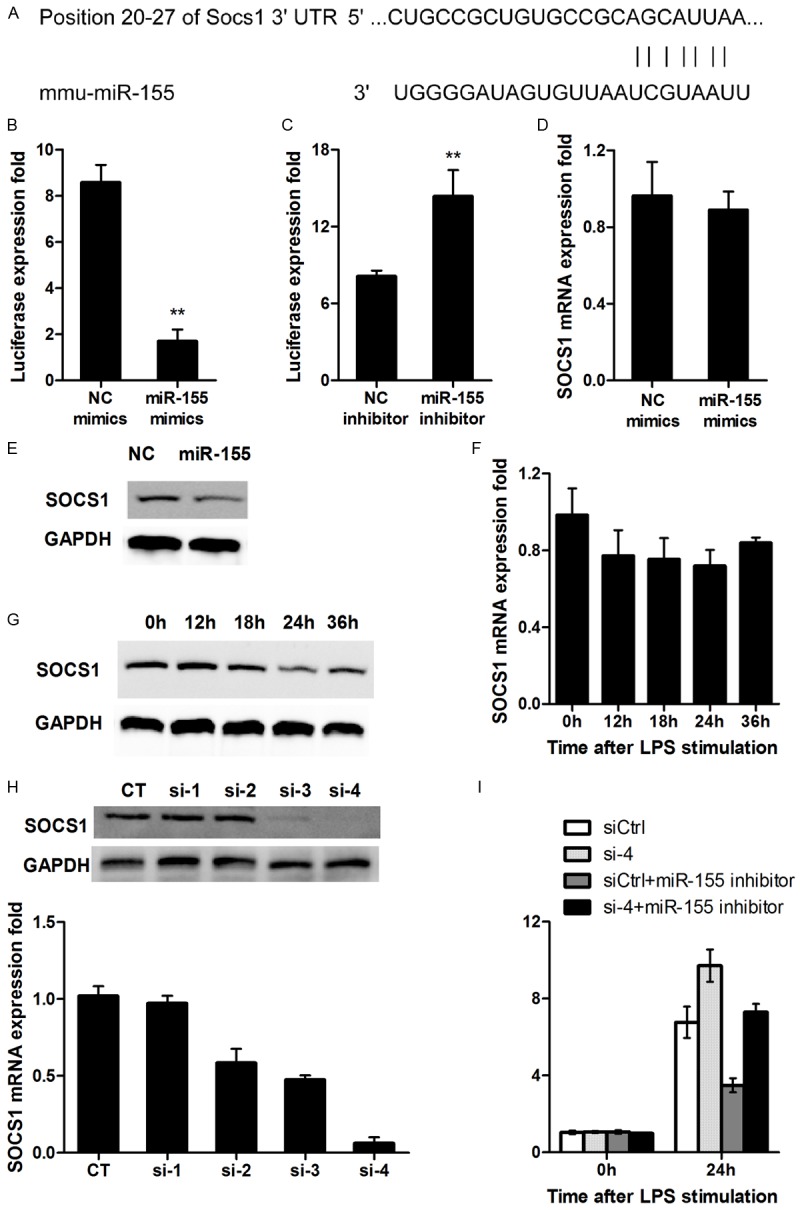
miR-155 targets SOCS1. (A) Mouse SOCS1 might be the molecular target of miR-155. Shown is a sequence alignment of miR-155 and its target sites in 3’-UTR of SOCS1, which was downloaded from TargetScan (www.targetscan. org). (B, C) HEK293 cells (1 × 104) were cotransfected with 80 ng of pMIR-SOCS1 3’-UTR luciferase reporter plasmids, 40 ng of pTK-Renilla-luciferase plasmids, together with control mimics or miR-155 mimics (C), control inhibitor or miR-155 inhibitor (D) at 20 nM. After 24 h, firefly luciferase activity was measured and normalized by Renilla luciferase activity. (D, E) DC2.4 cells were transfected as described in (A). After 48 h, SOCS1 mRNA and protein level were detected by qRT-PCR (D) and western blot (E) respectively. (F, G) Murine dendritic cells were stimulated by 100 ng/mL LPS for indicated times, SOCS1 mRNA and protein level was detected by qRT-PCR (F) and western blot (G) respectively. (H) DC2.4 cells were transfected with control RNA, four separate SOCS1 siRNAs as indicated. After 24 h, SOCS1 mRNA expression was measured by qRT-PCR and protein level was measured by western-blot. (I) DC2.4 cells were transfected with siCtrl, siSOCS1, miR-155 inhibitor combined with siCtrl, or miR-155 inhibitor combined with SOCS1 siRNA, respectively, as indicated. After 48 h, DCs were treated, stimulated, and detected as in Figure 2C, 2D. For (B-D, F, H), data are shown as mean ± SD (n = 3) of one representative experiment. Similar results were obtained in three independent experiments. **p < 0.01; *p < 0.05. For western blot in (E, G, H), data shown are representative of three separate experiments.
SOCS1 is a target for miR-155 in DCs
Bioinformatics analyses were next conducted by using the TargetScan and KEGG Pathway software, and SOCS1 was identified to be a potential target for miR-155 (Figure 3A). To validate this prediction, a dual-luciferase report plasmid with SOCS1 3’UTR was constructed and then co-transfected a miR-155 mimic or an inhibitor into HEK293 cells. Indeed, the miR-155 mimic significantly inhibited the expression of SOCS1 as defined by the luciferase activity (Figure 3B), while the miR-155 inhibitor significantly enhanced the fluorescence intensity (Figure 3C). Surprisingly, the miR-155 mimic, did not affect SOCS1 mRNA levels in DCs significantly (Figure 3D), but the protein levels for SOCS1 were downregulated (Figure 3E), suggesting that miR-155 may play a role for inhibition of SOCS1 protein translation. Studies in BMDCs with LPS stimulation further confirmed that SOCS1 mRNA was not significantly changed over time (Figure 3F), while SOCS1 protein levels were downregulated gradually (Figure 3G). Together, these data suggested that miR-155 directly regulates SOCS1 expression.
To demonstrate that miR-155 promotes DC maturation by targeting SOCS1, we next examined the role of SOCS1 in DC function by SOCS1 siRNAs, in which four SOCS1 siRNAs were designed as described, and siSOCS-4 manifested the highest efficiency for knockdown of SOCS1 expression (Figure 3H). DC2.4 cells were transfected with si-4, siCtrl+miR-155 inhibitor, si-4+miR-155 inhibitor, respectively. It was found that si-4 significantly upregulated LPS-induced production of IL-12p40 in DC2.4 cells, and an opposite effect was noted in DCs transfected with siCtrl+miR-155 inhibitor. Particularly, miR-155 inhibitor attenuated the effect of si-4 on SOCS1 expression (Figure 3I). Collectively, these results suggest that miR-155 regulates DC maturation at least in part by targeting SOCS1.
miR-155 regulates the capacity of DCs for antigen presentation and allostimulation
To further define the role of miR-155 in DC functionality, we assessed the capacity fo DCs for antigen presentation and allostimulation. It was noted that the miR-155 mimic significantly promoted OVA specific T cell proliferation, while the miR-155 inhibitor executed an opposite role (Figure 4B), indicating that miR-155 regulates the capability of DCs for antigen presentation. Similarly, DCs transfected with a miR-155 mimic manifested significantly higher capacity to stimulate allogeneic T cell proliferation (Figure 4C), while allogenic T cell proliferation was significantly attenuated once DCs treated with a miR-155 inhibitor (Figure 4D). In consistent with these results, the production of IL-2 (Figure 4E) and IFN-γ (Figure 4F) was significantly higher in MLR supernatants of DCs treated with a miR-155 mimic. Taken together, these data support that miR-155 also regulates the capability of DCs to stimulate allogenic T cell responses.
Figure 4.
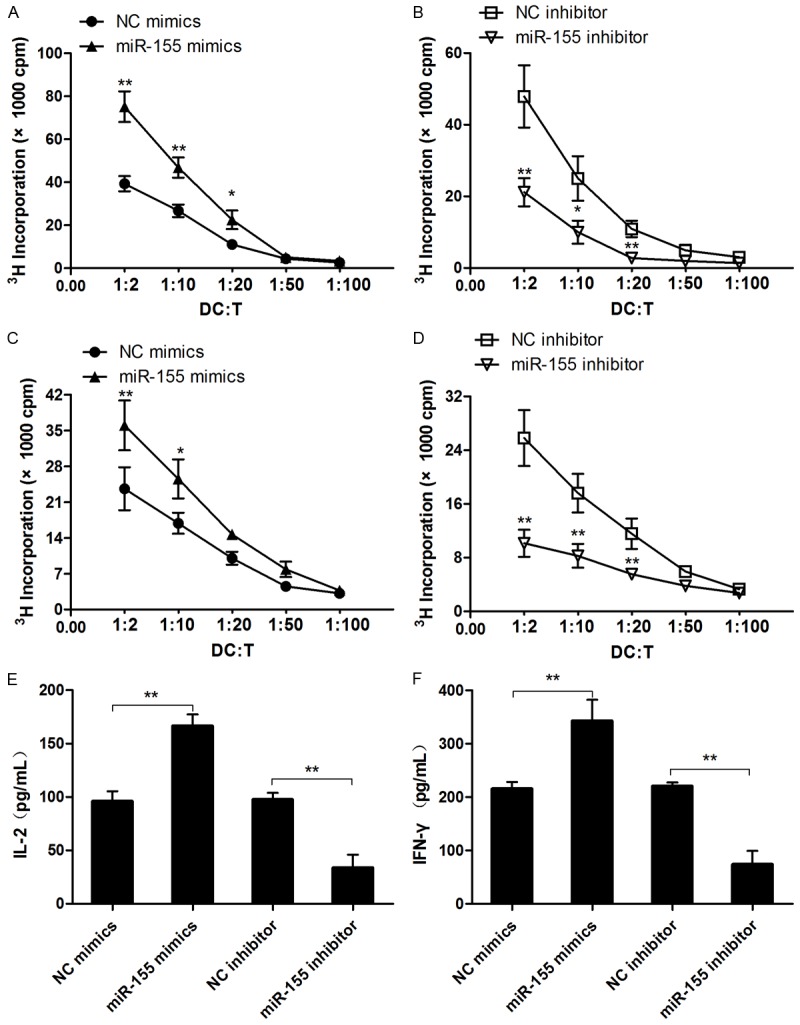
miR-155 promotes DC function mainly through targeting SOCS1. (A, B) DCs obtained from C57BL/6 mice were transfected with miR-155 mimics or inhibitor then pretreated with OVA (1 μg/mL), next co-cultured with CD4+ T-cells from OT-2 mice for 5 days at indicated radio. T cell proliferation was measured by pulsing with [3H]-thymidine. (C, D) DCs from C57BL/6 mice were transfected as in (A, B), then co-cultured with BALB/c splenic CD4+ T cells for 5 days. T cell proliferation was measured by pulsing with [3H]-thymidine. (E, F) IL-2 and IFN-γ in supernatant of (C, D) was measured by ELISA. Data are shown as mean ± SD (n = 3) of one representative experiment. Similar results were obtained in three independent experiments.
Inhibition of miR-155 prolonges cardiac allograft survival
The above data prompted us to examine the impact of miR-155 on allograft rejection, in which a murine cardiac transplantation model was employed for the study. For this purpose, we first treated mice with a miR-155 antagomir or a Ctrl antagomir at 100 nM by intravenous injection. A time-dependent attenuation of mir-155 expression was characterized with the observation of lowest expressions at day 7 of injection in the heart, peripheral blood mononuclear cells (PBMCs), and spleen (Figure 5A). Recipient mice after cervical heterotopic heart transplantion were next treated with 100 nM antagomiR-155 or Ctrl antagomir followed by monitoring allograft survival. Administration of antagomiR-155 markedly protected allografts from rejection as manifested the significantly prolonged mean allograft survival time (MST) (Figure 5B). Analysis of DCs originated from recipient mice 4 days after antagomiR-155 administration revealed that antagomiR-155 significantly enhanced SOCS1 expression (Figure 5C) along with attenuated surface markers such as CD40, CD80 and CD86 expressions (Figure 5D). All together, our data suggest that suppression of miR-155 in DCs provides protection for recipient mice against allograft rejection.
Figure 5.
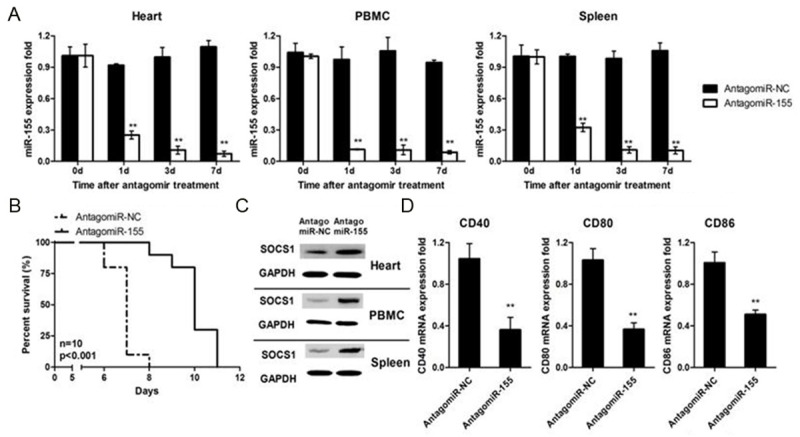
miR-155 protected grafts from rejection in vivo by impacting DCs maturation. (A) Mice i.v injected with AantagomiR-155 (100 nM) or its negative ctrl (NC). After indicated time, mice were sacrificed, and heart, PBMC and spleen were harvested to measure miR-93 expression. (B) Mice were treated as in (A), grafts (BALBc to C57BL/6) survival was evaluated for 12 days (Mann-Whitney test, n = 10). (C, D) Mice were treated as in (A), after four days PBMC, spleens and lymph nodes were harvested, then CD11c cells were sorted from PBMC or tissue suspension. SOCS1 protein expression was measured by western blot and CD40, CD80, CD86 mRNA level was detected by RT-PCR. Data are shown as the mean ± SD. (n = 3) (A, D) or representative of three separate experiments (C) with similar results. **p < 0.01.
Discussion
Given the role of miRNAs played in the regulation of gene expression, they have been recognized to be important in the setting of allograft rejection [5,20,21]. In 2009, Anglicheau group reported the changes of miRNA expression profiles of subjects undergone acute rejection after kidney transplantation [22]. By analysis of miRNA expression profiles with renal biopsy specimens after transplantation, they concluded that miRNAs can be used as biomarkers for acute rejection. Follow up studies by Asaoka and colleagues further suggested that miRNAs have the potential to serve as biomarkers for diagnosis of graft rejection and prognosis of anti-rejection therapy in small intestine transplantation [23]. Together, these studies demonstrate that miRNAs are likely to be involved in allograft rejection, but the exact mechanisms, particular how miRNAs affect antigen-presenting cells such as DCs during the course of rejection, are yet to be elucidated.
Antigen presenting cells (APCs), especially DCs, have long been recognized to play an essential role in the process of allograft rejection [24]. During transplantion related surgical process, ischemic insult would lead to the release of massive danger signals [25], while complement and sugar chains on heterologous cell surface would trigger a cascade of reactions, which would result in the production of cleaved fragments and compounds. Both of which would act on APCs through the pattern recognition receptors. Indeed, APCs are responsible for the uptake and presentation of antigens, and secrete large amounts of cytokines upon activation. The activation of APCs would strengthen acquired immune response by promoting both humoral rejection [26] and T cell-mediated rejection [27]. Particularly, DCs act as the most powerful antigen-presenting cells are implicated in antigen presentation and identification, initiation of immune responses and induction of graft rejection. DCs not only auto-secret TNF-α to promote their maturation and antigen presentation, but also produce cytokines such as IL-1β, IL-12, IL-23 to regulate T cell differentiation and activation [24,28]. As a result, DCs could be a viable target for prevention and treatment of allograft rejection in the setting of transplantation.
As a famous star miRNA, miR-155 has been identified to be a necessary molecule in the regulation of immune response. Mice deficient in miR-155 manifest poor resistance to infection featured by dysfunctional T, B and DC development [29]. Several genes have been reported to be miR-155 target relevant to immune pathways such as IKK, TNF, and FADD [30-32]. Studies in macrophages and DCs revealed that miR-155 can promote innate immune response by regulating NF-KB and MAPK pathway [33]. In line with these observation, abnormal miR-155 expression has been noted in mice with EAE and IBD mice [34,35]. Previously, we demonstrated evidence indicating that miRNA-155 upregulation promotes DC and several other APCs activation. We now obtained convincing data supporting that mir-155 regulates DC maturation by targeting SOCS1. Inhibition of miR-155 in DCs significantly impaired their capacity for antigen presentation and induction of alloreactive T cell proliferation along with suppressed production of IL-12 and IFN-γ. The results greatly prompted us to further determine the role of miR-155 in the regulation of allograft rejection. An antagomiR-155 complementary to the specific mir-155 targets was thus synthesized which blocks mir-155 from binding to the 3’UTR of its targeting genes [36]. The antagomiR-155 was then administered into recipient mice after cardiac allograft transplantation. Remarkably, administration of miR-155 antagomir significantly prevented cardiac allografts from rejection as manifested by the prolonged graft survival time. Therefore, miR-155 could be an important regulator in the setting of allograft rejection and suppression of miR-155 expression in DCs could be a viable strategy for prevention and treatment of graft rejection in clinical transplantation.
In conclusion, miR-155 is upregulated during DC maturation, which is dependent on the TLR4/Myd88/NF-κB signaling. miR-155 regulation of DC phenotype and its allostimulatory capability by inhibiting the expression of SOCS1. As a result, administration of antagomiR-155 provides protection for cardiac allografts from rejection. Together, our data support that miR-155 possesses the potential to be a therapeutic target for prevention and treatment of allograft rejection in the setting of organ transplantation.
Acknowledgements
This work was supported by grants from the Shanghai Committee of Science and Technology (10ZR1438700), National Natural Science Foundation of China (31370896, 81373171 and 30972792), the National High Technology Research and Development Program 863 (2014AA021601), the health inspection institute Minhang district task of Shanghai (2012MW03) and Shanghai Changzheng Hospital Foundation for Young Scientist (2012CZQN01).
Disclosure of conflict of interest
The authors declare no competing financial interests.
References
- 1.Rowley DA, Fitch FW. The road to the discovery of dendritic cells, a tribute to Ralph Steinman. Cell Immunol. 2012;273:95. doi: 10.1016/j.cellimm.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 2.Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol. 2007;7:610. doi: 10.1038/nri2132. [DOI] [PubMed] [Google Scholar]
- 3.Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci U S A. 2002;99:351. doi: 10.1073/pnas.231606698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monguio-Tortajada M, Lauzurica-Valdemoros R, Borras FE. Tolerance in organ transplantation: from conventional immunosuppression to extracellular vesicles. Front Immunol. 2014;5:416. doi: 10.3389/fimmu.2014.00416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 6.O’Connell RM, Rao DS, Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30:295. doi: 10.1146/annurev-immunol-020711-075013. [DOI] [PubMed] [Google Scholar]
- 7.Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008;9:839. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 8.Ma F, Xu S, Liu X, Zhang Q, Xu X, Liu M, Hua M, Li N, Yao H, Cao X. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nat Immunol. 2011;12:861. doi: 10.1038/ni.2073. [DOI] [PubMed] [Google Scholar]
- 9.Wang P, Hou J, Lin L, Wang C, Liu X, Li D, Ma F, Wang Z, Cao X. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J Immunol. 2010;185:6226. doi: 10.4049/jimmunol.1000491. [DOI] [PubMed] [Google Scholar]
- 10.Su X, Qian C, Zhang Q, Hou J, Gu Y, Han Y, Chen Y, Jiang M, Cao X. miRNomes of haematopoietic stem cells and dendritic cells identify miR-30b as a regulator of Notch1. Nat Commun. 2013;4:2903. doi: 10.1038/ncomms3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Zhang HX, Sun YP, Liu ZX, Liu XS, Wang L, Lu SY, Kong H, Liu QL, Li XH, Lu ZY, Chen SJ, Chen Z, Bao SS, Dai W, Wang ZG. Rig-I-/- mice develop colitis associated with downregulation of G alpha i2. Cell Res. 2007;17:858. doi: 10.1038/cr.2007.81. [DOI] [PubMed] [Google Scholar]
- 12.Fu H, Song S, Liu F, Ni Z, Tang Y, Shen X, Xiao L, Ding G, Wang Q. Dendritic cells transduced with SOCS1 gene exhibit regulatory DC properties and prolong allograft survival. Cell Mol Immunol. 2009;6:87. doi: 10.1038/cmi.2009.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun W, Wang Q, Zhang L, Pan J, Zhang M, Lu G, Yao H, Wang J, Cao X. TGF-beta(1) gene modified immature dendritic cells exhibit enhanced tolerogenicity but induce allograft fibrosis in vivo. J Mol Med (Berl) 2002;80:514. doi: 10.1007/s00109-002-0346-2. [DOI] [PubMed] [Google Scholar]
- 14.Wang Q, Liu Y, Wang J, Ding G, Zhang W, Chen G, Zhang M, Zheng S, Cao X. Induction of allospecific tolerance by immature dendritic cells genetically modified to express soluble TNF receptor. J Immunol. 2006;177:2175. doi: 10.4049/jimmunol.177.4.2175. [DOI] [PubMed] [Google Scholar]
- 15.Wang P, Gu Y, Zhang Q, Han Y, Hou J, Lin L, Wu C, Bao Y, Su X, Jiang M, Wang Q, Li N, Cao X. Identification of resting and type I IFN-activated human NK cell miRNomes reveals microRNA-378 and microRNA-30e as negative regulators of NK cell cytotoxicity. J Immunol. 2012;189:211. doi: 10.4049/jimmunol.1200609. [DOI] [PubMed] [Google Scholar]
- 16.Hou J, Wang P, Lin L, Liu X, Ma F, An H, Wang Z, Cao X. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol. 2009;183:2150. doi: 10.4049/jimmunol.0900707. [DOI] [PubMed] [Google Scholar]
- 17.Liu X, Zhan Z, Xu L, Ma F, Li D, Guo Z, Li N, Cao X. MicroRNA-148/152 impair innate response and antigen presentation of TLR-triggered dendritic cells by targeting CaMKIIalpha. J Immunol. 2010;185:7244. doi: 10.4049/jimmunol.1001573. [DOI] [PubMed] [Google Scholar]
- 18.Xu S, Liu X, Bao Y, Zhu X, Han C, Zhang P, Zhang X, Li W, Cao X. Constitutive MHC class I molecules negatively regulate TLR-triggered inflammatory responses via the Fps-SHP-2 pathway. Nat Immunol. 2012;13:551. doi: 10.1038/ni.2283. [DOI] [PubMed] [Google Scholar]
- 19.Mackern-Oberti JP, Vega F, Llanos C, Bueno SM, Kalergis AM. Targeting dendritic cell function during systemic autoimmunity to restore tolerance. Int J Mol Sci. 2014;15:16381. doi: 10.3390/ijms150916381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffiths-Jones S. The microRNA Registry. Nucleic Acids Res. 2004;32:D109. doi: 10.1093/nar/gkh023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chekulaeva M, Filipowicz W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr Opin Cell Biol. 2009;21:452. doi: 10.1016/j.ceb.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 22.Griffiths-Jones S. miRBase: the microRNA sequence database. Methods Mol Biol. 2006;342:129. doi: 10.1385/1-59745-123-1:129. [DOI] [PubMed] [Google Scholar]
- 23.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meltzer PS. Cancer genomics: small RNAs with big impacts. Nature. 2005;435:745. doi: 10.1038/435745a. [DOI] [PubMed] [Google Scholar]
- 25.Ruvkun G. Molecular biology. Glimpses of a tiny RNA world. Science. 2001;294:797. doi: 10.1126/science.1066315. [DOI] [PubMed] [Google Scholar]
- 26.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zamore PD, Haley B. Ribo-gnome: the big world of small RNAs. Science. 2005;309:1519. doi: 10.1126/science.1111444. [DOI] [PubMed] [Google Scholar]
- 28.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, Vetrie D, Okkenhaug K, Enright AJ, Dougan G, Turner M, Bradley A. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen DF, Gong BD, Xie Q, Ben QW, Liu J, Yuan YZ. MicroRNA155 is induced in activated CD4(+) T cells of TNBS-induced colitis in mice. World J Gastroenterol. 2010;16:854. doi: 10.3748/wjg.v16.i7.854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang HQ, Yu XD, Liu ZH, Cheng X, Samartzis D, Jia LT, Wu SX, Huang J, Chen J, Luo ZJ. Deregulated miR-155 promotes Fas-mediated apoptosis in human intervertebral disc degeneration by targeting FADD and caspase-3. J Pathol. 2011;225:232. doi: 10.1002/path.2931. [DOI] [PubMed] [Google Scholar]
- 32.Sullivan RP, Fogel LA, Leong JW, Schneider SE, Wong R, Romee R, Thai TH, Sexl V, Matkovich SJ, Dorn GN, French AR, Fehniger TA. Micro-RNA-155 tunes both the threshold and extent of NK cell activation via targeting of multiple signaling pathways. J Immunol. 2013;191:5904. doi: 10.4049/jimmunol.1301950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seddiki N, Brezar V, Ruffin N, Levy Y, Swaminathan S. Role of miR-155 in the regulation of lymphocyte immune function and disease. Immunology. 2014;142:32. doi: 10.1111/imm.12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, Kahn ME, Rao DS, Baltimore D. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 2010;33:607. doi: 10.1016/j.immuni.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Svrcek M, El-Murr N, Wanherdrick K, Dumont S, Beaugerie L, Cosnes J, Colombel JF, Tiret E, Flejou JF, Lesuffleur T, Duval A. Overexpression of microRNAs-155 and 21 targeting mismatch repair proteins in inflammatory bowel diseases. Carcinogenesis. 2013;34:828. doi: 10.1093/carcin/bgs408. [DOI] [PubMed] [Google Scholar]
- 36.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]