

Abstract
Adsorbing small charged nanoparticles onto liposome surfaces to stabilize them against fusion and payload leakage has resulted in a new class of liposomes capable of environment-responsive drug delivery. Herein, we engineered a liposome formulation with a lipid composition sensitive to bacterium-secreted phospholipase A2 (PLA2) and adsorbed chitosan-modified gold nanoparticles (AuChi) onto the liposome surface. The resulting AuChi-stabilized liposomes (AuChi-liposomes) showed prohibited fusion activity and negligible drug leakage. However, upon exposure to either purified PLA2 enzyme or PLA2 secreted by Helicobacter pylori (H. pylori) bacteria in culture, AuChi-liposomes rapidly released the encapsulated payloads and such responsive release was retarded by adding quinacrine dihydrochloride, a PLA2 inhibitor. When loaded with doxycycline, AuChi-liposomes effectively inhibited H. pylori growth. Overall, the AuChi-liposomes allowed for smart “on-demand” antibitoic delivery: the more enzymes or bacteria present at the infection site, the more drug will be released to treat the infection. Given the strong association of PLA2 with a diverse range of diseases, the present liposomal delivery technique holds broad application potential for tissue microenvironment-responsive drug delivery.
Keywords: liposome, gold nanoparticle, phospholipase, enzyme responsive, antimicrobial delivery
INTRODUCTION
Liposomes are an established drug carrier with well-documented advantages including highly biocompatible lipid materials, readily tunable formulation properties, and high drug carrying capacity1–3. Owing particularly to their distinguishable bilayer structure, liposomes are prone to fusion with bacterial membranes, making them a suitable delivery system for various antimicrobial treatments4–6. To further improve on the therapeutic efficacy of liposomal drugs, a myriad of environmentally responsive liposomal formulations have been developed that possess preferential liposome-bacterium fusion ability or triggered drug release at infection sites upon external stimulation7, 8. Common stimuli include temperature, pH, redox potential, and enzymatic activities, and these stimulus-responsive liposomal systems hold great promise to improve the current treatment regimes of bacterial infection9–12.
Recently, nanoparticle-stabilized liposomes have emerged as a new and robust liposomal delivery system that involves the attachment of small charged nanoparticles onto the outer surfaces of phospholipid liposomes for liposome stabilization and triggered cargo release13, 14. The nonspecific adsorption of charged nanoparticles onto phospholipid bilayers provided steric repulsion that inhibited liposome fusion. It also reduced liposome surface tension and thus further enhanced liposome stability15, 16. Intriguingly, the charge and charge density of both the nanoparticle stabilizers and the liposomes could be precisely tailored to enable stimulus-responsive binding and detachment of the nanoparticles, thereby allowing for an on-demand control over liposome fusion activity for smart drug delivery. For instance, cationic liposomes bound with negatively charged gold nanoparticles only fused with bacteria at acidic pH, which made them suitable for treating various skin pathogens that thrive in acidic infection sites such as the case with Propionibacterium acnes13. Conversely, anionic liposomes stabilized by positively charged gold nanoparticles were highly stable in gastric acid, but capable of fusing with bacteria at physiological pH, making them suitable to treat gastric pathogens such as Helicobacter pylori (H. pylori)14. Even in the absence of such stimulus-induced detachment of the nanoparticle stabilizers, these liposomes still had a substantial fraction of their surface areas exposed and highly accessible to membrane-targeting biochemical molecules such as bacterial toxins and enzymes. In a previous study, it has been shown that pore-forming toxins could effectively punch holes in the exposed lipid membranes to trigger antibiotic release from the nanoparticle-stabilized liposomes17. Herein, we report their responsiveness to hydrolytic enzymes secreted by bacteria and demonstrate that the enzyme-triggered drug release subsequently kills or inhibits the growth of the enzyme-secreting bacteria. While in principle the enzyme-triggered antibiotic release from nanoparticle-stabilized liposomes can be applied to a broad range of pathogens that secrete membrane-damaging enzymes, particular interest is focused on H. pylori in this work.
H. pylori infects nearly half of the world population and is of a significant public health concern. Infection with H. pylori is the primary cause of chronic gastritis, peptic ulcers, and gastric malignancy6, 18, 19. However, eradication of H. pylori is challenging regardless of the treatment regimens, owing partly to the rapid emergence of H. pylori strains resistant to the antibiotics20, 21. H. pylori bacteria are known to secrete phospholipase A2 (PLA2), a family of enzymes capable of hydrolyzing membrane phospholipids, causing mucosal damage and benefiting bacterial survival22, 23. Such enzymatic activity can be utilized as an environment cue to disrupt membrane integrity for triggered payload release from liposomes24, 25. In this study, we synthesized liposomes with lipid composition sensitive to PLA2 and stabilized them with small chitosan-modified gold nanoparticles (AuChi). The adsorbed AuChi were effective in preventing liposome fusion and drug leakage, while leaving a considerable fraction of liposome surfaces accessible to PLA2 enzyme. As shown in Figure 1, the cationic AuChi bind to the negatively charged liposome surfaces through electrostatic attraction and thus stabilize liposomes against fusion and avoid undesirable antibiotic leakage. When the stabilized liposomes are in the vicinity of H. pylori bacteria, the bacterium-secreted PLA2 degrades phospholipids, compromises the membrane integrity, and subsequently releases the antibiotic payload. Such on-site release of antibiotics enables localized and rapid killing of H. pylori bacteria. We first demonstrated liposome stabilization upon AuChi adsorption and then examined the payload release kinetics of the AuChi-stabilized liposome (AuChi-liposome) in the presence of both purified PLA2 and H. pylori culture, respectively. We further demonstrated that the released antibiotics from the liposomes in the presence of H. pylori were effective in inhibiting the growth of the bacteria.
Fig. 1.
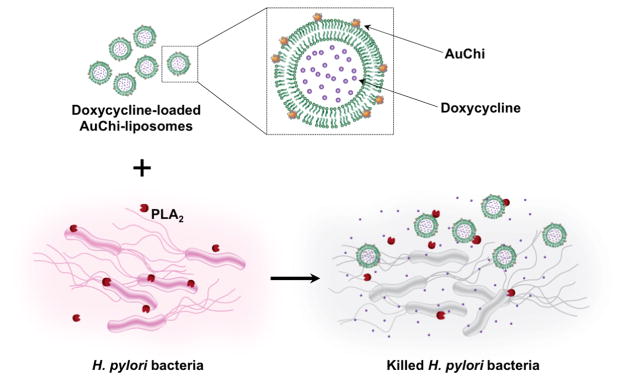
Schematic illustration of phospholipase A2 (PLA2)-triggered antibiotic release from liposomes stabilized by chitosan-modified gold nanoparticles (AuChi-liposome) to treat bacteria (e.g., H. pylori) that secrete the enzyme. Antibiotic (e.g. doxycycline)-loaded liposomes are prohibited from fusion by absorbing AuChi nanoparticles onto their surface. Once the AuChi-liposomes encounter bacteria-secreted PLA2, the enzyme cleaves the phospholipids that form the liposome membranes and thus release the encapsulated antibiotics, which subsequently kill or inhibit the growth of the bacteria.
RESULTS AND DISCUSSION
The preparation of AuChi-liposome can be divided into three steps. First, AuChi were synthesized by an ex-situ stabilization technique as previously described, where gold nanoparticles were made with a sodium borohydride reduction method and then stabilized by adding calculated amount of chitosan under ambient condition14, 17. Dynamic light scattering (DLS) measurements of AuChi showed a diameter of approximately 10 nm with a narrow size distribution and a strong positive surface charge of 35.5 ± 0.9 mV, indicating the presence of cationic amine groups of chitosan on gold surfaces. Second, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) and 1,2-dioctadecanoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (sodium salt) (DSPG) in a molar ration of 9:1 were used to formulate anionic liposomes following a standard extrusion method in deionized water6. DSPG has been found susceptible to PLA2-mediated degradation and liposomes made from DSPG are expected to be disrupted by PLA2, leading to drug release from the liposomes26, 27. DLS measurements of the liposomes showed a diameter of 84.7 ± 0.8 nm with polydispersity index of 0.12 ± 0.02 and a surface zeta potential of −54.7 ± 2.6 mV. Lastly, the anionic liposomes were mixed with cationic AuChi nanoparticles at a liposome-to-AuChi molar ratio of 1:300 under bath sonication for 10 min14, 17. The resulting AuChi-liposomes showed a diameter of 95.0 ± 2.7 nm in water, which remained stable in PBS at pH = 6.5 with a diameter of 97.1 ± 2.4 nm. The size increase of AuChi-liposomes compared to bare liposomes corresponds to the adsorption of AuChi onto the liposome surface. Liposome surface charge also switched from the strong negative value of the bare liposomes to 36.9 ± 1.3 mV of AuChi-liposomes, further confirming the binding of positively charged AuChi to the negatively charged liposomes through electrostatic interactions (Figure 2). Further studies showed that the size and surface zeta potential AuChi-liposome remained stable upon loading small molecule cargoes such as rhodamine B (RhB) and doxycycline into the liposomes.
Fig. 2.
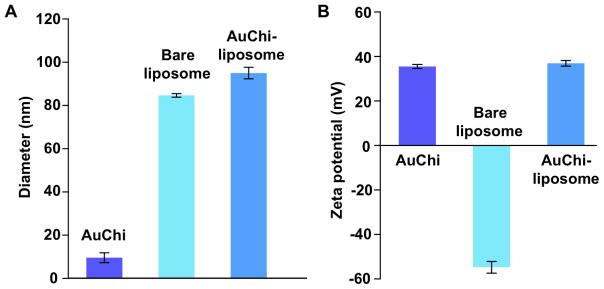
(A) Hydrodynamic size (diameter, nm) and (B) surface zeta potential of AuChi, bare liposome (without AuChi), and AuChi-liposome with an AuChi-to-liposome molar ratio of 300:1.
Adsorbed AuChi nanoparticles are expected to stabilize liposomes that are otherwise prone to fusion and drug leakage14, 17, 28. To test this, we first used a bacterium-liposome fusion assay6, 14. Specifically, 0.5 mM AuChi-liposomes containing 0.5 mol% DMPE-RhB were prepared and incubated with 5 × 108 CFU/mL H. pylori bacteria at 37°C for 30 min. Following the incubation, the bacteria pellets were collected and resuspended in PBS solution. The fusion ability of liposomes was then quantified by measuring the fluorescence intensity of the bacterial suspension. H. pylori bacteria incubated with bare liposomes without AuChi stabilizers showed strong fluorescence intensity In contrast, a much weaker florescence signal was detected from the bacteria incubated with AuChi-liposomes, indicating a significantly reduced liposome fusion activity upon the adsorption of small AuChi nanoparticles (Figure 3A). We next tested the inhibition of drug leakage by AuChi nanoparticles. For this study, rhodamine B (RhB) was used as a model drug. As shown in Figure 3B, bare liposomes released all the fluorescence dye in 48 hrs. In contrast, less than 2% of the encapsulated RhB was released in the same time span from AuChi-liposomes. Together, these results confirm strong stabilization effects conferred by adsorbing AuChi stabilizers onto the liposome surface.
Fig. 3.
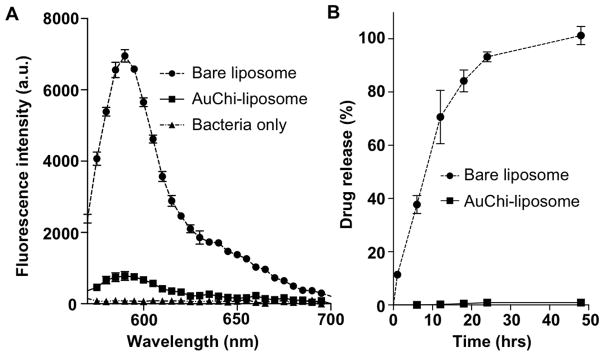
(A) Fusion ability of bare liposome and AuChi-liposome with H. pylori bacteria. Fluorescently labeled liposome (bare liposome or AuChi-liposome) was incubated with 5×108 CFU H. pylori bacteria at pH 6.5 for 10 min. After incubation, the bacteria pellet was collected and quantified for fluorescence intensity at the range of 550–700 nm. The same amount of bacteria without incubating with any liposome formulations was tested in parallel serving as background signal. (B) Accumulative drug release profile from bare liposome and AuChi-liposome. RhB was used as a model drug loaded inside the liposome. The released RhB was quantified by measuring the fluorescence intensity at 585 nm.
We next proceeded to examine whether the drug release from AuChi-liposomes could be triggered by PLA2. By adding purified PLA2 into the RhB-loaded AuChi-liposome solutions, we found that the drug release rates increased with the increase of PLA2 concentrations. When PLA2 concentration was at 1, 10, and 100 μg/mL, approximately 5, 50, and 67% of encapsulated RhB molecules were released within 24 hrs, respectively (Figure 4A). In addition, accumulative RhB release profiles showed gradual increases with time without a burst release, implying that drug release kinetics from AuChi-liposomes in the presence of PLA2 is dominated by diffusional liposome efflux 29, 30. Therefore, we attempted to use a diffusion-dominant Higuchi model to analyze the drug release profiles: Mt = Kt1/2, where Mt is drug release at time t in hours and K is the Higuchi constant 31, 32. Plotting the drug release percentage against the square root of time yielded linear fittings with R2 = 0.97 and 0.98 for 10 and 100 μg/mL PLA2, respectively (Figure 4B). The goodness of the fit indicates a diffusion-controlled liposome release mechanism. On the basis of this analysis, the Higuchi constants of drug release with 10 and 100 μg/mL of PLA2 were determined to be 10.57 ± 0.25, and 14.74 ± 0.26 h−1/2, respectively.
Fig. 4.
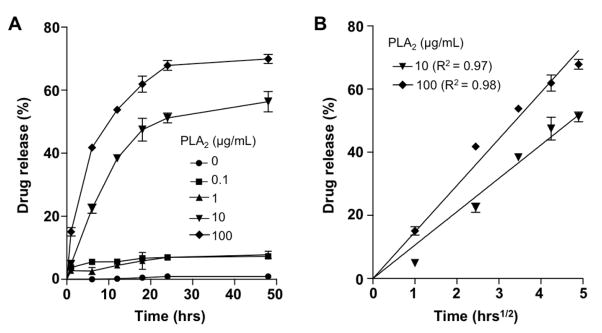
(A) Accumulative drug release kinetics from AuChi-liposome in the presence of various PLA2 enzyme concentrations. (B) The drug release percentage was plotted against the square root of time, which yielded linear fittings using a diffusion-dominant Higuchi model. The fitted line was determined from the first order polynomial fitting assuming the y-interception at zero. The slope was then calculated by minimizing the sum of the squares of the vertical distances between the point and the line.
To further verify that H. pylori-secreted PLA2 can indeed trigger drug release from AuChi-liposomes, we incubated RhB-loaded AuChi-liposomes with H. pylori culture and monitored the release of RhB from the liposomes33. As shown in Figure 5, when AuChi-liposomes were incubated in fresh culture medium without H. pylori bacteria, less than 5% RhB was released in 24 hrs, confirming that AuChi-liposomes were stable. However, when the AuChi-liposomes were incubated in bacterial culture containing 1×107 CFU/mL H. pylori bacteria, 9.8%, 15.5%, and 17.0% of RhB was released in 1, 12, and 24 hrs, respectively. The drug release rate further increased when the bacterial concentration was increased. Specifically, when the bacterial concentration was increased to 1×108 CFU/mL, AuChi-liposomes released 13.4%, 30.3% and 28.7% of RhB in 1, 12, and 24 hrs, respectively. It seemed that the drug release has reached a plateau within 12 hrs and the observed release difference between 12 and 24 hrs was likely due to experimental variation. To further confirm that PLA2 was indeed responsible for the accelerated drug release, 0.13 μM quinacrine dihydrochloride, a PLA2 inhibitor, was added to the culture containing 1×108 CFU/mL H. pylori. Under this condition, a reduced drug release rate was observed; AuChi-liposomes released 6.8%, 9.7% and 15.1% of RhB in 1, 12, and 24 hrs, respectively. The incomplete inhibition of RhB release in the presence of quinacrine dihydrochloride was likely due to other H. pylori-secreted virulence factors such as CagA, VacA, and TlyA, which are all known to damage phospholipid membranes through various mechanisms34–37.
Fig. 5.
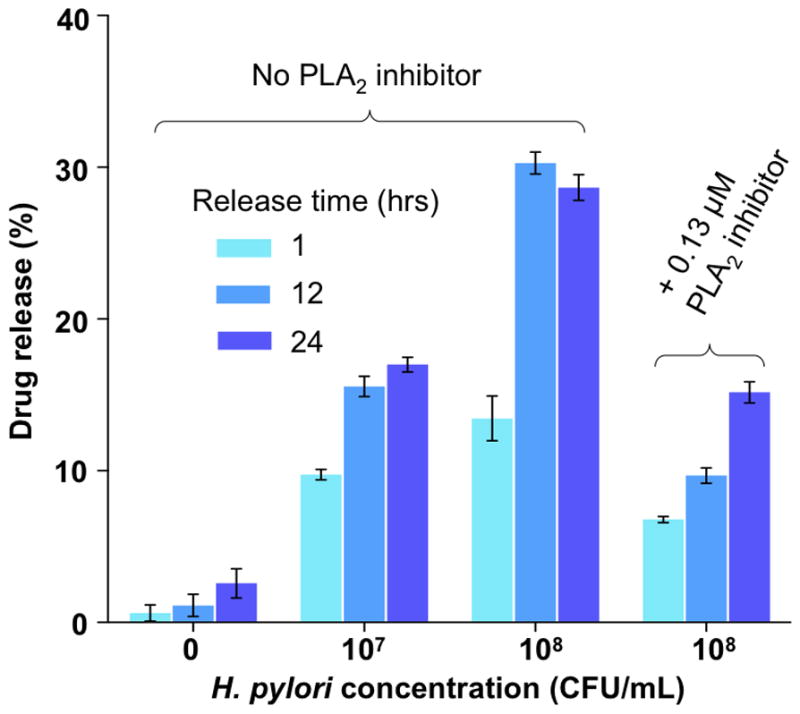
Drug release from AuChi-liposome at 1 hr, 12 hrs, and 24 hrs post incubation with 0, 1×107 and 1×108 CFU/mL H. pylori bacteria culture, respectively. As a control group, quinacrine dihydrochloride (0.13 μM), a PLA2 inhibitor, was added to the bacterial culture to inhibit PLA2 activity. Data represent mean ±SD (n = 3).
After having verified the responsive drug release from AuChi-liposomes in the presence of both purified PLA2 and PLA2 secreted by H. pylori bacteria, we finally tested the antimicrobial activity of doxycycline-loaded AuChi-liposome against H. pylori bacteria. In the study, doxycycline-loaded AuChi-liposomes with a doxycycline concentration of 0.2 mM were incubated with H. pylori bacteria (5×107 CFU/mL) in 5% BHI for 12 hrs, followed by serial dilution of each sample for bacterial colony enumeration. For comparison, the same concentration of free doxycycline was tested in parallel as a positive control, and empty AuChi–liposomes (without drug) and PBS (1X) served as negative controls. As shown in Figure 6, empty AuChi–liposome did not show any inhibitory effect against H. pylori, as their incubation with the bacteria resulted in a comparable colony formation to the PBS (1X) control, whereas free doxycycline resulted a complete bacterial killing under the experimental condition. In contrast, doxycycline-loaded AuChi–liposomes showed excellent antimicrobial efficacy against H. pylori and such anti-H. pylori efficacy was significantly weakened when PLA2 inhibitor (0.13 μM) was added to the bacterial culture. The incomplete killing of H. pylori by AuChi-liposomes was likely due to the partial release of doxycycline during the 12 hrs of incubation time. With bacterial enzyme-triggered drug release mechanism, the doxycycline-loaded AuChi-liposomes confer distinct advantages to treat bacterial infections. For example, with a high stability, the formulation improves on the shelf-time of the liposomal drug with minimum drug leakage prior to administration. In addition, AuChi-liposomes allow antibiotics to be delivered in a bacterium-targeted fashion: antibiotic payloads will only be released at the infection sites where the bacteria secrete hydrolytic enzymes. More importantly, by using cues from the target bacteria to trigger drug release, the dosage of the antibiotics is self-regulated by the severity of the infections: the more bacteria present at the infection site, the more drugs will be released to treat the bacteria.
Fig. 6.
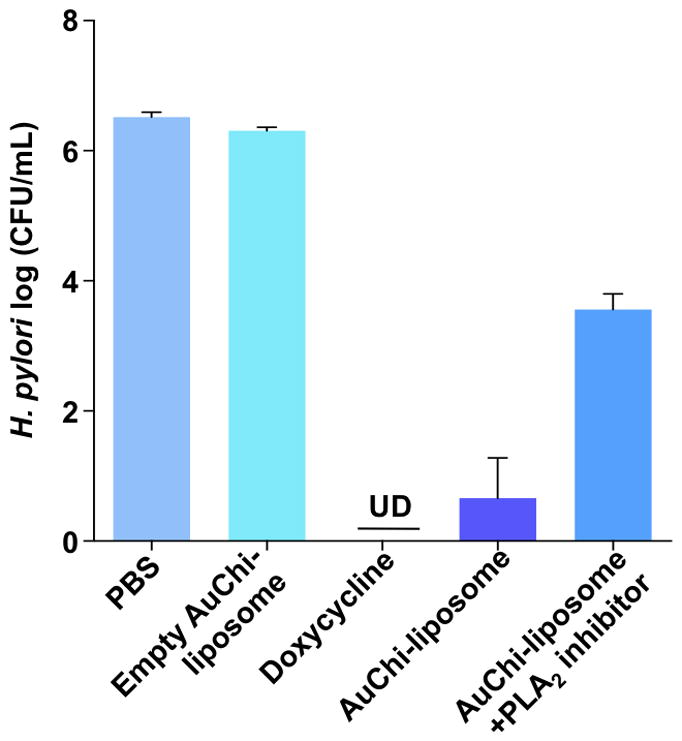
Antimicrobial activity of doxycycline-loaded AuChi-liposome against H. pylori bacteria. Doxycycline-loaded AuChi-liposome was incubated with H. pylori bacteria (5×107 CFU/mL) in 5% TSB for 24 hrs before the bacterium enumeration. To test the effect of PLA2 on the observed antimicrobial activity, PLA2 inhibitor (0.13 μM) was added the doxycycline-loaded AuChi-liposome and H. pylori mixture solution. Free doxycycline (0.2 mM) served as a positive control. Empty AuChi-liposome without doxycycline and PBS (pH = 6.5) served as two negative control groups. Data represent mean ±SD (n = 3).
CONCLUSION
In conclusion, we formulated a PLA2-degradable liposome formulation and further adsorbed AuChi nanoparticles onto the liposome surfaces. The resulting AuChi-liposomes were stable under storage conditions but were susceptible to PLA2 degradation at infection site. Such liposomal formulation effectively prevented undesirable liposome fusion and drug leakage. However, the presence of PLA2, either in purified form or in H. pylori culture, caused rapid drug release due to the enzymatic degradation of phospholipids and the subsequent damage of liposome integrity. When incubated with H. pylori bacteria in vitro, AuChi-liposomes effectively inhibited the bacterial growth. Although aimed for anti-H. pylori treatment in this particular study, the critical role played by PLA2 has been increasingly recognized in various disease pathogenesis including bacterial infections, viral infections, and cancer development38–41. Therefore, PLA2-responsive AuChi-liposomes hold great potential for preferential drug delivery with minimized side effects and targeted therapeutic efficacy to treat a wide range of diseases.
MATERIALS AND METHODS
Materials
1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dioctadecanoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (sodium salt) (DSPG), and 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-lissamine rhodamine B sulfonyl (DMPE-RhB) were purchased from Avanti Polar Lipids, Inc (Alabaster, AL). 8-aminonaphthalene-1,3,6-trisulfonic acid disodium salt (ANTS) and p-xylene-bis-pyridinium bromide (DPX) were purchased from Life Technology (Carlsbad, CA). Rhodamine B, doxycycline, phospholipase A2 (Apis mellifera), and quinacrine dihydrochloride were purchased from Sigma Aldrich (St Louis, MO). Brain-heart infusion (BHI) broth and Columbia agar were purchased from Becton Dickinson (Sparks, MD). Hydrogen tetrachloroaurate (HAuCl4) and sodium borohydride (NaBH4) were purchased from ACROS Organics (Geel, Belgium). Chitosan-50 was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan).
Preparation of chitosan-modified gold nanoparticles (AuChi)
AuChi nanoparticles were prepared by sodium borohydride reduction technique as previously described 13, 14. Briefly, an aqueous solution of HAuCl4 (0.1 mM, 50 mL) was first reduced by 5 mg of NaBH4 to form gold nanoparticles, followed by overnight incubation with 0.1% w/v chitosan that was pre-dissolved in 0.1 M acetic acid. The resulting AuChi nanoparticles were purified three times by using an Amicon Ultra-4 centrifugal filter with a molecular weight cut-off of 10 kDa and the final pH was adjusted to 6.5 by adding HCl. The nanoparticle size and surface zeta potential were determined by dynamic light scattering (DLS) measurements (Malvern Zetasizer ZS, Malvern Instruments, Worcestershire, UK).
Preparation of AuChi-stabilized liposomes (AuChi-liposomes)
Anionic liposomes were prepared by using a vesicle extrusion method. Briefly, DSPC, a zwitterionic phospholipid, and DSPG, an anionic phospholipid, were dissolved in chloroform and mixed at 9:1 molar ratio. The organic solvent was evaporated under a stream of nitrogen gas until the thin lipid film was formed. Then the dry lipid film was hydrated with deionized water, or 2 mM rhodamine B (RhB), or 20mM doxycycline, followed by 2 min of vortexing and 30 min of bath sonication (Fisher Scientific FS30D, Pittsburgh, PA) to produce multilamellar vesicles (MLVs). The solution was then sonicated for 1 min at 20 W by a titanium probe (Branson 450 sonifier, Danbury, CT) to produce unilamellar vesicles. Following the sonication, the solution was extruded through a 100 nm pore-sized polycarbonate membrane for 11 times at 60°C to form narrowly distributed small unilamellar vesicles (SUVs). The liposomes were purified by gel filtration through a Sephadex G-75 column. To prepare fluorescently labeled liposomes, DMPE-RhB (0.5% mol) was added to the lipid mixture prior to liposome preparation. To prepare AuChi-liposomes, the liposomes and AuChi were mixed at 1:300 molar ratio, followed by 12 hrs of vortexing. Hydrodynamic size, size distribution, and surface charge of the liposomes and AuChi-liposomes were characterized by DLS (Malvern Zetasizer ZS, Malvern Instruments, Worcestershire, UK). All measurements were repeated three times at 25°C.
Helicobacter pylori (H. pylori) bacterial culture
H. pylori Sydney strain 1 (SS1) were routinely maintained on Columbia agar supplemented with 5% laked horse blood at 37°C under microaerobic conditions (10% CO2, 85% N2, and 5% O2) 6. For experiments, broth cultures of H. pylori were prepared by sub-culturing fresh colonies from agar plates into BHI containing 5% fetal bovine serum (FBS) overnight at 37°C under microaerobic conditions with moderate reciprocal shaking.
Liposome stability assay
A fluorescence method was used to study the fusion of AuChi-liposome with H. pylori bacteria. Specifically, 5×108 CFU H. pylori bacteria (determined by OD600 value, OD600 = 1.0 corresponding to approximately 1×108 CFU/mL) was washed three times with PBS by repeated centrifugation at 4,000 ×g. The bacteria pellet was collected and then mixed with 0.5 mM fluorescently labeled AuChi-liposome (containing 0.5 mol% of DMPE-RhB) at pH 6.5. After 10 min incubation, the bacteria pellet was collected by centrifugation at 4,000 ×g for 5 min and then resuspended in 1 mL PBS. The bacteria were then measured for fluorescence intensity at the range of 550–700 nm (DMPE-RhB’s fluorescence emission range). DMPE-RhB-labeled bare liposomes (without AuChi stabilizer) were used as a control. The experiment was carried out in triplicate and average value was reported.
Phospholipase A2 (PLA2)-triggered drug release from AuChi-liposomes
RhB was used as a model drug for release study. RhB-loaded AuChi liposomes were formulated as described above and the unencapsulated RhB molecules were removed by gel filtration through a Sephadex G-75 column. The samples were added with PLA2 (at a final enzyme concentration of 0–100 μg/mL) and the mixtures were incubated at 37°C. At predetermined time points, released RhB was separated by filtration using an Amicon Ultra-4 centrifugal filter with a molecular weight cut-off of 10 kDa at 14,000 × g for 20 min. RhB emission intensity at 585 nm was measured. To obtain 100% drug release, freshly prepared RhB-loaded AuChi-liposome suspension was disrupted by Triton-X-100 (1% v/v) to completely release the encapsulated drug, followed by drug quantification. Percentage of released drug was defined as following: Percentage of released drug (%) = (IPLA2 − IPBS)/(ITriton-X-100 − IPBS)×100, in which IPLA2, IPBS, and ITriton-X-100 represent fluorescence emission intensity at 585 nm of the samples incubated with PLA2, PBS, and Triton-X-100, respectively. The experiment was performed in triplicate.
AuChi-liposome drug release in H. pylori bacterial culture
In the study, 190 μL of RhB-loaded AuChi-liposome was added with 10 μL of overnight broth from H. pylori cultures originally containing 1×107 CFU/mL and 1×108 CFU/mL bacteria, respectively. The mixture was incubated for 1, 12, and 24 hrs under gentle shaking. After incubation, released RhB was separated by the same filtration process as described above. Quinacrine dihydrochloride (final concentration 0.13 μM), a PLA2 inhibitor, was used to inhibit PLA2 activity. RhB-loaded AuChi-liposome incubated in 5% (v/v) fresh BHI broth without being used for H. pylori culture was taken as a negative control. To obtain 100% drug release, Triton-X-100 (1% v/v) was added to disrupt liposomes. The experiments were repeated there times.
Anti-H. pylori activity study
Doxycycline-loaded AuChi-liposomes were prepared as described. Free doxycycline molecules were removed by using a Sephadex G-75 column. Doxycycline concentration was determined by measuring the absorbance at 273 nm and a standard curve of doxycycline was made. To quatnify encapsulated doxycycline, liposomes were disrupted by Triton-X-100 (1% v/v), followed by absorbance measurement. Then 10 μL bacterial suspension containing 1×107 CFU H. pylori bacteria was added to 190 μL of doxycycline-loaded AuChi-liposome. The mixture was incubated with gentle shaking at 37°C under microaerobic conditions. After 12 hrs incubation, a series of 10-fold dilutions of the bacterial suspension (1:10 to 1:105) was prepared, and 5 μL from each diluted sample was inoculated onto a Columbia agar plate supplemented with 5% laked horse blood. The plates were cultured in the incubator for 4 days before colony counting. Free doxycycline served as a positive control, while empty AuChi-liposome (without doxycycline) and PBS (1X, pH=6.5) served as negative controls. All experiments were repeated three times.
Acknowledgments
This work is supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number R01DK095168.
References
- 1.Torchilin VP. Nat Rev Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 2.Allen TM, Cullis PR. Adv Drug Del Rev. 2013;65:36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 3.Gao W, Hu CMJ, Fang RH, Zhang L. J Mater Chem B. 2013;1:6569–6585. doi: 10.1039/C3TB21238F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang D, Pornpattananangkul D, Nakatsuji T, Chan M, Carson D, Huang CM, Zhang L. Biomaterials. 2009;30:6035–6040. doi: 10.1016/j.biomaterials.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang L, Pornpattananangkul D, Hu CM, Huang CM. Curr Med Chem. 2010;17:585–594. doi: 10.2174/092986710790416290. [DOI] [PubMed] [Google Scholar]
- 6.Obonyo M, Zhang L, Thamphiwatana S, Pornpattananangkul D, Fu V, Zhang L. Mol Pharm. 2012;9:2677–2685. doi: 10.1021/mp300243w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao W, Chan JM, Farokhzad OC. Mol Pharm. 2010;7:1913–1920. doi: 10.1021/mp100253e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mura S, Nicolas J, Couvreur P. Nat Mater. 2013;12:991–1003. doi: 10.1038/nmat3776. [DOI] [PubMed] [Google Scholar]
- 9.Gao W, Zhang L. Nat Chem. 2012;4:971–972. doi: 10.1038/nchem.1515. [DOI] [PubMed] [Google Scholar]
- 10.de la Rica R, Aili D, Stevens MM. Adv Drug Del Rev. 2012;64:967–978. doi: 10.1016/j.addr.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 11.Chan A, Orme RP, Fricker RA, Roach P. Adv Drug Del Rev. 2013;65:497–514. doi: 10.1016/j.addr.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 12.Ta T, Porter TM. J Controlled Release. 2013;169:112–125. doi: 10.1016/j.jconrel.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pornpattananangkul D, Olson S, Aryal S, Sartor M, Huang CM, Vecchio K, Zhang L. ACS Nano. 2010;4:1935–1942. doi: 10.1021/nn9018587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thamphiwatana S, Fu V, Zhu J, Lu D, Gao W, Zhang L. Langmuir. 2013;29:12228–12233. doi: 10.1021/la402695c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L, Granick S. Nano Lett. 2006;6:694–698. doi: 10.1021/nl052455y. [DOI] [PubMed] [Google Scholar]
- 16.Wang B, Zhang L, Bae SC, Granick S. Proc Natl Acad Sci USA. 2008;105:18171–18175. doi: 10.1073/pnas.0807296105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pornpattananangkul D, Zhang L, Olson S, Aryal S, Obonyo M, Vecchio K, Huang CM, Zhang L. J Am Chem Soc. 2011;133:4132–4139. doi: 10.1021/ja111110e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peek RM, Blaser MJ. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 19.Suerbaum S, Michetti P. New Engl J Med. 2002;347:1175–1186. doi: 10.1056/NEJMra020542. [DOI] [PubMed] [Google Scholar]
- 20.Iwanczak F, Iwanczak B. Adv Clin Exp Med. 2012;21:671–680. [PubMed] [Google Scholar]
- 21.Wu W, Yang Y, Sun G. Gastroenterol Res Pract. 2012;2012:Article ID 723183. doi: 10.1155/2012/723183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nardone G, Holicky EL, Uhl JR, Sabatino L, Staibano S, Rocco A, Colantuoni V, Manzo BA, Romano M, Budillon G, Franklin I, Cockerill R, Miller LJ. Infect Immun. 2001;69:5857–5863. doi: 10.1128/IAI.69.9.5857-5863.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lusini P, Figura N, Valassina M, Roviello F, Vindigni C, Trabalzini L, Nuti R, Lenzi C, Gonnelli C, Nardi M, Martelli P, Santucci A. Dig Liver Dis. 2005;37:232–239. doi: 10.1016/j.dld.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 24.Andresen TL, Davidsen J, Begtrup M, Mouritsen OG, Jorgensen K. J Med Chem. 2004;47:1694–1703. doi: 10.1021/jm031029r. [DOI] [PubMed] [Google Scholar]
- 25.Jorgensen K, Vermehren C, Mouritsen OG. Pharm Res. 1999;16:1491–1493. doi: 10.1023/a:1018931915924. [DOI] [PubMed] [Google Scholar]
- 26.Andresen TL, Jensen SS, Kaasgaard T, Jorgensen K. Curr Drug Del. 2005;2:353–362. doi: 10.2174/156720105774370203. [DOI] [PubMed] [Google Scholar]
- 27.Zhu G, Mock JN, Aljuffali I, Cummings BS, Arnold RD. J Pharm Sci. 2011;100:3146–3159. doi: 10.1002/jps.22530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao W, Vecchio D, Li J, Zhu J, Zhang Q, Fu V, Li J, Thamphiwatana S, Lu D, Zhang L. ACS Nano. 2014;8:2900–2907. doi: 10.1021/nn500110a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jensen SS, Andresen TL, Davidsen J, Hoyrup P, Shnyder SD, Bibby MC, Gill JH, Jorgensen K. Mol Cancer Ther. 2004;3:1451–1458. [PubMed] [Google Scholar]
- 30.Aili D, Mager M, Roche D, Stevens MM. Nano Lett. 2011;11:1401–1405. doi: 10.1021/nl1024062. [DOI] [PubMed] [Google Scholar]
- 31.Higuchi T. J Pharm Sci. 1961;50:874–875. doi: 10.1002/jps.2600501018. [DOI] [PubMed] [Google Scholar]
- 32.Siepmann J, Peppas NA. Int J Pharm. 2011;418:6–12. doi: 10.1016/j.ijpharm.2011.03.051. [DOI] [PubMed] [Google Scholar]
- 33.Berkland C, Kipper MJ, Narasimhan B, Kim KK, Pack DW. J Controlled Release. 2004;94:129–141. doi: 10.1016/j.jconrel.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 34.Dorrell N, Martino MC, Stabler RA, Ward SJ, Zhang ZW, McColm AA, Farthing MJG, Wren BW. Gastroenterology. 1999;117:1098–1104. doi: 10.1016/s0016-5085(99)70394-x. [DOI] [PubMed] [Google Scholar]
- 35.Martino MC, Stabler RA, Zhang ZW, Farthing MJG, Wren BW, Dorrell N. Infect Immun. 2001;69:1697–1703. doi: 10.1128/IAI.69.3.1697-1703.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hatakeyama M, Higashi H. Cancer Sci. 2005;96:835–843. doi: 10.1111/j.1349-7006.2005.00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones KR, Whitmire JM, Merrell DS. Front Microbiol. 2010;1:article 115. doi: 10.3389/fmicb.2010.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sitkiewicz I, Stockbauer KE, Musser JM. Trends Microbiol. 2007;15:63–69. doi: 10.1016/j.tim.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 39.Murakami M, Taketomi Y, Sato H, Yamamoto K. J Biochem. 2011;150:233–255. doi: 10.1093/jb/mvr088. [DOI] [PubMed] [Google Scholar]
- 40.Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G. Chem Rev. 2011;111:6130–6185. doi: 10.1021/cr200085w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murakami M, Lambeau G. Biochimie. 2013;95:43–50. doi: 10.1016/j.biochi.2012.09.007. [DOI] [PubMed] [Google Scholar]