

Abstract
Annotation of the Anopheles gambiae genome has revealed a large increase in the number of genes encoding cuticular proteins with the Rebers and Riddiford Consensus (the CPR gene family) relative to Drosophila melanogaster. This increase reflects an expansion of the RR-2 group of CPR genes, particularly the amplification of sets of highly similar paralogs. Patterns of nucleotide variation indicate that extensive concerted evolution is occurring within these clusters. The pattern of concerted evolution is complex, however, as sequence similarity within clusters is uncorrelated with gene order and orientation, and no comparable clusters occur within similarly compact arrays of the RR-1 group in mosquitoes or in either group in D. melanogaster. The dearth of pseudogenes suggests that sequence clusters are maintained by selection for high gene-copy number, perhaps due to selection for high expression rates. This hypothesis is consistent with the apparently parallel evolution of compact gene architectures within sequence clusters relative to single-copy genes. We show that RR-2 proteins from sequence–cluster genes have complex repeats and extreme amino-acid compositions relative to single-copy CPR proteins in An. gambiae, and that the amino-acid composition of the N-terminal and C-terminal sequence flanking the chitin-binding consensus region evolves in a correlated fashion.
Keywords: Anopheles gambiae, Aedes aegypti, Drosophila melanogaster, cuticular protein, concerted evolution, CPR family, Rebers and Riddiford Consensus
1. Introduction
Proteins with the Rebers and Riddiford Consensus “R&R Consensus” hereafter; Rebers and Riddiford, 1998) are a major component of insect cuticle (Andersen et al., 1995; Willis et al., 2005). These proteins, called CPR proteins, include two major evolutionary groups defined by variants of the R&R Consensus (RR-1 and RR-2). The RR-2 consensus sequence is approximately 64 amino acids long and is well conserved, whereas the RR-1 consensus is more variable in length and sequence. The two variants are readily distinguished by Hidden Markov Models (Karouzou et al., 2007). Although examples of both variants have been shown to bind chitin in vitro (Rebers and Willis, 2001; Togawa et al., 2004), Rebers and Willis (2001) also showed that the affinity of the R&R Consensus to chitin may be disrupted in vitro by nonconservative substitutions at particular positions. Such substitutions are actually present in known CPR proteins and it is reasonable to postulate that CPR proteins differ in their affinity to chitin under physiological conditions. Nonetheless, the vast majority of Anopheles gambiae CPR proteins have been detected in cuticle by proteomic analyses, including those with potentially disrupted chitin affinity (He et al., 2007; He personal communication). Thus, the presence of CPR proteins within cuticular structures appears to be a general feature of this gene family.
It is therefore remarkable that recent characterizations of the CPR gene family within the genomes of Drosophila melanogaster (Karouzou et al., 2007) and An. gambiae (Cornman et al., 2008; Togawa et al., 2008) have confirmed a large diversity of expressed CPR genes and identified very few pseudogenes. The complement of RR-2 genes is particularly large in mosquitoes. The PEST strain of An. gambiae has 101 annotated RR-2 CPR genes whereas only 35 were identified in D. melanogaster. Comparing the phylogeny and genomic organization of RR-2 genes in An. gambiae (Cornman et al., 2008) and D. melanogaster (Karouzou et al., 2007), it is clear that much of the difference in RR-2 gene number is due to the presence in An. gambiae of sets of highly similar genes that are often but not exclusively arranged in complex clusters within larger tandem arrays (Cornman et al., 2008) and which are co-expressed (Togawa et al., 2008).
Here we identify orthologous genes and syntenic regions between An. gambiae and both Ae. aegypti and D. melanogaster in order to investigate the origins and evolution of these RR-2 ‘sequence clusters.’ We further investigate whether sequence clusters are evolving in a non-independent manner through some combination of unequal crossing-over and intergenic gene conversion. This process is commonly referred to as concerted evolution and is a potentially important component of multigene family evolution (Ohta, 1983; Walsh, 1987; Innan, 2003). Nei and Rooney (2005) have argued that the contribution of concerted evolution to the maintenance of highly similar paralogs is generally weak; they posit that strong purifying selection and a background level of gene turnover are sufficient to explain most such cases (the ‘birth-and-death’ model). Nonetheless, gene conversion events among paralogs have been identified in a diverse array of taxa (Drouin, 2002a; Teshima and Innan, 2004) and appear to be relatively frequent at the genome scale in mice and rats (Ezawa et al., 2006), rice (Wang et al., 2007), and yeast (Drouin, 2002b) but less so in nematodes (Semple and Wolfe, 1999). Drouin et al. (1999) and Drouin (2002a) evaluated different approaches for detecting gene conversion in the absence of population-genetic data, i.e. when few alleles for each gene-family member are known from a given species (as is often the case). They found that several methods are effective and appear to give low rates of false-positives. We utilize several of these approaches to show that concerted evolution is more important than birth-and-death evolution in maintaining RR-2 sequence clusters in An. gambiae. Finally, to gain insight into the possible functional significance of these ‘sequence clusters’, we compared properties of these genes and their encoded proteins to ‘single copy’ RR-2 genes and to RR-1 genes.
2. Methods
2.1. Phylogeny, organization, and properties of RR-2 genes
In An. gambiae, some CPR genes occur in isolation, but most occur in tandem arrays in which genes are usually spaced a few kb apart and not more than 20 kb based on our operational cutoff. Many of these tandem arrays include sets of genes that are alignable across the length of the protein, which may be due to recent duplication, purifying selection, and/or gene conversion (see Results and Discussion). In this paper, we use the term ‘sequence cluster’ in contradistinction to ‘tandem array’ to denote a set of highly similar paralogs that are often but not exclusively contiguous. Following Koonin (2005) we use ‘co-orthologous region’ to describe a chromosomal region in two species that is syntenic and contains putatively orthologous genes although the orthology of individual genes is uncertain due to the differential gain or loss of genes or due to gene–conversion events. In this study, An. gambiae sequence clusters are named by their order on chromosomes, e.g. 2LA, 2LB, and 2LC are the three sequence clusters on chromosome 2L ordered by Ensembl coordinates (see Cornman et al., 2008 for details).
Most An. gambiae CPR genes are available from Ensembl (http://www.ensembl.org/index.html) and all of their conceptual translations are available from the cuticleDB database (http://bioinformatics.biol.uoa.gr/cuticleDB/; Magkrioti et al., 2004). Cornman et al. (2008) provide contig coordinates for all An. gambiae CPR genes as well as supporting evidence for their annotation. Ae. aegypti genes were obtained by examining all gene predictions of Ensembl v. 40 that contained the Pfam00379 domain (http://www.sanger.ac.uk/cgi-bin/Pfam/getacc?PF00379), which defines the extended R&R Consensus sequence, and identifying putative RR-2 genes (Supplementary File 1). We corrected twenty-six gene predictions for annotation errors that were evident based on alignment with other mosquito RR-2 genes (Supplementary File 2). We removed two predicted genes of Ae. aegypti (AAEL014925 and AAEL011037) from the analysis because of major annotation concerns that could not be resolved by inspection. Six Ae. aegypti CPR genes not annotated by Ensembl v. 40 were identified by BLAST searches or dot-plots (listed in Supplementary File 3 and named AeCPR A-F). Our confidence in the annotated An. gambiae RR-2 genes allows us to confirm the overall accuracy of the Ae. aegypti RR-2 genes used in this study with respect to the consensus region, despite uncertainties regarding the 5′ boundaries of some genes and whether nearly identical genes on different contigs are distinct genes. We included all such duplicates in this study because the intergenic regions were not similar when examined by dot-plot, which shows that these putative paralogs are unlikely to be alleles of a single locus. Exclusion of these sequences did not qualitatively change the results of any analysis. The annotation of the CPR family in D. melanogaster has been recently updated (Karouzou et al., 2007) and the sequences are available from Flybase (http://www.flybase.org) and the cuticleDB website given above.
We developed a phylogeny of all RR-2 genes of the three species based on the Pfam00379 domain alignment, but excluding the first five positions of that alignment because they do not align well across all three species. We used amino-acid sequence rather than nucleotide sequence because there is clear evidence of mutational saturation (pairwise synonymous substitution rates ≫ 1, see Cornman et al. [2008]). A neighbor-joining tree with 1000 bootstrap replicates was generated in MEGA3 (Kumar et al., 2004) using the JTT cost matrix (Jones et al., 1992) and performing pairwise deletion of indels. We assumed a gamma distribution of rates with β = 1.0, as an evolutionary model with rate variation among sites gives a significantly better fit to the data than does a single ratio of nonsynonymous to synonymous substitutions (Ka/Ks) for An. gambiae RR-2 genes (Cornman et al., 2008).
We identified all probable orthologs of An. gambiae RR-2 genes in Ae. aegypti and D. melanogaster. While the R&R Consensus is strongly conserved, flanking regions are often of low complexity and typically are not alignable among paralogs. However, these flanking regions are conserved among orthologs in An. gambiae and Ae. aegypti and often share sequence motifs within tandem arrays that aid in the identification of syntenic chromosomal regions in D. melanogaster (Cornman et al., 2008). We therefore used both consensus-region phylogenetic analysis and reciprocal BLAST scores of the full protein sequence, including the signal peptide, to identify putatively orthologous genes or co-orthologous regions. Importantly, conserved orthologous single-copy genes (whether CPR genes or otherwise) serve as genomic landmarks that provide strong support for the putative co-orthology of sequence clusters in the two species (see Results).
For two single-copy An. gambiae genes, CPR59 and CPR68, we identified two potential orthologs each in Ae. aegypti on different contigs; Ka and Ks estimates between An. gambiae and Ae. aegypti were obtained by averaging the values for the two potential orthologs. Three other An. gambiae genes, CPR47, CPR63, and CPR156 were ambiguous as to whether they belonged in sequence clusters and their placement in the phylogenetic tree was sensitive to changes in phylogenetic method. CPR47 and CPR156 are physically adjacent to the sequence clusters in question and CPR47 showed evidence of non-orthologous recombination with other sequences in 2LB (assessed with the RDP2 program, see below), but this was not true of CPR156 nor CPR63. We conservatively included CPR47 in the 2LB cluster and CPR156 in the 3RB cluster but excluded CPR63 from 2LC, although these placements do not materially affect our results.
2.2. Molecular evolution of sequence-cluster genes
To infer intergenic conversion as a mode of molecular evolution within sequence clusters, we used three approaches. We first examined patterns of synonymous polymorphisms and intron polymorphism to determine whether purifying selection or recent duplications are sufficient explanations of protein similarity. We used DnaSP (Rozas et al., 2003) to calculate levels of synonymous nucleotide polymorphism within the aligned R&R Consensus. Genomic DNA dot-plots were created with Dotter (Sonnhammer and Durbin, 1995). All statistical tests were performed using the software PAST (Hammer et al., 2001) unless stated otherwise.
We then used the RDP2 program (Martin et al., 2005) to implement a suite of statistical tests to detect recombination or conversion among a set of alleles, including the method of Sawyer (1989), which was shown to perform well for paralogous comparisons (Drouin, 2002a, 2002b). To determine whether the pattern of nucleotide polymorphism within a sequence cluster is consistent with intergenic recombination or conversion, we conservatively required that five of the six methods detect events at α = 0.01, adjusted by a statistical correction for multiple tests (the Bonferroni correction). For each sequence cluster, we used as much nucleotide sequence 5′ of the start codon and 3′ of the stop codon as could be reasonably aligned for at least four members of that cluster.
The third method we used to detect intergenic conversion was to compare the phylogenetic relationships of co-ortholgous sequence clusters in An. gambiae and Ae. aegypti. Extensive clustering of paralogs by species rather than the clustering of each An. gambiae gene with its ortholog in Ae. aegypti is consistent with concerted evolution as the predominant mode of molecular evolution. In fact, ‘quartet methods’ that compare alleles of duplicated genes from two species are statistical implementations of this concept (e.g. Balding et al., 1992; Ezawa et al., 2006), but these methods have not been generalized for large, co-orthologous sets of paralogs and we therefore limit ourselves to an appeal to parsimony.
To investigate the likelihood of pseudogenes occurring within sequence clusters under an assumption of redundancy, we used the codeml program of PAML (Yang, 1997) to reconstruct the most likely ancestral sequence of sequence cluster 2RA from An. gambiae and Ae. aegypti sequences. We then simulated sequence evolution of six copies of this sequence (corresponding to an assumed six genes in the common ancestor of the two species) with the evolver program of PAML. The evolver program generates mutations in an input sequence based on a given evolutionary model until the branch length between the initial and final sequence is that specified by the user. The evolutionary model was the Kimura two-parameter model with observed nucleotide frequencies and PAML-estimated transition-to-transversion ratio. The branch lengths of the evolved sequences were obtained from a neighbor-joining phylogeny constructed in MEGA3 (Kumar et al., 2004) using the Kimura two-parameter model and pairwise deletion of gaps, and using genes of both species. The Ae. aegypti sequences were then removed and the length of the An. gambiae stem branch was added to the tip-branch lengths of the An. gambiae genes. The sequences were evolved as a polytomy rather than along the inferred tree so as to simulate independent evolution of a cluster of functional genes since the divergence of the two species. We then computed the proportion of evolved sequences that had a stop codon in 10,000 replicate sets. The length of the sequences was constrained to 255 nucleotides, corresponding to the length of alignable sequence of the 2RA sequence cluster through the end of the R&R Consensus, because the functional significance of a stop codon after the R&R Consensus is unknown.
2.3. Protein properties
We calculated the amino-acid composition of all CPR proteins of An. gambiae with the predicted signal peptide removed. Correlations and principal components analysis were then computed on the percentage composition matrix after removing cysteine and methionine, which are very rare in mature RR-2 proteins and can therefore be removed to reduce the dimensions of the matrix without loss of information. We calculated a sequence repetition score with the program HHRep, which creates a Hidden Markov model of a sequence and any identified homologs and then computes the best suboptimal alignment of the model with itself (Söding et al., 2006). Protein dot-plots were created with BioEdit (Hall, 1999).
3. Results
3.1. Phylogeny, organization, and properties of RR-2 genes
The phylogeny we recovered of RR-2 proteins from all three Dipteran genomes is shown in Figure 1. Sequence clusters of highly similar paralogs are evident within the two mosquito genomes and are marked as they are referred to in the text (following Cornman et al., 2008). Importantly, previous results have shown that no sequence clusters occur within the RR-1 group of genes in mosquitoes, despite the fact that many occur in large and compact tandem arrays (Cornman et al., 2008). Thus, the organization of homologous CPR genes in compact tandem arrays is clearly not sufficient to generate sequence clusters.
Fig. 1.

Neighbor-joining phylogeny of the RR-2 group of CPR proteins from three Dipteran genomes, Ae. aegypti, An. gambiae, and D. melanogaster. The tree is based on the consensus region only as described in the text. The tree was created with MEGA3 (Kumar et al., 2004) using the JTT cost matrix (Jones et al., 1992) with pairwise deletion of gaps. A gamma distribution of rate variation among sites was assumed with β = 1.0. The tree is rooted with two An. gambiae RR-1 proteins, CPR14 and CPR28. Groups discussed in text are indicated.
Although tandem arrays of RR-2 genes in the three genomes are dynamic with respect to gene number, we were able to identify clearly co-orthologous groups in Ae. aegypti of all An. gambiae sequence clusters based on phylogeny and overall synteny, with one exception. We were not able to determine which genes in Ae. aegypti are co-orthologs of An. gambiae sequence cluster 2LB versus sequence cluster 2LC (Fig. 2), so instead we combined these two sequence clusters for comparisons with Ae. aegypti. However, since the 2LB and 2LC sequence clusters in An. gambiae are distinct by sequence and gene expression profile (Togawa et al., 2008), we continued to treat them separately for comparisons with other An. gambiae clusters.
Fig. 2.

Schematic of the An. gambiae 2LB-2LC sequence clusters and the co-orthologous regions of Ae. aegypti, and D. melanogaster. An. gambiae genes with light gray shading belong to sequence cluster 2LB and those with dark gray shading belong to sequence cluster 2LC (see text for details). Gene names are given in order along the chromosome, represented by horizontal black lines. For clarity, Ensembl gene names for Ae. aegyptii and An. gambiae are given without the full Ensembl prefixes AAEL and AGAP and leading zeroes. The positions of gene names above and below the line represent the gene orientation. Black lines connect putatively orthologous single-copy genes, whereas gray lines connect putatively co-orthologous groups. Genes with names in gray font do not code for cuticular proteins, whereas all other genes encode proteins of the RR-2 group.
Additional examples of syntenic regions containing RR-2 genes are detailed in Figures 3–4. The order and orientation of genes are indicated but the diagrams are not drawn to scale for clarity. Genes of different sequence clusters are generally not intermixed along chromosomes; the single exception is the presence of an additional gene (CPR149) of An. gambiae sequence cluster 3RB in the middle of the region containing sequence cluster 3RC (Fig. 4). Overall, genetic distance between gene pairs within a sequence cluster is not correlated with either gene order or gene orientation by Mantel test (r = −0.060 and r = −0.018, respectively, P > 0.1 for both coefficients) in An. gambiae. We can infer that these co-orthologous groups are on the order of 100 million years old in An. gambiae and Ae. aegypti based on current estimates of divergence time (Krzywinski et al., 2001). Properties of sequence clusters, which were originally delineated in An. gambiae on the basis of sequence similarity only, are summarized in Table 1 and alignments with co-orthologous sequences in Ae. aegypti are shown in Supplementary File 4.
Fig. 3.

Schematic of the An. gambiae 2RB sequence cluster and the co-orthologous regions of Ae. aegypti, and D. melanogaster. See Fig. 2 for explanation of symbols. Light gray shading indicates genes of the 2RB sequence cluster (see text for details). Question mark indicates a possible but uncertain orthologous relationship.
Fig. 4.

Schematic of the An. gambiae 3R sequence clusters and the co-orthologous regions of Ae. aegypti, and D. melanogaster. See Fig. 2 for explanation of symbols. Progressively darker shading represents sequence clusters 3RA, 3RB, and 3RC, respectively (see text for details). *4751 and 4745 are within a predicted intron of the non-CPR gene 4761. **A CPR gene that has been provisionally annotated by the authors but has not yet been named.
Table 1.
Summary of RR-2 sequence clusters in An. gambiae defined for this study. Bootstrap support is based on the phylogeny of all An. gambiae CPR proteins (Cornman et al., 2008). Values reflect similarity of the aligned R&R Consensus sequence only.
| Cluster | Bootstrap support | Average pairwise distance within cluster1 | Average distance to nearest outgroup1 | Number of genes in An. gambiae | Number of Ae.aegypti homologs in co-orthologous group |
|---|---|---|---|---|---|
| 2LA | 99 | 0.12 | 0.33 | 4 | 7 |
| 2LB2 | 36 | 0.14 | 0.39 | 9 | 59 |
| 2LC2 | 76 | 0.18 | 0.26 | 17 | |
| 2RA | 97 | 0.06 | 0.27 | 6 | 15 |
| 2RB | 97 | 0.09 | 0.27 | 9 | 3 |
| 3RA | 64 | 0.32 | 0.64 | 5 | 9 |
| 3RB | 99 | 0.02 | 0.64 | 9 | 7 |
| 3RC | 99 | 0.06 | 0.29 | 10 | 5 |
Distance measure is the expected fraction of accepted mutations using the JTT cost matrix (Jones et al., 1992), scaled such that a value of 1.0 equals 100 iterations of the matrix.
Bootstrap support is reduced due to the inclusion of a single, more divergent gene. This was done because in each case the gene in question was similar outside of the consensus region as well.
We identified only two tandem arrays in D. melanogaster that appear to correspond to RR-2 sequence clusters in An. gambiae. The tandemly arrayed genes Ccp84Aa-g in Drosophila appear to be co-orthologous to the 2RA sequence cluster and are peculiar because they interrupt the Antennapedia complex of Hox genes between labial and proboscipedia in apparently all Drosophila (Negre and Ruiz, 2007). The 2RA sequence cluster has putative orthologs in Apis and Tribolium as well, but the genes retain a higher level of similarity within the mosquito species than within these other genomes or D. melanogaster (Supplementary File 5). A second tandem array of CPR genes in D. melanogaster, Cpr64Aa-d, is co-orthologous to a tandem array in An. gambiae that includes the sequence cluster 2RB. Thus, sequence clusters of highly similar RR-2 genes occur in mosquitoes but not D. melanogaster, and most mosquito RR-2 sequence clusters have no recognizable co-orthologs in D. melanogaster at all. Supplementary File 6 summarizes putative orthologs of all An. gambiae CPRs in Ae. aegypti and D. melanogaster.
Genes within An. gambiae RR-2 sequence clusters have a highly compact architecture compared to other CPR genes (Fig. 5 and Table 2). The average lengths of the predicted protein (165 amino acids) and of all introns (80 nucleotides) were shorter than either single-copy RR-2 genes (226 amino acids and 883 nucleotides, respectively) or RR-1 genes (191 amino acids and 688 nucleotides). Sequence-cluster genes rarely have more than one intron, whereas having two or more is common among other CPR genes in An. gambiae. This pattern is found even though sequence clusters are more closely related phylogenetically to single-copy genes on the same chromosome than to other sequence clusters (Cornman et al., 2008). Thus, the compact organization of sequence clusters appears to have arisen independently multiple times.
Fig. 5.
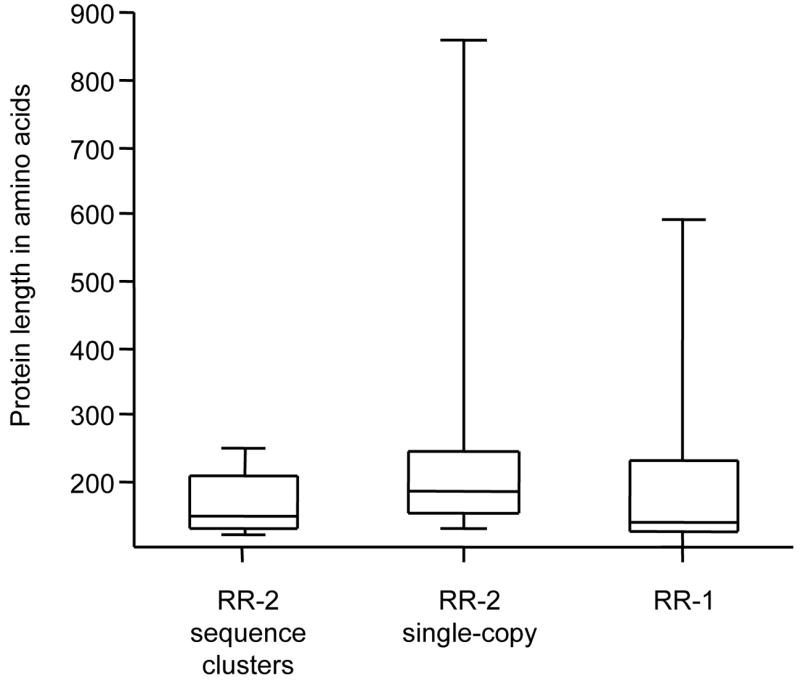
Box-plots of protein length (including signal peptide) for three groups of An. gambiae CPR proteins. Each box represents the 25%–75% range, with the minimum, maximum, and median values represented by lines. Proteins with the RR-2 version of the Consensus within sequence clusters (see text for details) have substantially shorter proteins than RR-2 proteins that are encoded single-copy genes or RR-1 proteins.
Table 2.
Percentage of genes with introns in each category for An. gambiae CPR genes. Only first and second introns are considered, as An. gambiae CPR genes with more than two introns are uncommon and do not occur in sequence-cluster genes.
| First intron1 | Second intron1 | |||||
|---|---|---|---|---|---|---|
| Size range of intron | RR-2 sequence cluster | RR-2 single copy | RR-1 | RR-2 sequence cluster | RR-2 single copy | RR-1 |
| none | 16.4 | 6.3 | 9.3 | 88.1 | 56.3 | 50.0 |
| ≤120 | 82.1 | 40.6 | 37.0 | 11.9 | 15.6 | 16.7 |
| 121–600 | 1.5 | 28.1 | 29.6 | 0 | 15.6 | 18.5 |
| >600 | 0 | 25.0 | 24.1 | 0 | 12.5 | 14.8 |
Equal proportions among classes rejected by contingency test at P < 0.001.
3.2. Molecular evolution of sequence-cluster genes
The average Ks within a sequence cluster of a single mosquito species is significantly lower than the Ks between co-orthologous sequence clusters in the two species (Fig. 6, P < 0.001), demonstrating substantially less divergence among paralogs within lineages than among co-orthologs between lineages. Since by definition co-orthologous groups had approximately equal amounts of synonymous variation at the time of divergence from their common ancestor, independent evolution of gene copies would result in similar levels of synonymous divergence within a species and between species. Reduced synonymous variation among paralogs within species is therefore inconsistent with the hypothesis that genes within each sequence cluster are equally old and evolving independently under purifying selection. Rather, either gene turnover or gene conversion within lineages has substantially reduced the average level of synonymous variation within sequence clusters.
Fig. 6.
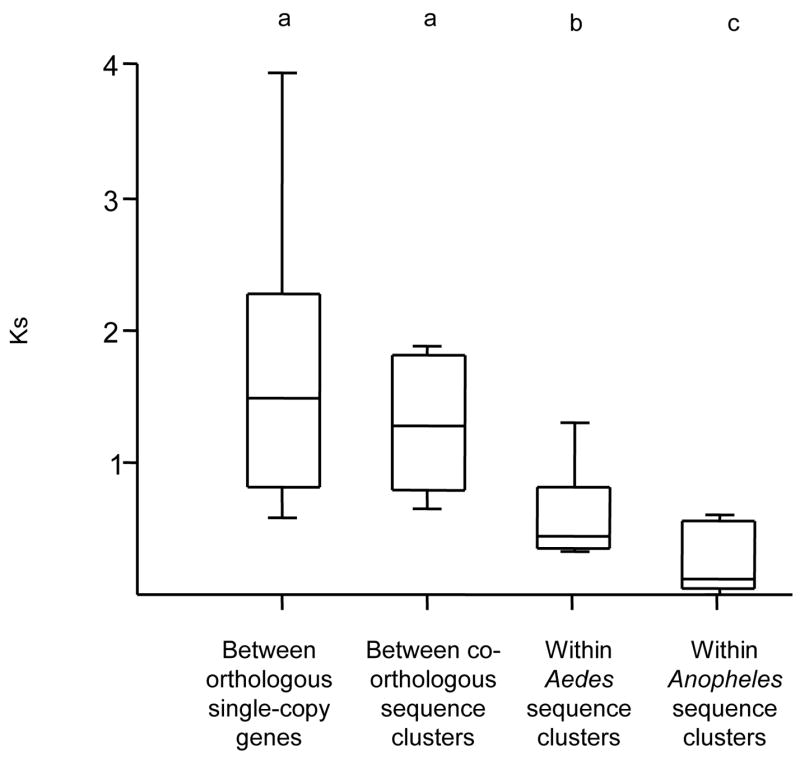
Box-plots of synonymous polymorphism (Ks) within the coding sequence of CPR genes of An. gambiae and Ae. aegypti. Each box represents the 25%–75% range, with the minimum, maximum, and median values represented by horizontal lines. For sequence clusters (see text for details), the mean Ks was calculated from all pairwise comparisons. Letters represent groups that are significantly different (P < 0.01) by ANOVA.
Since codon bias can constrain the pattern of synonymous polymorphism and produce misleading interpretations of Ks (Sharp and Li, 1986), we used the program INCA (Supek and Vlahovicek, 2004) to estimate the effective number of codons (ENC’; Novembre, 2002) for RR-2 sequence clusters and single-copy genes. Mean ENC’ was 60.3 for An. gambiae RR-2 sequence clusters as a group, which is near the maximum value of 61 and indicates approximately random codon usage, whereas single-copy genes collectively do show some codon-usage bias (ENC’ = 48.7). Thus, codon bias is excluded as an alternative explanation for reduced Ks within sequence clusters relative to single-copy genes.
Patterns of nucleotide similarity within introns also indicate recent gene duplication or gene conversion. Table 3 lists the number of nucleotide differences and percentage similarity of all pairwise combinations within sequence clusters for ClustalW-aligned intron sequence. Values were calculated for first introns because only sequence cluster 2RB has more than one intron. Five of the eight sequence clusters include gene pairs with very similar introns. Although introns may include regulatory sequences (Fedorova and Fedorov, 2003), strongly multimodal patterns of intron similarity that include highly similar and highly divergent sequences are not well explained by purifying selection.
Table 3.
Percentage identity (below diagonal, italicized) and number of nucleotide differences (above diagonal) for first introns of sequence-cluster genes.
| 2RA | CPR1 | CPR2 | CPR3 | CPR4 | CPR5 | CPR6 | |||||
| CPR1 | 44 | 46 | 44 | 43 | 42 | ||||||
| CPR2 | 32 | 3 | 0 | 42 | 44 | ||||||
| CPR3 | 29 | 95 | 3 | 43 | 44 | ||||||
| CPR4 | 32 | 100 | 95 | 42 | 44 | ||||||
| CPR5 | 34 | 35 | 34 | 35 | 44 | ||||||
| CPR6 | 35 | 32 | 32 | 32 | 32 | ||||||
| 2RB | CPR1541 | CPR115 | CPR117 | CPR118 | CPR119 | CPR120 | CPR121 | CPR122 | CPR123 | ||
| CPR1541 | - | - | - | - | - | - | - | - | |||
| CPR115 | - | 20 | 0 | 2 | 0 | 0 | 46 | 44 | |||
| CPR117 | - | 72 | 20 | 21 | 20 | 20 | 47 | 50 | |||
| CPR118 | - | 100 | 72 | 2 | 0 | 0 | 46 | 44 | |||
| CPR119 | - | 97 | 71 | 97 | 2 | 2 | 45 | 44 | |||
| CPR120 | - | 100 | 72 | 100 | 97 | 0 | 46 | 44 | |||
| CPR121 | - | 100 | 72 | 100 | 97 | 100 | 46 | 44 | |||
| CPR122 | - | 36 | 35 | 36 | 37 | 36 | 36 | 49 | |||
| CPR123 | - | 39 | 31 | 39 | 39 | 39 | 39 | 32 | |||
| 2LA | CPR17 | CPR18 | CPR19 | CPR20 | |||||||
| CPR17 | 41 | 43 | 38 | ||||||||
| CPR18 | 35 | 40 | 40 | ||||||||
| CPR19 | 32 | 37 | 35 | ||||||||
| CPR20 | 40 | 37 | 44 | ||||||||
| 2LB | CPR136 | CPR54 | CPR53 | CPR52 | CPR51 | CPR50 | CPR49 | CPR48 | CPR47 | ||
| CPR136 | 31 | 31 | 36 | 32 | 30 | 31 | 32 | 35 | |||
| CPR54 | 47 | 0 | 34 | 3 | 1 | 2 | 40 | 40 | |||
| CPR53 | 47 | 100 | 34 | 3 | 1 | 2 | 40 | 40 | |||
| CPR52 | 39 | 42 | 42 | 36 | 34 | 34 | 32 | 39 | |||
| CPR51 | 46 | 95 | 95 | 39 | 4 | 3 | 38 | 40 | |||
| CPR50 | 49 | 98 | 98 | 42 | 93 | 3 | 40 | 41 | |||
| CPR49 | 47 | 97 | 97 | 42 | 95 | 95 | 38 | 41 | |||
| CPR48 | 46 | 32 | 32 | 46 | 36 | 32 | 36 | 42 | |||
| CPR47 | 41 | 32 | 32 | 34 | 32 | 31 | 31 | 29 | |||
| 2LC2 | CPR72 | CPR37 | CPR66 | CPR65 | CPR34 | ||||||
| CPR72 | 37 | 42 | 35 | 45 | |||||||
| CPR37 | 40 | 36 | 40 | 42 | |||||||
| CPR66 | 32 | 42 | 47 | 42 | |||||||
| CPR65 | 44 | 35 | 24 | 42 | |||||||
| CPR34 | 24 | 32 | 32 | 32 | |||||||
| 3RA | CPR82 | CPR107 | CPR83 | CPR108 | CPR84 | ||||||
| CPR82 | 39 | 36 | 37 | 40 | |||||||
| CPR107 | 37 | 36 | 37 | 31 | |||||||
| CPR83 | 42 | 42 | 32 | 41 | |||||||
| CPR108 | 40 | 40 | 48 | 40 | |||||||
| CPR84 | 35 | 50 | 34 | 35 | |||||||
| 3RB | CPR156 | CPR148 | CPR86 | CPR87 | CPR88 | CPR89 | CPR90 | CPR91 | CPR149 | ||
| CPR156 | 33 | 34 | 35 | 29 | 31 | 34 | 33 | 32 | |||
| CPR148 | 42 | 2 | 2 | 27 | 4 | 1 | 2 | 3 | |||
| CPR86 | 40 | 96 | 2 | 28 | 6 | 3 | 4 | 5 | |||
| CPR87 | 39 | 96 | 96 | 29 | 6 | 3 | 4 | 5 | |||
| CPR88 | 49 | 53 | 51 | 49 | 27 | 28 | 29 | 28 | |||
| CPR89 | 46 | 93 | 89 | 89 | 53 | 5 | 4 | 1 | |||
| CPR90 | 40 | 98 | 95 | 95 | 51 | 91 | 3 | 4 | |||
| CPR91 | 42 | 96 | 93 | 93 | 49 | 93 | 95 | 3 | |||
| CPR149 | 44 | 95 | 91 | 91 | 51 | 98 | 93 | 95 | |||
| 3RC | CPR92 | CPR93 | CPR94 | CPR109 | CPR95 | CPR96 | CPR97 | CPR142 | CPR99 | CPR100 | |
| CPR92 | 2 | 2 | 10 | 16 | 15 | 10 | 0 | 1 | 11 | ||
| CPR93 | 98 | 4 | 10 | 16 | 15 | 11 | 2 | 3 | 13 | ||
| CPR94 | 98 | 95 | 11 | 17 | 16 | 10 | 2 | 3 | 11 | ||
| CPR109 | 89 | 89 | 87 | 7 | 6 | 15 | 10 | 11 | 16 | ||
| CPR95 | 82 | 82 | 80 | 92 | 1 | 11 | 16 | 17 | 12 | ||
| CPR96 | 83 | 83 | 82 | 93 | 99 | 10 | 15 | 16 | 11 | ||
| CPR97 | 89 | 87 | 89 | 83 | 87 | 89 | 10 | 11 | 3 | ||
| CPR142 | 100 | 98 | 98 | 89 | 82 | 83 | 89 | 1 | 11 | ||
| CPR99 | 99 | 97 | 97 | 87 | 80 | 82 | 87 | 99 | 12 | ||
| CPR100 | 87 | 85 | 87 | 82 | 86 | 87 | 97 | 87 | 86 |
Values not calculated for CPR154 because of N′s in the whole genome sequence
Excludes intron-less genes.
We then investigated whether ongoing segmental duplication could be a sufficient explanation of the reduced synonymous variation within sequence clusters. Segmental duplication via unequal crossing-over should generate roughly equal levels of sequence similarity between duplicated intergenic sequence as between synonymous sites within duplicated coding sequence. However, neither dot-plots of genomic regions containing sequence clusters nor levels of nucleotide diversity within and near genes support unequal crossing over alone as a sufficient mechanism for the maintenance of sequence clusters. For example, the sequence cluster 3RB has low synonymous nucleotide diversity within codons and a simple genomic organization in which all genes share the same orientation. These two features are prima facie indicators of tandem duplication by unequal crossing-over. Yet dot-plots of the 3RB sequence cluster under various threshold parameters settings do not resolve any residual sequence similarity in intergenic regions among gene copies (Supplementary File 7). This is true of other sequence clusters as well.
Furthermore, the rate of decline in nucleotide diversity in the 5′ and 3′ directions of sequence-cluster genes is rapid for most sequence clusters, as shown in Figure 7. Interestingly, the 3RB and 3RC sequence clusters do retain a high degree of similarity in the region immediately upstream of the start codon (marked on Figure 7). In 3RB, this region of similarity persists for approximately 100 bp and includes the core promoter. In 3RC, there are two distinct haplotype groups among the ten genes of this sequence cluster, corresponding with shared gene orientation, within which the 5′ regions upstream of the start codon are >90% similar for approximately 150 bp and 500 bp respectively. Thus, for these two sequence clusters similarity does in fact extend upstream of the coding region for a small distance relative to the intergenic distance. However, downstream of the stop codon similarity rapidly disappears. We do not know of a duplication mechanism that would generate such an asymmetric pattern of similarity with respect to the flanking genomic DNA. An alternative explanation for these particular cases is selectively favored gene-conversion events homogenizing 5′ regulatory sequences.
Fig. 7.

Average nucleotide diversity (π) of aligned PEST genomic sequence 500 bp 5′ and 3′ of genes in sequence clusters. The dashed box represents the gap associated with the coding sequence, and average values of π for synonymous sites in codons (black line) and first introns (gray line) are shown. Lines to the left and right of the box represent nucleotide diversity of flanking regions for each sequence cluster. Only the 5′ regions associated with sequence clusters 3RB and 3RC, which are discussed in the text, are indicated. The outlier in the 3′ region is sequence cluster 2RB, as indicated on the figure. Nucleotide diversity was calculated with DnaSP (Rozas et al., 2003) from ClustalW-aligned nucleotide sequence with default nucleotide gap penalties.
Several additional lines of evidence further show that concerted evolution is occurring within most sequence clusters and refute a birth-and-death model of gene duplication as a sufficient explanation. These lines of evidence are: 1) statistically significant patterns of recombination or conversion among different genes; 2) reciprocal monophyly of co-orthologous sequence clusters in each species; and 3) the dearth of pseudogenes in sequence clusters. We emphasize that these results do not imply an absence of gene turnover in the two mosquito lineages, only that birth-and-death processes are insufficient to explain the evolution and maintenance of RR-2 sequence clusters.
In all but one sequence cluster, at least five of the six methods implemented by RDP2 (Martin et al., 2005) identified multiple recombination or gene conversion events (Table 4). Events were identified between genes in the same as well as in opposite orientations. The sequence cluster that did not have sufficient evidence of recombination based on our conservative criteria was 3RA, the least compact of the sequence clusters we defined in An. gambiae. We note that while the mean number of potential events varies considerably among groups, the statistical power to identify events is a function of sequence diversity within a sequence cluster and not all potential events are necessarily compatible. Furthermore, the estimated number of events can be inflated by gene duplications occurring after a gene conversion. We therefore do not report numerical estimates but instead state whether events were identified at α = 0.01.
Table 4.
Detection of intergenic recombination or gene conversion within An. gambiae RR-2 sequence clusters. An ‘X’ indicates significant recombination or conversion events detected between loci. Evidence of recombination or gene conversion was considered significant if five out of the six algorithms implemented by the program RDP2 (Martin et al., 2005) detected events at α = 0.01.
| Cluster | Significant evidence of recombination? | RDP | GENE-CONV | Bootscan | MaxC hi | Chimaera | SiScan |
|---|---|---|---|---|---|---|---|
| 2LA | Yes | X | X | X | X | X | |
| 2LB | Yes | X | X | X | X | X | X |
| 2LC1 | Yes | X | X | X | X | X | X |
| 2RA | Yes | X | X | X | X | X | X |
| 2RB2 | Yes | X | X | X | X | X | X |
| 3RA | No | X | X | X | |||
| 3RB3 | Yes | X | X | X | X | X | X |
| 3RC | Yes | X | X | X | X | X | X |
To control for structural variation that might bias detection of gene conversion, only the intronless genes of this cluster were used
CPR154 was removed because the upstream region and beginning of gene sequence contained unresolved bases in the whole genome sequence
CPR149 was removed because the upstream region contained unresolved bases in the whole genome sequence
Several sequence clusters are reciprocally monophyletic with the co-orthologous group in Ae. aegyptii based on phylogenies derived from the complete protein sequence. These reciprocally monophyletic sequence clusters are 3RB, 3RC, 2LA, and 2RA (Fig. 1). While ongoing gene turnover and gene duplication coupled with purifying selection could in principle result in extensive within-species clustering, this process is not a plausible explanation of such extensive reciprocal monophyly because it requires complete loss of ancestral gene copies independently in both lineages, or else multiple parallel amplifications of similar single-copy genes in each lineage. On the other hand, the 3RA sequence cluster is not reciprocally monophyletic nor is there evidence of intergenic exchange based on the analysis presented above, and therefore that sequence cluster is likely evolving under a birth-and-death process. The 2RB sequence cluster contains only three genes in Ae. aegypti, all of which have their closest homologs in An. gambiae, whereas there are nine highly similar genes in An. gambiae. Thus, this sequence cluster has not been extensively amplified in Ae. aegypti, and it appears that the relative importance of concerted evolution versus birth-and-death evolution differs between the two mosquito species and among sequence clusters. This conclusion is supported by the fact that average Ks within sequence clusters is not as low in Ae. aegypti as it is in An. gambiae (Fig. 6).
Recognizable RR-2 pseudogenes are largely absent from RR-2 sequence clusters in the An. gambiae and Ae. aegypti genomes. One gene in the 3RC cluster of the sequenced PEST strain encodes a likely nonfunctional protein (Cornman et al., 2008), indicated by a truncated R&R Consensus and the presence of numerous cysteine residues that are otherwise rare in mature RR-2 proteins. A comparison with a sequenced genomic clone from the Sua strain of the same region shows that this pseudogene was created by a partial gene duplication event in PEST that resulted in no net change in functional gene number. Thus, this pseudogene is a gene fragment that does not derive from a formerly intact gene. No other gene in an An. gambiae RR-2 sequence cluster has been found to contain a frameshift or premature stop codon (i.e., prior to the end of the R&R Consensus), which is the strongest sequence-based evidence for a nonfunctional product in the event the gene is transcribed. (We note that other features such as the degeneration or apparent absence of a promoter are weak evidence of nonfunctionality. In fact, An. gambiae contains several CPR genes without evident TATA boxes that are nonetheless expressed [Cornman et al., 2008; Togawa et al., 2008].) Furthermore, all sequence-cluster genes for which unique primers could be designed have been shown by quantitative RT-PCR to be expressed in the G3 strain of An. gambiae (Togawa et al., 2008). Protein from all RR-2 sequence clusters has been identified in cuticle by shot-gun proteomics (He et al., 2007, He personal communication), although it is often not possible to distinguish products of individual genes within a sequence cluster due to the small number of diagnostic peptides. Within Ae. aegypti, we found two potential pseudogenes within sequence clusters as evidenced by premature stop codons and frameshifts (Supplementary File 2). If these unverified pseudogenes are not sequencing artifacts, they represent a small fraction (2 of 104) of the total Ae. aegypti genes that we identified occurring in sequence clusters. Of course, pseudogenes may be more difficult to identify than active genes. To minimize this bias, we performed extensive BLAST searches of the An. gambiae genome with full and partial R&R Consensus sequences in order to identify candidate genes or pseudogenes missed by gene-prediction software. No other pseudogenes were found.
The dearth of pseudogenes is not predicted under a birth-and-death model (Nei and Rooney 2005) without concomitantly strong selection for maintenance of all genes in a sequence cluster. This intuitive view is supported by our simple but biologically reasonable simulation (see Methods). The inferred ancestral sequences of the 2RA sequence cluster ‘evolved’ by simulation under neutral conditions acquired a nonsense mutation 83.4% of the time. This figure does not reflect the additional occurrences of radical amino-acid changes, such as to cysteine residues, nor were frameshift mutations allowed. We conclude that the virtual absence of pseudogenes within sequence clusters is incompatible with functional redundancy and implies natural selection in their maintenance. Of course, it remains possible but improbable that all genes in sequence clusters are selected independently for unique functions that remain to be identified. Furthermore, selection for high expression is also compatible with the birth-and-death model and could in principle prevent the fixation of pseudogenes in a lineage. However, the observed pattern of reciprocal monophyly implies high rates of ancestral gene loss under the birth-and-death model, which is difficult to reconcile with expression on selection.
3.3. Protein properties
Several investigations of CPR proteins have noted the prevalence of tandem amino-acid sequence repeats in the regions flanking the R&R Consensus; such sequence repeats are particularly common in An. gambiae sequence-cluster proteins. Dot-plots of representative sequences from each sequence cluster illustrate these repetitive regions (Supplementary File 8). These repeats do not have any consistent secondary structure among proteins of different sequence clusters based on the output of structure prediction programs (not shown). To quantify the level of sequence repetition in sequence clusters versus single-copy RR-2 proteins, we compared repetitive sequence scores computed with the HHrep tool (Söding et al., 2006). Because a single gene may have different repeats in the sequence N-terminal to the R&R Consensus versus the C-terminal sequence, we submitted each region separately to the HHrep server (signal peptide excluded). The results are summarized in Fig. 8. As a group, RR-2 proteins in sequence clusters have significantly higher HHrep scores than single copy RR-2 proteins (P < 0.01 by ANOVA of log-transformed data), although the two highest-scoring proteins were single-copy proteins. While these results provide a quantitative, whole-genome confirmation of previous qualitative descriptions of high amino-acid sequence repetition within arrays of similar RR-2 genes (Andersen et al., 1995; Dotson et al., 1998; Dombrovsky et al., 2003), there is no a priori expectation for why this should be. The implication is that genes that are amplified and retained at high copy number are under selection at least in part for properties relating to these repetitive amino-acid sequences.
Fig. 8.
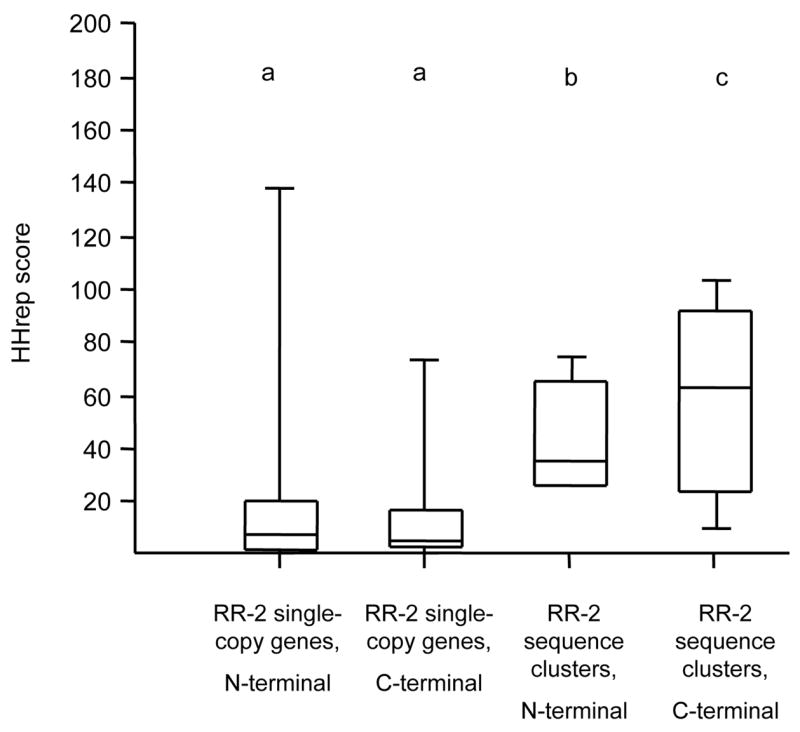
Box-plots of the distribution of scores representing the extent of repetitive amino-acid sequence in An. gambiae. Repetitive sequence scores were calculated with the HHrep tool (Söding et al., 2006). Each box represents the 25%–75% range, with the minimum, maximum, and median values represented by horizontal lines. Scores were calculated for single-copy and sequence-cluster RR-2 proteins separately (see text for details). The aminoacid sequence N-terminal and C-terminal to the Consensus was examined separately because different repeats are found within the two regions of the same protein. Letters indicate statistically significant differences in mean score (P < 0.01) by ANOVA of log-transformed data.
A hint as to the possible importance of this repetitive sequence comes from an examination of the amino-acid composition of An. gambiae sequence clusters. We performed a principal components analysis of the composition matrix of all RR-2 genes and have plotted all single-copy genes and sequence clusters along the first two principal component axes, which represent 63.6% of the total variation, in Fig. 9A. In all but one case, 3RA, sequence clusters fall along the edges of the distribution of points, indicating that these proteins are more extreme in amino-acid composition than single-copy genes, at least with respect to these two axes. The amino acids that contribute most to this variation are alanine, glycine, and histidine, as shown by the vectors in Fig. 9B. These amino acids are prominent components of repetitive sequence in RR-2 genes and, in principle. expansion of such repeats by unequal crossing over can cause pronounced biases in the amino-acid composition of individual sequence clusters. Importantly, the N-terminal and C-terminal flanking regions of the same sequence cluster typically have different repeats (Fig. 10A) but tend to have similar aminoacid compositions (Fig 10B). Figure 10B shows the degree to which the amino-acid composition in these two regions is correlated within RR-2 sequence clusters, RR-2 single-copy proteins, and RR-1 proteins. The mean correlation coefficient is significantly higher for sequence clusters than for single copy RR-2 genes, and both means are significantly greater than that of RR-1 genes (ANOVA of arcsine square-root transformed data, P < 0.01 for all comparisons). We infer from this that the observed composition biases are not merely a byproduct of repetitive sequence per se, because if they were, the similarity between the two flanking regions would not be any greater on average for the more repetitive sequence-cluster proteins than for the less repetitive single-copy proteins. Thus, the tendency for different repeats in the N-terminal and C-terminal flanking regions to have similar amino-acid compositions suggests that these amino-acid compositions are functionally important.
Fig. 9.

An. gambiae RR-2 proteins in sequence clusters (see text for details) have more extreme amino-acid compositions than do single-copy RR-2 proteins. (A) Scatterplot of An. gambiae RR-2 proteins based on their values for the first two principal components of the amino-acid composition matrix. Sequence clusters (see text for details) are marked according to the legend. The percentage of variation explained by each axis is also stated. (B) Vector representation of the contribution of the original amino acids to the distribution of points in the scatterplot, with four major amino acids labelled.
Fig. 10.

Correlated amino-acid compositions of N-terminal and C-terminal sequences flanking the R&R Consensus in RR-2 sequence-cluster proteins. The N- and C-terminal sequences flanking the R&R Consensus of sequence-cluster proteins are more similar to each other in amino-acid composition than are single-copy RR-2 or RR-1 proteins. (A) Examples of N-terminal and C-terminal sequence from a representative protein of each sequence cluster. All residues of the mature protein are shown that flank the extended R&R Consensus as defined in the Materials and Methods, rather than only the repetitive sequence identified by HHrep (Söding et al., 2006). (B) Box-plots of the correlation coefficients between the amino-acid composition of the regions N-terminal and C-terminal of the R&R Consensus within a given protein. Each box represents the 25%–75% range, with the minimum, maximum, and median values represented by horizontal lines. Three groups of proteins of An. gambiae were examined: RR-2 sequence-cluster genes (see text for details), RR-2 single-copy genes, and RR-1 genes. Letters indicate significant differences between the means (P < 0.01) by ANOVA of arcsine square-root transformed data.
The underlying mechanisms by which repetitive amino-acid sequence is generated include replication slippage and unequal crossing-over within genes. While both mechanisms may be important for CPR genes, striking examples of repeat expansion by unequal crossing are evident for individual CPR genes. We have identified a particularly telling case by comparing the protein sequences (Fig. 11A) and exon structures (Fig. 11B) of the D. melanogaster RR-1 gene Cpr47Ef (CG13214) and its orthologs in other Drosophila. This gene has two predicted transcripts, one of which (CG13214-RA) produces a protein with a long glycine-rich repetitive sequence C-terminal of the R&R Consensus. The other predicted transcript (CG13214-RB) terminates soon after the R&R Consensus and within an annotated intron of CG13214-RA and is probably an artefact. The glycine-rich repeats of CG13214-RB show a tendency to be more similar within species than they are among species. What is telling, however, is that although the orthologous proteins are similar in length, the D. melanogaster gene has 19 exons and the D. pseudoobscura gene has 15 exons but the orthologous genes in the other species have only three or four exons. For the two species that have many introns in the C-terminal repeat, all the introns and exons are very similar in size and sequence. These annotations (Flybase [http://www.flybase.org/] and Supplementary File 9) are supported by GenBank EST EL883355 from D. melanogaster, which is consistent only with CG13214-RA. The only plausible explanation for this rapid change in gene architecture is the propagation of an intron and exon by recurrent unequal crossing over, or else the propagation of an intron-loss event by the same mechanism. Note that this type of event must have occurred twice (Fig. 11B) based on the current phylogeny of these species (Drosophila 12 Genomes Consortium 2007).
Fig. 11.

Structure of the Drosophila CPR gene Cpr47Ef (CG13214) in seven species indicating the propagation of an exon-intron structure by unequal crossing over in the repetitive sequence C-terminal of the R&R Consensus. (A) Alignment of the conceptual translation of each gene from the R&R Consensus to the stop codon (the X at position 650). The R&R Consensus is marked. (B) Annotated exon structure of the complete gene in each species, although the first intron has not been identified in all cases. Exons are represented by gray boxes and introns by gaps; the schematic is not drawn to scale. The approximate location of the R&R Consensus is indicated by dark gray. The species phylogeny follows that of the Drosophila 12 Genomes Consortium (2007) but is given without branch lengths.
3.4. Culex pipiens genome further supports concerted evolution of RR-2 sequence clusters
Culex pipiens is the third mosquito species targeted for full genome sequencing. While the current genome assembly is not sufficient to fully incorporate C. pipiens into the analyses reported here, automated gene predictions are available that provided a means of assessing the generality of our observations. We obtained all predicted proteins from the Broad Institute annotation version CpipJ1.0_5 (http://cpipiens.vectorbase.org/GetData/Downloads) and identified proteins co-orthologous to all sequence clusters except the 2LB - 2LC supercluster by reciprocal Blast searches of whole protein sequence. We deemed the 2LB - 2LC supercluster to be too large and complicated to adequately investigate for this analysis. We then created three-mosquito phylogenies of ClustalW-aligned sequence (with default gap penalties for proteins) for each sequence cluster. We used similar methods as above but with full protein sequences; we also assumed rate homogeneity so as to avoid applying a gamma distribution to regions with frequent indels. The resulting phylogenies are shown in Supplementary File 10 (a–f). The addition of C. pipiens fully supports and strengthens our conclusions regarding evolutionary modes, as the phylogenetic pattern is strikingly similar to what was observed with An. gambiae and Ae. aegypti alone. For example, with the addition of C. pipiens sequences, the phylogeny of sequence cluster 3RA continues to show little clustering by species, whereas the 3RB and 3RC sequence clusters show very strong clustering by species (Supplementary File 10d vs. e,f). The sequence clusters 2LA and 2RA also show very strong clustering by species (Supplementary File 10a, b), but sequence cluster 2RB seems to have expanded only in the Anopheles lineage (Supplementary File 10c). All of these conclusions are the same as for the phylogeny of RR-2 genes in the two mosquito species in Figure 1.
4. Discussion
4.1. Overview
In this study, we have found that sets of RR-2 genes are amplified within the mosquito lineage relative to Drosophila and are undergoing extensive concerted evolution (tens of genes across all sequence clusters). We have concluded that most RR-2 sequence clusters evolve primarily in a concerted fashion rather than by a birth-and-death process based on multiple lines of evidence: 1) reduced synonymous and intron nucleotide polymorphism within sequence clusters without concomitant evidence of segmental duplication, 2) statistical evidence for intergenic recombination or conversion identified by the program RDP2 (Martin et al., 2005), 3) reciprocal monophyly of putatively co-orthologous sequence clusters in the two mosquito species, and 4) a near absence of pseudogenes. Although concerted evolution is known to occur among small subsets of CPR genes in Drosophila tandem arrays (Steinemann et al., 1996; Charles et al., 1997; Cornman unpublished data), the apparent scale of concerted evolution in mosquitoes is striking.
We have found that genes in sequence clusters have repeatedly acquired compact architectures relative to single-copy RR-2 genes, a pattern that suggests selection for transcriptional and translational efficiency. Furthermore, RR-2 sequence-cluster proteins as a group are more repetitive and have extreme amino-acid compositions relative to other CPR proteins in An. gambiae, and that the amino-acid composition of the N-terminal and C-terminal sequence flanking the chitin-binding consensus evolves in a correlated fashion. Our results suggest that sequence clusters derive from genes that have been preferentially amplified because they have acquired favorable characteristics within the rapidly evolving, low-complexity regions that flank the consensus.
4.2. Organization of sequence clusters requires a complex model of concerted evolution
While concerted evolution seems evident, the pattern of RR-2 gene-family evolution is difficult to reconcile with simple models of gene conversion (e.g., models without selection for homogeneity). For example, Teshima and Innan (2004), building on the work of Walsh (1987), have modeled concerted evolution as a by-product of local gene amplification. Under this model, tandem arrays of homologous sequence proceed through distinct phases that can be parameterized by level of sequence similarity. The length of the period of concerted evolution depends on chance loss of sequence similarity at the edges of homologous units, which gradually reduces the likelihood and extent of gene conversion events. Additionally, gross mutations that disrupt tandem arrays cause an abrupt cessation of concerted evolution. In contrast to this model, RR-2 sequence clusters are not characterized by an erosion of sequence similarity within the coding sequence, but generally experience rapid decline in flanking nucleotide sequence (Figure 7 and Supplementary File 7; an even more striking case was found for An. albimanus heat shock proteins by Benedict et al., [1996]). We also do not observe an escape from gene conversion caused by the accumulation of barriers to non-orthologous recombination within large tandem arrays of CPR genes. Instead, sequence clusters are frequently interrupted by changes in gene orientation and sometimes by other genes (Figs. 2–4). On the other hand, we did find that in some sequence clusters there is a single gene at the edge of a contiguous group that is notably more divergent (discussed in Methods), presumably due to reduced intergenic exchange. As stated previously, RR-1 genes do not show evidence of substantial concerted evolution despite the fact that many occur in similarly compact arrays and have comparable tracts of sequence homology (Cornman et al., 2008). Furthermore, there are three cases in which different RR-2 sequence clusters are closely adjacent within the same tandem array without any known gene functioning as a barrier. Finally, neither gene order nor gene orientation is correlated with genetic distance within clusters, which further suggests a complex pattern of gene conversion.
Thomas (2006) also found little correlation between proximity or orientation and sequence similarity within concertedly evolving sequence clusters of Caenorhabditis species, and suggested that inversion within tandem arrays help stabilize gene copy number by inhibiting unequal crossing over. Wang et al. (1999) observed similarly complicated patterns of gene conversion within an array of trypsin genes in D. melanogaster. On the other hand, Mondragon-Palomino and Gaut (2005) did find such correlations in a survey of disease-resistance genes in Arabidopsis thaliana. Interestingly, Drouin (2002b) found evidence of gene conversions occurring through cDNA intermediates, which obviates the need for chromatin-level interactions. As emphasized by Innan (2003), our understanding of the mechanisms of gene conversion is inadequate and we believe that the formulation and evaluation of models will be an ongoing challenge. In addition, statistical methods for generating null distributions for rates of gene duplication (e.g. Hahn et al., 2005) or synonymous divergence among duplicated genes (Innan, 2003; Teshima and Innan, 2004) may help clarify the relative importance of different evolutionary modes.
4.3. Possible functional significance of sequence clusters
Kondrashov et al. (2002) argued that the assumption of complete redundancy of duplicated genes in evolutionary models is probably not generally valid, especially for genes encoding secreted proteins that interact with the environment. Those authors instead posit that gene duplicates that achieve fixation generally do so because of natural selection acting on a duplicated-gene phenotype, presumably an increase in gene product. Such a view is consistent with the pattern of duplication, concerted evolution without evident pseudogene formation, and co-expression that we have identified within sequence clusters. It seems plausible that selection for high levels of gene expression during particular developmental stages or contributing to particular structures would both favor gene duplication and also eliminate pseudogenes and disruptive amino-acid substitutions that might otherwise be sheltered from purifying selection by gene redundancy. Indeed, Li (1997), expanding on Gojobori and Nei (1984), noted that higher levels of concerted evolution should be found for gene families that are under selection for rapid production of functionally equivalent transcripts and cited ribosomal RNA genes and histone gene families as examples. Ribosomal genes have long been known to evolve in a concerted fashion (Arnheim, 1983) whereas a major role of concerted evolution in the maintenance of histone gene similarity has been disputed by Nei and Rooney (2005). The compact gene architectures within RR-2 sequence clusters are interesting in this regard, because evidence of selection for shorter genes has been found with respect to both transcriptional efficiency (Castillo-Davis et al., 2002) and translational efficiency (Akashi, 2003). In comparison, the coding sequence and introns of orthologous single-copy genes are much longer (Fig. 5 and Table 2). On the other hand, the near maximal ENC’ of RR-2 genes in clusters does not suggest selection for transcription efficiency based on optimal codon usage, a common signature among highly expressed genes (Akashi, 2003). However, the response to selection from gene duplication is likely to be much greater than from codon optimization. One indication of translational efficiency was found; alignment of the Kozak consensus regions revealed that sequence cluster genes had a slightly better match to the Drosophila consensus (Cavener and Ray, 1991) than the other CPR genes (data not shown).
The properties of sequence-cluster proteins provide further hints as to the possible selective advantage of their amplification. Previous work (Cornman et al., 2008) showed that RR-2 proteins as a group have lower rates of evolution within the R&R Consensus but greater diversity of amino-acid composition than do RR-1 proteins in An. gambiae. Here we have found that in general, RR-2 sequence clusters have more extreme amino-acid compositions than do single-copy RR-2 genes and are significantly more repetitive. Furthermore, aminoacid compositions in the N-terminal and C-terminal region are more strongly correlated within sequence clusters than within single-copy genes despite the fact that the repeats differ between the two regions of the same gene. These findings suggest that the proportions of key amino acids are being selectively maintained and that the independent expansion of convergent amino-acid repeats is a response to such selection. A consequence of such repeats may also be an accelerated evolutionary rate, exemplified by the rapid gain or loss of introns in the Drosophila gene Cpr47Ef. Interestingly, Hayashi and Lewis (2000) identified an almost identical pattern of repeated exon-intron ‘modules’ within flagelliform silk genes of orb-weaving spiders and concluded that these repeats were concertedly evolving based on multi-species comparisons. Johannesson et al. (2005) also identified a process of repeat-homogenization (including intron sequence) by concerted evolution within a cell-surface protein gene of a human pathogenic fungus that they speculated was adaptive for evading host immune response. These studies support the hypothesis that the evolutionary dynamics of low-complexity regions within CPR proteins can drive the adaptive diversification of paralogs, such as is implied by the rapid divergence of these regions among paralogous CPR proteins but generally strong conservation of these sequences among orthologous CPR proteins, at least within mosquitoes (Cornman et al., 2008) and within Drosophila (Cornman unpublished data).
Supplementary Material
Supplementary file 1 - List of Ensembl Ae. aegypti genes used in or excluded from this study.
Supplementary file 2 - List of revised Ensembl Ae. aegypti genes used in this study.
Supplementary file 3 - List of Ae. aegypti genes not annotated by Ensembl used in this study.
Supplementary file 4 - Alignment of all sequence-cluster proteins we defined in Anopheles gambiae and the co-orthologous regions in Ae. aegypti.
Supplementary file 5 - Phylogeny of the 2RA sequence cluster in An. gambiae and co-orthologous sequences in Ae. aegypti, D. melanogaster, Apis mellifera, and Tribolium castaneum.
Supplementary file 6 - Table of An. gambiae CPR genes and putative orthologs in Ae. aegypti and D. melanogaster.
Supplementary file 7 - Representative dot plot of PEST genome sequence showing patterns of sequence similarity within sequence clusters. The dot plot is a self-alignment of a region on 3R that includes most of the genes in sequence cluster 3RB. Gene names are given above the horizontal axis. Intergenic regions show low sequence similarity relative to the genic regions. Figure created with the program Dotter (Sonnhammer and Durbin, 1995).
Supplementary file 8 - Dot plots of amino-acid sequence aligned with itself for a representative gene of each sequence cluster, which illustrate regions of sequence repetition. Figures created with BioEdit using a window size of one and with only identities shaded (Hall, 1999).
Supplementary file 9 - Coordinates of orthologs of the D. melanogaster gene Cpr47Ef from six Drosophila species.
Supplementary file 10 - Unrooted phylogenies of full protein sequence from mosquito sequence clusters, incorporating C. pipiens proteins predicted by the Broad Institute’s annotation version CpipJ1.0_5 (http://cpipiens.vectorbase.org/GetData/Downloads/). Sequences were aligned by ClustalW with default protein gap penalties. Neighbor-joining trees were created with MEGA3 (Kumar et al., 2004) using the JTT cost matrix (Jones et al., 1992) and pairwise deletion of gaps, with 1000 bootstrap replicates.
Acknowledgments
We thank Reed Cartwright, Jeffrey Ross-Ibarra, Russell Malmberg, Monica Poelchau, Toru Togawa and anonymous reviewers for helpful advice or comments on previous versions of the manuscript. This work was supported by a grant from the National Institutes of Health (AI55624) to JHW.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
R Scott Cornman, Email: scornman@uga.edu.
Judith H Willis, Email: jhwillis@cb.uga.edu.
References
- Akashi H. Translational selection and yeast proteome evolution. Genetics. 2003;164:1291–1303. doi: 10.1093/genetics/164.4.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen SO, Hojrup P, Roepstorff P. Insect cuticular proteins. Insect Biochem Mol Biol. 1995;25:153–176. doi: 10.1016/0965-1748(94)00052-j. [DOI] [PubMed] [Google Scholar]
- Arnheim N. Concerted evolution of multigene families. In: Nei M, Koehn RK, editors. Evolution of genes and proteins. Sinauer Associates; Sunderland, Mass.: 1983. pp. 38–61. [Google Scholar]
- Balding DJ, Nichols RA, Hunt DM. Detecting gene conversion: primate visual pigment genes. Proc R Soc B. 1992;249:275–280. doi: 10.1098/rspb.1992.0114. [DOI] [PubMed] [Google Scholar]
- Benedict MQ, Levine BJ, Ke ZX, Cockburn AF, Seawright JA. Precise limitation of concerted evolution to ORFs in mosquito Hsp82 genes. Insect Biochem Mol Biol. 1996;5:73–79. doi: 10.1111/j.1365-2583.1996.tb00042.x. [DOI] [PubMed] [Google Scholar]
- Castillo-Davis C, Mekhedov SL, Hartl DL, Koonin EV, Kondrashov FA. Selection for short introns in highly expressed genes. Nat Genet. 2002;31:415–418. doi: 10.1038/ng940. [DOI] [PubMed] [Google Scholar]
- Cavener DR, Ray SC. Eukaryotic start and stop translation sites. Nucleic Acids Res. 1991;19:3185–3192. doi: 10.1093/nar/19.12.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles JP, Chihara C, Nejad S, Riddiford LM. A cluster of cuticle protein genes of Drosophila melanogaster at 65A: sequence, structure and evolution. Genetics. 1997;147:1213–1224. doi: 10.1093/genetics/147.3.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornman RS, Togawa T, Dunn WA, He N, Emmons AC, Willis JH. Annotation and analysis of a large cuticular protein family with the R&R Consensus in Anopheles gambiae. BMC Genomics. 2008;9:22. doi: 10.1186/1471-2164-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrovsky A, Huet H, Zhang H, Chejanovksy N, Raccah B. Comparison of newly isolated cuticular protein genes from six aphid species. Insect Biochem Mol Biol. 2003;33:709–715. doi: 10.1016/s0965-1748(03)00065-1. [DOI] [PubMed] [Google Scholar]
- Dotson EM, Cornel AJ, Willis JH, Collins FH. A family of pupal-specific cuticular protein genes in the mosquito Anopheles gambiae. Insect Biochem Molec Biol. 1998;28:459–472. doi: 10.1016/s0965-1748(98)00016-2. [DOI] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Drouin G. Testing claims of gene conversion between multigene family members: examples from echinoderm actin genes. J Mol Evol. 2002a;54:138–139. doi: 10.1007/s00239-001-0026-9. [DOI] [PubMed] [Google Scholar]
- Drouin G. Characterization of the gene conversions between multigene family members of the yeast genome. J Mol Evol. 2002b;55:14–23. doi: 10.1007/s00239-001-0085-y. [DOI] [PubMed] [Google Scholar]
- Drouin G, Prat F, Ell M, Clarke GDP. Detecting and characterizing gene conversions between multigene family members. Mol Biol Evol. 1999;16:1369–1390. doi: 10.1093/oxfordjournals.molbev.a026047. [DOI] [PubMed] [Google Scholar]
- Ezawa K, Ota S, Saitou N. Genome-wide search of gene conversions in duplicated genes of mouse and rat. Mol Biol Evol. 2006;23:927–940. doi: 10.1093/molbev/msj093. [DOI] [PubMed] [Google Scholar]
- Fedorova L, Fedorov A. Introns in gene evolution. Genetica. 2003;118:123–131. [PubMed] [Google Scholar]
- Gojobori T, Nei M. Concerted evolution of the immunoglobin VH gene family. Mol Biol Evol. 1984;1:195–212. doi: 10.1093/oxfordjournals.molbev.a040311. [DOI] [PubMed] [Google Scholar]
- Hahn MW, De Bie T, Stajich JE, Nguyen C, Cristianini N. Estimating the tempo and mode of gene family evolution from comparative genomic data. Genome Res. 2005;15:1153–1160. doi: 10.1101/gr.3567505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Hammer Ø, Harper DAT, Ryan PD. PAST: paleontological statistics software package for education and data analysis. Palaeont Elect. 2001;4:1–9. [Google Scholar]
- Hayashi CY, Lewis RV. Molecular architecture and evolution of a modular spider silk protein gene. Science. 2000;287:1477–1479. doi: 10.1126/science.287.5457.1477. [DOI] [PubMed] [Google Scholar]
- He N, Botelho JMC, McNall RJ, Belozerov V, Dunn WA, Mize T, Orlando R, Willis JH. Proteomic analysis of cast cuticles from Anopheles gambiae by tandem mass spectrometry. Insect Biochem Mol Biol. 2007;37:135–146. doi: 10.1016/j.ibmb.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Innan H. The coalescent and infinite-site model of a small multigene family. Genetics. 2003;163:803–810. doi: 10.1093/genetics/163.2.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannesson H, Townsend JP, Hung CY, Cole GT, Taylor JW. Concerted evolution in the repeats of an immunomodulating cell surface protein, SOWgp, of the human pathogenic fungi Coccidioides immitis and C. posadasii. Genetics. 2005;171:109–117. doi: 10.1534/genetics.105.040923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Computer Applications in BioSciences. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- Karouzou MV, Spyropoulos Y, Iconomidou VA, Cornman RS, Hamodrakas SJ, Willis JH. Drosophila cuticular proteins with the R&R Consensus: annotation and classification with a new tool for discriminating RR-1 and RR-2 sequences. Insect Biochem Mol Biol. 2007;37:754–760. doi: 10.1016/j.ibmb.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Kondrashov FA, Rogozin IB, Wolf YI, Koonin E. Selection in the evolution of gene duplications. Genome Biol. 2002;3:RESARCH0008.1 –0008.9. doi: 10.1186/gb-2002-3-2-research0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV. Orthologs, paralogs, and evolutionary genomics. Annu Rev Genet. 2005;39:309–338. doi: 10.1146/annurev.genet.39.073003.114725. [DOI] [PubMed] [Google Scholar]
- Krzywinski J, Wilkerson RC, Besansky NJ. Toward understanding Anopheline (Diptera, Culicidae) phylogeny: insights from nuclear single-copy genes and the weight of evidence. Syst Biol. 2001;50:540–556. [PubMed] [Google Scholar]
- Kumar S, Tamura K, Nei M. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- Li W-H. Molecular Evolution. Sinauer Associates; Sunderland, Mass.: 1997. pp. 330–331. [Google Scholar]
- Magkrioti CK, Spyropoulos IC, Iconomidou VA, Willis JH, Hamodrakas SJ. cuticleDB: a relational database of arthropod cuticular proteins. BMC Bioinform. 2004;5:138–143. doi: 10.1186/1471-2105-5-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Williamson C, Posada D. RDP2: recombination detection and analysis from sequence alignments. Bioinformatics. 2005;21:260–262. doi: 10.1093/bioinformatics/bth490. [DOI] [PubMed] [Google Scholar]
- Mondragon-Palomino M, Gaut BS. Gene conversion and the evolution of three leucine-rich repeat gene families in A. thaliana. Mol Biol Evol. 2005;22:2444–2456. doi: 10.1093/molbev/msi241. [DOI] [PubMed] [Google Scholar]
- Negre B, Ruiz A. HOM-C evolution in Drosophila: is there a need for Hox gene clustering? Trends in Genetics. 2007;23:55–59. doi: 10.1016/j.tig.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Nei M, Rooney AP. Concerted and birth-and-death evolution of multigene families. Annu Rev Genet. 2005;39:121–152. doi: 10.1146/annurev.genet.39.073003.112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novembre JA. Accounting for background nucleotide composition when measuring codon usage bias. Mol Biol Evol. 2002;19:1390–1394. doi: 10.1093/oxfordjournals.molbev.a004201. [DOI] [PubMed] [Google Scholar]
- Ohta T. On the evolution of multigene families. Theoret Pop Biol. 1983;23:216–240. doi: 10.1016/0040-5809(83)90015-1. [DOI] [PubMed] [Google Scholar]
- Rebers JE, Riddiford LM. Structure and expression of a Manduca sexta larval cuticle gene homologous to Drosophila cuticle genes. J Mol Biol. 1998;203:411–423. doi: 10.1016/0022-2836(88)90009-5. [DOI] [PubMed] [Google Scholar]
- Rebers JE, Willis JH. A conserved domain in arthropod cuticular proteins binds chitin. Insect Biochem Mol Biol. 2001;31:1083–1093. doi: 10.1016/s0965-1748(01)00056-x. [DOI] [PubMed] [Google Scholar]
- Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Sawyer S. Statistical tests for detecting gene conversion. Mol Biol Evol. 1989;6:526–538. doi: 10.1093/oxfordjournals.molbev.a040567. [DOI] [PubMed] [Google Scholar]
- Semple C, Wolfe KH. Gene duplication and gene conversion in the Caenorhabditis elegans genome. J Mol Evol. 1999;48:555–564. doi: 10.1007/pl00006498. [DOI] [PubMed] [Google Scholar]
- Sharp PM, Li W-H. Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’ codons. Nuc Acids Res. 1986;14:7737–7749. doi: 10.1093/nar/14.19.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söding J, Remmert M, Biegert A. HHrep: de novo protein repeat detection and the origin of TIM barrels. Nucl Acid Res. 2006;34:W137–W142. doi: 10.1093/nar/gkl130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer ELL, Durbin R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene. 1995;167:1–10. doi: 10.1016/0378-1119(95)00714-8. [DOI] [PubMed] [Google Scholar]
- Steinemann M, Steinemann S, Pinsker W. Evolution of the larval cuticle proteins coded by the secondary sex chromosome pair: X2 and Neo-Y of Drosophila miranda: I. comparison at the DNA sequence level. J Mol Evol. 1996;43:405–412. [PubMed] [Google Scholar]
- Supek F, Vlahovicek K. INCA: synonymous codon usage analysis and clustering by means of self-organizing map. Bioinformatics. 2004;20:2329–2330. doi: 10.1093/bioinformatics/bth238. [DOI] [PubMed] [Google Scholar]
- Teshima KM, Innan H. The effect of gene conversion on the divergence between duplicated genes. Genetics. 2004;166:1553–1560. doi: 10.1534/genetics.166.3.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JH. Concerted evolution of two novel protein families in Caenorhabditis species. Genetics. 2006;172:2269–2281. doi: 10.1534/genetics.105.052746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togawa T, Nakato H, Izumi S. Analysis of the chitin recognition mechanism of cuticle proteins from the soft cuticle of the silkworm, Bombyx mori. Insect Biochem Mol Biol. 2004;34:1059–1067. doi: 10.1016/j.ibmb.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Togawa T, Dunn WA, Emmons AC, Nagao J, Willis JH. Developmental expression patterns of cuticular protein genes with R&R Consensus from Anopheles gambiae. Insect Biochem Mol Biol. 2008 doi: 10.1016/j.ibmb.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh JB. Sequence-dependent gene conversion: can duplicated genes diverge fast enough to escape conversion? Genetics. 1987;117:543–557. doi: 10.1093/genetics/117.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Magoulas C, Hickey D. Concerted evolution within a trypsin gene cluster in Drosophila. Mol Biol Evol. 1999;16:1117–1124. doi: 10.1093/oxfordjournals.molbev.a026202. [DOI] [PubMed] [Google Scholar]
- Wang X, Tang H, Bowers JE, Feltus FA, Paterson AH. Extensive concerted evolution of rice paralogs and the road to regaining independence. Genetics. 2007;177:1753–1763. doi: 10.1534/genetics.107.073197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis JH, Iconomidou VA, Smith RF, Hamodrakas SJ. Comprehensive Insect Science. In: Gilbert LI, Iatrou K, Gill S, editors. Cuticular proteins. Vol. 4. Elsevier; Oxford: 2005. pp. 79–109. [Google Scholar]
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Computer Applications in BioSciences. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file 1 - List of Ensembl Ae. aegypti genes used in or excluded from this study.
Supplementary file 2 - List of revised Ensembl Ae. aegypti genes used in this study.
Supplementary file 3 - List of Ae. aegypti genes not annotated by Ensembl used in this study.
Supplementary file 4 - Alignment of all sequence-cluster proteins we defined in Anopheles gambiae and the co-orthologous regions in Ae. aegypti.
Supplementary file 5 - Phylogeny of the 2RA sequence cluster in An. gambiae and co-orthologous sequences in Ae. aegypti, D. melanogaster, Apis mellifera, and Tribolium castaneum.
Supplementary file 6 - Table of An. gambiae CPR genes and putative orthologs in Ae. aegypti and D. melanogaster.
Supplementary file 7 - Representative dot plot of PEST genome sequence showing patterns of sequence similarity within sequence clusters. The dot plot is a self-alignment of a region on 3R that includes most of the genes in sequence cluster 3RB. Gene names are given above the horizontal axis. Intergenic regions show low sequence similarity relative to the genic regions. Figure created with the program Dotter (Sonnhammer and Durbin, 1995).
Supplementary file 8 - Dot plots of amino-acid sequence aligned with itself for a representative gene of each sequence cluster, which illustrate regions of sequence repetition. Figures created with BioEdit using a window size of one and with only identities shaded (Hall, 1999).
Supplementary file 9 - Coordinates of orthologs of the D. melanogaster gene Cpr47Ef from six Drosophila species.
Supplementary file 10 - Unrooted phylogenies of full protein sequence from mosquito sequence clusters, incorporating C. pipiens proteins predicted by the Broad Institute’s annotation version CpipJ1.0_5 (http://cpipiens.vectorbase.org/GetData/Downloads/). Sequences were aligned by ClustalW with default protein gap penalties. Neighbor-joining trees were created with MEGA3 (Kumar et al., 2004) using the JTT cost matrix (Jones et al., 1992) and pairwise deletion of gaps, with 1000 bootstrap replicates.