

Abstract
Objective:
To elucidate the genetic cause of a rare recessive ataxia presented by 2 siblings from a consanguineous Turkish family with a nonprogressive, congenital ataxia with mental retardation of unknown etiology.
Methods:
Whole-exome sequencing was combined with homozygosity mapping, linkage, and expression analysis to identify candidate genes, confirmed by Sanger sequencing. Reverse transcription–PCR and immunoblotting were used to determine the functional consequences of the gene variant. A zebrafish model was developed using morpholino-mediated knockdown.
Results:
We identified a homozygous mutation at the invariant +1 position (c.964+1G>A) in intron 9 of the CWF19L1 (complexed with cdc5 protein 19-like 1) gene. This mutation is absent in >6,500 European and African American individuals and 200 Turkish control DNAs. The mutation causes exon skipping, reduction in messenger RNA levels, and protein loss in cell lines of affected individuals. Morpholino-mediated knockdown in a zebrafish model demonstrates that loss of the evolutionarily highly conserved CWF19L1, whose normal biological function is unknown, alters cerebellar morphology and causes movement abnormalities.
Conclusions:
Our results suggest that CWF19L1 mutations may be a novel cause of recessive ataxia with developmental delay. Our research may help with diagnosis, especially in Turkey, identify causes of other ataxias, and may lead to novel therapies.
Autosomal recessive cerebellar ataxias are a clinically and genetically heterogeneous group of neurologic disorders characterized by deficiencies in the coordination of movements, most prominently the limbs, trunk, and eyes. While most forms of ataxia are individually rare, recessive ataxias are cumulatively not uncommon, with an estimated frequency of 1/20,000 that varies between countries.1,2 Most suspected recessive ataxia cases test negative for the 21 ataxia genes that are routinely included in clinical genetic testing,2 suggesting that most recessive ataxia genes are still unknown. Identifying additional recessive ataxia genes may help in diagnosis and prognosis and the identification of novel ataxia pathways,3 which in turn may lead subsequently to novel drug development.4,5
Next-generation sequencing has recently been used to identify genes involved in rare neurologic disorders,6 including ataxia,2,7–9 often with the help of consanguinity,2 as homozygosity further narrows down the linkage evidence,10 and homozygous mutations are easier to detect than 2 compound heterozygotes. Here, we identified a novel splice mutation by next-generation sequencing and homozygosity mapping in a small consanguineous family that leads to ataxia, developmental delay, and mental retardation in humans, and abnormalities in cerebellar morphology and movement in a zebrafish model with the same splicing defect.
METHODS
Standard protocol approvals, registrations, and patient consents.
Informed consent was obtained from participants, and the institutional review board of the University of Michigan Medical School approved this study. Heparin (green) blood from the affected individuals was separated by density centrifugation and transformed with Epstein-Barr virus.11 After growth initiation, aliquots were frozen and grown as needed.
Exome sequencing and homozygosity mapping.
Homozygosity mapping was performed by hybridizing DNA from both affected individuals to high-density Sentrix Human Hap 550 genotyping chips (Illumina, San Diego, CA). Linkage analysis was performed by hybridizing DNA from both affected individuals to Infinium HumanLinkage-12 genotyping chips (Illumina) and data were analyzed using Merlin. Note that these linkage chips are no longer being sold.
Exome capture was performed with the NimbleGen SeqCap EZ Exome Library v1.0 kit (Roche, Indianapolis, IN). The exon-enriched DNA from both affected individuals was sequenced with an Illumina HiSeq2000 instrument at the University of Michigan DNA Sequencing Core to an average depth of coverage of 20×. We filtered the exome data to variants that were (1) in the homozygosity regions, (2) homozygous in both individuals, and (3) predicted to change the protein sequence or expression.
PCR.
CWF19L1 sequences were obtained from the National Center for Biotechnology Information using NC_000010.11 for DNA and NM_018294 for RNA. DNA was extracted from EDTA (lavender) blood samples using the Puregene Blood Core Kit (Qiagen, Valencia, CA). RNA was extracted from lymphoblastoid cell lines (LCLs) using TRIzol reagent (Life Technologies, Grand Island, NY) according to the manufacturer's instructions. RNA was subjected to DNase I treatment (Ambion, Grand Island, NY) and reverse transcribed using the Invitrogen (now Life Technologies, Grand island, NY) SuperScript II reverse transcription kit using Oligo dTs and random hexamers.
Microarray and quantitative RT-PCR.
RNA was extracted from LCLs of the affected siblings and 28 affected with other ataxias and control individuals. Complementary RNA was prepared by standard methods and hybridized to Illumina human genome expression micro-array (RefSeq8). Data were analyzed using Illumina BeadStudio. Quantitative reverse transcription (qRT)-PCR was performed on an iQ5 cycler (BioRad, Hercules, CA). Assays were performed in 20-μL reaction mixtures, using a SYBR Green Master Kit, following the manufacturer's protocol. All measurements were done in triplicate. The threshold cycle value for each product was determined and normalized to that of the internal controls, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and β-actin. qRT-PCR results were analyzed using MyQ (BioRad).
Western blotting.
Protein was extracted from LCLs of affected and control individuals. Commercial tissue lysates for liver, stomach, cerebellum (left and right), spleen, heart, whole brain, frontal lobe, kidney, skeletal muscle, and lung were obtained from ProSci Incorporated (Poway, CA). Commercial normal tissue brain lysates blot (15 µg protein/lane; catalog no. 1526) and normal tissue lysates blot (15 µg protein/lane, catalog no. 1521) were obtained from ProSci Incorporated. C19L1 commercial antibody was obtained from Sigma (St. Louis, MO). β-Tubulin was used as a loading control (Abcam, Cambridge, MA).
Zebrafish experimentation.
Wild-type (AB strain) adult zebrafish were maintained in accordance with Institutional Animal Care and Use Committee–approved standards. Adult ABs were mated to generate embryos for all subsequent analyses. A morpholino was designed to the exon 9 splice acceptor and donor sites of the zebrafish cwf19l1 gene. All studies were done as a comparison between the cwf19l1 morpholino and a standardized control morpholino (Gene Tools, Inc., Philomath, OR). The sequence of the cwf19l1 exon 9 splice acceptor morpholino is TGCTGGTTCTTCCTGATCAAAGAGA, and the sequence of the cwf19l1 exon 9 splice donor morpholino is AGAGTGCATGTGAATGGACTCACGT. The standard control morpholino sequence is CCTCTTACCTCAGTTACAATTTATA. Morpholinos were injected into 1- to 2-cell stage embryos. Increasing concentrations were injected and screened by RT-PCR to determine efficacy regarding interrupting splicing. The minimal dose for effect was 0.15 mM for the splice acceptor and 0.45 mM for the splice donor and was used for all subsequent experimentation. Characterization of the morphant zebrafish involved behavioral analysis techniques that have been previously described.12,13
Whole-mount immunostaining.
Immunostaining was performed using the protocol of Bae et al.14 Anti-zebrin II primary antibody (1/200, hybridoma supernatant),15,16 Alexa Fluor 488 goat anti-mouse (1/1,000 dilution, Molecular Probes [Invitrogen, Grand Island, NY]), and DAPI (Thermo Fisher, Rockford, IL) were used.
For each test, 50 to 100 embryos were injected and scored by 2 investigators who were blinded to the injection cocktail. All experiments were performed 2 to 3 times, and statistical significance was calculated using χ2 test.
RESULTS
Exome sequencing identifies mutation in CWF19L1 in affected siblings.
The consanguineous family (parents are first cousins) has been previously described.17 Two siblings, but not their parents or their unaffected sibling, are affected with hypotonia, developmental delay, mental retardation, and nonprogressive truncal and extremity ataxia. MRI demonstrated hypoplasia in the vermis and cerebellar hemispheres.17 To identify candidate genes causing the ataxia–mental retardation syndrome in this family, we used whole-exome sequencing filtered by homozygosity. Homozygosity mapping identified 13 regions >500 kb, spanning 71 Mb, approximately 2% of the genome, as homozygous and shared between the siblings, reaching LOD (logarithm of the odds) scores of 1.0 to 1.8 (see figure 1A for chromosome 10). These regions contained 485 candidate genes, but no previously known ataxia genes.
Figure 1. Exome sequencing identifies mutation in CWF19L1.

(A) LOD scores on chromosome 10 from Merlin, using the Illumina Infinium HumanLinkage-12 Genotyping panel. (B) Flow chart variants obtained by exome sequencing data after each filtering step. (C) Sequence reads from whole-exome sequencing of one of the affected individuals indicating a homozygous C>T change. The sequence is in reverse orientation, thus there is a G>A change in a canonical splice donor site after exon 9. (D) Sanger chromatogram demonstrating mutation of the +1 site following exon 9 in affected individuals (arrowhead). Mother and father are heterozygous for the mutation (arrow). LOD = logarithm of the odds.
Exome sequencing variants were filtered by being on target (in or near exons), the homozygosity regions, and predicted damaging function (nonsense, missense, and splice variants; see figure 1B). A total of 4,969 variants were shared between the 2 affected individuals, 713 were homozygous, 28 of which mapped to the shared homozygosity regions, with 3 of these in gene regions, which were also absent in the EVS server (National Heart, Lung, and Blood Institute Exome Variant Server [NHLBI GO Exome Sequencing Project ESP], Seattle, WA [URL: http://evs.gs.washington.edu/EVS/; October 2011, similar results January 2014]). These 3 variants were predicted to be potentially damaging by POLYPHEN and SIFT18,19: 2 missense mutations in conserved amino acids and one a splice mutation (figure 1, C and D).
Of the 3 candidate gene mutations, 2 were unlikely to cause ataxia: a missense mutation in CDC73, as other mutations in this gene cause hyperparathyroidism, jaw syndromes, and cancer,20,21 and a missense mutation in GSTO1, as mutations in GSTO1 in humans and loss of GSTO1 in mice and drosophila does not cause any discernible phenotype.22–24 The third mutation was a mutation in CWF19L1 in an obligatory splice site (c.964+1G>A, i.e., the first base of intron 9, always G, is changed to an A; figure 1D). Sanger sequencing confirmed this mutation in the affected siblings and demonstrated that it is absent in 200 Turkish individuals without neurologic disorder. The parents are each heterozygous for this mutation, and the unaffected sibling is homozygous for the reference allele (figure 1D). We also tested 64 other subjects with unexplained ataxia and detected no damaging homozygous or compound heterozygous mutations in any of the exons or flanking intronic regions of CWF19L1.
Splice mutation causes exon skipping, decreased messenger RNA levels, and loss of protein.
The identification of a splice donor site mutation after exon 9 is predicted to have deleterious impact on messenger RNA (mRNA) stability and translation. To test whether the mutation affects gene expression, we extracted RNA from LCLs of the affected siblings and 28 other individuals, prepared complementary RNAs, and hybridized them to Illumina human genome expression micro-arrays (RefSeq8). The level of expression of CWF19L1 in LCLs from the 2 affected children was 2- to 8-fold less when compared with 28 other individuals (ranked within the 10 largest expression changes for these 2 affected individuals) while expression of GSTO1 and CDC73 was unchanged (not shown). To further quantify the expression changes, we performed qRT-PCR using primers designed to CWF19L1. Normalized against GAPDH and ACTIN-β, mRNA from the affected individuals is approximately 6-fold reduced compared with that of control (n = 3) individuals (data not shown).
RT-PCR of RNA from LCLs derived from the 2 affected siblings and control individuals, using primers in exon 7 and exon 11, showed the expected 421-bp product in control individuals, but a shorter, 302-bp, fragment in affected individuals (figure 2A). Sanger sequencing confirmed that the shorter complementary DNA misses all 119 bases of exon 9 (schematic figure 2B). Skipping this exon is predicted to lead to an out-of-frame stop codon after 60 aberrant amino acids. Hence, nonsense-mediated decay may explain the reduction in mRNA level seen in the LCLs.
Figure 2. RT-PCR and sequencing demonstrate excision of exon 9.

(A) RT-PCR from lymphoblastoid cell–derived RNA using primers spanning exons 7 to 11 of the CWF19L1 gene. PCR product from cDNA from control individuals (lanes 2–4) amplifies a product in the expected range (421 bp), while cDNA from affected individuals (lanes 5–6) amplifies a product of approximately 119 bp, shorter by the size of exon 9 (302 bp). Negative control shown in lane 1. (B) Schematic of exons 7 to 11 of CWF19L1. Control individuals show normal processing of these exons, while affected individuals show abnormal mRNA products that splice out exon 9. Sanger sequencing of products in panel A confirmed this model (data not shown). cDNA = complementary DNA; gDNA = genomic DNA; RT = reverse transcription.
Western blot analysis of C19L1, the protein encoded by CWF19L1, using protein extracts from LCLs, shows 2 protein bands of approximate molecular weight of 61 and 46 kDa in control individuals, both of which are missing in the patients (figure 3A). There are 3 predicted protein isoforms from the various splice variants of CWF19L1. Because the antiserum epitope spans exons 5 to 7, we expected to detect 2 of the 3 predicted isoforms (schematic figure 3B). The absence of protein bands in the patients demonstrates specificity of this antiserum. In affected individuals, there was no evidence for presence of normal or the predicted truncated (344 amino acid) protein, even when overloading or overexposing the blot (data not shown).
Figure 3. Immunoblot shows distribution of C19L1 in brain and protein deficiency in LCLs of affected individuals.

(A) Western blot using antiserum against the N-terminus of C19L1 (the protein product of CWF19L1) detects 2 protein bands in control individuals. No protein bands are detected in patients (42 and 43). Thirty micrograms of protein was loaded; β-tubulin was used as loading control. Arrow indicates the size of a predicted truncated protein product, if it was made. (B) Schematic of predicted (from complementary DNA sequences) C19L1 protein isoforms. The C19L1 epitope against which the antiserum was raised is indicated by a gray box. (C) Western blot using antiserum against N-terminus of C19L1 detects C19L1 in most brain regions including cerebellum. GAPDH = glyceraldehyde 3-phosphate dehydrogenase; LCL = lymphoblastoid cell line.
Western blot of C19L1, using commercial tissue lysates, detected expression of C19L1 throughout many brain regions, including the cerebellum (figure 3C, and figure e-1 on the Neurology® Web site at Neurology.org). Immunoblot revealed only the canonical protein band in brain tissues, suggesting that this is the isoform present in the brain, although we cannot exclude undetectable levels of the other predicted isoforms of C19L1.
Morpholino-mediated knockdown of cwf19l1 demonstrates abnormal motor behavior and cerebellar development in zebrafish.
Although these results clearly demonstrate a detrimental mutation in CWF19L1 leading to absence of the protein in a linked and homozygous region, it is not sufficient. Private null mutations exist in approximately 20% of humans,25 and a function of CWF19L1 in ataxia is not immediately apparent. Therefore, we could not exclude that this could be a rare Turkish variant without pathologic implications. To test the effect of this mutation in vivo, we used a morpholino knockdown strategy to the homologous intron-exon junction in a zebrafish model. The zebrafish cwf19l1 gene has 14 exons, shares 65% protein identity with the human CWF19L1 gene,26,27 and has the same predicted intron-exon structure. The cwf19l1 gene is expressed ubiquitously in zebrafish, with higher expression in blood and head.28 We designed 2 different morpholinos to the exon 9 splice acceptor and donor site (cwf19l1 I8E9 and E9I9 MOs) to target splicing out of exon 9, as in the affected individuals (schematic figure 4A). MOs were injected into 1- to 2-cell stage embryos and RNA was extracted at 3 days postfertilization. RT-PCR, using primers to exon 6 and exon 10, revealed the expected band of approximately 450 bp in fish injected with a standard control morpholino (Gene Tools). However, RT-PCR from morphant fish injected with the cwf19l1 MOs demonstrated knockdown of the RT-PCR product with increasing morpholino concentration (data not shown). In addition, Western blots performed with a C19L1 antiserum (Novus Biologicals, Littleton, CO) demonstrated knockdown and loss of the c19l1 protein with increasing MO concentration (figure 4B), indicating deficiency of the protein in morphant fish.
Figure 4. Morpholino-mediated knockdown shows protein deficiency and abnormal motor behavior in morphant fish.
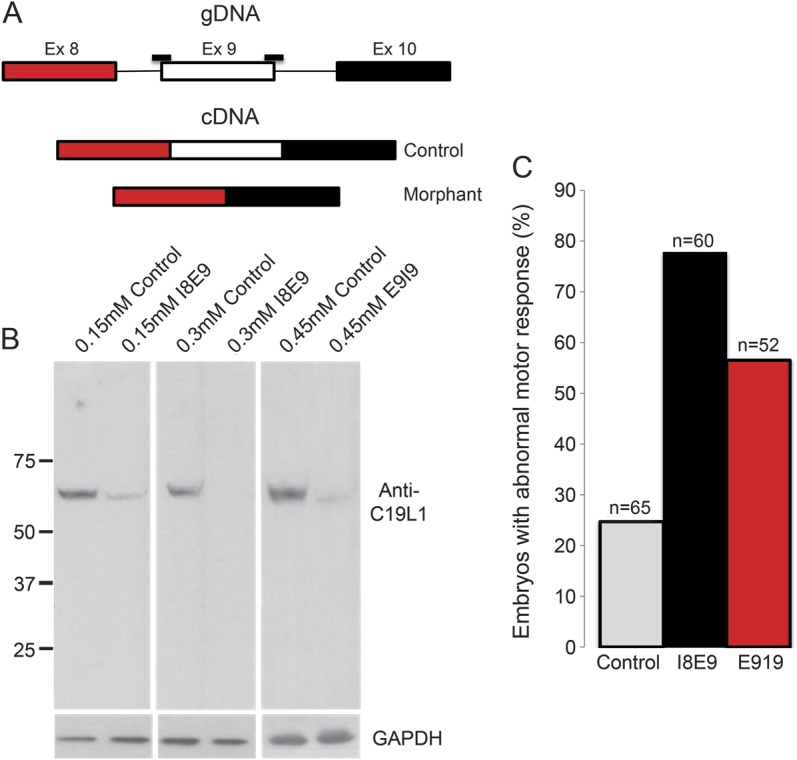
(A) Schematic of morpholino (MO) target sites (black bars). (B) Western blot with antiserum that recognizes 3′ end of c19l1 protein demonstrates knockdown and loss of protein in morphant fish with increasing MO dose. (C) cwf19l1 morphants (I8E9 and E9I9 MOs) had abnormal motor behavior. The touch-evoked escape response of 0.15 mM I8E9 and 0.45 mM E9I9 fish are shown in the graph (see also videos 1–4) (n = 5 independently injected clutches with a minimum of 10 embryos assessed per clutch). cDNA = complementary DNA; GAPDH = glyceraldehyde 3-phosphate dehydrogenase; gDNA = genomic DNA.
Behavioral analysis of cwf19l1 morphant embryos (as compared with control morphants) revealed reduced swim speed and abnormal touch-evoked escape response, a stereotyped behavior that is prominent at 3 days postfertilization (see figure 4C and videos 1–4). As this response was elicited with both cwf19l1 morpholinos, this suggests that knockdown of cwf19l1 causes the abnormal motor behavior in these fish. Because the E9I9 MO required a higher dose to show morpholino effect, touch-evoked escape response was only quantified for comparison of 0.15 mM I8E9 and 0.45 mM E9I9 morphant fish. Doses above 0.5 mM are associated with an increase in mortality rate in control MO-injected fish and potential off-target effects of the individual morpholinos,29,30 therefore doses above 0.5 mM were not used for the E9I9 morpholino.
Because the affected individuals showed cerebellar hypoplasia, we tested whether knockdown of cwf19l1 in morphant fish altered cerebellar morphology. Immunostaining with the zebrin II monoclonal antibody, which labels aldolase C-positive Purkinje cells,15,16 demonstrated altered/diminished cerebellar staining in I8E9 morphants that worsened with increasing MO concentration (figure 5, A–D), suggestive of a defect in cerebellar structure. E9I9 morphant fish had similar results (not shown). These results corroborate that c19l1 protein deficiency is associated with a neurologic movement disorder and with cerebellar abnormalities.
Figure 5. Zebrin II staining shows abnormal staining in cwf19l1 morphant fish.
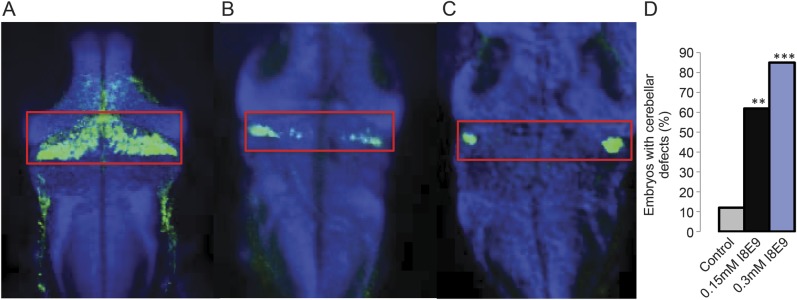
Zebrin II staining in 5 days postfertilization (A) control morpholino (MO) fish, (B) 0.15 mM I8E9 MO fish, and (C) 0.3 mM I8E9 MO fish demonstrating gradually diminished staining in cerebellum with increasing MO dose (D) quantification (n = 20 fish per condition, ** p < 0.01, *** p < 0.001).
DISCUSSION
Using a combination of homozygosity mapping and next-generation sequencing, we have identified a novel gene associated with a rare, recessive ataxia–mental retardation syndrome. It is estimated that 40% of recessive ataxias still have unknown etiology,2 therefore, discovery of this gene may lead to the identification of novel pathways in ataxia or may uncover a role for this gene in current ataxia pathways.
The identification of a mutation in CWF19L1 in siblings with congenital ataxia, confirmed by morpholino knockdown in zebrafish leading to cerebellar and movement abnormalities, suggests that this gene has a role in neuronal development. CWF19L1 encodes several protein isoforms. Our data suggest that the mutation leads to a null allele, because no protein could be detected even by overexposure. However, we can not differentiate between a severe hypomorph and a null allele, because we can not exclude the possibility that some normal splicing persists, and a small amount of normal protein below our level of detection limit may still be made. In addition, immunoblot of human brain tissue lysates demonstrated expression of C19L1 throughout the brain, suggesting it is important for proper brain function.
The function of the CWF19L1-encoded C19L1 proteins is unknown. Based on in vitro interaction experiments, it was postulated to have a role in cell-cycle control31,32 or in endosomal trafficking.33,34 While one yeast ortholog, cwf19, was identified as having a role in mRNA processing, the role for CWF19L1 may be different in humans, because CWF19L1 has been shown in a large human yeast 2-hybrid screen to interact with TOM1L1, a Src-activating and signaling molecule, which is not present in yeast. Preliminary data in our laboratory show nuclear localization of the C19L1 protein. In addition, the C19L1 protein has a metallophosphatase domain, which is found in RNA lariat debranching enzymes, and in proteins involved in double-strand break repair, including MRE11.35–37 Taken together, these results suggest a role in mRNA processing; however, because the in vitro interactions have not been experimentally verified, further studies will be needed to determine the role of the C19L1 protein in brain development and function.
While the function of this gene is unknown, its conservation from yeast to humans indicates its evolutionary importance. One strategy to determine the functional importance of a gene in an organism is to decrease or abolish expression of the gene to determine the effects of reduction or loss of the gene product. Zebrafish animal models have recently been used to study mutations in ataxia genes.38–40 In our study, knockdown of cwf19l1 in fish caused abnormal motor behavior and alteration of cerebellar structure. These data represent functional validation of the whole-exome sequencing results, demonstrating that loss of cwf19l1 is associated with an abnormal motor phenotype. These similarities in phenotype between the affected subjects with CWF19L1 mutations and the zebrafish with knockdown of cwf19l1 could, however, be coincidental. Additional families with the same mutation and phenotype, which may be found in Turkey due to a possible founder mutation or ataxia families with other mutations in CWF19L1, will be needed to confirm our results, and are required before CWF19L1 can be used for clinical diagnosis of ataxia and prognosis.
There are currently no therapies for ataxia; therefore, determining new pathways or targets for therapy are important. Recently, mouse models for spinocerebellar ataxia 1 and Friedreich ataxia have led to potential drug targets and clinical trials.4,5 In the future, this zebrafish model may also be used to understand the function of C19L1, analyze and confirm cell experiments, and ultimately test potential therapies for this form of ataxia.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the family participating in this study, Elzbieta Sliwerska, Angela Busta, and Linda Gates for technical assistance, and Richard Hawkes, University of Calgary, Canada, for the anti-zebrin II antiserum. The authors also acknowledge the University of Michigan DNA Sequencing Core (Director: Robert Lyons) for next-generation sequencing, Dr. Miriam Meisler, University of Michigan, and Dr. Mustafa Cengiz Yakicier (ACIBADEM University, Turkey, Department of Medical Biology and Genetic Diagnostic Center) for congenital ataxia and Turkish control DNA samples, respectively.
GLOSSARY
- CWF19L1
complexed with cdc5 protein 19-like 1
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- LCL
lymphoblastoid cell line
- MO
morpholino oligonucleotide
- mRNA
messenger RNA
- qRT
quantitative reverse transcription
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
R. Burns: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, statistical analysis. K. Majczenko: acquisition or interpretation of data. J. Xu: data analysis. W. Peng: interpretation of data. J.J. Dowling: contribution of vital reagents/tools, analysis or interpretation of data. Z. Yapici: contribution of vital clinical materials. J.Z. Li: study concept or design, analysis or interpretation of data, study supervision, and obtaining funding. M. Burmeister: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, study supervision, and obtaining funding.
STUDY FUNDING
This work was supported by NIH, National Institute on Neurological Disorders grants R21-DC010074, R01-NS078560, and NS078560S1 to M.B. R.B. was supported by NIH T-32-GM007315.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Finsterer J. Ataxias with autosomal, X-chromosomal or maternal inheritance. Can J Neurol Sci 2009;36:409–428. [DOI] [PubMed] [Google Scholar]
- 2.Sailer A, Houlden H. Recent advances in the genetics of cerebellar ataxias. Curr Neurol Neurosci Rep 2012;12:227–236. [DOI] [PubMed] [Google Scholar]
- 3.Guan Y, Gorenshteyn D, Burmeister M, et al. Tissue-specific functional networks for prioritizing phenotype and disease genes. PLoS Comput Biol 2012;8:e1002694. 10.1371/journal.pcbi.1002694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watase K, Gatchel JR, Sun Y, et al. Lithium therapy improves neurological function and hippocampal dendritic arborization in a spinocerebellar ataxia type 1 mouse model. PLoS Med 2007;4:e182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rai M, Soragni E, Jenssen K, et al. HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS One 2008;3:e1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foo JN, Liu JJ, Tan EK. Whole-genome and whole-exome sequencing in neurological diseases. Nat Rev Neurol 2012;8:508–517. [DOI] [PubMed] [Google Scholar]
- 7.Doi H, Yoshida K, Yasuda T, et al. Exome sequencing reveals a homozygous SYT14 mutation in adult-onset, autosomal-recessive spinocerebellar ataxia with psychomotor retardation. Am J Hum Genet 2011;89:320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li M, Pang SYY, Song Y, Kung MHW, Ho SL, Sham PC. Whole exome sequencing identifies a novel mutation in the transglutaminase 6 gene for spinocerebellar ataxia in a Chinese family. Clin Genet 2013;83:269–273. [DOI] [PubMed] [Google Scholar]
- 9.Lee YC, Durr A, Majczenko K, et al. Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann Neurol 2012;72:859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lander ES, Botstein D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science 1987;236:1567–1570. [DOI] [PubMed] [Google Scholar]
- 11.Doyle A. Establishment of lymphoblastoid cell lines. In: Pollard J, Walker J, editors. Animal Cell Culture. Clifton, NJ: Humana Press; 1990:43–47. [Google Scholar]
- 12.Telfer WR, Busta AS, Bonnemann CG, Feldman EL, Dowling JJ. Zebrafish models of collagen VI-related myopathies. Hum Mol Genet 2010;19:2433–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dowling JJ, Low SE, Busta AS, Feldman EL. Zebrafish MTMR14 is required for excitation-contraction coupling, developmental motor function and the regulation of autophagy. Hum Mol Genet 2010;19:2668–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bae YK, Kani S, Shimizu T, et al. Anatomy of zebrafish cerebellum and screen for mutations affecting its development. Dev Biol 2009;330:406–426. [DOI] [PubMed] [Google Scholar]
- 15.Lannoo MJ, Brochu G, Maler L, Hawkes R. Zebrin II immunoreactivity in the rat and in the weakly electric teleost Eigenmannia (gymnotiformes) reveals three modes of Purkinje cell development. J Comp Neurol 1991;310:215–233. [DOI] [PubMed] [Google Scholar]
- 16.Lannoo MJ, Ross L, Maler L, Hawkes R. Development of the cerebellum and its extracerebellar Purkinje cell projection in teleost fishes as determined by zebrin II immunocytochemistry. Prog Neurobiol 1991;37:329–363. [DOI] [PubMed] [Google Scholar]
- 17.Yapici Z, Eraksoy M. Non-progressive congenital ataxia with cerebellar hypoplasia in three families. Acta Paediatr 2005;94:248–253. [DOI] [PubMed] [Google Scholar]
- 18.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 2003;31:3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flanagan SE, Patch AM, Ellard S. Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genet Test Mol Biomarkers 2010;14:533–537. [DOI] [PubMed] [Google Scholar]
- 20.Frank-Raue K, Haag C, Schulze E, et al. CDC73-related hereditary hyperparathyroidism: five new mutations and the clinical spectrum. Eur J Endocrinol 2011;165:477–483. [DOI] [PubMed] [Google Scholar]
- 21.Enomoto K, Uchino S, Ito A, et al. The surgical strategy and the molecular analysis of patients with parathyroid cancer. World J Surg 2010;34:2604–2610. [DOI] [PubMed] [Google Scholar]
- 22.Chowdhury UK, Zakharyan RA, Hernandez A, Avram MD, Kopplin MJ, Aposhian HV. Glutathione-S-transferase-omega [MMA(V) reductase] knockout mice: enzyme and arsenic species concentrations in tissues after arsenate administration. Toxicol Appl Pharmacol 2006;216:446–457. [DOI] [PubMed] [Google Scholar]
- 23.Hsu LI, Chen WP, Yang TY, et al. Genetic polymorphisms in glutathione S-transferase (GST) superfamily and risk of arsenic-induced urothelial carcinoma in residents of southwestern Taiwan. J Biomed Sci 2011;18:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim K, Kim SH, Kim J, Kim H, Yim J. Glutathione S-transferase omega 1 activity is sufficient to suppress neurodegeneration in a Drosophila model of Parkinson disease. J Biol Chem 2012;287:6628–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacArthur DG, Balasubramanian S, Frankish A, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 2012;335:823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujita PA, Rhead B, Zweig AS, et al. The UCSC genome browser database: update 2011. Nucleic Acids Res 2011;39(suppl 1):D876–D882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramachandran S, Ruef B, Pich C, Sprague J. Exploring zebrafish genomic, functional and phenotypic data using ZFIN. Curr Protoc Bioinformatics [Internet] 2002. Available at: http://dx.doi.org/10.1002/0471250953.bi0118s31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thisse B, Heyer V, Lux A, et al. Spatial and temporal expression of the zebrafish genome by large-scale in situ hybridization screening. Methods Cell Biol 2004;77:505–519. [DOI] [PubMed] [Google Scholar]
- 29.Bill BR, Petzold AM, Clark KJ, Schimmenti LA, Ekker SC. A primer for morpholino use in zebrafish. Zebrafish 2009;6:69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eisen JS, Smith JC. Controlling morpholino experiments: don't stop making antisense. Development 2008;135:1735–1743. [DOI] [PubMed] [Google Scholar]
- 31.Ohi MD, Link AJ, Ren L, Jennings JL, McDonald WH, Gould KL. Proteomics analysis reveals stable multiprotein complexes in both fission and budding yeasts containing Myb-related Cdc5p/Cef1p, novel pre-mRNA splicing factors, and snRNAs. Mol Cell Biol 2002;22:2011–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ren L, McLean JR, Hazbun TR, et al. Systematic two-hybrid and comparative proteomic analyses reveal novel yeast pre-mRNA splicing factors connected to Prp19. PLoS One 2011;6:e16719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stelzl U, Worm U, Lalowski M, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell 2005;122:957–968. [DOI] [PubMed] [Google Scholar]
- 34.Wang T, Liu NS, Seet LF, Hong W. The emerging role of VHS domain-containing Tom1, Tom1L1 and Tom1L2 in membrane trafficking. Traffic 2010;11:1119–1128. [DOI] [PubMed] [Google Scholar]
- 35.NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 2014;42:D7–D17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Regal JA, Festerling TA, Buis JM, Ferguson DO. Disease-associated MRE11 mutants impact ATM/ATR DNA damage signaling by distinct mechanisms. Hum Mol Genet 2013;22:5146–5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyamoto R, Morino H, Yoshizawa A, et al. Exome sequencing reveals a novel MRE11 mutation in a patient with progressive myoclonic ataxia. J Neurol Sci 2014;337:219–223. [DOI] [PubMed] [Google Scholar]
- 38.Mahmood F, Mozere M, Zdebik AA, et al. Generation and validation of a zebrafish model of EAST (epilepsy, ataxia, sensorineural deafness and tubulopathy) syndrome. Dis Model Mech 2013;6:652–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aspatwar A, Tolvanen MEE, Jokitalo E, et al. Abnormal cerebellar development and ataxia in CARP VIII morphant zebrafish. Hum Mol Genet 2013;22:417–432. [DOI] [PubMed] [Google Scholar]
- 40.Yanicostas C, Barbieri E, Hibi M, Brice A, Stevanin G, Soussi-Yanicostas N. Requirement for zebrafish ataxin-7 in differentiation of photoreceptors and cerebellar neurons. PLoS One 2012;7:e50705. 10.1371/journal.pone.0050705. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.