

Abstract
The pharmacokinetics of non-renally cleared drugs in patients with chronic kidney disease is often unpredictable. Some of this variability may be due to alterations in the expression and activity of extra-renal drug metabolizing enzymes and transporters, primarily localized in the liver and intestine. Studies conducted in rodent models of renal failure have shown decreased mRNA and protein expression of many members of the cytochrome P450 enzyme (CYP) gene family and the ATP-Binding Cassette (ABC) and Solute Carrier (SLC) gene families of drug transporters. Uremic toxins interfere with transcriptional activation, cause down-regulation of gene expression mediated by proinflammatory cytokines, and directly inhibit the activity of the cytochrome P450s and drug transporters. While much has been learned about the effects of kidney disease on non-renal drug disposition, important questions remain regarding the mechanisms of these effects, as well as the interplay between drug metabolizing enzymes and drug transporters in the uremic milieu. In this review, we have highlighted the existing gaps in our knowledge and understanding of the impact of chronic kidney disease on non-renal drug clearance, and identified areas of opportunity for future research.
Introduction
Chronic kidney disease (CKD) is a public health problem that affects more than 20 million people in the US.1 Currently, almost 500,000 patients require chronic hemodialysis.2 An average dialysis patient may require more than 12 medications.3 A pooled analysis identified 1,593 medication-related problems in 385 dialysis patients, with over- or under- dosing errors accounting for 20.4% of these issues.4 Despite the large number of patients affected and the devastating consequences of medication related problems, our understanding of the impact of kidney disease on drug disposition is incomplete, particularly for those drugs eliminated primarily by non-renal pathways. Obviously, clearance of drugs that depend primarily on the kidneys for elimination is reduced, but significant changes also occur in drug exposure with medications that are eliminated by the liver, intestine, and possibly other organs.
In 2009, the FDA published a survey of New Drug Applications (NDA) approved between January 2003 and July 2007 that assessed the impact of renal impairment on systemic exposure of new molecular entities.5 In this analysis, NDA sponsors for 37 orally administered drugs included renal impairment studies as part of their submission; 23 (62%) of these were eliminated by non-renal pathways (defined as fraction eliminated via renal route <15). Despite being cleared non-renally, 13 of these 23 new drugs (57%) showed an average 1.5-fold increase in area under the plasma concentration-time curve (AUC) in renally impaired patients compared with health controls. In fact, the change in drug exposure for five drugs cleared mainly by hepatic metabolism and/or transport were of a magnitude (viz. duloxetine ΔAUC +2.0-fold, tadalafil ΔAUC +2.7- to 4.1-fold, rosuvastatin ΔCplasma +3-fold, telithromycin ΔAUC +1.9-fold, solifenacin ΔAUC +2.1-fold) that required labeling recommendations for dose adjustment in renally impaired patients. Seven other drugs showed an effect of renal impairment on drug exposure but did not require dosage adjustment (aliskiren, alfuzosin, aprepitant, ranolazine, vardenafil, darifenacin, and lanthanum). These data along with a large body of earlier literature suggest that CKD alters the pharmacokinetics of drugs that are cleared by non-renal mechanisms; however, the underlying molecular mechanisms accounting for these pharmacokinetic changes remain poorly defined (reviewed by Nolin, LeBlond and others) 6-9. The purpose of the present mini-review is to highlight the present gaps in our understanding of the impact of CKD on non-renal drug clearance involving metabolism and transport processes and to identify areas of opportunity for future research.
Drug Metabolism and Transport Processes
The following is a brief introduction to the key drug-metabolizing enzymes and drug transporters whose function is known to be altered in CKD. Phase I drug metabolism, involving oxidation, reduction, and hydrolysis, generally converts drug molecules to more polar or water soluble metabolites that are readily excreted by the kidneys or via the biliary system. Drug oxidation, which is particularly known to be altered in CKD, is catalyzed by two large families of enzymes, namely the cytochrome P450 (CYPs) and flavin-containing monoxygenases (FMOs).10 Many of the CYPs exhibit genetic polymorphisms which range from gene duplication resulting in gene overexpression to null mutations producing a non-functional enzyme. The recent focus of CYP research is on enzymes expressed in the liver and the intestinal mucosa, which govern the oral bioavailability (i.e., first-pass metabolism) and systemic metabolic clearance of drug molecules. Human hepatic cytochrome P450s include CYP3A4 and 3A5 (40% of total liver P450 content), CYP2Cs (25%), CYP1A2 (18%), CYP2E1 (9%), CYP2A6 (2%), CYP2D6 (2%) and CYP2B6 (<1%), as well as FMO3. Human intestinal CYPs that are functionally important include CYP1A1, CYP3A4, CYP3A5, and CYP2J2.11 CYP1A1, CYP1A2, CYP3A5, CYP4A1, and FMO1 are also expressed in human kidneys, but at levels much lower than in the liver and intestine.11, 12 Many drugs or their Phase I metabolites also undergo conjugation reactions mediated by Phase II enzymes.10 In particular, N-acetylation and O-glucuronidation of drugs or drug metabolites are known to be altered in CKD13. The liver and intestinal mucosa are the major sites for the biotransformation of drugs and drug metabolites by Phase II enzymes (Figure 1). It should be noted that the products of Phase I and Phase II metabolism are not always pharmacologically inactive or less toxic than the parent drug.
Figure 1.
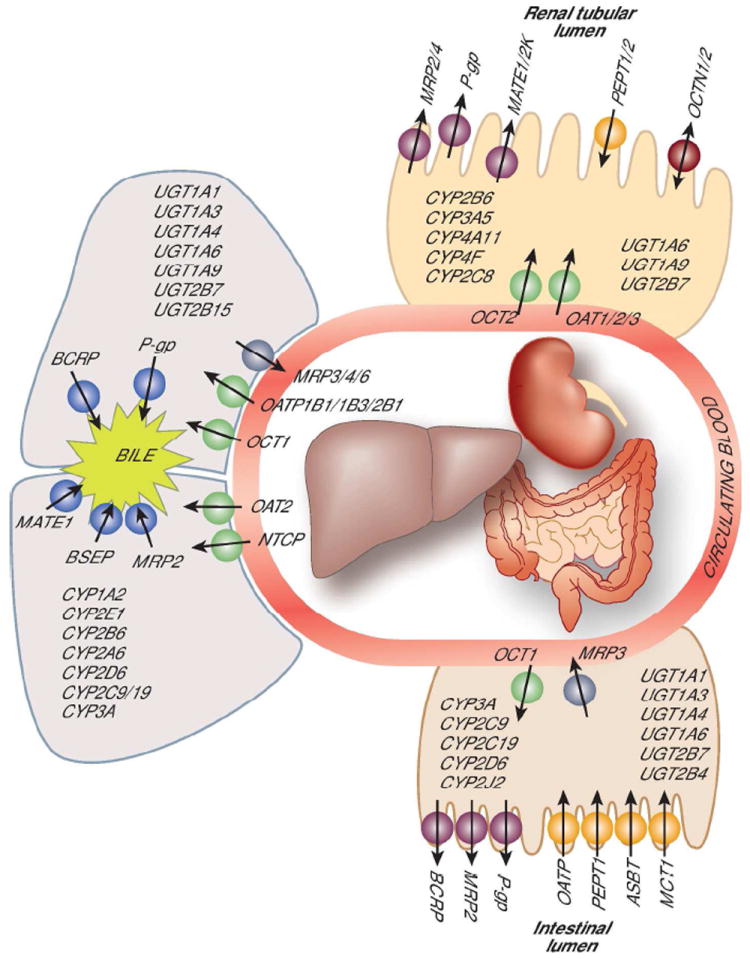
Current knowledge of drug metabolizing enzymes and drug transporters that operate in the human liver, kidney and intestine
Drug transporters also play a critical role in controlling drug exposure. Transporters are transmembrane proteins facilitating the passage of both drugs and other xenobiotics across biological barriers encountered during drug absorption, tissue distribution, and excretion. Transporters, like drug-metabolizing enzymes, are expressed differentially across body tissues and are characterized as either uptake or influx transporters (transport into the cellular barrier) or efflux transporters (transport out of the cellular barrier). The importance of transporters in governing the intestinal absorption of drugs and nutrients and renal tubular secretion or reabsorption of drugs or their metabolites is increasingly being recognized. On the other hand, the role of hepatic sinusoidal transporters in regulating the access of drug substrates to the hepatocellular enzymes and that of canalicular transporters in biliary excretion of drugs and/or their conjugate metabolites are not as widely appreciated. Recent studies in experimental models of CKD have demonstrated altered expression and/or activities of intestinal and hepatic drug transporters that could modulate the respective intestinal absorption and hepatic uptake and metabolism of drugs.7, 14, 15
Drug Metabolism in CKD (summarized in Figure 2)
Figure 2.
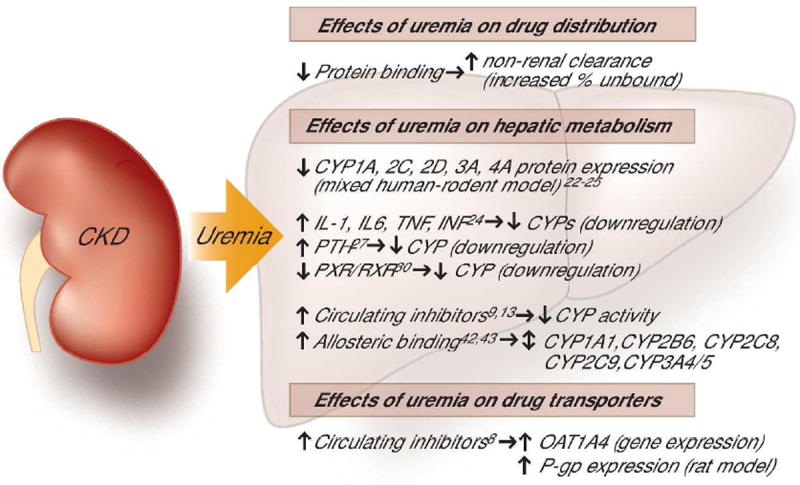
Summary of the recognized effects of uremia on drug metabolism and transport
More than 75 commonly used drugs have been reported to exhibit altered non-renal clearance in patients with CKD (see Table 1 for compilation). Most of these drugs are eliminated by CYP-mediated, oxidative metabolism. Only a few are subject to primary Phase II metabolism, namely O-glucuronidation (diacerein, morphine, oxprenolol, and zidovudine) and N-acetylation (isoniazid and procainamide). In almost all cases, reduced non-renal clearance, along with an increase in oral bioavailability in some cases (especially for drugs that undergo first-pass metabolism in the intestinal mucosa and/or liver), was observed in CKD. A case in point is the diminished non-renal clearance of nimodipine, which could result in as much as a 7-fold increase in its AUC,16 although the increase in drug exposure is usually more modest (1.5-3.0-fold), variable across patients, and dependent upon the degree of renal impairment and the dialysis regimen in patients near or at the end-stage of renal disease. Increased clearance has been reported for a handful of drugs, including phenytoin, fosinopril, cefpiramide, nifedipine, bumetanide, and sulfadimidine.17, 18 At least in the case of phenytoin, the apparent acceleration in non-renal clearance is attributed to reduced binding of phenytoin to albumin in uremic serum resulting in a higher fraction of circulating drug being available for uptake and metabolism by the liver.
Table 1. Currently used drugs reported to exhibit reduced non-renal clearance and/or increased oral bioavailability in CKD patientsa.
| Acyclovir | Dihydrocodeineb,c | Nortriptyline |
| Aliskiren | Desmethyldiazepam | Oxprenololb,c |
| Alfuzosin | Duloxetine | Procainamided |
| Aprepitant | Encainide | Propoxypheneb |
| Aztreonam | Eprosartan | Propranololb |
| Bupropion | Erythromycinb | Quinapril |
| Captopril | Felbamate | Raloxifene |
| Capsofungin | 5-Fluorouracil | Ranolazine |
| Carvedilol | Guanadrel | Reboxetine |
| Cefepime | Imipenem | Repaglinide |
| Cefmenoxime | Isoniazidd | Rosuvastatin |
| Cefmetazole | Ketoprofen | Roxithromycin |
| Cefonicid | Ketorolac | Simvastatin |
| Cefotaxime | Lanthanum | Solifenacin |
| Ceftibuten | Lidocaine | Sparfloxacin |
| Ceftizoxime | Lomefloxacin | Tacrolimus |
| Cefsulodin | Losartan | Tadalafil |
| Ceftriaxone | Lovastatin | Telithromycin |
| Cibenzolin | Metoclopromide | Valsartan |
| Cilastatin | Minoxidil | Vancomycin |
| Cimetidine | Morphinec | Vardenafil |
| Ciprofloxaxin | Moxalactam | Verapamilb |
| Cyclophophamide | Nefopam | Warfarin |
| Darifenacin | Nicardipineb | Zidovudinec |
| Diacereinc | Nimodipine | |
| Didanosine | Nitrendipine |
Except where noted, nearly all the listed drugs undergo oxidative metabolism mediated by CYPs.
Drugs known to exhibit an increase in oral bioavailability as well as a reduced non-renal clearance.
Drugs that mainly undergo O-glucuronidation.
Drugs that mainly undergo N-acetylation.
A number of mechanisms have been hypothesized for the impairment of drug metabolism in CKD, particularly metabolic pathways mediated by CYP enzymes. The supporting evidence is drawn largely from experimental studies in animal models of acute and chronic renal failure. The proposed mechanisms include: alterations in gene transcription and protein translation, reduced CYP expression due to inhibition of hemoprotein biosynthesis and/or increased enzyme degradation, depletion of co-factors (e.g., supply of NADPH), and direct competitive inhibition of CYP enzyme by circulating uremic constituents.13, 18
Supply of δ-aminolevulinic acid is recognized as a rate-limiting step in the hepatic synthesis of cytochrome P450 hemoproteins. Total microsomal cytochrome P450 content is consistently reduced in various experimental models of renal failure, and mitochondrial δ-aminolevulinic synthetase activity is depressed in the two-step 5/6th nephrectomy model in rats.19 Leber et al.20 reported that intraperitoneal supplementation of δ-aminolevulinic acid in rats following subtotal nephrectomy normalized the level of cytochrome P450 in the liver, but did not reverse the reduction in CYP activities; hence, interference of hemoprotein synthesis is not a major mechanism of uremia’s effect on CYP functioning in the rat. In addition, there is no experimental evidence in support of a diminished pool of hepatic NADPH/NADH+ in renal failure, thereby limiting microsomal oxidation reactions.
Currently, the two most likely mechanisms are transcriptional and/or translational modifications and direct competitive inhibition of the CYP enzymes. Nolin et al.13 have provided a thorough summary of experimental studies conducted over the past decade, which clearly demonstrated reduced expression of CYP genes and gene products (i.e., reduced mRNA and protein, or reduced protein with no change in mRNA) in several animal models of CKD. The precise mechanism(s) of the down-regulation of CYP genes in these CKD models remains unknown. Also, there is no prima facie evidence that transcriptional and/or translational modifications in CYP genes involved in drug metabolism occur in humans with CKD. The only available human data come from ex vivo studies of uremic serum obtained from patients with end-stage renal disease (ESRD),21-23 which showed that incubating rat hepatocytes in primary culture with uremic human serum led to a decrease in protein expression and activity for all the major xenobiotic-metabolizing CYPs (i.e., 1A, 2C, 2D, 3A and 4A families), except for CYP2E1.
Michaud et al.23 observed that, while pre-hemodialysis serum caused significant reductions in CYP protein expression compared to serum from healthy controls, post-dialysis serum showed no effect. Fractionation of uremic serum by ultrafiltration and size-exclusion HPLC revealed that the inhibitory constituents have a molecular weight range between 10 and 15 kDa. These investigators postulated that proinflammatory cytokines and parathyroid hormone, which have the requisite molecular size and are known to be elevated in CKD,24 could mediate the down-regulation of CYPs in CKD.25, 26 Indeed, a follow-up study by the same group provided strong evidence that parathyroid hormone was a major component in uremic rat serum responsible for CYP down-regulation, and parathyroidectomy abolished the alteration in CYP transcription and translation.27 Possible mechanisms of parathyroid hormone’s effect on CYP gene regulation include increased cAMP production, elevations in intracellular calcium, and/or activation of the NF-κB pathway. Down-regulation of CYP gene expression in response to proinflammatory cytokines and other mediators of acute phase response (e.g., interleukin-1, interleukin-6, tumor necrosis factor-α, interferon) are well established.28 It is also possible that circulating uremic constituents interfere with signaling of nuclear receptors involved in transcriptional activation of CYP genes, such as Pregnane-X-Receptor (PXR) and Constitutive Androstane Receptor (CAR). Also, uremic plasma ultrafiltrate and peritoneal dialysate have been shown to inhibit VDR-RXR hetero-dimerization and attenuate activation of vitamin D responsive genes, which include CYP3A4. 29, 30
The ex vivo evidence for uremia-induced transcriptional and translational modifications appears convincing, but there are caveats. Interspecies differences exist in the binding and activation of nuclear receptors (e.g., PXR) that regulate transcription of CYP genes, particularly between rodents and humans.31 In addition, the human orthologs of rat CYP enzymes often have vastly different drug substrate selectivity.32 The degree of uremia in animal models often exceeds that observed in stage III CKD subjects and may not be generalizable to this population; however, it may mimic the uremic state observed in patients with stage IV CKD or in sub-optimally dialyzed stage V subjects. Hence, caution should be exercised in extrapolating genomic and functional data gathered from studies with rat hepatocytes and severely uremic animal models to the clinical context in CKD patients. It would be of considerable interest to repeat the ex vivo studies of uremic human serum on short term CYP regulation using a three-dimensional human hepatocyte culture system (vide infra).
The presence of circulating, competitive inhibitor(s) of CYP enzymes in uremic blood or plasma was demonstrated in some of the early experimental studies.6, 33 As early as 1985, Terao et al.34 showed a reduced extraction of S-propranolol—a high intrinsic clearance CYP substrate, by livers isolated from normal rats perfused with uremic blood. Furthermore, livers from acute renal failure rats showed no reduction in S-propranolol extraction when perfused with normal blood from control rats. This set of cross-perfusion experiments presented the first evidence of a rapidly acting inhibitory factor(s) in uremic blood directly affecting the functioning of hepatic CYP enzymes. A later study by the same laboratory showed that a low molecular weight ultrafiltrate fraction (<10 kDa) of uremic plasma obtained from ESRD patients was capable of inhibiting the oxidative metabolism of S-propranolol in human liver microsomes mediated by CYP2D6 and CYP1A2.35 Corroborating evidence of circulating uremic inhibitors have also been provided by Yoshitani et al.36 who showed that uremic sera from experimental models of acute renal failure in rats were capable of inhibiting oxidative metabolism of losartan in rat liver microsomes. Likewise, Taburet et al.37 reported that uremic plasma from ESRD patients inhibited the metabolism of the CYP2C9 probe tolbutamide and the CYP3A probe midazolam in human liver microsomes from donors with normal renal function. Another piece of indirect evidence comes from pharmacokinetic studies in ESRD patients undergoing hemodialysis; where the metabolic clearance of both propranolol38 and telithromycin39 following oral administration was partially or completely normalized when the drug was given shortly after a regular dialysis session compared to before dialysis. The rapid reversibility of uremia’s effect is more consistent with dialytic removal of competitive enzyme inhibitors than reversal of uremia-induced down-regulation of CYP. While the latter process normally takes several days to achieve in accordance to the turnover half-life of cytochrome P450s (≥ 24 hours), downregulation in the expression of one or more CYP proteins cannot be completely excluded.40 Until now, no systematic investigation has been undertaken to identify the putative CYP inhibitors in uremic blood of CKD patients.
Unfortunately, our incomplete understanding of the effect of uremia on drug metabolism means that we are unable to predict which drug substrates and under what clinical circumstance would we expect to encounter a significant perturbation in metabolic clearance that warrants dosage adjustment. Early on, it was hypothesized that uremic inhibition of metabolic clearance may be confined to drug substrates of select CYP subfamilies if competitive enzyme inhibition by circulating uremic inhibitors is CYP specific. Soon it became apparent that uremic inhibition is observed in members of nearly every major drug-metabolizing CYP subfamilies; more puzzling is the fact that inhibition is inconsistent in its manifestation across substrates for the same CYP isoenzyme.41 For example, most of the lipophilic β-adrenergic blockers are metabolized to a large extent by CYP2D6. Whereas the first-pass and systemic clearance of orally administered propranolol and bufuralol are significantly reduced in CKD patients resulting in 3- to 5-fold increases in AUC, no such changes are observed with metoprolol and propafenone.41 The same is observed with substrates of CYP3A: first-pass metabolism and systemic clearance of midazolam are not affected by renal dysfunction;8 in contrast, increased oral bioavailability and decreased systemic clearance have been reported for other CYP3A-selective substrates, such as the antidepressant—reboxetine and the dihydropyridine calcium channel blockers—nicardipine, nimodipine, and nitrendipine.13, 25 Thus, until we have identified the uremic constituents that modulate CYP-mediated metabolism, we will not begin to appreciate the exact nature and complexity of the effects of kidney disease on drug metabolism.
For CYP enzymes exhibiting allosteric behavior, such as CYP1A1, CYP2B6, CYP2C8, CYP2C9, and CYP3A4/5, heterotropic cooperativity induced by two substrates or a substrate-inhibitor pair can lead to either apparent activation or inhibition in metabolism.42, 43 The outcome of interactions become even harder to predict when multiple inhibitors are present, which is the likely scenario in uremia; such complex uremia retention solute-drug interactions will require meticulous enzyme kinetic studies. For those substrates whose first-pass metabolism is inhibited, we will need to delineate the separate effects of uremia on drug extraction at the intestinal mucosa versus that at the liver. Leblond et al.44, 45 have shown that uremia-induced down-regulation of some CYPs is tissue-specific; for example, chronic renal failure in rats induced by two-stage, 5/6th nephrectomy resulted in a significant down-regulation in CYP1A2 in the intestine but not in the liver, whereas the opposite is observed with CYP2C11. Some of the variability in the effects of renal disease on drug metabolism undoubtedly reflects differences in the stage of kidney disease and severity of uremia of patients between studies. There is always the problem of unrecognized confounders, such as differences in diet and nutritional support that may give rise to variations in composition and levels of uremic toxins, and the ever present problem of assessing drug-drug interactions.
Drug Transport in CKD
There is increasing awareness that uptake transport of drugs across the sinusoidal membrane of hepatocytes regulates the access of drug substrates to hepatocelluar enzymes as well as canalcular transport into the bile canaliculi, and can be a rate-limiting step in the overall process of hepatic drug clearance.46 In 1984, Bowmer, Yates and their colleagues47, 48,47, 48 reported that the hepatic uptake of two anionic dyes (indocyanin green and bromosulfophthalein) that are non-selective OATP substrates were reduced in acute renal failure rats. The functional and clinical significance of these findings did not become evident until a series of recent investigations showed that inhibition of uptake transport into the liver may explain to a large extent the reduced non-renal clearance observed in CKD patients for several commonly used drugs that are moderately good to high affinity substrates of human hepatic OATPs: erythromycin,9, 49 eprosartan,50 fexofenadine,8 and digoxin.51
The inhibitory mechanism(s) of hepatic drug uptake was explored to a limited extent in the above referenced studies. Nolin et al.8 incubated normal rat hepatocytes with uremic serum drawn from patients with ESRD and showed a 29% decrease in OATP1A4 expression and a 37% increase in P-gp expression in rat hepatocytes exposed to uremic serum compared with those exposed to healthy serum; the effect on OATP is consistent with the in vivo finding of a 63% decrease in the oral clearance of fexofenadine. A subsequent in vitro investigation with digoxin by Tsujimoto et al.51 also yielded similar findings. It would be important to replicate these findings in human hepatocytes since the complement of human sinusoidal OATPs (OATP1B1, OATP1B3, and OATP2B1) are not functional orthologs of rat sinusoidal OATPs. It is also relevant to note that among the various anionic uremic toxins tested, 3-carboxy-4-methyl-5-propyl-2-furanpropionic acid (CMPF) was consistently shown to be the most potent inhibitor of rOATP or hOATP for the uptake of erythromycin, eprosartan, and digoxin, with a Ki in the order of 20 to 50 μM, which is within the plasma concentration range reported in uremic patients (median 254 μM, maximum 392 μM).52 Another uremic constituent, p-cresol was also shown to inhibit digoxin uptake into isolated human hepatocytes.
It is important to recognize that sinusoidal uptake transport and intracellular processing by hepatocellular metabolism and canalicular secretion into bile are coupled kinetic processes; moreover, either or both transport and metabolic steps can be altered by uremia to produce the net effect of reduced hepatic clearance. Hence, in vitro modeling of functional disruption of hepatocellular processes must be evaluated in a cell-based system that maintains the coupling of transport at the sinusoidal membrane and enzymatic function at the endoplasmic reticulum. All in vitro studies conducted to date in this area have used hepatocytes isolated from rat or human livers and maintained in short-term, conventional monolayer culture; as a result, the hepatocytes do not retain cell polarity (i.e., sinusoidal versus canalicular membrane domain) and rapidly de-differentiate. Over the past decade, the sandwich-cultured human hepatocyte model (SCH) has gained favor in order to study the complex interplay between metabolic enzymes and transporters. When cultured on a substrate of BioCoat with an overlay of Matrigel or collagen, primary hepatocytes develop a cuboidal three-dimensional structure with intact bile canaliculi and proper localization of efflux transporters (e.g., OATPs, P-gp, MRPs, and concentrative/equilibrative nucleoside transporters), while maintaining functional metabolic enzymes (e.g., CYPs, UDP-glucuronosyltransferases).53-55 This system features the connecting between sinusoidal uptake, intracellular metabolism, and efflux into the bile canaliculi, and allows assessment of the net effects of these sequential processes on heaptic drug processing. Exposing SCH to uremic serum or its derived fractions could provide a more realistic in vitro model for investigating the alterations in drug metabolism and transport observed in CKD patients.
An emerging novel approach to enhancing in vitro tissue or organ system models relevant to altered drug metabolism is the development of microphysiological systems, or “organs on a chip.” Microphysiological systems utilize microfluidic technologies, can incorporate three dimensional architecture and mimic physiological fluid shear stress.56 These microphysiological systems frequently use human cell sources to overcome concerns about species differences in drug transport and metabolism. Human microphysiological systems are currently under development at the University of Washington and other institutions, and should expand our ability to explore the interactions between kidney disease and hepatic drug metabolism and transport. Microphysiological systems can also feature microvascular endothelial cells and perivascular cells cultured on a 3-dimensional scaffold that form luminal or microvasular structures;57 hence, the systems can recapitulate normal perfusion flow and the resulting physiological dynamics of solute transport. It is conceivable that eventually we will be able to capture the impact of impairment in kidney function on hepatocyte function through coupling of vascularized microphysiological systems based on human kidney and liver.
Great strides have been made in understanding the effects of CKD on drug disposition in the past 15 years, particularly on how uremia affects hepatic drug metabolism and coupled transport. Nonetheless, as highlighted in this review, important questions remain regarding the underlying uremic mechanisms. Until we fully understand uremia’s impact on drug metabolism and transport at the cellular and molecular level, it will be difficult to develop a rational strategy for drug dosing in the CKD population. It is also clear that progress will depend on the application of novel techniques and methodologies to delineate the complex and multiplex details of uremia’s effect on the drug disposition system.
Acknowledgments
Sources of support: CKY was supported by NCATS Grant KL2 TR000421. The authors also acknowledge NCATS 1UH2 TR000504
Footnotes
Disclosure
All authors report no competing interests.
References
- 1.Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA : the journal of the American Medical Association. 2007;298:2038–2047. doi: 10.1001/jama.298.17.2038. [DOI] [PubMed] [Google Scholar]
- 2.MEDPAC. A Data Book: Healthcare spending and the Medicare program. 2009 Jun [Google Scholar]
- 3.Manley HJ, Cannella CA. Nondialysis (home) medication utilization and cost in diabetic and nondiabetic hemodialysis patients. Nephrology news & issues. 2005;19:27–28. 33–24, 36–28. [PubMed] [Google Scholar]
- 4.Manley HJ, Cannella CA, Bailie GR, et al. Medication-related problems in ambulatory hemodialysis patients: a pooled analysis. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2005;46:669–680. doi: 10.1053/j.ajkd.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Zhang L, Abraham S, et al. Assessment of the impact of renal impairment on systemic exposure of new molecular entities: evaluation of recent new drug applications. Clin Pharmacol Ther. 2009;85:305–311. doi: 10.1038/clpt.2008.208. [DOI] [PubMed] [Google Scholar]
- 6.Dreisbach AW, Lertora JJ. The effect of chronic renal failure on drug metabolism and transport. Expert Opin Drug Metab Toxicol. 2008;4:1065–1074. doi: 10.1517/17425255.4.8.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naud J, Nolin TD, Leblond FA, et al. Current understanding of drug disposition in kidney disease. J Clin Pharmacol. 2012;52:10S–22S. doi: 10.1177/0091270011413588. [DOI] [PubMed] [Google Scholar]
- 8.Nolin TD, Frye RF, Le P, et al. ESRD impairs nonrenal clearance of fexofenadine but not midazolam. Journal of the American Society of Nephrology : JASN. 2009;20:2269–2276. doi: 10.1681/ASN.2009010082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun H, Huang Y, Frassetto L, et al. Effects of uremic toxins on hepatic uptake and metabolism of erythromycin. Drug Metab Dispos. 2004;32:1239–1246. doi: 10.1124/dmd.104.000521. [DOI] [PubMed] [Google Scholar]
- 10.Bauer LA. Clinical Pharmacokinetics and Pharmacodynamics. In: DiPiro JT, Talbert RL, Yee GC, Matzke GR, et al., editors. Pharmacotherapy: A Pathophysiologic Approach. 6. McGraw-Hill; New York: 2005. pp. 51–73. [Google Scholar]
- 11.Paine MF, Hart HL, Ludington SS, et al. The human intestinal cytochrome P450 “pie”. Drug Metab Dispos. 2006;34:880–886. doi: 10.1124/dmd.105.008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yeung CK, Lang DH, Thummel KE, et al. Immunoquantitation of FMO1 in human liver, kidney, and intestine. Drug Metab Dispos. 2000;28:1107–1111. [PubMed] [Google Scholar]
- 13.Nolin TD, Naud J, Leblond FA, et al. Emerging evidence of the impact of kidney disease on drug metabolism and transport. Clin Pharmacol Ther. 2008;83:898–903. doi: 10.1038/clpt.2008.59. [DOI] [PubMed] [Google Scholar]
- 14.Dreisbach AW. The influence of chronic renal failure on drug metabolism and transport. Clin Pharmacol Ther. 2009;86:553–556. doi: 10.1038/clpt.2009.163. [DOI] [PubMed] [Google Scholar]
- 15.Sun H, Frassetto L, Benet LZ. Effects of renal failure on drug transport and metabolism. Pharmacol Ther. 2006;109:1–11. doi: 10.1016/j.pharmthera.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 16.Kirch W, Ramsch KD, Duhrsen U, et al. Clinical pharmacokinetics of nimodipine in normal and impaired renal function. Int J Clin Pharmacol Res. 1984;4:381–384. [PubMed] [Google Scholar]
- 17.Dreisbach AW, Lertora JJ. The effect of chronic renal failure on hepatic drug metabolism and drug disposition. Semin Dial. 2003;16:45–50. doi: 10.1046/j.1525-139x.2003.03011.x. [DOI] [PubMed] [Google Scholar]
- 18.Elston AC, Bayliss MK, Park GR. Effect of renal failure on drug metabolism by the liver. Br J Anaesth. 1993;71:282–290. doi: 10.1093/bja/71.2.282. [DOI] [PubMed] [Google Scholar]
- 19.Uchida N, Kurata N, Shimada K, et al. Changes of hepatic microsomal oxidative drug metabolizing enzymes in chronic renal failure (CRF) rats by partial nephrectomy. Jpn J Pharmacol. 1995;68:431–439. doi: 10.1254/jjp.68.431. [DOI] [PubMed] [Google Scholar]
- 20.Leber HW, Schutterle G. Oxidative drug metabolism in liver microsomes from uremic rats. Kidney Int. 1972;2:152–158. doi: 10.1038/ki.1972.85. [DOI] [PubMed] [Google Scholar]
- 21.Guevin C, Michaud J, Naud J, et al. Down-regulation of hepatic cytochrome p450 in chronic renal failure: role of uremic mediators. Br J Pharmacol. 2002;137:1039–1046. doi: 10.1038/sj.bjp.0704951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michaud J, Dube P, Naud J, et al. Effects of serum from patients with chronic renal failure on rat hepatic cytochrome P450. Br J Pharmacol. 2005;144:1067–1077. doi: 10.1038/sj.bjp.0706138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michaud J, Nolin TD, Naud J, et al. Effect of hemodialysis on hepatic cytochrome P450 functional expression. Journal of pharmacological sciences. 2008;108:157–163. doi: 10.1254/jphs.08042fp. [DOI] [PubMed] [Google Scholar]
- 24.Miyamoto T, Carrero JJ, Stenvinkel P. Inflammation as a risk factor and target for therapy in chronic kidney disease. Curr Opin Nephrol Hypertens. 2011;20:662–668. doi: 10.1097/MNH.0b013e32834ad504. [DOI] [PubMed] [Google Scholar]
- 25.Nolin TD. Altered nonrenal drug clearance in ESRD. Curr Opin Nephrol Hypertens. 2008;17:555–559. doi: 10.1097/MNH.0b013e3283136732. [DOI] [PubMed] [Google Scholar]
- 26.Pichette V, Leblond FA. Drug metabolism in chronic renal failure. Curr Drug Metab. 2003;4:91–103. doi: 10.2174/1389200033489532. [DOI] [PubMed] [Google Scholar]
- 27.Michaud J, Naud J, Chouinard J, et al. Role of parathyroid hormone in the downregulation of liver cytochrome P450 in chronic renal failure. J Am Soc Nephrol. 2006;17:3041–3048. doi: 10.1681/ASN.2006010035. [DOI] [PubMed] [Google Scholar]
- 28.Morgan ET, Goralski KB, Piquette-Miller M, et al. Regulation of drug-metabolizing enzymes and transporters in infection, inflammation, and cancer. Drug Metab Dispos. 2008;36:205–216. doi: 10.1124/dmd.107.018747. [DOI] [PubMed] [Google Scholar]
- 29.Schmiedlin-Ren P, Thummel KE, Fisher JM, et al. Induction of CYP3A4 by 1 alpha,25-dihydroxyvitamin D3 is human cell line-specific and is unlikely to involve pregnane X receptor. Drug Metab Dispos. 2001;29:1446–1453. [PubMed] [Google Scholar]
- 30.Toell A, Degenhardt S, Grabensee B, et al. Inhibitory effect of uremic solutions on protein-DNA-complex formation of the vitamin D receptor and other members of the nuclear receptor superfamily. J Cell Biochem. 1999;74:386–394. doi: 10.1002/(sici)1097-4644(19990901)74:3<386::aid-jcb7>3.3.co;2-t. [DOI] [PubMed] [Google Scholar]
- 31.Jones SA, Moore LB, Shenk JL, et al. The pregnane X receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol Endocrinol. 2000;14:27–39. doi: 10.1210/mend.14.1.0409. [DOI] [PubMed] [Google Scholar]
- 32.Bogaards JJ, Bertrand M, Jackson P, et al. Determining the best animal model for human cytochrome P450 activities: a comparison of mouse, rat, rabbit, dog, micropig, monkey and man. Xenobiotica. 2000;30:1131–1152. doi: 10.1080/00498250010021684. [DOI] [PubMed] [Google Scholar]
- 33.Nolin TD, Frye RF, Matzke GR. Hepatic drug metabolism and transport in patients with kidney disease. Am J Kidney Dis. 2003;42:906–925. doi: 10.1016/j.ajkd.2003.07.019. [DOI] [PubMed] [Google Scholar]
- 34.Terao N, Shen DD. Reduced extraction of I-propranolol by perfused rat liver in the presence of uremic blood. J Pharmacol Exp Ther. 1985;233:277–284. [PubMed] [Google Scholar]
- 35.Roskos LK. Ph D thesis. University of Washington; Seattle: 1994. Inhibition of the hepatic metabolism of (S)-propranolol by uremic blood constituents. [Google Scholar]
- 36.Yoshitani T, Yagi H, Inotsume N, et al. Effect of experimental renal failure on the pharmacokinetics of losartan in rats. Biol Pharm Bull. 2002;25:1077–1083. doi: 10.1248/bpb.25.1077. [DOI] [PubMed] [Google Scholar]
- 37.Taburet AM, Vincent I, Perello L, et al. Impairment of drug biotransformation in renal disease: An in vitro model. Clin Pharmacol Ther. 1996;59:136. [Google Scholar]
- 38.Bianchetti G, Graziani G, Brancaccio D, et al. Pharmacokinetics and effects of propranolol in terminal uraemic patients and in patients undergoing regular dialysis treatment. Clin Pharmacokinet. 1976;1:373–384. doi: 10.2165/00003088-197601050-00004. [DOI] [PubMed] [Google Scholar]
- 39.Shi J, Montay G, Chapel S, et al. Pharmacokinetics and safety of the ketolide telithromycin in patients with renal impairment. J Clin Pharmacol. 2004;44:234–244. doi: 10.1177/0091270003262952. [DOI] [PubMed] [Google Scholar]
- 40.Yang J, Liao M, Shou M, et al. Cytochrome p450 turnover: regulation of synthesis and degradation, methods for determining rates, and implications for the prediction of drug interactions. Curr Drug Metab. 2008;9:384–394. doi: 10.2174/138920008784746382. [DOI] [PubMed] [Google Scholar]
- 41.Touchette MA, Slaughter RL. The effect of renal failure on hepatic drug clearance. DICP. 1991;25:1214–1224. doi: 10.1177/106002809102501111. [DOI] [PubMed] [Google Scholar]
- 42.Korzekwa KR, Krishnamachary N, Shou M, et al. Evaluation of atypical cytochrome P450 kinetics with two-substrate models: evidence that multiple substrates can simultaneously bind to cytochrome P450 active sites. Biochemistry. 1998;37:4137–4147. doi: 10.1021/bi9715627. [DOI] [PubMed] [Google Scholar]
- 43.Tang W, Stearns RA. Heterotropic cooperativity of cytochrome P450 3A4 and potential drug-drug interactions. Curr Drug Metab. 2001;2:185–198. doi: 10.2174/1389200013338658. [DOI] [PubMed] [Google Scholar]
- 44.Leblond F, Guevin C, Demers C, et al. Downregulation of hepatic cytochrome P450 in chronic renal failure. J Am Soc Nephrol. 2001;12:326–332. doi: 10.1681/ASN.V122326. [DOI] [PubMed] [Google Scholar]
- 45.Leblond FA, Petrucci M, Dube P, et al. Downregulation of intestinal cytochrome p450 in chronic renal failure. J Am Soc Nephrol. 2002;13:1579–1585. doi: 10.1097/01.asn.0000017575.50319.77. [DOI] [PubMed] [Google Scholar]
- 46.Fenner KS, Jones HM, Ullah M, et al. The evolution of the OATP hepatic uptake transport protein family in DMPK sciences: from obscure liver transporters to key determinants of hepatobiliary clearance. Xenobiotica. 2012;42:28–45. doi: 10.3109/00498254.2011.626464. [DOI] [PubMed] [Google Scholar]
- 47.Bowmer CJ, Yates MS. Pharmacokinetics and biliary excretion of bromosulphophthalein, [3H]-ouabain and [3H]-taurocholic acid in rats with glycerol-induced acute renal failure. Br J Pharmacol. 1984;83:773–782. doi: 10.1111/j.1476-5381.1984.tb16232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yates MS, Bowmer CJ, Emmerson J. Effect of acute renal failure on the clearance and biliary excretion of indocyanine green in perfused rat liver. Biochem Pharmacol. 1984;33:1695–1696. doi: 10.1016/0006-2952(84)90297-1. [DOI] [PubMed] [Google Scholar]
- 49.Sun H, Frassetto LA, Huang Y, et al. Hepatic clearance, but not gut availability, of erythromycin is altered in patients with end-stage renal disease. Clin Pharmacol Ther. 2010;87:465–472. doi: 10.1038/clpt.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun H, Huang Y, Okochi H, et al. Uremic toxins inhibit hepatic uptake of eprosartan. Clin Pharmacol Ther. 2005;77:2. [Google Scholar]
- 51.Tsujimoto M, Kinoshita Y, Hirata S, et al. Effects of uremic serum and uremic toxins on hepatic uptake of digoxin. Ther Drug Monit. 2008;30:576–582. doi: 10.1097/FTD.0b013e3181838077. [DOI] [PubMed] [Google Scholar]
- 52.Vanholder R, De Smet R, Glorieux G, et al. Review on uremic toxins: classification, concentration, and interindividual variability. Kidney Int. 2003;63:1934–1943. doi: 10.1046/j.1523-1755.2003.00924.x. [DOI] [PubMed] [Google Scholar]
- 53.Govindarajan R, Endres CJ, Whittington D, et al. Expression and hepatobiliary transport characteristics of the concentrative and equilibrative nucleoside transporters in sandwich-cultured human hepatocytes. American journal of physiology Gastrointestinal and liver physiology. 2008;295:G570–580. doi: 10.1152/ajpgi.00542.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoffmaster KA, Turncliff RZ, LeCluyse EL, et al. P-glycoprotein expression, localization, and function in sandwich-cultured primary rat and human hepatocytes: relevance to the hepatobiliary disposition of a model opioid peptide. Pharmaceutical research. 2004;21:1294–1302. doi: 10.1023/b:pham.0000033018.97745.0d. [DOI] [PubMed] [Google Scholar]
- 55.LeCluyse EL, Fix JA, Audus KL, et al. Regeneration and maintenance of bile canalicular networks in collagen-sandwiched hepatocytes. Toxicology in vitro : an international journal published in association with BIBRA. 2000;14:117–132. doi: 10.1016/s0887-2333(99)00096-x. [DOI] [PubMed] [Google Scholar]
- 56.Huh D, Torisawa YS, Hamilton GA, et al. Microengineered physiological biomimicry: organs-on-chips. Lab Chip. 2012;12:2156–2164. doi: 10.1039/c2lc40089h. [DOI] [PubMed] [Google Scholar]
- 57.Zheng Y, Chen J, Craven M, et al. In vitro microvessels for the study of angiogenesis and thrombosis. Proc Natl Acad Sci U S A. 2012;109:9342–9347. doi: 10.1073/pnas.1201240109. [DOI] [PMC free article] [PubMed] [Google Scholar]