

Abstract
The iron content of livers from 57Fe-enriched C57BL/6 mice of different ages were investigated using Mössbauer spectroscopy, electron paramagnetic resonance (EPR), electronic absorption spectroscopy and inductively coupled plasma mass spectrometry (ICP-MS). About 80% of the Fe in an adult liver was due to blood; thus removal of blood by flushing with buffer was essential to observe endogenous liver Fe. Even after exhaustive flushing, ca. 20% of the Fe in anaerobically dissected livers was typical of deoxy-hemoglobin. The concentration of Fe in newborn livers was the highest of any developmental stage (~ 1.2 mM). Most was stored as ferritin, with little mitochondrial Fe (consisting primarily of Fe/S clusters and haems) evident. Within the first few weeks of life, about half of ferritin Fe was mobilized and exported, illustrating the importance of Fe release as well as Fe storage in liver function. Additional ferritin Fe was used to generate mitochondrial Fe centres. From ca. 4 weeks of age to the end of the mouse’s natural lifespan, the concentration of mitochondrial Fe in liver was essentially invariant. A minor contribution from nonhaem high-spin FeII was observed in most liver samples and was also invariant with age. Some portion of these species may constitute the labile iron pool. Livers from mice raised on an Fe-deficient diet were highly Fe depleted; they were devoid of ferritin and contained 1/3 as much mitochondrial Fe as found in Fe-sufficient livers. In contrast, brains of the same Fe-deficient mice retained normal levels of mitochondrial Fe. Livers from mice with inflammatory hepatitis and from IRP2(−/−) mice hyper-accumulated Fe. These livers had high ferritin levels but low levels of mitochondrial Fe.
Introduction
The liver plays a central role in mammalian iron (Fe) metabolism, storage and regulation.1,2,3 Hepatocytes, the parenchymal cells of the liver, occupy ~80% of the liver’s volume, contain the vast majority of its Fe, and perform most of its functions. These cells generate hepcidin, the master regulator of Fe in the body.4 They secrete this peptide hormone into the blood. Hepcidin blocks the import of Fe into the blood by promoting the degradation of ferroportin, a membrane-bound protein mainly expressed in intestinal enterocytes, hepatocytes and macrophages. Numerous factors regulate the expression of the hepcidin gene, with the BMP-SMAD pathway playing a major role. Dysregulation of hepcidin is associated with the pathogenesis of iron-overload diseases such as hereditary hemochromatosis.
Hepatocytes store Fe as ferritin, a spherically-shaped protein complex with a hollow core that can be filled with FeIII oxyhydroxide aggregates.5 Cytosolic FeII ions enter ferritin through channels that catalyse O2-dependent oxidation; the resulting FeIII ions incorporate into the core. The process is reversible such that under Fe-deficient conditions, FeII ions are released into the cytosol. This contributes to a labile Fe pool that has been detected only indirectly using fluorescent probes.6,7
Blood flows to the liver through the hepatic artery and portal vein. Portal blood carries digested nutrients from the gastrointestinal tract and spleen. The oxygen-rich arterial blood and oxygen-poor, nutrient-rich portal blood drain into the hepatic sinusoids. Kupffer cells are resident macrophages that line these sinusoids and are important in iron metabolism. They are part of the recticuloendothelial system in which erythrocytes are recycled.8 They degrade haemoglobin contained within these cells using haem oxygenase-1, a highly-inducible isozyme involved in Fe homeostasis and protection against oxidative stress.9 Kupffer cells import Fe which stimulates NF-κB activation10 and induces reactions associated with a burst of respiration and generation of ROS/RNS.11 Kupffer cells sequester Fe as ferritin and export it via NRAMP1, a homolog of the membrane-bound divalent metal transporter DMT1.12 In hemochromatosis, the concentration of Fe in Kupffer cells is low because an unusually low concentration of hepcidin promotes exodus of Fe from these cells.8
Iron regulatory protein 2 (IRP2) regulates expression of particular mRNA transcripts associated with Fe regulation.13 In Fe-deficient cells, IRP2 promotes cellular Fe import by increasing levels of the transferrin receptor (TfR). It also discourages cellular Fe export by inhibiting ferroportin synthesis, and it inhibits cellular Fe storage by decreasing ferritin synthesis. IRP2(−/−) mice are anaemic and functionally starved of Fe because they produce too much ferritin and not enough TfR.14 They experience Fe-dependent neurodegeneration with an increase of ferric ion staining in the brain, as well as mitochondrial dysfunction and decline of Fe/S cluster protein levels.15 Hentze and coworkers16 found that IPR1-and-IRP2-deficient hepatocytes overexpress ferritin (nearly 50-fold greater expression, relative to controls). The concentration of Fe in IRP1/2(−/−) livers is ~ 50% higher than controls. Activities of the respiratory complexes are reduced. This is probably due to lower than normal mitochondrial Fe levels, lower haem levels, and impaired Fe/S cluster biosynthesis.
Mössbauer spectroscopy (MBS) is arguably the most powerful tool to investigate the speciation of iron in complex biological samples17, yet relatively few investigations of the iron content of liver using MBS have been reported. Chorazy et al.18 used MBS to identify both ferritin and hemosiderin. Hemosiderin is a degradation product of ferritin. The Fe in human23 and chicken24 livers is dominated by ferritin. Whitnall et al.19 found that mouse livers lacking frataxin, a protein involved in mitochondrial Fe/S-cluster metabolism, are loaded with ferritin. Chua-anusorn et al.20 used MBS to identify the forms of Fe in livers. Spectra were dominated by ferrihydrite, with some deoxy-haemoglobin and met-haemoglobin evident. Hemosiderin-like features were not detected. Rimbert et al.21 used MBS to examine Fe-overloaded livers in β-thalassemia patients. Spectra of normal human livers were typical of ferritin/hemosiderin whereas livers with hemosiderosis also exhibited another HS FeIII species with super-paramagnetic behaviour. Ward et al.22 mimicked hemochromatosis by feeding rats Fe-overloaded chow for 2–6 wks. Their livers contained > 100 mM Fe, much of which was present as ferritin and hemosiderin. The corresponding MB spectra exhibited super-paramagnetic behaviour typical of ferrihydrite. The majority of this Fe was in lysosomes and cytosol. In nutritionally-created Fe-overloaded animals, only hepatocytes become overloaded, whereas in hemosiderosis (overload of hemosiderin) both Kupffer cells and hepatocytes are overloaded with Fe.36
Mitochondria are the major traffic hubs of cellular Fe metabolism25,26, and the liver contains high levels of this organelle. Thus, we were surprised that all previous MB spectra of livers were devoid of features due to mitochondria. We hypothesized that the dominance of ferritin and/or hemosiderin in diseased and Fe-overloaded livers obscured the contribution of mitochondria and other minor forms of Fe. Thus, we performed MBS and other related physical methods (EPR and UV-vis) on healthy mouse livers that were fully enriched in 57Fe. 57Fe enriched samples have much higher signal-to-noise ratio than do unenriched samples. Here, we present MB spectra that include both mitochondrial Fe and mononuclear nonhaem high-spin (NHHS) FeII features, neither of which has been reported previously. We also observed substantial spectral intensity arising from blood in these samples. We examined developmental changes in these Fe components, as well as changes associated with Fe deficiency, with an absence of IRP2, and with inflammatory hepatitis which is associated with Fe overload.27 Our results provide substantial new insights into liver iron metabolism.
Experimental
All procedures involving mice were approved by the Animal Care and Use Committee at Texas A&M University. C57BL/6 mice were a gift from Louise Abbott (Texas A&M University). IRP2(−/−) mice were a gift from Tracey Rouault (National Institutes of Health). Both colonies were raised in an iron-free environment as described28, and fed Fe-deficient chow (Harlan Teklad, Madison, WI; ID# 80396) enriched with 50 mg 57Fe (Isoflex USA, San Francisco CA) citrate per Kg chow. Fe-deficient mice were treated in the same way but their chow lacked added 57Fe. Mice were euthanized at various ages, using ketamine and xylazine. Immediately thereafter, bodies were imported into a refrigerated (5 °C) argon-atmosphere glove box maintained at ~ 3 ppm O2 as monitored by a Teledyne model 310 analyser. Unless otherwise stated, blood was removed from animals > 1 wk old. Blood from newborn and 1 wk mice could not be flushed. Flushing was performed by puncturing the heart with a needle, clipping the caudal vena cava and passing Ringer’s buffer (5 min per 10 gm animal mass at 0.7 mL/min) through the needle into the mouse. Some blood samples were transferred to a MB cup and frozen in LN2. Flushed livers and other organs were isolated by dissection, weighed, and either placed in a MB cup and frozen in LN2, or homogenized using a tissue grinder and plastic knife as described.28 A density of 1.06 mg/mL was assumed. Homogenates were either packed into EPR tubes, or transferred to a UV-vis cuvette. MBS, EPR, and UV-vis spectroscopies were performed as described.28 Other samples were transferred to plastic screw-top tubes and incubated in 3.5% trace-metal grade HNO3 overnight. Concentrations of metals in these samples were determined by ICP-MS. Mitochondria were isolated as described in Supplementary Information.
Results
Iron concentrations during liver development
MB spectra of un-flushed newborn and 1 wk livers exhibited a quadrupole doublet with parameters typical of the high-spin (HS) FeII haem centres found in deoxy-haemoglobin (δ ~ 0.96 mm s−1, ΔEQ ~ 2.3 mm s−1). The same haem doublet (HD) was observed in blood (Fig. 1B). By quantifying the relative spectral intensity of the HD, the proportions of Fe due to blood in the newborn and 1 wk livers were estimated to be 40% and 30%, respectively.
Figure 1. 5 K, 0.05 T Mössbauer spectra of 96 wk flushed mouse liver (A) and mouse blood (B).

C is the difference spectrum, A minus B; D – F are simulations using the parameters mentioned in the text: D, Ferritin; E, CD; F, NHHS FeII; The red line in B is a simulation with parameters of HS haems. The field was applied parallel to the gamma radiation.
Livers of buffer-flushed animals also contained significant amounts of blood that were not removed despite extensive flushing, as evidenced by a significant HD in the corresponding MB spectra. The low-temperature, low-applied-field spectra of a flushed 96 wk liver is shown in Fig. 1A. Flushed livers older than 2 wks exhibited a HD representing 20% ± 5% of overall spectral intensity.
Some of this intensity arose from HS FeII haemcentres endogenous to liver tissue. To retain these contributions in the spectrum, but remove the exogenous blood contribution, we compared the MB spectrum of Fig. 1A to that of human T-lymphocytes (Jurkat cells) grown under Fe-sufficient conditions (Fig. 5A of ref 25). Both contained the same components, in approximately the same relative percentages except for the HD which was less intense in Jurkat cell spectra. The HD exhibited by Jurkat cells arose exclusively from endogenous HS FeII haems in these cells, not from blood. We assumed that the same percentage of Fe in liver arose from endogenous haemcentres relative to other spectral features that were common to both samples. Thus, we subtracted a HD simulation from the 96 wk liver spectrum such that the remaining HD intensity matched that observed in Jurkat cell spectra. The resulting difference spectrum (Fig. 1C) included a small HS FeII HD that should reflect the endogenous haemcentres in liver tissue. Similar subtractions were performed for the rest of the spectra included in this report. This procedure may not have been perfectly accurate because the liver probably contains a higher concentration of mitochondria than do Jurkat cells. However, any error introduced by this assumption would be modest.
This procedure allowed us to estimate the endogenous concentration of Fe in livers (designated [FeL]) of different ages, ranging from newborn to the end of their natural lifespan, ca. 100 wks. [FeL] in newborn livers ranged from 1.1 – 1.8 mM (Fig. 2, 0 wks, square), whereas the total Fe concentration ([FeL] + [Feblood]; Fig. 2, 0 wks, circle) was nearly 2.4 mM. These concentrations were significantly higher than in WT healthy livers at any other age (Fig. 2). Immediately after birth, [FeL] declined sharply, reaching a minimum of ca. 300 μM at 3 wks. This corresponded to an exodus of > 1 mM Fe from the liver during the first wk of life! The endogenous FeL concentration increased gradually thereafter such that in a 96 wk liver it was 650 μM. Thus, the liver of a healthy young adult, raised on Fe-sufficient chow, only accumulated a few hundred μM of Fe during the rest of its life. This differed from our expectation that the liver would accumulate large amounts of Fe over the lifetime of the animal. Perhaps this would have occurred had the mice been fed a high-Fe diet. These changes also highlight the importance of flushing blood from the animals. The observed changes in [FeL] would have been difficult to detect had unflushed livers or the uncorrected Fe concentrations of flushed livers been used. The overall Fe concentration of an unflushed 16 wk liver was 3.8 mM, more than 6-fold higher than [FeL] at that age. Small variations in the proportion of blood in such livers could obscure age-dependent variations in [FeL].
Figure 2. Iron concentration of flushed mouse liver.
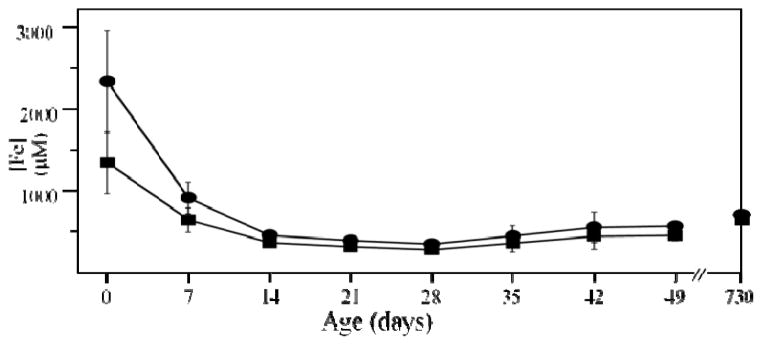
Circles, with blood contribution included; squares, with blood contribution re moved. Concentrations are listed in Table S1.
Ninety-six wk liver
After removing the blood contribution to the spectrum of a 96 wk liver, the dominant component in the difference spectrum (Fig. 1C), representing 65% of the intensity, was a sextet that could be simulated with parameters of δ = 0.5 mm s−1, ΔEQ = −0.3 mm s−1, and Heff = 480 kG (simulated in Fig. 1D). These parameters are typical of the FeIII oxyhydroxide core of ferritin29, and so we assigned the feature as such. The same sextet dominates the low-temperature spectrum of Jurkat cells.26,27 We doubt that other super-paramagnetic forms of FeIII, including hemosiderin and mitochondrial ferritin, contributed significantly to the observed sextet, as the 70 K MB spectrum lacked a sextet (Fig. S1, B and D). Hemosiderin exhibits a sextet at that temperature, so its absence indicated that this degradation product of ferritin was absent in our mouse livers. This result was expected, as the animals were healthy and tissues were prepared under refrigerated and anaerobic conditions immediately after euthanization. The MB spectrum of isolated liver mitochondria (Fig. 3, upper panel) also lacked a significant sextet, indicating that mitochondrial ferritin was not expressed in high amounts.
Figure 3.

5 K, 0.05 T Mössbauer spectrum (upper) and EPR spectrum (lower) of isolated liver mitochondria. The solid line in the upper panel is a simulation with parameters given in the text and in Table S2. EPR simulations were performed using Spin count (http://www.chem.cmu.edu/groups/hendrich/facilities/) and the g values listed in the text.
The next-most-intense feature in the 5 K, 0.05 T MB spectrum of the 96 wk liver (Fig. 1C), corresponding to 25% overall intensity, was a quadrupole doublet with δ = 0.45 mm s−1 and ΔEQ = 1.15 mm s−1. These parameters are typical of S = 0 [Fe4S4]2+ clusters and low spin FeII haemcentres; such centres cannot be distinguished independently by MB spectroscopy. This so-called Central Doublet (CD), simulated in Fig. 1E, is the same as the CD observed in liver mitochondria (Fig. 3, upper panel), brain28, Jurkat cells26,27 and yeast cells.30,31,32 The CD in whole cells and tissues arises primarily from mitochondrial respiratory centres though such centres in other parts of the cell undoubtedly contribute.
The MB spectrum of Fig. 1C also contained a 3% intensity contribution due to a quadrupole doublet with parameters (δ ~ 1.3 mm s−1, ΔEQ ~ 3.3 mm s−1). These parameters are typical of NHHS FeII species coordinated by 5–6 O/N donor ligands. Such a feature, simulated in Fig. 1F, is also observed, at about the same percentage intensity, in MB spectra of yeast and human cells, and in mouse brain. As mentioned above, the 96 wk liver difference-spectrum also included a small contribution of an FeII HD due to endogenous haemcentres.
The UV-vis spectrum of the flushed 96 wk mouse liver homogenate provided additional evidence for the endogenous haem centres in this sample. The spectrum (Fig. S2, D) exhibited α and β bands arising from FeII haemcentres, most of which were probably associated with mitochondrial respiratory complexes.
Low-temperature X-band EPR spectra of the 96 wk liver homogenate exhibited numerous overlapping signals in both the g = 2 and g = 6 – regions, many of which originated from mitochondrial proteins (Fig. S3, D). Resonances at g = 1.93 and 1.86 probably arose, respectively, from the S = ½ [Fe2S2]+ clusters of succinate dehydrogenase and the Rieske Fe/S protein associated with cytochrome bc1.30 Resonances at g = 2.4, 2.25, 1.92 originated from the low-spin FeIII haem centre of mitochondrial cytochrome P450.33,34 Resonances at g = 6.4 and 5.4 were due to rhombic H.S FeIII haem a3 centres in cytochrome c oxidase.30,31 The weak signal at g = 4.3 arose from mononuclear rhombic NHHS FeIII ions. The origin of this signal is uncertain, but a similar signal was observed in the EPR of brain homogenates.28 The weak signal at g = 6 probably arose from axial HS FeIII haems. The g = 2.00 signal probably arose from organic radicals. At low temperatures, the EPR-active Fe-containing species exhibit Mössbauer features with magnetic hyperfine interactions, but the percentage of such Fe in the sample is insignificant.
Three wk liver
The 5 K, 0.05 T Mössbauer spectrum of a 3 wk mouse liver (Fig. 4C) was dominated by the CD. Thus, the majority of Fe (ca. 70%) was present in respiration-related [Fe4S4]2+ clusters (e.g. respiratory complexes, aconitase) and LS FeII haemcentres (e.g. cytochrome c) in the mitochondria. The CD corresponded to ca. 180 μM (0.7 × 250 μM), about the same concentration as found in 96 wk livers. This implies that, apart from the first few weeks of life, the concentration of mitochondrial Fe in the liver is essentially invariant throughout the natural lifespan of the mouse. The ferritin-associated sextet that dominated the spectrum of the 96 wk old liver was less intense (< 20% spectral intensity), indicating that 3 wk livers contained little of this Fe-storage protein complex. This implies that the liver at 3 wks is Fe-deficient despite feeding the mice an Fe-sufficient diet. The quadrupole doublet due to NHHS FeII was also present, but at an intensity (ca. 8%, 20 μM) corresponding to about half of the absolute concentration found in the 96 wk liver (40 μM). With age, ferritin Fe, and perhaps NHHS FeII, accumulated to 2–3 times the levels present in young adult animals.
Figure 4. Mössbauer spectra of flushed livers isolated at different ages.

A, 1 dy; B, 1 wk; C, 3 wk; D, 3 wk Fe-deficient; E, 63 wk IRP2(−/−); F, 56 wk diseased. Red lines are simulations using parameters in the text and percentages in Table S2. Spectra were collected at 5 K and 0.05 T field applied parallel to the γ radiation.
EPR and electronic absorption spectra of homogenates from 3 wk livers were slightly less intense relative to those of 96 wk livers. The electronic absorption spectrum of the former (Fig. S2, C) exhibited peaks indicating reduced FeII haem centres.32 The EPR spectrum of a 3 wk liver homogenate (Fig. S3, C) exhibited about the same signals and (slightly greater) intensities relative to that of the 96 wk liver homogenate. Overall, this provides further evidence that the concentrations of mitochondria in 96 and 3 wk livers are similar. One difference was a hyperfine-split MnII EPR signal in the spectrum of the 3 wk liver and its absence in that of the 96 wk liver. This difference probably reflects variations among individuals rather than systematic age-dependent differences, but further studies are required to establish this.
Mitochondria isolated from mice livers exhibited intense EPR signals in the g = 6 and g = 2 regions (Fig. 3, lower panel). This included signals from FeIII cytochrome P450, an [Fe2S2]+1 cluster (probably from succinate dehydrogenase), the Rieske [Fe2S2]+1 cluster, and the mixed-valence FeIIIa3:CuIb cytochrome c oxidase active site. These same signals were also observed in whole mouse liver (Fig. S3), confirming that most EPR-active species in the tissue arise from mitochondria. The EPR spectrum of isolated liver mitochondria was devoid of the hyperfine-split MnII signal observed in whole liver homogenates, indicating that these MnII ions are not located in mitochondria. A hyperfine-split MnII signal was found in liver33, yeast cells31 and in the mouse brain28, but not in human Jurkat cells.25,26
About 80% of the corresponding MB spectral intensity of liver mitochondria belonged to the CD (Fig. 3, upper panel). The spectrum also showed two minor doublets due to HS haem centres and NHHS FeII, each representing ~ 5% of spectral intensity. Given that mitochondria occupy a modest percentage of cell (and organ) volume, these results indicate that most of the NHHS FeII and HS FeII haem centres observed in liver homogenates are not located in mitochondria.
One wk liver
The 5 K, 0.05 T MB spectrum of a 1 wk mouse liver (Fig. 4B) included the ferritin sextet, the CD, and the NHHS FeII doublet. The CD accounted for ca. 30% of the overall spectral intensity, corresponding to 100 μM (0.3 × 350 μM). This is ca. 60% of the CD concentration found in livers from older mice. The corresponding UV-vis spectrum of a 1 wk mouse liver (Fig. S2, B) was largely devoid of haem-related signals. However, the contribution from blood was considerable because the sample could not be flushed; thus, α and β bands of haem centres were barely detectable. The EPR spectrum of 1 wk liver (Fig. S3, B) was similar to those of 3 and 96 wk livers (Fig. S3, C and D), albeit with lower overall intensity (also the hyperfine-split MnII signal was not evident). We conclude that the concentration of mitochondria in 1 wk liver was lower by a factor of 2 – 4 relative to that in young adult or senior livers.
Newborn liver
Due to our inability to flush the blood from newborn mice, the raw 5 K, 0.05 T MB spectrum of their livers exhibited an intense doublet due to haemoglobin. After removing that contribution, ca. 96% of the resulting spectrum (Fig. 4A) was due to the ferritin sextet. This spectrum showed only minor contributions (ca. 4% collectively) from the CD and NHHS FeII ions. The corresponding UV-vis spectrum (Fig. S2, A) was dominated by haemoglobin features, with no cytochrome features evident. The liver homogenate from newborn mice exhibited EPR signals (Fig. S3, A) similar to those of older livers, but the overall intensity was significantly reduced. Collectively, these data indicate that the livers of newborn mice contain low concentrations of mitochondria. With an endogenous Fe concentration of 1.1 mM in the newborn liver, the concentration of mitochondrial Fe, as reflected by the CD was only ca. 30 μM (0.03 × 1.1 mM). In contrast, the ferritin Fe concentration was 1.1 mM! Our analysis demonstrates that during the first week of life, the majority of this ferritin Fe is mobilized and exported from the liver. Simultaneously, some ferritin Fe is converted into mitochondrial Fe.
Iron-deficient liver
The diet of a pregnant female was shifted from iron-sufficient (50 mg/kg) to Fe-deficient (no added Fe) ~ 4 days before she gave birth. While on the Fe-deficient diet, she nursed the pups for 3 wks, becoming increasingly Fe-deficient during this period. At 3 wks, the pups were euthanized and their livers were dissected. MBS of these livers (Fig. 4D) exhibited the same features as were observed in 3 wk livers from Fe-sufficient animals (Fig. 4C). Spectra were dominated (80% of overall intensity) by the CD, with little or no intensity due to the ferritin-associated sextet. The 3 wk Fe-deficient liver exhibited a NHHS FeII doublet with ca. 15% spectral intensity, higher percentage wise (but lower in absolute concentration) relative to the same doublet in spectra of the Fe-sufficient liver. The most dramatic difference was that the overall spectral intensity of the Fe-deficient liver was ca. 3-fold less than that of the Fe-sufficient liver. This decline correlated with a 4-fold reduction in [FeL] (Table S1).
IRP2(−/−) and diseased livers
IRP2(−/−) livers accumulate high amount of iron16, with an upregulation of ferritin. However, the form of Fe that accumulates has not been established directly. The [FeL] of a flushed 63 wk old IRP2(−/−) liver was 3.3 mM, more than 5 fold higher than in comparable WT livers. At 5 K and 0.05 T, MB spectra (Fig. 4E) exhibited an intense sextet due to ferritin, representing 95% of overall spectral intensity. Less than 3% of the intensity belonged to the CD. Given the concentration of Fe in the sample, this corresponded to a mitochondrial Fe concentration of < 100 μM (normal is ~ 180 μM). Thus, the vast majority of the Fe in the IRP2(−/−) liver is ferritin. This implies that in these livers, the rate of Fe import into the cell is increased (due to the lack of regulation), with the increased amount of cytosolic Fe stored as ferritin. Parenthetically, we also examined the brain of the same IRP2(−/−) mouse using MB spectroscopy. The spectrum (Fig. S4) was similar to those of WT brains. There was no Fe overload, consistent with an earlier study.16
Three livers that appeared by visual inspection to be diseased were examined by MBS. All were visibly discoloured and had a grainy texture. Histological investigation of one sample indicated inflammatory hepatitis with abundant fibrosis and necrotic patches (Fig. 5, upper panel). The MB spectrum of this sample (Fig. 4F) was dominated (> 90% of the overall intensity) by the ferritin-associated sextet. Less than 5% of the intensity was due to the CD. For this particular sample, the Fe concentration was 1.0 mM, corresponding to < 50 μM CD-associated Fe, at least 3 fold less than in WT samples.
Figure 5. Diseased liver (upper) and model of liver development (lower).

The examined liver section was stained with haematoxylin and eosin. Dark small punctate structures are inflammatory response cells. In the model, the newborn liver (0 wk) contains a high concentration of Fe, primarily in the form of ferritin (brown circles); a small amount of mitochondria (yellow and brown ovals) is also present. During the first few weeks of life, there is a massive exodus of Fe occurring while the organ is both growing and generating mitochondria. This causes the concentration of Fe in the organ to decline, achieving a minimum at ~3 wks. The Fe concentration gradually recovers over time. The distribution of that Fe is essentially invariant for the majority of the animal’s adult life.
Discussion
Importance of Flushing
Failing to remove blood from livers before determining the endogenous Fe concentration of this organ overestimates that concentration by a factor of ca. 6! This is a tremendous error. Moreover, extensive flushing is not completely effective, such that ~ 20% of the measured Fe in flushed livers arises from haem. For our analysis, we assumed that this component arose from blood, but a portion might have arisen from erythrocytes that had been phagocytized by Kupffer cells. If these effects of blood are not taken into account, many physiological changes in endogenous liver Fe concentrations could easily go undetected. Previous results in which the Fe concentration of unflushed livers were determined need to be reinterpreted in light of these considerations. Flushing blood from organs and correcting for residual blood contributions is essential in studies where endogenous liver Fe concentrations are considered.
At our level of resolution, the Fe-sufficient liver, brain and cultured human Jurkat T-lymphocytes cells are similar in terms of Fe speciation. The Fe speciation of adult mouse liver includes ca. 60% ferritin, 25% mitochondria, 10% NHHS FeII and 5% HS FeII haemcentres, similar to what we observed for brain and Jurkat cells. In absolute concentrations, the liver contains more ferritin and mitochondria than these other systems. The endogenous concentration of Fe in the adult liver (from Fe-sufficient mice) is ~ 0.6 mM, only about double that of these other cellular samples (depending on age and growth conditions of those cells).
Exodus of Fe from the newborn liver
We discovered a dramatic decline in the Fe concentration of Fe-replete newborn livers during the first few weeks of life, associated with a loss of ferritin Fe. Was this due to an exodus of mobilized ferritin Fe from the growing liver, or did the Fe concentration decline merely because the volume of the liver increased during that period? To distinguish these, we multiplied the volumes of the liver during the first few weeks of life by the corresponding Fe concentrations. The volumes of the newborn and 1 wk old livers were 49 ± 11 μL and 100 ± 17 μL, respectively (n = 4).35 Using the concentration data of Fig. 2 and Table S1, we calculated that the newborn liver contained 66 nmol of Fe whereas the 1 wk old liver contained 47 nmol of Fe. Thus, nearly 30% of the total Fe in the organ exited during the first week of life! This and other developmental changes are illustrated in Fig. 5, lower panel. The volume of the 3 wk liver was 360 ± 80 μL corresponding to ca. 100 nmol of Fe.35 This calculation indicates that there was a net import of Fe during weeks 2–3. That import rate must have been slower than the dilution rate caused by organ volume growth (since the organ remained Fe deficient at that age). As animals aged and the rate of liver growth declined, the Fe deficiency was relieved and ferritin levels recovered. However, at no age did the Fe concentration in the liver return to what it was as a newborn.
We hypothesize that the fetal liver stores sufficient Fe (as ferritin) to support the growth and development of other organs immediately after birth. The exodus of mobilized ferritin Fe from the newborn liver is synchronized with growth and development. We suspect that there is an Fe-associated developmental signal, triggered soon after birth, that either increases the rate of ferroportin biosynthesis or decreases its rate of degradation. By this mechanism, the newborn animal is ensured of having sufficient Fe for organ development, regardless of the Fe status of the mother’s milk. One exception might be the brain, which appears to store sufficient Fe (again as ferritin) to accommodate its own growth and mitochondriogenesis during the first few wks of life28, and not rely on Fe expelled by the liver. The Fe deficiency evident in the brain at 3 wks appears to be due to the growth in the volume of that organ rather than to an exodus of Fe.
Different response of Brain vs. Liver to Fe deficiency
Under Fe-sufficient conditions, the newborn liver loses its Fe stores during the first few weeks of life but maintains a normal (healthy) concentration of mitochondria. Under Fe-deficient conditions, the young liver not only loses its Fe stores, it also lacks the burst of mitochondriogenesis that occurs in young healthy livers. The concentration of mitochondria in the Fe-deficient liver was 3-fold less than in healthy Fe-sufficient animals of the same age. This lack of ferritin should not negatively impact cellular function but the lack of mitochondria likely creates an energy-starved condition in Fe-deficient livers which might impact energy-dependent liver functions. Indeed, under Fe-deficient conditions, there is a shift from aerobic to anaerobic metabolism36, an increase in plasma glucose, a reduction in fatty acid beta oxidation, an increase in lipids in the liver, and a 2.3 fold decline in aconitase. All of these changes are consistent with a decreased reliance on mitochondria under Fe-deficient conditions.
The brain handles Fe deficiency differently. During Fe-deficiency, the brain depletes its stores of ferritin Fe but the concentration of mitochondria is similar to that of Fe-sufficient brains.28 The implication is that the mouse protects the brain from Fe deficiency at the expense of the liver (and perhaps of other organs).
Hyper-accumulation of Fe in liver of diseased mice
When animals were fed an Fe-sufficient diet, liver ferritin gradual accumulated in the liver as the adult animals aged. However, Fe did not hyper-accumulate as long as the animal remained healthy. The situation was different for diseased states where hyper-accumulation of liver Fe was observed. In each sample investigated, the endogenous Fe concentration of the diseased livers was 2–3 times higher than that of healthy livers of approximately the same age. The additional Fe was in the form of ferritin. This supports previous results which show that diseased livers (associated with hepatic steatosis, chronic hepatitis C) are overloaded with Fe.37 Ferritin loading in the liver (and other organs) might be a property of many types of diseases. In the cases we examined, Fe loading was caused, not by a conversion of existing Fe in the tissue from others forms into ferritin, but by the import of new Fe into the diseased tissue that either was ferritin or was ultimately converted into ferritin.
The rate of Fe import into the tissue probably increases as a result of signals associated with the diseased tissue. Fe could be imported into diseased hepatocytes or Kupffer cells. Alternatively, the additional Fe might arrive in cells that localize to the liver as an immune response to the disease. In the sample examined by both MBS and microscopy, cells other than hepatocytes were observed in the tissue. Further studies are required to establish the choreography of events that results in Fe accumulating as ferritin in diseased livers.
Cytosolic Labile Fe Pool
We routinely detected NHHS FeII species in liver samples of all ages. The concentration of such species was only 20 - 40 μM. In samples containing large quantities of ferritin, the presence of this form of Fe could only be observed at high temperatures such that the magnetic features of ferritin have collapsed. Nonhaem mononuclear high-spin FeII is potentially important metabolically as this is the type of Fe that may have labile ligands and undergo Fenton chemistry. The “labile Fe pool” plays a critical role in hepatic Fe homeostasis10 as it influences Fe import, export and storage. Further studies are required to determine whether the species that we have observed here for the first time might be involved in these processes.
Comparison to previous studies
Most previous MB studies of the liver involved unenriched samples, leading to poor signal-to- noise (S/N) ratios in spectra in which ferritin was the only form of Fe that was evident. The poor S/N may have hindered the identification of minor, albeit important spectral features, including those arising from mitochondria and NHHS FeII. The challenges of obtaining 57Fe-enriched rodent organs are more than counter balanced by the ability to detect and analyse minor Fe-containing species. Although the mitochondrial respiratory complexes and what may be the labile Fe pool are indeed minor species, they play critical roles in mammalian metabolism, on par with the dominant form of Fe that functions to store and release Fe.
Conclusions
In this Mössbauer-centred study of the iron content of livers from 57Fe-enriched mice of different ages, we found that the majority of Fe in unflushed liver is due to blood; thus removal of blood by flushing is essential to quantifying and characterizing endogenous liver Fe. The concentration of Fe in newborn livers was the highest of any developmental stage, with most present as ferritin. Shortly after birth, nearly half of ferritin Fe was mobilized and exported from the liver; some of the remainder was used in mitochondriogenesis. From a month of age to the end of the mouse’s natural lifespan, the concentration of mitochondrial Fe was largely unchanged, while the total endogenous Fe concentration increased marginally. A small proportion of Fe in liver was nonhaem high-spin FeII. This form of Fe was present throughout the animal’s natural lifespan; some of it might constitute the labile Fe pool. Livers of Fe-deficient mice were devoid of ferritin and contained far less mitochondrial Fe relative to that in Fe-sufficient livers. Livers from mice with inflammatory hepatitis and from IRP2(−/−) mice hyper-accumulated Fe. These livers had high ferritin and low mitochondrial Fe levels.
Supplementary Material
Acknowledgments
We thank Dr. Aline Rodrigues Hoffmann (College of Veterinary Medicine, Texas A&M University) for collecting and interpreting microscopic images. The National Institutes of Health (GM084266 and GM46441) and the Robert A Welch Foundation (A1170) sponsored this study.
Abbreviations
- CD
central doublet
- EPR
electron paramagnetic resonance
- [FeL]
endogenous Fe concentration in the liver
- HD
heme doublet
- ICP-MS
inductively coupled plasma mass spectrometry
- IRP2
iron regulatory protein 2
- MB
Mössbauer
- MBS
Mössbauer spectroscopy
- NHHS
non-haem high-spin
- ROS/RNS
reactive oxygen species/reactive nitrogen species
- TfR
transferrin receptor
Footnotes
Electronic Supplementary Information (ESI) available: Details of mitochondria isolation; Table S1, Iron concentration of mice livers; Figure S1, 0.05 T Mössbauer spectra of 1 wk and 16 wk mouse liver; Figure S2, UV-vis spectra of livers isolated at different ages. Figure S3, EPR spectra of packed liver homogenates from mice of different ages; Table S2, Percentage spectral contribution and corresponding concentration the different spectral features obtained by Mössbauer and ICP-MS analysis; Figure S4, 5K, 0.05 T Mössbauer spectrum of the brain from a 63 wk IRP2(−/−) mouse. See DOI: 10.1039/b000000x/
Notes and references
- 1.Ganz T. Systemic Iron Homeostasis. Physiol Rev. 2013;93:1721–1741. doi: 10.1152/physrev.00008.2013. [DOI] [PubMed] [Google Scholar]
- 2.Meynard D, Babitt JL, Lin HY. The Liver: Conductor of systemic iron balance. Blood. 2014;123:168–176. doi: 10.1182/blood-2013-06-427757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson GJ, Frazer DM. Hepatic Iron Metabolism. Seminars in Liver Disease. 2005;25:420–432. doi: 10.1055/s-2005-923314. [DOI] [PubMed] [Google Scholar]
- 4.Ganz T, Vaulont S. Molecular Regulation of Systemic Iron Metabolism. In: Anderson GJ, McLaren GD, editors. Iron Physiology and Pathophysiology in Humans. Humana Press; NJ: 2012. pp. 173–190. [Google Scholar]
- 5.Andrews SC. The Ferritin-like superfamily: evolution of the biological iron storeman from a rubrerythrin-like ancestor. Biochem Biophys Acta General Subjects. 2010;1800:691–705. doi: 10.1016/j.bbagen.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 6.Li Y, Deng Y, Tang Y, Yu H, Gao C, Liu L, Liu L, Yao P. Quercetin protects rat hepatocytes from oxidative damage induced by ethanol and iron by maintaining the intercellular labile iron pool. Human and Experimental Toxicology. 2014;33:534–541. doi: 10.1177/0960327113499168. [DOI] [PubMed] [Google Scholar]
- 7.Au-Yeung HY, Chan J, Chantarojsiri T, Chang CJ. Molecular imaging of labile iron(II) pools in living cells with a turn-on fluorescent probe. J Am Chem Soc. 2013;135:15165–15173. doi: 10.1021/ja4072964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gammella E, Buratti P, Cairo G, Recalcati S. Macrophages: central regulators of iron balance. Metallomics. 2014 doi: 10.1039/c4mt00104d. [DOI] [PubMed] [Google Scholar]
- 9.Bauer M, Bauer I. Heme oxygenase-1: Redox regulation and role in the hepatic response to oxidative stress. Antioxidant and Redox Signaling. 2002;4:749–758. doi: 10.1089/152308602760598891. [DOI] [PubMed] [Google Scholar]
- 10.She H, Xiong S, Lin M, Zandl E, Giulivi C, Tsukamoto H. Iron activates NF-kappa-B in Kupffer cells. Am J Physiol Gastrointest Liver Physiol. 2002;283:G719–G726. doi: 10.1152/ajpgi.00108.2002. [DOI] [PubMed] [Google Scholar]
- 11.Videla LA, Fernandez V, Tapia G, Varela P. Oxidative stress-mediated hepatotoxicity of iron and copper: Role of Kupffer cells. Biometals. 2003;16:103–111. doi: 10.1023/a:1020707811707. [DOI] [PubMed] [Google Scholar]
- 12.Wyllie S, Seu P, Gao FQ, Gros P, Goss JA. Disruption of the Nramp1 (also known as Slc11a1) gene in Kupffer cells attenuates early-phase, warm ischemia-reperfusion injury in the mouse liver. J Leukocyte Biol. 2002;72:885–897. [PubMed] [Google Scholar]
- 13.Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nature chemical Biology. 2006;2:406–414. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 14.LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, Switzer R, Grinberg A, Love P, Tresser N, Rouault TA. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative diseases in mice. Nature Genetics. 2001;27:209–214. doi: 10.1038/84859. [DOI] [PubMed] [Google Scholar]
- 15.Jeong SY, Crooks DR, Wilson-Ollivierre H, Ghosh MC, Sougrat R, Lee J, Cooperman S, Mitchell JB, Beaumont C, Rouault TA. Iron Insufficiency comprises motor neurons and their mitochondrial function in Irp2-null mice. PLOS one. 2011;6:e25404. doi: 10.1371/journal.pone.0025404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galy B, Ferring D, Minana B, Bell O, Janser HG, Muckenthaler M, Schümann K, Hentze MW. Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2) Blood. 2005;106:2580–2589. doi: 10.1182/blood-2005-04-1365. [DOI] [PubMed] [Google Scholar]
- 17.Chakrabarti M, Lindahl PA. The Utility of Mössbauer Spectroscopy in Eukaryotic Cell Biology and Animal Physiology. In: Rouault Tracey., editor. Iron-sulfur clusters. DeGruyter Publisher; Berlin Germany: 2014. in press. [Google Scholar]
- 18.Chorazy M, Meczynskiand R, Panek TJ. Mössbauer effect study of liver tissues. J Radioanalytical and Nuclear Chem lett. 1988;128:201–206. [Google Scholar]
- 19.Whitnall M, Rahmanto YS, Huang MLH, Saletta F, Lok HC, Gutierrez L, Lazaro FJ, Fleming AJ, St Pierre TG, Mikhael MR, Ponka P, Richardson DR. Identification of nonferritin mitochondrial iron deposits in a mouse model of Friedrich ataxia. Proc Natl Acad Sci USA. 2012;109:20590–20595. doi: 10.1073/pnas.1215349109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chua-anusorn W, St Pierre TG, Webb J, Macey DJ, Yansukon P, Pootrakul P. Mössbauer spectroscopic study of the forms of iron in normal human liver and spleen tissue. Hyperfine Interactions. 1994;91:905–910. [Google Scholar]
- 21.Rimbert JN, Dumas F, Kellershohn C, Girot R, Brissot P. Mössbauer spectroscopy study of iron overloaded livers. Biochimie. 1985;67:773–668. doi: 10.1016/s0300-9084(85)80209-1. [DOI] [PubMed] [Google Scholar]
- 22.Ward RJ, Florence AL, Baldwin D, Abiaka C, Roland F, Ramsey MH, Dickson DPE, Peters TJ, Crighton RR. Biochemical and Biophysical investigations of the ferrocene-iron-loaded rat – an animal model of primary hemochromatosis. Eur J Biochem. 1991;202:405–410. doi: 10.1111/j.1432-1033.1991.tb16389.x. [DOI] [PubMed] [Google Scholar]
- 23.Oshtrakh MI, Alenkina IV, Vinogradov AV, Konstantinova TS, Kuzmannand E, Semionkin VA. Mössbauer spectroscopy of the iron cores in human liver ferritin in normal human spleen and ferritin in spleen from patient with primary myelofibrosis: preliminary results of comparative analysis. Biometals. 2013;26:229–239. doi: 10.1007/s10534-012-9602-2. [DOI] [PubMed] [Google Scholar]
- 24.Alenkina IV, Oshtrakh MI, Klepova YV, Dubiel SM, Sadovnikov NV. Comparative study of the iron cores in human liver ferritin, its pharmaceutical models and ferritin in chicken liver and spleen tissues using Mössbauer spectroscopy with a high velocity resolution. Spectrochimica ACTA Part A: Molecular and Biomolecular Spectroscopy. 2013;100:88–93. doi: 10.1016/j.saa.2012.02.083. [DOI] [PubMed] [Google Scholar]
- 25.Jhurry ND, Chakrabarti M, McCormick SP, Holmes-Hampton GP, Lindahl PA. Biophysical Investigation of the Ironome of Human Jurkat Cells and Mitochondria. Biochemistry. 2012;51:5276–5284. doi: 10.1021/bi300382d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jhurry ND, Chakrabarti M, McCormick SP, Gohil VM, Lindahl PA. Mössbauer Study and Modeling of Iron Import and Trafficking in Human Jurkat Cells. Biochemistry. 2013;52:926–7942. doi: 10.1021/bi401015t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edwards CQ, Griffin LM, Kushner JP. Coincidental hemochromatosis and viral hepatitis. Am J of the Medical Sciences. 1991;1:50–54. doi: 10.1097/00000441-199101000-00009. [DOI] [PubMed] [Google Scholar]
- 28.Holmes-Hampton GP, Chakrabarti M, Cockrell AL, McCormick SP, Abbott LC, Lindahl LS, Lindahl PA. Changing Iron Content of the Mouse Brain during Development. Metallomics. 2012;4:761–779. doi: 10.1039/c2mt20086d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chasteen ND, Harrison PM. Mineralization in Ferritin: An Efficient Means of Iron Storage. J Structural Biol. 1999;126:182–194. doi: 10.1006/jsbi.1999.4118. [DOI] [PubMed] [Google Scholar]
- 30.Hudder BN, Garber-Morales J, Stubna AA, Münck E, Hendrich MP, Lindahl PA. Electron paramagnetic resonance and Mössbauer spectroscopy of intact mitochondria from respiring Saccharomyces cerevisiae. J Biol Inorg Chem. 2007;12:1029–1053. doi: 10.1007/s00775-007-0275-1. [DOI] [PubMed] [Google Scholar]
- 31.Holmes-Hampton GP, Miao R, Garber-Morales J, Guo Y, Münck E, Lindahl PA. A Nonheme High-Spin Ferrous Pool in Mitochondria Isolated from Fermenting Saccharomyces cerevisiae. Biochemistry. 2010;49:4227–4234. doi: 10.1021/bi1001823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garber-Morales J, Holmes-Hampton GP, Miao R, Guo Y, Münck E, Lindahl PA. Biophysical Characterization of Iron in Mitochondria Isolated from Respiring and Fermenting Yeast. Biochemistry. 2010;49:5436–5444. doi: 10.1021/bi100558z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morita H, Yoshikawa H, Takizawa T, Shirai M, Akahori F, Yoshimura T. The formation of g=2.49 species of cytochrome P450 in the rat liver by PCB126 oral administration: identification of heme axial ligands by EPR spectroscopy. Biosci Biotech And Biochem. 2006;70:2974–2981. doi: 10.1271/bbb.60367. [DOI] [PubMed] [Google Scholar]
- 34.Omura T. Mitochondrial P450s. Chemico-Biological Interactions. 2006;163:86–93. doi: 10.1016/j.cbi.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 35.Chakrabarti M, Barlas MN, McCormick SP, Lindahl LS, Lindahl PA. Kinetics of Iron Import into Developing Mouse Organs Determined by a Pup-Swapping Method. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davis MR, Rendina E, Peterson SK, Lucas EA, Smith BJ, Clarke SL. Enhanced expression of lipogenic genes may contribute to hyperglycemia and alternations in plasma lipids in response to dietary iron deficiency. Genes Nutr. 2012;7:415–425. doi: 10.1007/s12263-011-0278-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Welch KD, Hall JO, Davis TZ, Aust SD. The effect of copper deficiency on the formation of hemosiderin in Sprague-dawley rats. Biometals. 2007;20:829–839. doi: 10.1007/s10534-006-9046-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.