

Abstract
Background
Dilated and hypertrophic cardiomyopathy mutations in troponin can blunt effects of protein kinase A (PKA) phosphorylation of cardiac troponin I (cTnI), decreasing myofilament Ca2+-sensitivity; however this effect has never been tested for restrictive cardiomyopathy (RCM) mutants. This study explores whether an RCM cardiac troponin T mutant (cTnT-ΔE96) interferes with convergent PKA regulation and if TnT instability contributes to greatly enhanced Ca2+-sensitivity in skinned fibers.
Methods and Results
A decrease of −0.26 and −0.25 pCa units in Ca2+-sensitivity of contraction after PKA incubation was observed for skinned fibers incorporated with WT or cTnT-ΔE96, respectively. To further assess whether cTnT-ΔE96 interferes solely with transmission of cTnI phosphorylation effects, skinned fibers were reconstituted with PKA pseudo-phosphorylated cTnI (cTnI-SS/DD.cTnC). Fibers displaced with cTnT-WT, reconstituted with cTnI-SS/DD.cTnC decreased Ca2+-sensitivity of force (pCa50 = 5.61) compared to control cTnI-WT. cTnC (pCa50 = 5.75), similarly affecting cTnT-ΔE96 (pCa50 = 6.03) compared to control cTnI-WT.cTnC (pCa50 = 6.14). Fluorescence studies measuring cTnCIAANS Ca2+-affinity changes due to cTnT-ΔE96 indicated higher complexity (thin filament) better recapitulates skinned fiber Ca2+ sensitive changes. Circular Dichroism revealed reduced α-helicity and earlier thermal unfolding for cTnT-ΔE96 compared to WT.
Conclusions
1) although ineffective in decreasing myofilament Ca2+-sensitivity to normal levels, cTnT-ΔE96 does not interfere with PKA cTnI phosphorylation mediated effects; 2) cTnT-ΔE96 requires actin to increase cTnC Ca2+-affinity; and 3) deletion of E96 reduces cTnT stability, likely disrupting crucial thin filament interactions.
General Significance
The pathological effect of cTnT-ΔE96 is largely manifested by dramatic myofilament Ca2+-sensitization which still persists even after PKA phosphorylation mediated Ca2+-desensitization.
Keywords: cardiac troponin T, restrictive cardiomyopathy, skinned fibers, troponin I phosphorylation, fluorescence, circular dichroism
1. INTRODUCTION
Restrictive Cardiomyopathy (RCM) is an uncommon cardiomyopathic disorder that is characterized by abnormal diastolic function that results from impaired ventricular filling, increased ventricular end-diastolic pressures, and dilated atria. RCM patients generally maintain systolic function, however dysfunction may occur in late stages thus leading to heart failure. Pediatric cases of RCM typically have poor prognosis and the treatment endpoint is often transplantation [1–5].
A number of mutations have been found in the genes encoding proteins that make up the cardiac troponin complex (cTn), the resulting mutant proteins have been shown to be significant causes of genetic based cardiomyopathies [6, 7]. To date, troponin-linked RCM mutations have been identified in the TNNI3 and TNNT2 genes. The first RCM mutation reported in the cTnT (TNNT2 gene) was a deletion of glutamic acid found at position 96 (cTnT-ΔE96) in a pediatric patient [1]. Cardiac Tn has an important role in regulating cardiac contractility, therefore amino acid deletions or substitutions that disrupt its function can lead to dysregulation of interactions between the thin and thick filaments [8, 9]. The cTn complex is constituted by three subunits: troponin C (cTnC), confers the Ca2+ sensitive properties to striated muscle; cTnI, prevents interactions of myosin with actin at subthreshold Ca2+ levels; cTnT, has a key role in activation of muscle contraction and physically links the Tn complex with tropomyosin (Tm) in the thin filament [10–12]. A more refined view of cTnT function has been derived from studying cardiomyopathic mutations in cTnT which appears to have additional nuanced roles in muscle contraction including modulation of actomyosin ATPase activity and the kinetics of contraction, Ca2+ sensitivity of contraction as well as maximal force [6, 13, 14].
Previously, our group has performed in vitro studies that elucidated the functional defects caused by the TNNT2 associated RCM mutation, the cTnT-ΔE96. Functional parameters of the mutant cTnT-ΔE96 were greatly altered, along with early presentation in the proband, both indicating the severity of the disease phenotype [1, 15]. Skinned fibers reconstituted with the cTnT-ΔE96 mutant protein showed a large increase in Ca2+ sensitivity of force and an inability to fully relax; reconstituted assays also revealed that the mutant troponin complex was unable to fully inhibit myosin-actin-tropomyosin ATPase activity [15]. Our findings were further corroborated by another study that recapitulated the increased Ca2+ sensitivity of contraction in skinned fibers containing cTnT-ΔE96, using a different protocol for incorporation of exogenous proteins [16]. Although little is known about the pathological mechanisms underlying TNNT2 RCM mutations, it has been previously suggested that the pathogenesis associated with TNNI3 RCM mutations involves drastic sensitization of the myofilament to Ca2+ [17, 18].
Developmentally important is the switching of TnI isoforms, from the fetal (slow skeletal TnI) to the the adult isoform (cTnI) during embryogenesis and postnatal development [19–21]. Since the patient had a severe onset of disease shortly after birth, we evaluated whether the deletion of amino acid E96 in cTnT futher altered regulatory mechanisms that modulate the contractile response in cTnI that contains the PKA target sites in the N-terminal extension. During β-adrenergic stimulation of the heart, cTnI is phosphorylated at serines 23 and 24 by PKA which decreases the Ca2+ sensitivity of contraction and enhances the relaxation rate of the heart [22–24]. Therefore, sarcomeric protein phosphorylation is a prominent mechanism for maintenance of cardiac function and homeostasis [25]. The rationale to study the effects of PKA phosphorylation in the presence of a RCM mutant is that recent reports indicate that sarcomeric mutants linked to dilated and hypertrophic cardiomyopathies interfere with the Ca2+ desensitizing effect of cTnI PKA phosphorylation [26–32]. However, this effect has never been tested for an RCM mutant protein incorporated into the thin filament.
This study was designed to elucidate the additional factors that could contribute to the severe disease demonstrated by the proband. The goals were twofold: 1) to test whether the RCM cTnT-ΔE96 mutant affected convergent regulation by cTnI PKA phosphorylation in skinned porcine fibers 2) to determine whether stability of the TnT underlies the mechanism causing greatly enhanced Ca2+ sensitivity of contraction seen in skinned fibers.
2. EXPERIMENTAL PROCEDURES
2.1. Cloning, Expression, and Purification of Human Cardiac Troponin, Cardiac Troponin T Isoforms WT and RCM Mutant
The cDNAs for human cTnI, and cTnC were cloned as previously described [33]. The pseudo-phosphorylated human cTnI-SS/DD was produced by overlapping mutagenic primers that replaced the two adjacent Ser at position 23/24, to aspartic acid (D). Standard laboratory protocols were utilized for expression and purification of human cTnC, cTnI (WT and SS/DD), and cTnT (WT and cTnT-ΔE96) [15, 33]. The porcine cardiac Tropomyosin (Tm) and rabbit skeletal actin were prepared as previously [33].
2.2. Cardiac Skinned Fiber Studies
2.2.1. Fiber Preparation
Porcine papillary muscle was isolated from porcine hearts and prepared according to the following methods [15]. The left ventricular papillary muscles were cut into strips and skinned overnight in a pCa 8.0 solution containing 50% glycerol and 1% Triton X-100 at 4 °C. Afterwards, the muscle strips were transferred to pCa 8.0 containing 50% glycerol, without Triton X-100 and stored up to 2 months at −20 °C.
2.2.2. Tn-displaced Skinned Cardiac Fibers
The effects of the RCM cTnT-ΔE96 mutant on Ca2+-dependent parameters of muscle contraction were determined upon displacement of endogenous porcine cTn with either the cTnT-WT or cTnT-ΔE96 mutant to be studied (for further details, see [15]. The fibers were then reconstituted with either binary complex: cTnI-WT. cTnC or cTnI-SS/DD.cTnC.
2.2.3. Steady State and Calcium Dependence of Force Development
Fibers were mounted on tweezer clips connected to a force transducer on one side and submerged in a 1.3-ml cuvette containing pCa 8.0 solution (10−8 M Ca2+, 1 mM Mg2+, 7 mM EGTA, 2.5 mM MgATP2−, 20 mM MOPS, pH 7.0, 20 mM creatine phosphate, and 15 units/ml creatine phosphokinase, ionic strength 150 mM). The Ca2+ sensitivity of contraction was measured by exposing the fibers to Ca2+-containing solutions of increasing Ca2+ concentrations ranging from pCa 8.0 to 4.0. Data were analyzed using the equation % change in force = 100 X [Ca]n/([Ca2+]n + [Ca2+ 50] n), where [Ca2+ 50] is the free Ca2+ concentration producing 50% force, and n is the Hill coefficient. For PKA measurements the skinned fibers were incubated with 500 units/ml PKA catalytic subunit (Sigma P2645) for 30 min in pCa 8.0.
2.3. Formation of Ternary and Binary Complexes
The troponin complexes were formed as previously described [15]. The correct stoichiometry of the binary or ternary complexes was verified by SDS-PAGE before storage of complexes at −80 °C.
2.4. Fluorescence Labeling of cTnC
For cTn and cTn including tropomyosin, the fluorescence measurements monitoring binding of Ca2+ to cTnC utililized the double label configuration with 2-(4′-(Iodoacetomido)aniline)Naphthalene-6-Sulfonic Acid (IAANS) located at Cys-35 and Cys-84 (prior to complex formation). For fluorescence measurements of the thin filament, cTnC had Cys-35 mutated to Ser (C35S) and were labeled with IAANS only at Cys-84. In this case, the troponin complexes were formed with only IAANS labeled cTnC C35S. IAANS was obtained from MolecularProbes, Plano, TX. Fluorescent labeling was performed according to established methods [34, 35].
2.5. Determination of Apparent Ca2+ Affinities by Fluorescence
Thin filaments were constructed using the protocol established in our laboratory [34]. Steady state fluorescence measurements were performed in a Jasco 6500 spectrofluorometer where IAANS fluorescence was excited at 330 nm and emission was detected at 450 nm. The protein concentrations used for the cTn, cTn with tropomyosin and thin filaments were 0.25μM, 0.54μM and 0.025 mg/ml, respectively. The concentration of free Ca2+ and amounts of titrated Ca2+ were calculated using the pCa calculator program [36]. The data were fitted to a version of the Hill equation that accounted for the spectral changes that occur at a low Ca2+ concentration.
2.6. Circular Dichroism Measurements
Far-UV CD spectra were collected using a 1-mm-path quartz cell in a Jasco J-720 spectropolarimeter. Spectra were recorded at 195–250 nm with a bandwidth of 1 nm at a speed of 50 nm/min, and a resolution of 0.5 nm at room temperature (20 °C). Ten scans were averaged, and no numerical smoothing was applied. The optical activity of the buffer was subtracted from relevant protein spectra. Mean residue ellipticities ([θ]MRE in millidegree.cm2/dmol) for the spectra were calculated using Jasco system software and the following equation: where [θ] is the measured ellipticity in millidegrees, Cr is the mean residue molar concentration, and L is the path length in cm. Protein concentrations were determined by the biuret reaction using bovine serum albumin as a standard. The experimental protein concentration for the cTnT-WT and cTnT-ΔE96 was 0.2 mg/ml. The buffer used contained 10 mM sodium phosphate pH 7.0, 0.5 M NaF and 1 mM DTT. For the thermal denaturation studies the wavelength was set at 222 nm (which represents the α-helical content) and the temperature was successively increased from 20–80°C.
2.7. Statistical Analysis
The experimental results are reported as mean ± S.E. and were analyzed for significance using Student’s t test at p< 0.05 (paired or unpaired depending on the experimental design).
3. RESULTS
3.1. Cardiac Skinned Fiber Experiments
3.1.1. PKA Incubation
Skinned fibers were displaced with cTnT-WT or cTnT-ΔE96, reconstituted with the binary complex cTnI.cTnC and the Ca2+ sensitivity of contraction was measured before and after PKA catalytic subunit incubation. Similar to what was previously published by our group [15], cTnT-ΔE96 sensitized the myofilament 0.38 pCa units compared to cTnT-WT (pCa50 6.11 ± 0.02 vs 5.73 ± 0.02). These same fibers, after PKA incubation, displayed a 0.26 and 0.25 pCa unit rightward shift in the Ca2+ sensitivity of force for the cTnT-WT and cTnT-ΔE96, respectively (Figure 1A and 1B and Table 1). Prior to PKA treatment, the cooperativity of thin filament activation (nH) was decreased in fibers displaced with the cTnT-ΔE96 mutant compared to cTnT-WT (Table 1). However, after PKA phosphorylation the nH only significantly increased in fibers containing cTnT-WT (Table 1). Note that even after PKA incubation, the fibers displaced with cTnT-ΔE96 still displayed increased myofilament Ca2+ sensitivity of 0.39 pCa units compared to cTnT-WT.
FIGURE 1. Normalized pCa force relationship in skinned cardiac muscle fibers before and after PKA incubation.

The Ca2+ dependence of force development was measured before (filled symbols) and after (open symbols) PKA catalytic subunit incubation. A) Fibers displaced with cTnT-WT; B) Fibers displaced with cTnT-ΔE96. Data are expressed as mean ± S.E.
Table 1. Summary of pCa-force relationship curves before and after PKA incubation in fibers reconstituted with cardiac TnI.TnC complex at pH 7.0.
The pCa50, nH and % Ca2+ unregulated force values are the average of many independent fiber experiments, and the errors are reported as S.E. values. The Ca2+ unregulated force was calculated using the following equation: , where the FpCa8 and FpCa4 are the force at pCa 8.0 and pCa 4.0 solutions, respectively.
| Before PKA treatment
|
After PKA treatment
|
||||||
|---|---|---|---|---|---|---|---|
| cTnT | pCa50 | Hill coefficient, nH | pCa50 | Hill coefficient, nH | ΔpCa50a | % Ca2+ Unregulated Force | N |
| cTnT-WT | 5.73 ± 0.02 | 2.95 ± 0.08 | 5.47 ± 0.02b | 3.73 ± 0.19b | −0.26 | 87.88 ± 4.95 | 6 |
| cTnT–ΔE96 | 6.11 ± 0.02c | 1.66 ± 0.05c | 5.86 ± 0.03b,d | 1.98 ± 0.17d | −0.25 | 96.19 ± 2.59 | 6 |
ΔpCa50 = pCa50 before PKA − pCa50 after PKA
p<0.05 same cTnT before vs after PKA treatment
p<0.05: cTnT–ΔE96 vs cTnT-WT before PKA treatment
p<0.05: cTnT–ΔE96 vs cTnT-WT after PKA treatment
3.1.2. PKA Phosphorylation Mimetic cTnI
Since PKA has been shown to have several targets in the myofilament including myosin binding protein C and troponin T, which could in turn affect myofilament Ca2+ sensitivity; we looked at effects of the cTnT-ΔE96 mutant in the presence of a pseudo-phosphorylated cTnI (cTnI-SS/DD) where the two serine sites 23 and 24 were replaced by aspartic acid. Skinned porcine fibers displaced with recombinantly expressed cTnT-WT and replaced with phosphorylation mimetic binary complex cTnI-SS/DD.cTnC recapitulated the effects of β-adrenergic stimulation, a decrease in Ca2+ sensitivity of force development ΔpCa50 = −0.14 was seen compared to fibers replaced with cTnI-WT.cTnC (See Figure 2A and Table 2). When the cTnT-ΔE96 mutant displaced cardiac skinned fibers were replaced with the PKA phosphorylation mimetic cTnI-SS/DD.cTnC, a similar decrease in Ca2+ sensitivity of force development ΔpCa50 = −0.11 was achieved (Figure 2B and Table 2). Whereas, displacement of endogenous cTnT with the cTnT-ΔE96 mutant led to decreased cooperativity indicated by the Hill coefficient (nH) in the presence of the phosphomimetic cTnI as well (Figure 2 and Table 2). Note that the difference in Ca2+ sensitivity of contraction between fibers displaced with cTnT-WT and cTnT-ΔE96 and reconstituted with cTnI-SS/DD.cTnC remained the same (+0.42 pCa units). No statistical differences in maximal force recovery (%) were found for the cTnT-ΔE96 mutant compared to cTnT-WT in the presence of cTnI-WT or cTnI-SS/DD (See Supplemental Figure 1).
FIGURE 2. Normalized pCa force relationship in skinned cardiac muscle fibers in the presence of PKA pseudo-phosphorylated cTnI.

The Ca2+ dependence of force development was measured in each preparation after cTnT displacement and binary complex reconstitution. In A) The pCa force relationship of fibers displaced with cTnT-WT and reconstituted with either cTnI-WT.cTnC (filled symbols) or PKA phosphorylation mimetic cTnI-SS/DD.cTnC complex (open symbols). Where in B) the skinned fibers were displaced with cTnT-ΔE96 and reconstituted with either cTnI-WT.cTnC (filled symbols) or the cTnI-SS/DD.cTnC (open symbols) complex. Data are expressed as mean ± S.E.
Table 2. Summary of pCa-force relationship curves in fibers reconstituted with different cardiac TnI.TnC complexes at pH 7.0.
The pCa50, nH and % Ca2+ unregulated force values are the average of many independent fiber experiments, and the errors are reported as S.E. values. The Ca2+ unregulated force was calculated by the following equation: , where the FpCa8 and FpCa4 are the force at pCa 8.0 and pCa 4.0 solutions, respectively.
| cTnT | cTnI | pCa50 | Hill coefficient, nH | ΔpCa50a | % Ca2+ unregulated Force | N |
|---|---|---|---|---|---|---|
| cTnT-WT | WT | 5.76 ± 0.02 | 2.68 ± 0.11 | - | 90.3 ± 3.4 | 8 |
| cTnT-WT | SS/DD | 5.62 ± 0.01b | 2.88 ± 0.22 | −0.14 | 82.2 ± 4.7 | 6 |
| cTnT–ΔE96 | WT | 6.14 ± 0.03c | 1.52 ± 0.05c | - | 95.5 ± 3.1 | 8 |
| cTnT–ΔE96 | SS/DD | 6.03 ± 0.06b,c | 1.63 ± 0.09c | −0.11 | 99.7 ± 0.3c | 5 |
ΔpCa50: cTnT + cTnI-SS/DD pCa50 − cTnT + cTnI-WT pCa50
p<0.05 cTnI-SS/DD vs cTnI-WT with the same cTnT
p<0.05 cTnT–ΔE96 vs cTnT-WT with the same cTnI
N = number of experiments
3.2. IAANS Fluorescence Measurements
The Ca2+ affinity measurements for cTnT-ΔE96 mutant were compared to that of cTnT-WT at different levels of thin filament complexity and changes of fluorescence signal could be detected due to changes in the conformation/environment of the extrinsic IAANS probe(s) bound to cTnC. The Ca2+ affinity of the cTn complex containing cTnI-WT was slightly decreased but significant for the cTn-ΔE96 mutant (pCa50 6.66 ± 0.01) versus cTn-WT (pCa50 6.69 ± 0.01) (Figure 3A and Table 3). When cTnT-ΔE96 was included in the cTn complex containing the pseudo-phosphorylated cTnI, there was a large decrease in Ca2+ affinity (ΔpCa50 = − 0.17) of the cTnC.cTnI-SS/DD.cTnT-ΔE96 mutant complex compared to cTnC.cTnI-SS/DD.cTnT-WT (Table 3). When porcine cardiac Tropomyosin (Tm) was added, the Ca2+ affinity of cTn was similar for either the cTn-ΔE96 mutant or cTn-WT (Figure 3B and Table 3). However, a large increase (ΔpCa50 = + 0.23) in Ca2+ affinity was detected for thin filaments containing the cTn-ΔE96 mutant versus cTn-WT (Figure 3C and Table 3). When the cTnT-ΔE96 mutant was incorporated into the thin filament containing cTnI-SS/DD an even greater increase in the Ca2+ affinity was seen (ΔpCa50 = + 0.30), this result is consistent with the skinned fiber data that showed that the cTnT-ΔE96 mutant did not ablate the Ca2+ sensitivity of contraction (Table 2 and 3).
FIGURE 3. Determination of the apparent Ca2+ affinities of troponin complexes containing cTnT-ΔE96 by fluorescence.
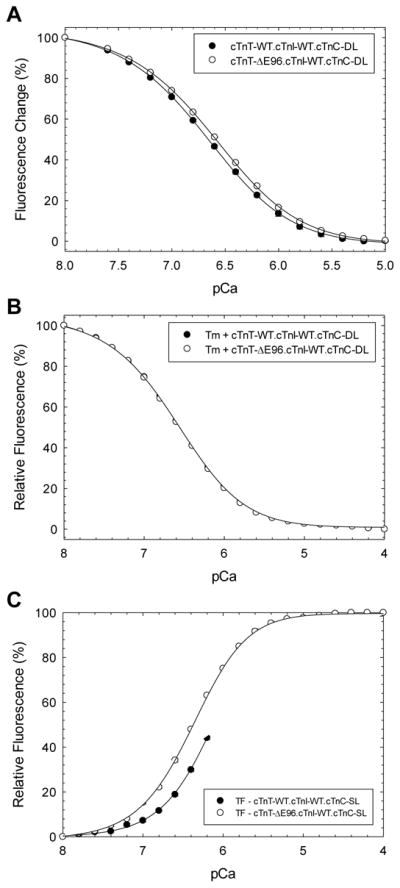
Steady state fluorescence measurements (see methods for details). A) troponin complex; B) tropomyosin and troponin; and C) thin filament. Data are expressed as mean ± S.E.
Table 3.
Summary of the fluorescence experiments.
| Troponin (cTnI-WT)
|
Tropomyosin + Troponin (cTnI-WT)
|
Thin Filament (cTnI-WT)
|
||||
|---|---|---|---|---|---|---|
| cTn-WT | cTn-ΔE96 | cTn-WT | cTn-ΔE96 | cTn-WT | cTn-ΔE96 | |
|
|
||||||
| pCa50 | 6.69 ± 0.01 | 6.66 ± 0.01a | 6.58 ± 0.01 | 6.59 ± 0.01 | 6.15 ± 0.02 | 6.38 ± 0.01a |
| nHill | 0.99 ± 0.02 | 1.05 ± 0.01a | 1.00 ± 0.01 | 1.04 ± 0.01a | 1.34 ± 0.04 | 1.28 ± 0.02 |
| Troponin (cTnI-SS/DD)
|
Tropomyosin + Troponin (cTnI-SS/DD)
|
Thin Filament (cTnI-SS/DD)
|
||||
|---|---|---|---|---|---|---|
| cTn-WT | cTn-ΔE96 | cTn-WT | cTn-ΔE96 | cTn-WT | cTn-ΔE96 | |
|
|
||||||
| sspCa50 | 6.71 ± 0.01 | 6.54 ± 0.01a,b | n/a | n/a | 6.03 ± 0.02c | 6.33 ± 0.01a,c |
| nHill | 1.16 ± 0.01b | 0.96 ± 0.01a,b | n/a | n/a | 1.39 ± 0.05 | 1.37 ± 0.05 |
p<0.05 RCM cTnT-ΔE96 mutant vs cTnT-WT within the same complex system.
p<0.05 troponin containing cTnT-WT.cTnI-WT vs troponin containing cTnT-WT.cTnI-SS/DD, or troponin containing cTnT-ΔE96.cTnI-WT vs troponin containing cTnT-ΔE96.cTnI-SS/DD.
p<0.05 thin filament containing cTnT-WT.cTnI-WT vs thin filament containing cTnT-WT.cTnI-SS/DD, or thin filament containing cTnT-ΔE96.cTnI-WT vs thin filament containing cTnT-ΔE96.cTnI-SS/DD.
n/a = not measured
Data are presented as mean ± s.e. n=4–5
3.3. Stability of the RCM cTnT Mutant
3.3.1. Circular Dichroism Analysis
The secondary structural characteristics of isolated cTnT-ΔE96 mutant were compared to cTnT-WT and it was found that the mutant had lower β-sheet content [θ]MRE = −12138.93 ± 265.52 at λ =222 than cTnT-WT with −14494.21 ± 138.99 at 21°C (Figure 4).
FIGURE 4. Determination of the secondary structural characteristics of the cTnT-ΔE96 mutant versus cTnT-WT.

Far-UV Circular Dichroism spectra was recorded at 195–250 nm at room temperature (20°C). Data are expressed as mean S.E. and n=7 performed for cTnT-WT and n=6 for cTnT-ΔE96.
3.3.2. Thermal Denaturation
Circular Dichroism was used to further assess the structural stability MRE% of the cTnT-ΔE96 mutant compared to the cTnT-WT, the proteins were subjected to thermal denaturation by incrementally increasing the temperature over a range of 20–80°C. The cTnT-ΔE96 mutant was physically less stable and had a lower melting temperature, T50 of 39.21 ± 0.93 °C compared to the cTnT-WT, T50 of 43.49 ± 0.63 °C (Figure 5A and 5B). This represents a loss of structural stability due to the deletion of glutamic acid 96 in cTnT, which could be the transmitted to the rest of the cTn complex and the adjoining thin filament. The alterations in TM or slope of a transition indicate that thermodynamic stability of the proteins is altered [37].
FIGURE 5. Circular Dichroism thermal denaturation curve monitored at a fixed wavelength.

The data was collected at λ=222nm. A) The thermal denaturation curve ([θ]MRE) of the RCM mutant cTnT-ΔE96 mutant versus cTnT-WT. B) Normalized graph of the thermal denaturation curve for the cTnT-ΔE96 RCM mutant versus cTnT-WT. Data are expressed as mean S.E. and n=4 performed for cTnT-WT and cTnT-ΔE96.
4. DISCUSSION
Sarcomeric protein mutations linked to HCM and DCM have been shown to uncouple the Ca2+ sensitivity from β-adrenergic mediated regulation of the myofilament [26–32, 38]. The lusiotropic effects of PKA phosphorylation of cTnI modulates cardiac contractility by increasing the rate of Ca2+ dissociation from the N-domain of cTnC [23, 24, 39–41]. Importantly, it has been shown that amino acid substitutions in other cTn subunits may cause the thin filament to become refractory to signal transduction by β-adrenergic pathways, as seen particularly with the phosphorylation of serines 23 and 24 in cTnI. Therefore, we evaluated the effects of the RCM cTnT-E96 deletion on the ability of cTnI to desensitize the myofilament to Ca2+ upon PKA phosphorylation. One of the questions that we addressed is whether an RCM mutant in the cTn complex has the same ability as HCM and DCM-linked mutants to impair cTnI PKA phosphorylation function at serines 23 and 24. Fibers incorporated with the RCM cTnT-ΔE96 mutant maintained the large increase in Ca2+ sensitivity compared to the WT-cTnT replaced fibers in every condition tested; therefore, the E96 deletion does not affect the ability of cTnI to modulate the Ca2+ sensitivity of contraction post PKA phosphorylation or in the presence of pseudo-phosphorylated cTnI. The phosphorylation-induced enhancement of the Ca2+ dissociation rate appears to be associated with global conformational changes in cTnI as shown by fluorescence anisotropy [42] and FRET measurements [43]. Additional functional changes induced by cTnI phosphorylation may be related to altered protein-protein interactions within the cTn complex [44–46].
The results obtained for the Ca2+ sensitivity of contraction and the cooperative activation of the myofilament, obtained using the phosphorylation mimetic cTnI-SS/DD, were consistent with that found when skinned fibers were incubated with PKA. The reduction in the pCa50 was less pronounced using this method compared to PKA incubation. This was not unexpected since PKA has numerous targets within the thin filament which would be phosphorylated when the entire skinned fiber was exposed to the PKA catalytic subunit. Therefore, the PKA-incubated skinned fibers more accurately portray what is happening in the myofilament during β-adrenergic stimulation since PKA phosphorylation of additional sites alter myofilament function [25, 47]. Since impairment of the PKA phosphorylation effects on myofilament Ca2+ sensitivity has not yet been studied in the presence of an RCM associated mutant; then, how would this lead to diastolic dysfunction? In this case, the degree of desensitization imposed by PKA phosphorylation may not effectively induce lusitropy due to the substantial Ca2+ sensitization caused by the RCM mutant. Furthermore, RCM is characterized by restrictive ventricular filling, therefore the properties at the myofilament level including increased basal force [15, 18] and enhanced contractility may result from ineffective modulation by β-adrenergic stimulation, contributing to ventricular stiffness and the severe diastolic dysfunction associated with RCM. Exploration of the thin filament hierarchy to determine the source of altered Ca2+ affinity of cTnC, due to the cTnT-ΔE96 mutant, revealed that only subtle changes occurred at the level of the troponin complex, with a small decrease in Ca2+ affinity for the mutant containing complex, with the phenotypic manifestations more pronounced in the thin filament. This finding is similar to what was shown by others [48–51]. In our study, the addition of Tm to Tn complex containing the (cTnT-WT or the cTnT-ΔE96 mutant) did not alter cTnC Ca2+ affinity. The Ca2+ affinity became increased in the more complex system (addition of actin) for the RCM-containing thin filament, indicating that the deletion of glutamic acid in cTnT, which lies at the TnT-Tm interface, alters the cTn-Tm interaction with actin. The location of the E96 deletion in the hypervariable N-terminal cTnT tail underlies its significant functional effects since this portion of cTnT is important for maintenance of diastolic function. As shown by Tobacman et al. that this portion of cTnT (1–153) was able to establish the blocked state without the presence of cTnI [13]. Transition from the blocked to closed state, is a known mechanism of increasing actomyosin ATPase activity at low Ca2+ and this RCM mutation has been previously shown to impair the ability of the troponin complex to inhibit actomyosin ATPase activity in the absence of Ca2+ [9, 15, 52]. Therefore, increased Ca2+ affinity of cTnC in the presence of the RCM deletion mutant may interfere with maintenance of the blocked state and subsequently alter interactions with actin. In addition, mutations (within the region 92–110) were shown to alter the Tm-dependent functions of the TnT fragment 70–170, such as binding to actin [53]. This data allows us to conclude that the E96 deletion in TnT indirectly affects Ca2+ binding to the cTnC N-terminal domain, most likely through direct effects on tropomyosin binding to actin. The fluorescence studies provided insight on the origin of altered Ca2+ affinity, that it was altered at the tropomyosin:actin interface.
In regards to cardiac function, a number of studies have shown that cardiomyopathy-associated mutants globally affect the troponin tail domain, in contrast to local effects manifested at the site of the substitution. To assess whether the ΔE96 deletion in TnT, altered its function in the thin filament, we determined whether the altered physical properties of cTnT-ΔE96 protein could be due to increased flexibility/decreased thermal stability. Two different groups found that the stability and/or flexibility of the TnT1 (tail domain) are crucial for the regulatory properties of tropomyosin and actin. In reference to our current study, it was previously shown that introduction of the mutants R92W, R94L, A104V and F110I into cTnT, changed the peptide Tm between −0.6 to −5.9 degrees Celsius, lower than the WT protein [53]. Although, these amino acid substitutions (between 92–110) introduced seemingly small changes in the physical properties of cTnT, the mutants have a diminished ability to stabilize the tropomyosin head-to-tail overlap complex. In another study, Hinkle and Tobacman showed that the R92Q (−1.8 degrees) and A104V (−4.2 degrees) mutants decreased the thermal stability of the TnT1 peptide (1–156) [54]. Since the troponin tail domain makes its primary interactions with Tm, any actin associated effects are thought to be indirect [54]. This results in weaker binding of Tn to Tm for most mutants in this region [53], as well as super-normal or sub-normal folding stability of the mutant TnT [54]. From this and previous studies we can suggest that altered interactions by TnT due to increased flexibility may be a common feature of cardiomyopathy-linked mutants located at the Tn-Tm interface. These changes in the properties of TnT, thus alters its ability to precisely regulate the transition from the active to inactive state during systole and diastole, respectively. An emerging concept is that mutations in tropomyosin that increase its flexibility are correlated with increased myofilament Ca2+ sensitivity [55–57], while tropomyosin mutations that decrease its flexibility are associated with decreased Ca2+ sensitivity [58–60]. These results have also been confirmed in mouse models [61]. Our study as well as others suggest that the flexibility of the troponin T tail may control/modulate Ca2+ sensitization in the same manner described for tropomyosin.
5. CONCLUSION
This study has explored the physical properties of the mutant cTnT-ΔE96 and effects of β-adrenergic regulation of the cTn complex containing the deletion. The severe phenotype manifested (early development of cardiac impairment) demonstrated by patient data including severe diastolic dysfunction and our functional data suggests that the cTnT-ΔE96 deletion mutant has substantial deleterious consequences that warranted further investigation. In summary, we have found that this deletion in cTnT contributes to disease development through altered protein stability which compromises its function, the heightened Ca2+ sensitivity in skinned fibers is improperly regulated by PKA mediated pathways which though not refractory, are ineffective at decreasing the Ca2+ sensitivity to the normal range, thus interfering with relaxation. Therefore, instability of cTnT caused by deletions of amino acid substitutions may be a major determinant leading to development of cardiomyopathy. Furthermore, it needs to be investigated whether ineffective PKA phosphorylation impairment is a trend or common phenotype for RCM mutations.
Supplementary Material
Highlights.
The RCM cTnT-ΔE96 mutant did not abolish effects of TnI phosphorylation
The malignant effects exerted by RCM cTnT-ΔE96 require the thin filament
Source of greatly enhanced myofilament calcium sensitivity was actin:Tm interface
The deletion of E96 led to an overall loss in stability of cTnT
Among cardiomyopathies, RCM Tn mutants may display distinct molecular phenotypes
Acknowledgments
MP received support from the American Heart Association Postdoctoral Fellowship 09POST2300030 and JRP from NIH HL103840 and the James & Esther King Biomedical Foundation grant 1KN13-34001.
We thank Michelle A. Jones and Jingsheng Liang for their excellent technical assistance during this project.
ABBREVIATIONS
- cTnT
cardiac Troponin T
- RCM
restrictive cardiomyopathy
- PKA
protein kinase A
- cTnT-ΔE96
cardiac troponin T with glutamic acid 96 deleted
- cTnI-SS/DD
cardiac troponin I with serines 23 and 24 mutated to aspartic acid
- IAANS
2-(4′-(Iodoacetomido)aniline)Naphthalene-6-Sulfonic Acid
Footnotes
The content of this study is solely the responsibility of the authors, and does not necessarily represent the official view of the awarding organization.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Peddy SB, Vricella LA, Crosson JE, Oswald GL, Cohn RD, Cameron DE, et al. Infantile restrictive cardiomyopathy resulting from a mutation in the cardiac troponin T gene. Pediatrics. 2006;117:1830–3. doi: 10.1542/peds.2005-2301. [DOI] [PubMed] [Google Scholar]
- 2.Kimberling MT, Balzer DT, Hirsch R, Mendeloff E, Huddleston CB, Canter CE. Cardiac transplantation for pediatric restrictive cardiomyopathy: presentation, evaluation, and short-term outcome. J Heart Lung Transplant. 2002;21:455–9. doi: 10.1016/s1053-2498(01)00400-4. [DOI] [PubMed] [Google Scholar]
- 3.Rivenes SM, Kearney DL, Smith EO, Towbin JA, Denfield SW. Sudden death and cardiovascular collapse in children with restrictive cardiomyopathy. Circulation. 2000;102:876–82. doi: 10.1161/01.cir.102.8.876. [DOI] [PubMed] [Google Scholar]
- 4.Kaski JP, Syrris P, Burch M, Tome-Esteban MT, Fenton M, Christiansen M, et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart. 2008;94:1478–84. doi: 10.1136/hrt.2007.134684. [DOI] [PubMed] [Google Scholar]
- 5.Yang SW, Hitz MP, Andelfinger G. Ventricular septal defect and restrictive cardiomyopathy in a paediatric TNNI3 mutation carrier. Cardiol Young. 2010;20:574–6. doi: 10.1017/S1047951110000715. [DOI] [PubMed] [Google Scholar]
- 6.Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, Potter JD. Mutations in Troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function? Journal of molecular and cellular cardiology. 2010;48:882–92. doi: 10.1016/j.yjmcc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 7.Parvatiyar MS, Pinto JR, Dweck D, Potter JD. Cardiac troponin mutations and restrictive cardiomyopathy. J Biomed Biotechnol. 2010;2010:350706. doi: 10.1155/2010/350706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tardiff JC. Thin filament mutations: developing an integrative approach to a complex disorder. Circulation research. 2011;108:765–82. doi: 10.1161/CIRCRESAHA.110.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chalovich JM. Disease causing mutations of troponin alter regulated actin state distributions. Journal of muscle research and cell motility. 2012;33:493–9. doi: 10.1007/s10974-012-9305-x. [DOI] [PubMed] [Google Scholar]
- 10.Potter JD, Sheng Z, Pan BS, Zhao J. A direct regulatory role for troponin T and a dual role for troponin C in the Ca2+ regulation of muscle contraction. J Biol Chem. 1995;270:2557–62. doi: 10.1074/jbc.270.6.2557. [DOI] [PubMed] [Google Scholar]
- 11.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 12.Farah CS, Reinach FC. The troponin complex and regulation of muscle contraction. Faseb J. 1995;9:755–67. doi: 10.1096/fasebj.9.9.7601340. [DOI] [PubMed] [Google Scholar]
- 13.Tobacman LS, Nihli M, Butters C, Heller M, Hatch V, Craig R, et al. The troponin tail domain promotes a conformational state of the thin filament that suppresses myosin activity. J Biol Chem. 2002;277:27636–42. doi: 10.1074/jbc.M201768200. [DOI] [PubMed] [Google Scholar]
- 14.Gomes AV, Barnes JA, Harada K, Potter JD. Role of troponin T in disease. Mol Cell Biochem. 2004;263:115–29. doi: 10.1023/B:MCBI.0000041853.20588.a0. [DOI] [PubMed] [Google Scholar]
- 15.Pinto JR, Parvatiyar MS, Jones MA, Liang J, Potter JD. A troponin T mutation that causes infantile restrictive cardiomyopathy increases Ca2+ sensitivity of force development and impairs the inhibitory properties of troponin. J Biol Chem. 2008;283:2156–66. doi: 10.1074/jbc.M707066200. [DOI] [PubMed] [Google Scholar]
- 16.Bai F, Caster HM, Pinto JR, Kawai M. Analysis of the molecular pathogenesis of cardiomyopathy-causing cTnT mutants I79N, DeltaE96, and DeltaK210. Biophysical journal. 2013;104:1979–88. doi: 10.1016/j.bpj.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yumoto F, Lu Q-W, Morimoto S, Tanaka H, Kono N, Nagata K, et al. Drastic Ca2+ sensitization of myofilament associated with a small structural change in troponin I in inherited restrictive cardiomyopathy. Biochemical and Biophysical Research Communications. 2005;338:1519–26. doi: 10.1016/j.bbrc.2005.10.116. [DOI] [PubMed] [Google Scholar]
- 18.Gomes AV, Liang J, Potter JD. Mutations in Human Cardiac Troponin I That Are Associated with Restrictive Cardiomyopathy Affect Basal ATPase Activity and the Calcium Sensitivity of Force Development. J Biol Chem. 2005;280:30909–15. doi: 10.1074/jbc.M500287200. [DOI] [PubMed] [Google Scholar]
- 19.Westfall MV, Borton AR, Albayya FP, Metzger JM. Myofilament Calcium Sensitivity and Cardiac Disease: Insights From Troponin I Isoforms and Mutants. Circ Res. 2002;91:525–31. doi: 10.1161/01.res.0000034710.46739.c0. [DOI] [PubMed] [Google Scholar]
- 20.Sasse S, Brand NJ, Kyprianou P, Dhoot GK, Wade R, Arai M, et al. Troponin I gene expression during human cardiac development and in end-stage heart failure. Circ Res. 1993;72:932–8. doi: 10.1161/01.res.72.5.932. [DOI] [PubMed] [Google Scholar]
- 21.Perry SV. Troponin I: inhibitor or facilitator. Mol Cell Biochem. 1999;190:9–32. [PubMed] [Google Scholar]
- 22.Kranias EG, Solaro RJ. Phosphorylation of troponin I and phospholamban during catecholamine stimulation of rabbit heart. Nature. 1982;298:182–4. doi: 10.1038/298182a0. [DOI] [PubMed] [Google Scholar]
- 23.Zhang R, Zhao J, Mandveno A, Potter JD. Cardiac Troponin I Phosphorylation Increases the Rate of Cardiac Muscle Relaxation. Circ Res. 1995;76:1028–35. doi: 10.1161/01.res.76.6.1028. [DOI] [PubMed] [Google Scholar]
- 24.Robertson SP, Johnson JD, Holroyde MJ, Kranias EG, Potter JD, Solaro RJ. The effect of troponin I phosphorylation on the Ca2+-binding properties of the Ca2+-regulatory site of bovine cardiac troponin. J Biol Chem. 1982;257:260–3. [PubMed] [Google Scholar]
- 25.Solaro RJ. Multiplex kinase signaling modifies cardiac function at the level of sarcomeric proteins. The Journal of biological chemistry. 2008;283:26829–33. doi: 10.1074/jbc.R800037200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biesiadecki BJ, Kobayashi T, Walker JS, John Solaro R, de Tombe PP. The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circ Res. 2007;100:1486–93. doi: 10.1161/01.RES.0000267744.92677.7f. [DOI] [PubMed] [Google Scholar]
- 27.Gomes AV, Harada K, Potter JD. A mutation in the N-terminus of Troponin I that is associated with hypertrophic cardiomyopathy affects the Ca2+-sensitivity, phosphorylation kinetics and proteolytic susceptibility of troponin. Journal of Molecular and Cellular Cardiology. 2005;39:754–65. doi: 10.1016/j.yjmcc.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 28.Pinto JR, Siegfried JD, Parvatiyar MS, Li D, Norton N, Jones MA, et al. Functional characterization of TNNC1 rare variants identified in dilated cardiomyopathy. J Biol Chem. 2011;286:34404–12. doi: 10.1074/jbc.M111.267211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Pinto JR, Solis RS, Dweck D, Liang J, Diaz-Perez Z, et al. Generation and Functional Characterization of Knock-in Mice Harboring the Cardiac Troponin I-R21C Mutation Associated with Hypertrophic Cardiomyopathy. J Biol Chem. 2012;287:2156–67. doi: 10.1074/jbc.M111.294306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Memo M, Leung MC, Ward DG, dos Remedios C, Morimoto S, Zhang L, et al. Familial dilated cardiomyopathy mutations uncouple troponin I phosphorylation from changes in myofibrillar Ca(2)(+) sensitivity. Cardiovasc Res. 2013;99:65–73. doi: 10.1093/cvr/cvt071. [DOI] [PubMed] [Google Scholar]
- 31.Bayliss CR, Jacques AM, Leung MC, Ward DG, Redwood CS, Gallon CE, et al. Myofibrillar Ca(2+) sensitivity is uncoupled from troponin I phosphorylation in hypertrophic obstructive cardiomyopathy due to abnormal troponin T. Cardiovasc Res. 2013;97:500–8. doi: 10.1093/cvr/cvs322. [DOI] [PubMed] [Google Scholar]
- 32.van Dijk SJ, Paalberends ER, Najafi A, Michels M, Sadayappan S, Carrier L, et al. Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ Heart Fail. 2012;5:36–46. doi: 10.1161/CIRCHEARTFAILURE.111.963702. [DOI] [PubMed] [Google Scholar]
- 33.Gomes AV, Guzman G, Zhao J, Potter JD. Cardiac troponin T isoforms affect the Ca2+ sensitivity and inhibition of force development: insights into the role of troponin T isoforms in the heart. J Biol Chem. 2002;277:35341–49. doi: 10.1074/jbc.M204118200. [DOI] [PubMed] [Google Scholar]
- 34.Pinto JR, Parvatiyar MS, Jones MA, Liang J, Ackerman MJ, Potter JD. A functional and structural study of troponin C mutations related to hypertrophic cardiomyopathy. The Journal of biological chemistry. 2009;284:19090–100. doi: 10.1074/jbc.M109.007021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Putkey JA, Liu W, Lin X, Ahmed S, Zhang M, Potter JD, et al. Fluorescent probes attached to Cys 35 or Cys 84 in cardiac troponin C are differentially sensitive to Ca(2+)-dependent events in vitro and in situ. Biochemistry. 1997;36:970–8. doi: 10.1021/bi9617466. [DOI] [PubMed] [Google Scholar]
- 36.Dweck D, Reyes-Alfonso A, Jr, Potter JD. Expanding the range of free calcium regulation in biological solutions. Anal Biochem. 2005;347:303–15. doi: 10.1016/j.ab.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 37.Greenfield NJ. Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nat Protoc. 2006;1:2527–35. doi: 10.1038/nprot.2006.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song W, Dyer E, Stuckey D, Leung MC, Memo M, Mansfield C, et al. Investigation of a transgenic mouse model of familial dilated cardiomyopathy. Journal of molecular and cellular cardiology. 2010;49:380–9. doi: 10.1016/j.yjmcc.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 39.Takimoto E, Soergel DG, Janssen PM, Stull LB, Kass DA, Murphy AM. Frequency- and afterload-dependent cardiac modulation in vivo by troponin I with constitutively active protein kinase A phosphorylation sites. Circulation research. 2004;94:496–504. doi: 10.1161/01.RES.0000117307.57798.F5. [DOI] [PubMed] [Google Scholar]
- 40.Yasuda S, Coutu P, Sadayappan S, Robbins J, Metzger JM. Cardiac transgenic and gene transfer strategies converge to support an important role for troponin I in regulating relaxation in cardiac myocytes. Circulation research. 2007;101:377–86. doi: 10.1161/CIRCRESAHA.106.145557. [DOI] [PubMed] [Google Scholar]
- 41.Sakthivel S, Finley NL, Rosevear PR, Lorenz JN, Gulick J, Kim S, et al. In vivo and in vitro analysis of cardiac troponin I phosphorylation. The Journal of biological chemistry. 2005;280:703–14. doi: 10.1074/jbc.M409513200. [DOI] [PubMed] [Google Scholar]
- 42.Liao R, Wang CK, Cheung HC. Coupling of calcium to the interaction of troponin I with troponin C from cardiac muscle. Biochemistry. 1994;33:12729–34. doi: 10.1021/bi00208a026. [DOI] [PubMed] [Google Scholar]
- 43.Dong WJ, Chandra M, Xing J, She M, Solaro RJ, Cheung HC. Phosphorylation-induced distance change in a cardiac muscle troponin I mutant. Biochemistry. 1997;36:6754–61. doi: 10.1021/bi9622276. [DOI] [PubMed] [Google Scholar]
- 44.Finley N, Abbott MB, Abusamhadneh E, Gaponenko V, Dong W, Gasmi-Seabrook G, et al. NMR analysis of cardiac troponin C-troponin I complexes: effects of phosphorylation. FEBS Lett. 1999;453:107–12. doi: 10.1016/s0014-5793(99)00693-6. [DOI] [PubMed] [Google Scholar]
- 45.Ababou A, Desjarlais JR. Solvation energetics and conformational change in EF-hand proteins. Protein Sci. 2001;10:301–12. doi: 10.1110/ps.33601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmidtmann A, Lohmann K, Jaquet K. The interaction of the bisphosphorylated N-terminal arm of cardiac troponin I-A 31P-NMR study. FEBS letters. 2002;513:289–93. doi: 10.1016/s0014-5793(02)02340-2. [DOI] [PubMed] [Google Scholar]
- 47.Sadayappan S, Gulick J, Osinska H, Barefield D, Cuello F, Avkiran M, et al. A critical function for Ser-282 in cardiac Myosin binding protein-C phosphorylation and cardiac function. Circulation research. 2011;109:141–50. doi: 10.1161/CIRCRESAHA.111.242560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kobayashi T, Solaro RJ. Increased Ca2+ Affinity of Cardiac Thin Filaments Reconstituted with Cardiomyopathy-related Mutant Cardiac Troponin I. J Biol Chem. 2006;281:13471–7. doi: 10.1074/jbc.M509561200. [DOI] [PubMed] [Google Scholar]
- 49.Robinson P, Griffiths PJ, Watkins H, Redwood CS. Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res. 2007;101:1266–73. doi: 10.1161/CIRCRESAHA.107.156380. [DOI] [PubMed] [Google Scholar]
- 50.Liu B, Tikunova SB, Kline KP, Siddiqui JK, Davis JP. Disease-related cardiac troponins alter thin filament Ca2+ association and dissociation rates. PloS one. 2012;7:e38259. doi: 10.1371/journal.pone.0038259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dweck D, Hus N, Potter JD. Challenging current paradigms related to cardiomyopathies. Are changes in the Ca2+ sensitivity of myofilaments containing cardiac troponin C mutations (G159D and L29Q) good predictors of the phenotypic outcomes? The Journal of biological chemistry. 2008;283:33119–28. doi: 10.1074/jbc.M804070200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McKillop DF, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palm T, Graboski S, Hitchcock-DeGregori SE, Greenfield NJ. Disease-causing mutations in cardiac troponin T: identification of a critical tropomyosin-binding region. Biophys J. 2001;81:2827–37. doi: 10.1016/S0006-3495(01)75924-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hinkle A, Tobacman LS. Folding and function of the troponin tail domain. Effects of cardiomyopathic troponin T mutations. J Biol Chem. 2003;278:506–13. doi: 10.1074/jbc.M209194200. [DOI] [PubMed] [Google Scholar]
- 55.Li XE, Suphamungmee W, Janco M, Geeves MA, Marston SB, Fischer S, et al. The flexibility of two tropomyosin mutants, D175N and E180G, that cause hypertrophic cardiomyopathy. Biochemical and biophysical research communications. 2012;424:493–6. doi: 10.1016/j.bbrc.2012.06.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Loong CK, Zhou HX, Chase PB. Familial hypertrophic cardiomyopathy related E180G mutation increases flexibility of human cardiac alpha-tropomyosin. FEBS letters. 2012;586:3503–7. doi: 10.1016/j.febslet.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heller MJ, Nili M, Homsher E, Tobacman LS. Cardiomyopathic tropomyosin mutations that increase thin filament Ca2+ sensitivity and tropomyosin N-domain flexibility. The Journal of biological chemistry. 2003;278:41742–8. doi: 10.1074/jbc.M303408200. [DOI] [PubMed] [Google Scholar]
- 58.Sumida JP, Wu E, Lehrer SS. Conserved Asp-137 imparts flexibility to tropomyosin and affects function. The Journal of biological chemistry. 2008;283:6728–34. doi: 10.1074/jbc.M707485200. [DOI] [PubMed] [Google Scholar]
- 59.Matyushenko AM, Artemova NV, Shchepkin DV, Kopylova GV, Bershitsky SY, Tsaturyan AK, et al. Structural and functional effects of two stabilizing substitutions, D137L and G126R, in the middle part of alpha-tropomyosin molecule. The FEBS journal. 2014 doi: 10.1111/febs.12756. [DOI] [PubMed] [Google Scholar]
- 60.Mirza M, Robinson P, Kremneva E, Copeland O, Nikolaeva O, Watkins H, et al. The effect of mutations in alpha-tropomyosin (E40K and E54K) that cause familial dilated cardiomyopathy on the regulatory mechanism of cardiac muscle thin filaments. The Journal of biological chemistry. 2007;282:13487–97. doi: 10.1074/jbc.M701071200. [DOI] [PubMed] [Google Scholar]
- 61.Yar S, Chowdhury SA, Davis RT, 3rd, Kobayashi M, Monasky MM, Rajan S, et al. Conserved Asp-137 is important for both structure and regulatory functions of cardiac alpha-tropomyosin (alpha-TM) in a novel transgenic mouse model expressing alpha-TM-D137L. The Journal of biological chemistry. 2013;288:16235–46. doi: 10.1074/jbc.M113.458695. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.