

Systemic juvenile idiopathic arthritis (sJIA) is a disease characterized by arthritis in children <16 years of age. JIA occurs in approximately 11/100,000 children annually in Europe and the United States, with systemic onset in 6–11%.1,2 sJIA is associated with serious complications including growth failure, osteoporosis, and musculoskeletal deformities; the main causes of sJIA-related deaths are infection and macrophage activation syndrome.3–5 Because interleukin-1 (IL-1) is a key mediator of the inflammatory cascade contributing to sJIA pathogenesis,6,7 this pro-inflammatory cytokine represents a therapeutic target.
Rilonacept (IL-1 Trap) is an IL-1 neutralizer incorporating into 1 molecule the extra-cellular domain of 2 human cytokine receptors required for IL-1 signaling, combined with the Fc portion of human IgG1.8 Rilonacept binds to IL-1β with high affinity and specificity, and blocks inflammation caused by overproduction of IL-1. This novel therapeutic molecule is therefore hypothesized to be effective against auto-immune diseases such as sJIA.
The pharmacokinetics (PK) of rilonacept have been evaluated in adults and children in a small study. In adults, the terminal apparent elimination half-life (T1/2) was approximately 1 week (154–184 hours) irrespective of dose, route of administration, or disease status.8 In children, a mean apparent T1/2 of 151 hours was observed.8 Rilonacept PK is not affected by renal failure as evidenced by similar PK observed in patients with end-stage renal disease on hemodialysis.9
Three clinical trials evaluating rilonacept in adults with rheumatoid arthritis have been completed: 2 phase I studies (combined N=137 patients) and 1 phase II trial (N=201). In both, rilonacept was generally well tolerated. The most frequently reported adverse event was injection site reaction, and no serious adverse events were felt to be drug-related. These studies suggested the best dose for children is 2–4 mg/kg/week subcutaneously (SC).
Recently, the Randomized, Placebo Phase Study of Rilonacept in sJIA (RAPPORT) study evaluated the efficacy and safety of rilonacept in children with sJIA.10 Dosing data in this trial were informed by review of data from the Regeneron pilot study, where there was no difference in efficacy in children who received a 4.4mg/kg loading dose/2.2mg/kg maintenance dose vs a loading dose of 8.8mg/kg and a maintenance dose of 4.4mg/kg.11 The current report presents the PK analysis of rilonacept conducted in this study.
Methods
PK samples for this analysis were obtained during routine study visits of the Randomized Placebo Phase study Of Rilonacept in the Treatment of sJIA (RAPPORT, 2007–2012, NCT00534495).10 RAPPORT was a multicenter, prospective, randomized, controlled, placebo-phase, double-blind study of rilonacept in 75 children 1–18 years of age with active sJIA. Two treatment arms were randomized 1:1 to the following groups—Group A: rilonacept loading SC dose of 4.4 mg/kg given at day 0 and then once weekly at a dose of 2.2 mg/kg throughout the 24-week study; Group B: placebo SC doses at days 0, 7, 14, and 21 followed by a loading dose of rilonacept (4.4 mg/kg) on day 28, and then rilonacept at 2.2 mg/kg once weekly thereafter until completion of 24 weeks. This design was meant to assess rilonacept efficacy by measuring a potential difference in time to response in Group B who received the active drug 4 week later than Group A. The study was approved by the institutional review boards at each institution, and informed consent was obtained from a parent or guardian prior to enrollment. Assent was obtained when possible in children ≥7 years of age.
Sparse PK sampling occurred at 3 time points; within 2 hours prior to administration of study drug at weeks 4, 14, and 24 post-randomization. Exact timing of rilonacept administration was not recorded and was assumed to occur 2 hours after PK sample collection. Dosing time was therefore imputed from the time of PK sample collection. Blood was collected in a 4.5 mL CTAD tube and centrifuged at 1,500 X g and 4°C for 10 minutes immediately prior to freezing at the study sites. PK samples were sent to Quest Diagnostics for storage at −70°C prior to analysis.
PK samples were analyzed at Regeneron Pharmaceuticals with an enzyme-linked immunosorbent assay. The inter- and intra-day accuracy and precision of the assay were ±25% and ≤25% coefficient variation, respectively. The assay was linear across the dosing range (0.08–0.5 ng/mL), and the lower limit of quantification was 0.08 ng/mL.
PK data were analyzed with a nonlinear mixed-effect modeling approach using NONMEM (version 7.2, Icon Solutions) in conjunction with Pirana (version 2.7.0b), Perl-speaks-NONMEM, and WINGS for NONMEM version 7.03 (Auckland, NZ). NONMEM output was summarized using STATA 12 (College Station, TX, USA). The first-order conditional estimation method with interaction was used for all model runs. The drug concentration data were transformed to mg/L prior to analysis. One- and 2-compartment structural PK models were evaluated. Inter-individual (IIV) random effects were evaluated on clearance (CL/F), distributional clearance (Q), and volume (V/F) parameters. An exponential model for IIV variance was used, and a proportional plus additive error model was deemed appropriate to describe residual variability. The potential effect of clinical covariates on PK parameters was explored if a relationship was suggested by visual inspection of scatter and box plots (continuous and categorical variables, respectively) of the individual deviations from the population-typical value PK parameters (ETAs) against covariates. The following covariates were explored: age, postmenstrual age (assumes gestational age of 40 weeks for all subjects), alanine aminotransferase level, aspartate aminotransferase level, serum albumin, serum bilirubin (conjugated and total), C-reactive protein, serum creatinine, serum ferritin, serum globulins, hematocrit, white blood cell count, sex, race, ethnicity, use of steroids, use of methotrexate, and use of naproxen. Body weight was assumed to be a significant covariate for clearance and volume and was included in the base model prior to assessment of other potential covariates. The relationship between body weight and PK parameters was characterized using an allometric relationship between weight and clearance parameters and a linear-scale for volume parameters (Equations 1 and 2).12,13 Scaling was based on a 70 kg standardized adult weight to allow direct comparison of our results with prior adult studies.13
| Equation 1 |
| Equation 2 |
Where CLstd represents the population parameter estimate for apparent drug clearance in a 70 kg adult; WTi is the individual weight; and Vstd is the population parameter estimate for apparent volume of distribution in a 70 kg adult. These same relationships were applied for all clearance (i.e., CL, Q) and volume of distribution (i.e., VC, VP) parameters. Once covariates were identified by visual inspection and physiologic plausibility, incorporation of covariates into the model was planned via standard forward addition backward elimination methods. Covariates that reduced the objective function by more than 3.84 (P<~0.05) during univariable analysis were included in a subsequent multivariable analysis. In the multivariable step, a reduction of 6.64 (P<~0.01) was required for retention of a covariate in the final model. Continuous covariates were scaled to their median values. Covariates that exhibited time-dependent changes (e.g., weight) were permitted to change with time. Missing body weights and laboratory values were imputed with closest value carried forward or back filled. Empirical Bayesian estimates of individual PK parameters were generated from the final model using the post-hoc subroutine and were summarized using descriptive statistics.
During the population PK model-building process, models were evaluated based on successful minimization, goodness-of-fit plots, plausibility of parameter estimates, objective function and shrinkage values, and precision of parameter estimates. The precision of the final population PK model parameter estimates were evaluated using non-parametric bootstrapping (1000 replicates) to generate the 95% confidence intervals for parameter estimates. For the visual predictive check, the final model was used to generate 1000 Monte Carlo simulation replicates per time point of rilonacept exposure, and simulated results were compared with those observed in the study. The number of observed concentrations outside of the 90% prediction interval for each time point was quantified. The dosing and covariate values used to generate the simulations in the visual predictive check were the same as those used in the study population.
We explored the relationship between rilonacept response and systemic exposure, as measured by the area under the concentration-time curve from time 0 to infinity (AUC0–inf). Similarly to the primary endpoint in the efficacy trial, rilonacept response was defined as a composite of improvement in the American College of Rheumatology Pediatric 30 (ACR Pedi 30) criteria,14 the absence of fever ≥38.5ºC in the previous 2 weeks, and at least 10% taper in the dose of systemic corticosteroids from baseline in patients receiving corticosteroids.10 We compared AUC0-inf between subjects who responded to rilonacept at 12 weeks vs subjects who did not respond using a Wilcoxon rank-sum test. This statistical analysis was performed using STATA 12 (College Station, TX). The statistical test was 2-sided with significance defined as p <0.05.
Results
Seventy-one children were included in the original datasets, of whom 66 were included in the PK analysis. Patients were excluded from the PK analysis due to missing dosing information (N=5), missing PK samples (N=1), and all concentration records equal to zero (N=3). The overall median (range) age and weight were 11 (1–18) years and 33 (11–120) kg, respectively. Rilonacept doses included in the analysis totaled 1351, with median dosing of 2.1 (1.3–5.4) mg/kg. Median serum creatinine and albumin were 0.5 (0.2–1.0) mg/dL and 4.1 (3.0–5.0) g/dL, respectively. The majority of subjects were female (65%). Sixty-seven percent were white, 17% were black, and 17% were Hispanic. The median concentration was 19.5 (0.03–53.3) mcg/mL. A total of 147 PK samples were included in the analysis with an average of 2.6 samples per subject (range 1–3).
A 1-compartment model with first-order absorption appropriately characterized the PK data as evidenced by goodness-of-fit plots (Figure 1). Lack of PK samples around the absorption phase prevented estimation of Ka, and therefore this parameter was fixed at the adult observed parameter of 0.5 day−1.15 During the univariable analysis of the population PK model-building process, rilonacept CL/F was significantly associated with the following covariates: serum albumin (change in the objective function value [ΔOFV] of −21.2 relative to the base model with weight), age (years) (ΔOFV −7.8), and aspartate aminotransferase (ΔOFV −6.6) (Supplemental Table S1). However, only serum albumin and age remained significant in the multivariable analysis (ΔOFV −7.7 relative to the albumin model). Rilonacept CL/F was therefore inversely related to serum albumin and directly related to body weight and age: CL/F = 1.495*(WT/70)0.75*(Albumin/4)−2.72*(Age/40)0.23. High shrinkage (>50%) in IIV estimates for V prevented accurate estimation of this random effect.
Figure 1.

Final population PK model diagnostic plots, showing observed versus population predictions (A), and individual predictions (B), and weighted residuals versus population predictions (C), and time (D). In panels A and B, the line of identity (solid black line) is included as a reference, and the dotted line represents the linear regression fit. For weighted residuals, a solid line at y = 0 is included as a reference and the dotted line represents the loess line.
For the final PK model, relative standard errors (RSE) of most parameter estimates were ≤30%, and the percent difference between model and bootstrapped median parameter estimates was <10% for fixed effects and <20% for random effects; 99% of bootstrap runs minimized successfully and, in 95%, the covariance step ran successfully (Supplemental Table S2). Changes in the assumed Ka value (e.g., 0.3, 0.7 day−1) did not result in changes in PK parameter estimates (data not shown). Goodness-of-fit diagnostic plots for the final model are shown in Figure 1. Eleven observed concentrations (7.5%) were outside of the visual predicted check 90% prediction interval, indicating good predictive performance of the final model (Supplemental Figure S1).
Because a 2-compartment model has been reported as a good fit for rilonacept PK data generated in adult patients with cryopyrin-associated periodic syndrome (CAPS),16 this model was also evaluated for goodness of fit in these sparse pediatric data. The 2-compartment model did not provide an improved fit over the 1-compartment model. RSEs of most parameter estimates for this model were ≤30%; however, the estimate for Vc was relatively imprecise (RSE >50%), and bootstrap estimates for Q were near parameter boundaries (data not shown); 94% of bootstrap runs minimized successfully and, in 51%, the covariance step ran successfully.
The median (5th, 95th percentile) CL/F, AUC0–inf, and T1/2 individual empirical Bayesian estimates from the final model were 0.557 L/day (0.151, 1.671), 158.1 mcg*days/mL (79.2, 239.8), and 8.9 days (3.8, 161.1), respectively (Supplemental Table S3). CL/F and T1/2 increased and decreased with age, respectively; an inverse relationship between CL/F and serum albumin was also observed (Figure 2).
Figure 2.
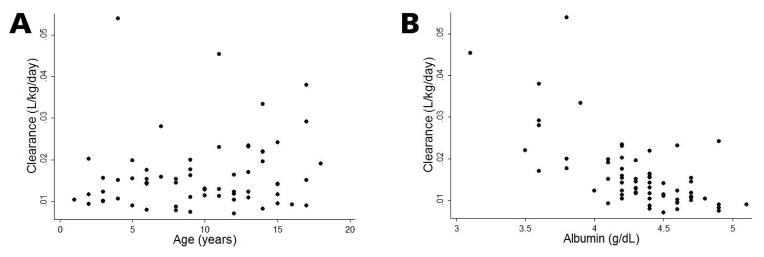
Rilonacept clearance (CL/F) vs. age (panel A) and albumin (panel B)
Values are individual empirical Bayesian CL/F estimates at first PK sample.
Among the 66 subjects included in the PK analysis, 48 responded to rilonacept at 12 weeks. Four subjects had missing response status because of early termination due to withdrawal of consent (n=2), serious adverse event (n=1), or loss to follow-up (n=1). Thus, 62 subjects were included in the analysis comparing systemic exposure by response status. Median AUC0-inf in the responder group was similar to that in the non-responder group (169.84 mcg*days/mL [25th, 75th percentile: 112.32, 225.69], vs. 136.42 mcg*days/mL [106.93, 179.64], p=0.06).
Discussion
In this analysis, rilonacept CL/F was directly associated with body weight and age and inversely related to serum albumin. Association of CL/F with these covariates was also observed in a rilonacept population PK model of the combined data from adults with CAPS and rheumatoid arthritis, and healthy volunteers.8 However, only weight had an effect that was clinically significant in the adult population PK model.8 In the present study, children with serum albumin levels <4.0 g/dL had predicted rilonacept exposure (AUC0–inf) ~50% lower than children with higher albumin levels. This finding could be related to higher availability of rilonacept to clearance mechanisms resulting in lower exposure in the setting of high inflammation of which low albumin is a surrogate marker. The exact mechanism of rilonacept clearance is not well understood, but intracellular proteolytic catabolism by the reticuloendothelial system (RES) is expected to play a role, similarly to other large proteins (rilonacept molecular weight, 252 kDa). It is believed that the Brambell receptor (FcRn) increases SC bioavailability of IgG-like drugs by protecting them from intracellular catabolism.17,18 However, this mechanism may be saturated at high endogenous antibody concentrations which can be the case in the setting of high inflammation.19 Thus, in severe inflammatory disease as indicated by low albumin, rilonacept could be more available to proteolytic catabolism due to saturation of the protection mechanism. Even though hypoalbuminemia was a statistically significant predictor of rilonacept CL, predicted exposure in children in the present study (median [5th, 95th percentile] AUC0–inf of 158.1 [79.2, 239.8] mcg*days/mL) was comparable to adults with CAPS receiving weekly SC 160 mg of rilonacept (AUC0–inf was 198 mcg*days/mL).20 Therefore, based on the PK data alone, no dosing modifications are warranted in children.
The population CL/F estimate in children with sJIA when scaled to an average 70 kg adult (1.50 L/day) was comparable to CL/F in healthy volunteers and adults with rheumatoid arthritis (1.81 L/day and 1.59 L/day, respectively).8 In a smaller pediatric study in which 21 children received rilonacept (2.2–4.4 mg/kg), CL/F was higher than the CL/F estimate in children with sJIA in the present study (0.83 L/day vs. 0.55 L/day, respectively).8,11 Similarly, the rilonacept T1/2 was shorter when compared with the present analysis (8.9 vs. 6.9 days). These differences could relate to differing demographic characteristics (e.g., age, weight) in the patient population included in these studies but are unlikely to result in dosing modifications. Therefore, children with sJIA should be dosed according to the efficacy results of this trial in which rilonacept demonstrated efficacy in active sJIA.10 The present study is the first published report on population PK of rilonacept in children. Our study is limited by sparse trough sampling, which prevented estimation of Ka. This parameter was therefore fixed based on adult values (0.5 day−1)15 which could differ from Ka in children. However, as expected, a parameter sensitivity analysis on Ka (fixing at different values) did not change the point estimates for the other PK parameters. Another limitation was the lack of exact record of dosing time. Based on the study protocol, dosing was assumed to occur 2h after PK sampling, but actual dosing time may have differed. However, because of the prolonged T1/2 of rilonacept and the comparability of our PK parameter estimates with prior reports, we do not believe that inaccuracies in dosing time would have a significant impact on the findings.
Systemic exposure in the responder group trended higher than in the non-responders, however, the difference was not statistically significant. Combining these data with prior data in children may help to better understand the rilonacept exposure-response relationship.
Conclusion
A population PK model was successfully developed using sparse and opportunistic PK data collected in children with sJIA. Rilonacept CL/F increased with increasing body weight and age and decreased with increasing serum albumin. In spite of the influence of these covariates on CL/F, the range of rilonacept exposures after a loading dose of 4.4 mg/kg and maintenance weekly doses of 2.2 mg/kg were comparable to therapeutic exposures observed in adults.
Supplementary Material
{kind=link}
Figure 3. Rilonacept systemic exposure vs. response.

Rilonacept response at 12 weeks is defined as a composite of improvement in the American College of Rheumatology Pediatric 30 (ACR Pedi 30) criteria, the absence of fever ≥38.5ºC in the previous 2 weeks, and at least 10% taper in the dose of systemic corticosteroids from baseline in patients receiving corticosteroids.
AUC0-inf indicates area under the concentration time curve from 0 to infinity.
Acknowledgments
Funding: Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award number UL1TR001117 and by the National Institute for Arthritis and Musculoskeletal Diseases N01AR700015. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
RAPPORT Investigators
Laura E. Schanberg, Duke University Medical Center, Durham, NC; Melissa Elder, University of Florida, Gainesville, FL; Diana Milojevic, University of California, San Francisco, CA; James W. Verbsky, Medical College of Wisconsin, Milwaukee, WI; Steven J. Spalding, The Cleveland Clinic, Cleveland, OH; Yukiko Kimura, Joseph M. Sanzari Children’s Hospital, Hackensack University Medical Center, Hackensack, NJ; Lisa F. Imundo, Morgan Stanley Children’s Hospital of New York-Presbyterian, Columbia University Medical Center, New York, NY; Marilynn G. Punaro, Texas Scottish Rite Hospital, Dallas, TX; David D. Sherry, Children’s Hospital of Philadelphia, PA; Stacey E. Tarvin and Kathleen O’Neil, Riley Hospital for Children, Indianapolis, IN; Lawrence S. Zemel, Connecticut Children’s Medical Center, Hartford, CT; James D. Birmingham, Michigan State University College of Human Medicine, East Lansing, MI; Beth S. Gottlieb, Steven and Alexandra Cohen Children’s Hospital, New York, NY; Michael L. Miller, Ann and Robert H. Lurie Children’s Hospital, Chicago, IL; Natasha M. Ruth, Medical University of South Carolina, Charleston, SC; Carol A. Wallace, Seattle Children’s Hospital & Research Institute, Seattle, WA; Nora G. Singer, Metro Health Medical Center, Cleveland, OH; Christy I. Sandborg, Stanford University, Stanford, CA; Kristi Prather and Yuliya Lokhnygina, Duke Clinical Research Institute.
Footnotes
Disclosures: Michael Cohen-Wolkowiez receives support for research from the National Institutes of Health (NIH) (1K23HD064814), the National Center for Advancing Translational Sciences of the NIH (UL1TR001117), the Food and Drug Administration (1U01FD004858-01), the Biomedical Advanced Research and Development Authority (BARDA) (HHSO100201300009C), the nonprofit organization Thrasher Research Fund (www.thrasherresearch.org), and from industry for drug development in adults and children (www.dcri.duke.edu/research/coi.jsp). Norman Ilowite receives funding from the National Institute for Arthritis and Musculoskeletal and Skin Diseases (NIAMS NO1 AR 700015) and has received funds for consulting from Novartis, Genentech and Janssen. Dr. Autmizguine has no potential conflicts to report. Regeneron provided rilonacept and placebo as well as performed serum rilonacept levels for the trial.
References
- 1.Gare BA, Fasth A. Epidemiology of juvenile chronic arthritis in southwestern Sweden: a 5-year prospective population study. Pediatrics. 1992 Dec;90(6):950–958. [PubMed] [Google Scholar]
- 2.Peterson LS, Mason T, Nelson AM, O'Fallon WM, Gabriel SE. Juvenile rheumatoid arthritis in Rochester, Minnesota 1960–1993. Is the epidemiology changing? Arthritis Rheum. 1996 Aug;39(8):1385–1390. doi: 10.1002/art.1780390817. [DOI] [PubMed] [Google Scholar]
- 3.Woo P. Systemic juvenile idiopathic arthritis: diagnosis, management, and outcome. Nat Clin Pract Rheumatol. 2006 Jan;2(1):28–34. doi: 10.1038/ncprheum0084. [DOI] [PubMed] [Google Scholar]
- 4.Duffy CM. Health outcomes in pediatric rheumatic diseases. Curr Opin Rheumatol. 2004 Mar;16(2):102–108. doi: 10.1097/00002281-200403000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007 Mar 3;369(9563):767–778. doi: 10.1016/S0140-6736(07)60363-8. [DOI] [PubMed] [Google Scholar]
- 6.Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005 May 2;201(9):1479–1486. doi: 10.1084/jem.20050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sikora KA, Grom AA. Update on the pathogenesis and treatment of systemic idiopathic arthritis. Curr Opin Pediatr. 2011 Dec;23(6):640–646. doi: 10.1097/MOP.0b013e32834cba24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Regeneron Pharmaceuticals I. Investigator's Brochure 070913. 10. 2007. [Google Scholar]
- 9.Radin A, Marbury T, Osgood G, Belomestnov P. Safety and pharmacokinetics of subcutaneously administered rilonacept in patients with well-controlled end-stage renal disease (ESRD) J Clin Pharmacol. 2010 Jul;50(7):835–841. doi: 10.1177/0091270009351882. [DOI] [PubMed] [Google Scholar]
- 10.Ilowite NT, Prather K, Lokhnygina Y, et al. The randomized placebo phase study of rilonacept in the treatment of systemic juvenile idiopathic arthritis (RAPPORT) Arthritis Rheumatol. 2014 May 16; doi: 10.1002/art.38699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lovell DJ, Giannini EH, Reiff AO, et al. Long-term safety and efficacy of rilonacept in patients with systemic juvenile idiopathic arthritis. Arthritis Rheum. 2013 Sep;65(9):2486–2496. doi: 10.1002/art.38042. [DOI] [PubMed] [Google Scholar]
- 12.Anderson BJ, Holford NH. Understanding dosing: children are small adults, neonates are immature children. Arch Dis Child. 2013 Sep;98(9):737–744. doi: 10.1136/archdischild-2013-303720. [DOI] [PubMed] [Google Scholar]
- 13.Holford NH. A size standard for pharmacokinetics. Clin Pharmacokinet. 1996 May;30(5):329–332. doi: 10.2165/00003088-199630050-00001. [DOI] [PubMed] [Google Scholar]
- 14.Giannini EH, Ruperto N, Ravelli A, Lovell DJ, Felson DT, Martini A. Preliminary definition of improvement in juvenile arthritis. Arthritis Rheum. 1997 Jul;40(7):1202–1209. doi: 10.1002/1529-0131(199707)40:7<1202::AID-ART3>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 15.Regeneron Pharmaceuticals I. Arcalyt (Rilonacept) Briefing document for the Arthritis Drugs Advisory Committee. 2012 May 8; [Google Scholar]
- 16.Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 2008 Aug;58(8):2443–2452. doi: 10.1002/art.23687. [DOI] [PubMed] [Google Scholar]
- 17.Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clinical pharmacology and therapeutics. 2008 Nov;84(5):548–558. doi: 10.1038/clpt.2008.170. [DOI] [PubMed] [Google Scholar]
- 18.Brambell FW, Hemmings WA, Morris IG. A THEORETICAL MODEL OF GAMMA-GLOBULIN CATABOLISM. Nature. 1964 Sep 26;203:1352–1354. doi: 10.1038/2031352a0. [DOI] [PubMed] [Google Scholar]
- 19.Mortensen DL, Walicke PA, Wang X, et al. Pharmacokinetics and pharmacodynamics of multiple weekly subcutaneous efalizumab doses in patients with plaque psoriasis. J Clin Pharmacol. 2005 Mar;45(3):286–298. doi: 10.1177/0091270004270260. [DOI] [PubMed] [Google Scholar]
- 20.Regeneron Pharmaceuticals I. Rilonacept [EMA Summary of product characteristics] 2012 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.