

Supplemental digital content is available in the text.
Key Words: anticonvulsants, alcohol dependence, cognition
Abstract
The anticonvulsant topiramate not only decreases ethanol consumption in alcohol dependence (AD) but also may produce several adverse events including cognitive impairment. Zonisamide is a structurally related anticonvulsant that is a promising agent for the treatment of AD and may have greater tolerability than topiramate. This study evaluated the effects of zonisamide (400 mg/d) on alcohol consumption and its neurotoxic effects in subjects with AD. A double-blind placebo-controlled clinical trial was conducted using 2 comparator anticonvulsant drugs, topiramate (300 mg/d) and levetiracetam (2000 mg/d), which does not impair cognition. Study medications were administered for 14 weeks, including a 2-week taper period. Medication adherence was facilitated using Brief Behavioral Compliance Enhancement Treatment. The neurotoxicity of the study drugs was assessed using neuropsychological tests and the AB-Neurotoxicity Scale. Compared with placebo, both zonisamide and topiramate produced significant reductions in the drinks consumed per day, percent days drinking, and percent days heavy drinking. Only the percent days heavy drinking was significantly decreased in the levetiracetam group. The topiramate cell was the only group that had a significant increase on the mental slowing subscale of the Neurotoxicity Scale compared with placebo at study weeks 11 and 12. Topiramate and zonisamide both produced modest reductions in verbal fluency and working memory. These findings indicate that zonisamide may have efficacy in the treatment of AD, with effect sizes similar to topiramate. Both of these drugs produced similar patterns of cognitive impairment, although only the topiramate group reported significant increases in mental slowing.
The results of several clinical trials, including 1 multisite study, indicate that the broad-spectrum, sulfamate-substituted anticonvulsant topiramate has a therapeutic effect size in the moderate range for the treatment of alcohol use disorders (AUDs).1–3 Some of the most common problematic adverse effects associated with topiramate administration involve the impairment of cognition that may include impaired verbal fluency and working memory.4–6 There is research that suggests that the cognitive adverse effects that occur with topiramate treatment of AUDS are similar to those reported for the treatment of epilepsy, migraine headaches, and obesity.5,7 Cognitive impairment induced by topiramate is a common reason for study dropout and drug discontinuation in clinical settings.5 In addition to the effects on cognition, topiramate administration also produces a range of other adverse effects, the most serious of which is metabolic acidosis, resulting from the inhibition of carbonic anhydrase.8
Several sulfamide and sulfonamide compounds have been identified, which share some of the structural features of topiramate and which have been shown to have actions as broad-spectrum anticonvulsants in animal models of seizure disorders.9–12 Of these compounds, only zonisamide is currently approved in the United States as a medication for use as a broad-spectrum anticonvulsant.13 This drug shares other therapeutic actions with topiramate, namely, antimigraine effects14,15 and the facilitation of weight loss.16 There is also evidence that zonisamide administration may help to promote reduced alcohol consumption. Ethanol intake has been found to be lowered by the administration of zonisamide to either mice or rats in limited access models of drinking.17 In humans, the administration of zonisamide decreased ethanol self-administration in a laboratory setting.18 Ethanol consumption was markedly reduced by the administration of zonisamide in both open-label19,20 and placebo-controlled clinical trials.21
Zonisamide, like topiramate, is a carbonic anhydrase inhibitor, but reports in patients with seizure suggest that it has a lower incidence of both paresthesias (2.5%–11.5% vs ≥22% for topiramate)22–25 and metabolic acidosis.8 With regard to the latter, in a study conducted by Mirza et al,8 7% of patients treated with zonisamide had low serum bicarbonate levels, an indicator of acidosis, whereas 29% of individuals receiving topiramate had low levels of this ion.
The effects of zonisamide on cognition have not been studied as extensively as have those associated with topiramate. In patients with seizure, after 1 year of monotherapy with zonisamide, participants had a significant decline from baseline performance on measures of verbal fluency and attention.26 A pilot study of patients with seizure showed impaired performance on the Wechsler Memory Scale and delayed recall tasks for logical memory and verbal paired associates.27 Caution must be exercised in interpreting extant studies because some diseases, such as epilepsy, are associated with greater adverse effects than in obese patients or patients with AUDS. Subjects with AUDS have reported cognitive problems when receiving zonisamide.21 In preliminary investigative trials conducted in our clinic, we found that in alcohol-dependent subjects, the cognitive impairing effects of zonisamide were less severe compared with those resulting from topiramate administration.19,28 Formal neuropsychological testing, however, has not been used to evaluate the effects of zonisamide on individuals with AUDS in placebo-controlled clinical trials.
The objectives of the present study were to evaluate the effects of zonisamide administration on ethanol intake and on cognitive functioning in subjects with moderate-to-severe AUDS, specifically those meeting Diagnostic Statistical Manual Fourth Edition criteria for alcohol dependence (AD). Cognitive functioning was assessed using a battery of neuropsychological tests that measured several aspects of cognitive functioning, including working memory, language function, executive function, as well as visual processing and psychomotor performance. The A-B Neurotoxicity Scale was administered in the present study to obtain subjects’ reports concerning their experiences of the neurotoxic actions of anticonvulsants. The effects of zonisamide were compared with those of placebo. Topiramate was also administered to a separate group of subjects as a positive control. The anticonvulsant levetiracetam was used as an additional comparator agent. Levetiracetam administration seems to produce few adverse effects on cognition.29,30 This drug had shown initial promise as a medication for the treatment of AD,31 but its use for this purpose has not since been supported by findings in recent studies.32,33
MATERIALS AND METHODS
Subjects
Eighty-five participants (37 women) aged 21 to 65 years who met Diagnostic Statistical Manual Fourth Edition criteria for AD were admitted into this study. Characteristics of the subjects are provided in Table 1. Eligibility criteria specified that during the 90-day period preceding screening, men drank 35 or more standard drinks per week, whereas women consumed 28 or more standard drinks per week during at least a 4-week-long consecutive period. Subjects had to have had a score of greater than 8 on the Alcohol Use Disorder Identification Test34 and were required to express a desire to stop drinking or reduce their intake of alcohol. Female subjects were required to have been using appropriate birth control procedures before randomization and during the period in which study medications were being administered or to be sterile or to have entered menopause. Pregnant women were excluded from the study.
TABLE 1.
Subject Description
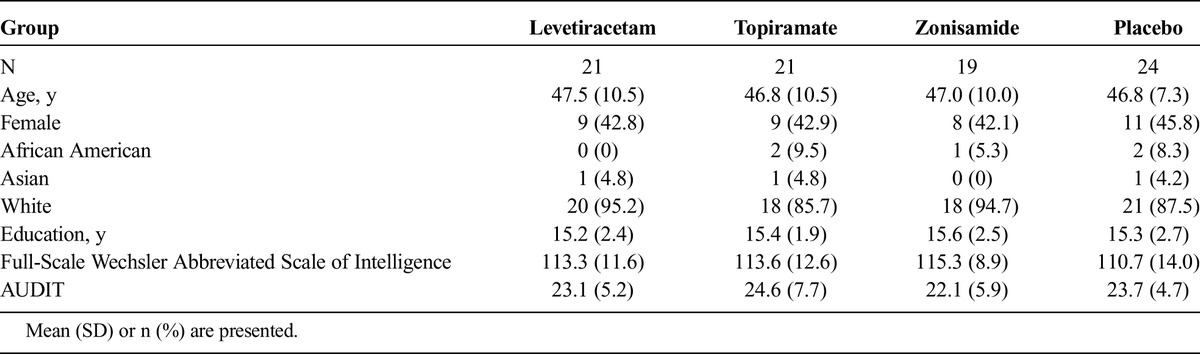
Exclusion criteria for this study included dependence on substances other than alcohol, nicotine, or caffeine; a score of 10 or greater on the Clinical Institute Withdrawal Assessment for Alcohol-Revised35 scale during screening; current treatment with acamprosate, disulfiram, or naltrexone; or the use of any of these drugs less than 2 weeks before randomization. Additional exclusion criteria were current treatment with sedative hypnotics, opioids (ie, required chronic opioids treatment), psychomotor stimulants, or antipsychotic, antimanic, or anticonvulsant medications.
All subjects provided written informed consent, when not intoxicated (ie, blood alcohol concentration, 0.00%), before entry into the study. This study was undertaken with the approval of the Boston University Medical Center’s Institutional Review Board.
Study Design and Procedures
This placebo-controlled study followed a double-blind, parallel group design. An adaptive randomization procedure was followed in this study with sex and heavy drinking (10 drinks per day for men and 8 drinks per day for women) for all previously randomized subjects used as variable levels. Subjects were randomized into one of the following 4 treatment groups: (1) levetiracetam, (2) placebo, (3) topiramate, or (4) zonisamide.
The flow diagram for this study can be found in Supplemental Figure 1 (in Supplemental Digital Content 1, http://links.lww.com/JCP/A256). Subjects were recruited by advertisements placed on the radio or in newspapers. A telephone interview was used to identify subjects who were qualified to undergo further screening. Subjects were evaluated during screening sessions to determine whether they met criteria for study admission. Those meeting the appropriate criteria were randomized to 1 of the 4 treatment groups. Subjects could have blood alcohol concentration no greater than 0.02% to be allowed to participate in any of the postscreening sessions. The study plan called for randomized subjects to visit the clinic on a weekly basis for 15 consecutive weeks. Before the administration of the first dose of study medication, baseline assessments were obtained. Subjects received medications for 14 weeks. This included a 7-week period of gradual dose increases, 5 weeks of treatment at the target maintenance dose levels, followed by a 2-week period for tapering of medications (see study medication dosing schedule in Supplemental Table 1, in Supplemental Digital Content 2, http://links.lww.com/JCP/A257). Target maintenance doses were 2000 mg/d for levetiracetam, 300 mg/d for topiramate, and 400 mg/d for zonisamide. Target maintenance doses were selected based on the results from previous clinical trials of the efficacy of levetiracetam,31 topiramate,2 and zonisamide19 in alcohol-dependent subjects. In addition to receiving study medication, every subject attended a 15-minute Brief Behavioral Compliance Enhancement Treatment36 session during every session attended between study weeks 1 and 14. The Brief Behavioral Compliance Enhancement Treatment is a psychosocial adherence enhancement procedure that emphasizes that medication adherence is important in the reduction of participants’ drinking behavior.
The 4 study medications were prepared, stored, and dispensed by the Boston Medical Center’s Investigational Drug Service. These medications were dispensed in identically appearing dark blue capsules. If possible, subjects received the same number of capsules for each corresponding day during the treatment period. The study psychiatrist was permitted to slow the rate at which medication doses were increased during the induction phase or to reduce the dose administered if subjects were unable to tolerate their medications at any point in the treatment period.
Subjects’ self-reports concerning the amount of ethanol they consumed were collected using the Time-Line Followback methods37 throughout the study. γ-glutamyl transpeptidase (GGT) was measured in blood at screening and study weeks 4, 8, 10, 12, and 15, as a biomarker of alcohol consumption.38 Symptoms of withdrawal were measured during screening and in each subsequent encounter using the Clinical Institute Withdrawal Assessment for Alcohol-Revised scale.35 Blood alcohol concentrations, as measured using a Breathalyzer, and vital signs were collected during each session. Alcohol craving was assessed using the 14-item Obsessive Compulsive Drinking Scale (OCDS), a reliable self-rating instrument that measures cognitive aspects of alcohol craving.39 Depression was evaluated with the Montgomery Asberg Depression Scale (MADRS),40 whereas anxiety levels were determined using the Hamilton Anxiety (HAM-A) scale.41 The HAM-A scale, MADRS, and OCDS were administered in study weeks 1, 4, 6, 8, 10, 12, and 15. Latency to sleep onset and hours of sleep per night were assessed using the Sleep Scale for Medical Outcomes (MOS).42 The A-B Neurotoxicity Scale43 was used to obtain subjects’ rating of their experience of adverse effects related to anticonvulsant-induced neurotoxicity. This scale and the MOS were administered on study weeks 1, 4, 8, 12, and 15.
Neuropsychological tests were administered on study week 1, before the start of drug administration, and study week 12, at the end of maintenance therapy, to evaluate cognitive functioning at baseline and in the last week of maintenance therapy. The Wechsler Abbreviated Scale of Intelligence was used to determine full-scale intelligence quotients.44 Working memory was assessed using the Spatial and Digit Span tests from the Wechsler Memory Scales-Third Edition (WMS-III).45 The Rey Audio Visual Learning Test46 was administered to examine verbal memory, whereas visual memory was evaluated through the use of the Rey Complex Figure Memory and Recognition Tests.47 Verbal fluency was assessed using the Controlled Word Association Test (COWAT).48 The Stroop Color-Word Test,49 Trail Making Tests50 and Wisconsin Card Sort Test51 were administered to evaluate executive function. Psychomotor function, attention, and visual-motor processing were assessed using part A of the Trail Making Test, the Symbol Digit Modalities Test,52 and the Grooved Pegboard Test.53
Statistical Analysis
Unless otherwise indicated, data are presented as mean ± SE values. Baseline group data for drinking and other measures were compared using 1-way analysis of variance, except for comparisons of proportional data, for which χ2 tests were used. An intent-to-treat approach was used to analyze all repeated outcome measures except those for the neuropsychological testing. Data for the neuropsychological tests were available for only study weeks 1 and 12 and were analyzed only for subjects for whom week 12 data were available. To assess the effects of subject dropout, an additional sensitivity analysis was conducted on data for the drinking measures using a last observation carried forward approach. This approach entailed last observation carried forward values being used to replace data missing after the last observation in the mixed models analyses described later. The same approach was used to analyze results for the COWAT, which were used as representative samples of data for the neuropsychological tests. The data from the COWAT were further analyzed using an intent-to-treat approach to see if the results were comparable with those obtained for the completers’ data set.
Drinking measures derived from the Time-Line Followback data included the percent days drinking, the number of drinks consumed per day, and the percent days heavy drinking. Heavy drinking was defined as 4 or more drinks per day for women and 5 or more drinks per day for men. Alcohol consumption measures were analyzed using repeated-measures mixed models analysis using SAS PROC MIXED (version 9.3; SAS Institute, Cary, NC) with baseline values for these measures used as covariates. Comparisons were made for data obtained for the 12-week treatment period for all 4 groups. An additional analysis was conducted on paired comparisons between values for the placebo group and each of the active medication groups. For the paired comparisons, the SLICE option available in PROC MIXED was used to determine differences between pairs of groups for study weeks 10, 11, and 12. The SLICE option offers a means for performing a partitioned analysis of the least square means for an interaction. Also known as the analysis of simple effects, the SLICE analysis can provide results for the paired comparisons of least square means for any given unit of time, for example, treatment week. During weeks 10, 11, and 12, drug blood concentrations should be at steady-state levels. This was based on the assumption that zonisamide, which has a half-life in the range of 50 to 60 hours,54 the longest of any of the study medications, would attain steady-state levels at 2 weeks after the initiation of administration of the maintenance dose in week 8.
Data collected for the A-B Neurotoxicity Scales, HAM-A scale, MADRS, OCDS, and Sleep-MOS, were also analyzed with repeated-measures mixed models analysis, with baseline values for these measures used as covariates. A similar approach was used to analyze GGT data, which were first transformed to natural logarithm values to reduce excessive skewness and kurtosis detected in the initial examination of the GGT values. The mental slowing and memory subscales of the Neurotoxicity Scales were also analyzed because they assess aspects of cognition that are likely to be negatively influenced by topiramate. Because subjects were not always able to attend sessions as originally scheduled, 2 weeks long rather than 1 week long, time bins were used to classify the time of assessment for measures that were collected on a less-than-weekly basis in an effort to provide a more accurate representation of the times at which these data were collected. That is to say, the 12-week treatment period was broken into six 2-week segments, with, for example, assessments being obtained for weeks 11 and 12 being placed into the segment 6 assessment period. If a particular time segment contained 2 values, the mean of these values was used in the analysis.
α levels of less than 0.05 were considered to be significant. An exception was when a value of less than 0.017, based on a Bonferroni correction, was regarded as being significant when 3 paired comparisons between the placebo and each of the active medication groups were made. In an effort to control for multiple comparisons, a second exception was also made for the neuropsychological test results. For these tests, differences from placebo values were considered to be significant only when the α value was less than 0.01 for the group-by-time interaction for comparisons with placebo. A P value of 0.05 was taken to be significant for SLICE effects analysis of paired comparisons between the placebo group and other individual active medication groups. The results of the SLICE analysis, consequently, are best considered as only an exploratory examination of the direction differences between these paired groups during the treatment period.
RESULTS
Demographic data for subjects are presented in Table 1, along with the findings for the AUDIT, WAIS Full-Scale IQ scores, and years of education completed. Group differences were not significant with respect to any of these variables. The percentage of subjects in each treatment group who were able to complete assessments for week 12 of the study were 81% for the levetiracetam group, 79% for the placebo group, 71% for the topiramate group, and 79% for the zonisamide group (see Supplemental Figure 1, in Supplemental Digital Content 1, http://links.lww.com/JCP/A256 for the number of subjects during each phase of the study). Based on pill count results, the mean ± SE percentage of prescribed drugs used by subjects while in the treatment period was 93.1% ± 4.7% for the levetiracetam group, 87.6% ± 5.7% for the placebo group, 95.4% ± 2.5% for the topiramate group, and 90.5% ± 4.4% for the zonisamide group. These percentages did not differ significantly among these groups.
Data obtained at baseline and during the 12-week treatment period for the number of drinks consumed per day and the percent days heavy drinking are presented in Figure 1. Findings for the weekly percent days drinking appear in Figure 2. No group differences were found for any of the 3 measures of drinking obtained at baseline. Treatment effects were significant for the percent days drinking (F3,81.2 = 6.7, P = 0.0005), the number of drinks consumed per day (F3,81.4 = 4.8, P = 0.004), and the percent days heavy drinking (F3,84.4 = 5.5, P = 0.002). The group-by-time interactions were not significant for any of these measures. For the pairwise comparisons between the placebo and topiramate groups, significant treatment effects were seen for weekly percent days drinking (F1,41.6 = 19.8, P < 0.0001), percent days heavy drinking (F1,41.8 = 19.4; P < 0.0001), and drinks consumed per day (F1,41 = 13.5, P = 0.0007). SLICE effects showed that values for all 3 drinking measures were significantly lower in the topiramate group as compared with the placebo group for weeks 10 to 12. For the placebo and zonisamide groups comparisons, treatment effects were significant for the percent days drinking (F1,42.8 = 8.4, P = 0.006), percent days heavy drinking (F1,43 = 10.8, P = 0.002), and drinks consumed per day (F1,40.8 = 7.5, P = 0.009) measures. Values for the percent days drinking and percent days heavy drinking were significantly less for the zonisamide group than for the placebo for weeks 10 to 12. For the number of drinks consumed per day, the values for the zonisamide group were only significantly lower than those for the placebo group for week 11. When the levetiracetam and placebo groups were compared, significant treatment effects were found only for the percent days heavy drinking (F1,43.2 = 7.4, P = 0.009), with values for the levetiracetam group being significantly less than those obtained for the placebo group during weeks 10 to 12. Drinking measure values that were found to be significant for paired comparisons in the intent-to-treat data set were also found to be significant in the sensitivity analysis, with the exception of those for the percent days drinking for the comparison of the zonisamide and placebo groups. The treatment effect P value for this comparison increased from 0.006 in the intent-to-treat analysis to 0.0176 in the sensitivity analysis.
FIGURE 1.
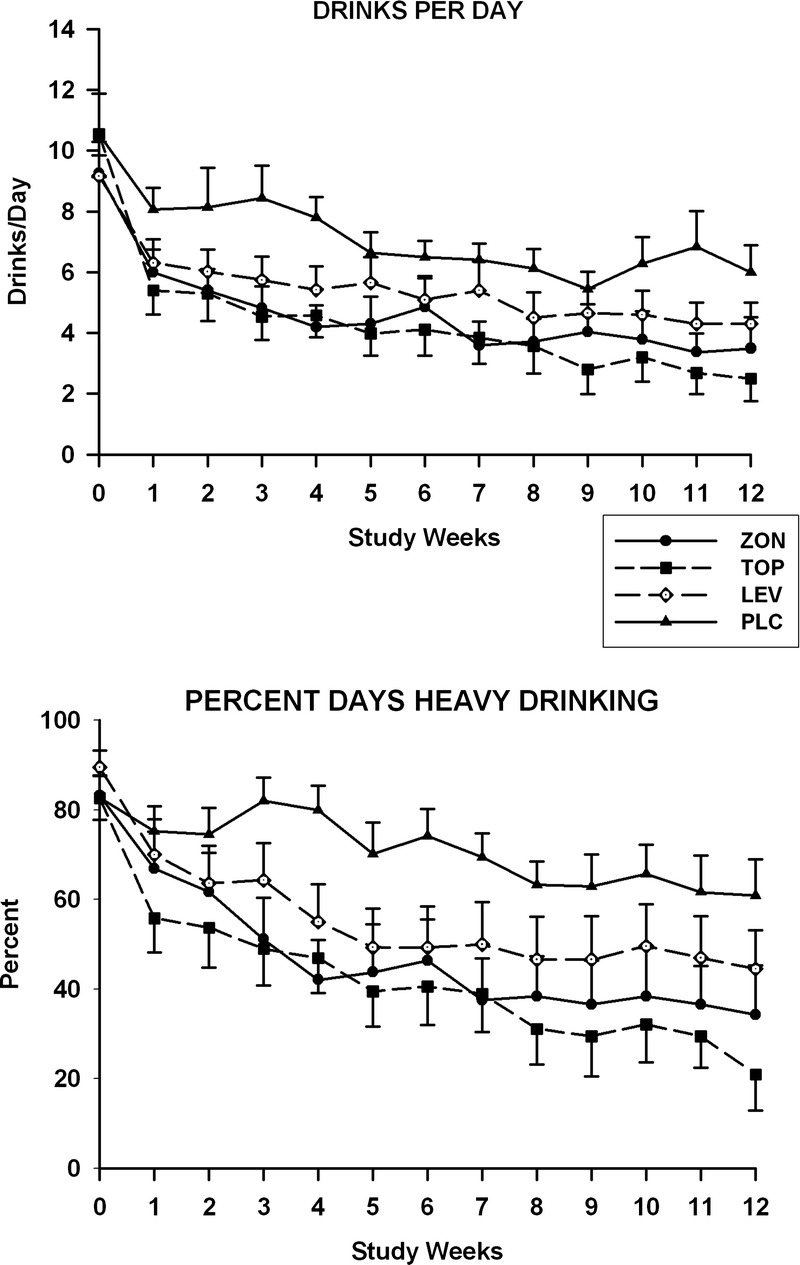
Ethanol consumption for subjects in the levetiracetam (LEV), placebo (PLC), topiramate (TOP), and zonisamide (ZON) groups. Mean ± SE weekly values are presented for drinks per day (top) and percent days heavy drinking (bottom) obtained during the prescreening (week 0), titration (weeks 1–7), and maintenance phases of the study (weeks 8–12).
FIGURE 2.
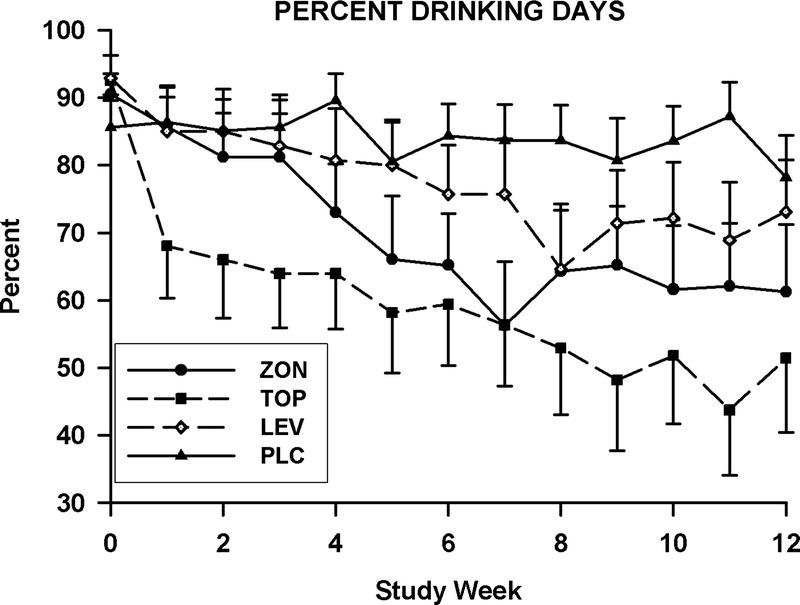
Mean weekly values for percent days drinking for subjects in the levetiracetam (LEV), placebo (PLC), topiramate (TOP), and zonisamide (ZON) groups. These values are presented for the percent days drinking obtained during the prescreening (week 0), titration (weeks 1–7), and maintenance phases of the study (weeks 8–12).
Least square means values obtained for the follow-up evaluation session were significantly lower for the topiramate group as compared with the placebo group for the mean drinks consumed per day (P = 0.02) and the mean percent days drinking (P = 0.02). This comparison was not significant for the mean value of the percent heavy drinking days. None of the values for the posttreatment evaluation were found to be significant when values for the placebo group were compared with those obtained for either the levetiracetam or zonisamide groups.
The mean GGT blood concentrations obtained at screening were higher than the upper limit for normal values (ie, 58 U/L) for all but the zonisamide group. For log-transformed GGT concentrations obtained at screening and for study weeks 3 to 12, the group-by-time interaction was found to be significant (F12,180 = 3.0, P = 0.0009). The pairwise comparison ln GGT values for the topiramate and placebo groups showed a significant group-by-time interaction (F4,127 = 5.0, P = 0.0009). Least square means ln (GGT) values for the topiramate group [(Seg-5 = 3.5 (0.2); Seg 6 = 3.3 (0.2)] were significantly lower than those obtained for the placebo group in the week 9 and 10 [Seg 5 = 3.8 (0.2)] and week 11 and 12 segments [Seg 6 = 3.8 (0.2)]. The treatment and group-by-time effects were not significant for the comparisons of ln (GGT) values for the zonisamide and placebo values and for levetiracetam and placebo.
Mean values obtained for the OCDS during the treatment period are shown in Figure 3. Repeated-measures analysis of these values obtained for total OCDS scores revealed a significant group-by-time interaction (F15,102 = 1.9, P = 0.032). Only the pairwise comparison between the topiramate and placebo groups showed a significant group-by-time interaction (F5,51.9 = 3.6, P = 0.007) for OCDS values measured during the treatment period. Least square means OCDS scores for the topiramate group were significantly lower than those obtained for the placebo group in the week 9 and 10 segment and the week 11 and 12 segment. Scores for this measure followed a similar trend for both the levetiracetam and zonisamide groups; however, no significant effects were found for the paired comparisons of values obtained for these 2 groups with those found for the placebo group. Treatment effects and group-by-time interactions were not found for any of the comparisons made for the HAM-A scale score with the exception of the group-by-time effect (F5,76.4 = 3.9, P = 0.003) for the comparison between the topiramate and placebo groups (Fig. 4). SLICE effects showed topiramate group HAM-A scale scores to be significantly higher than placebo group scores for the week 1 and 2 segment (P = 0.01). None of comparisons obtained for the MADRS scores were found to be significant.
FIGURE 3.
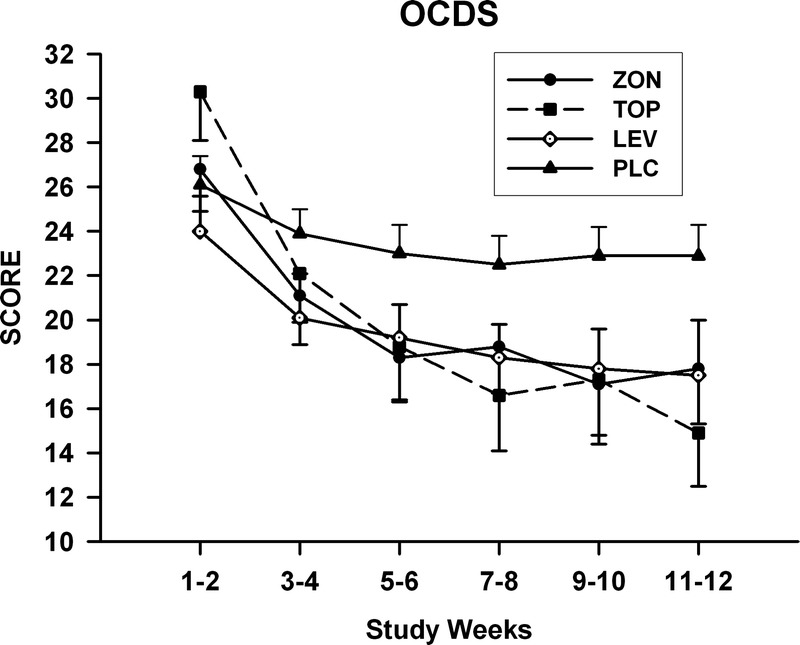
Mean ± SE total OCDS scores for each treatment group obtained during the titration and maintenance phases of the study.
FIGURE 4.
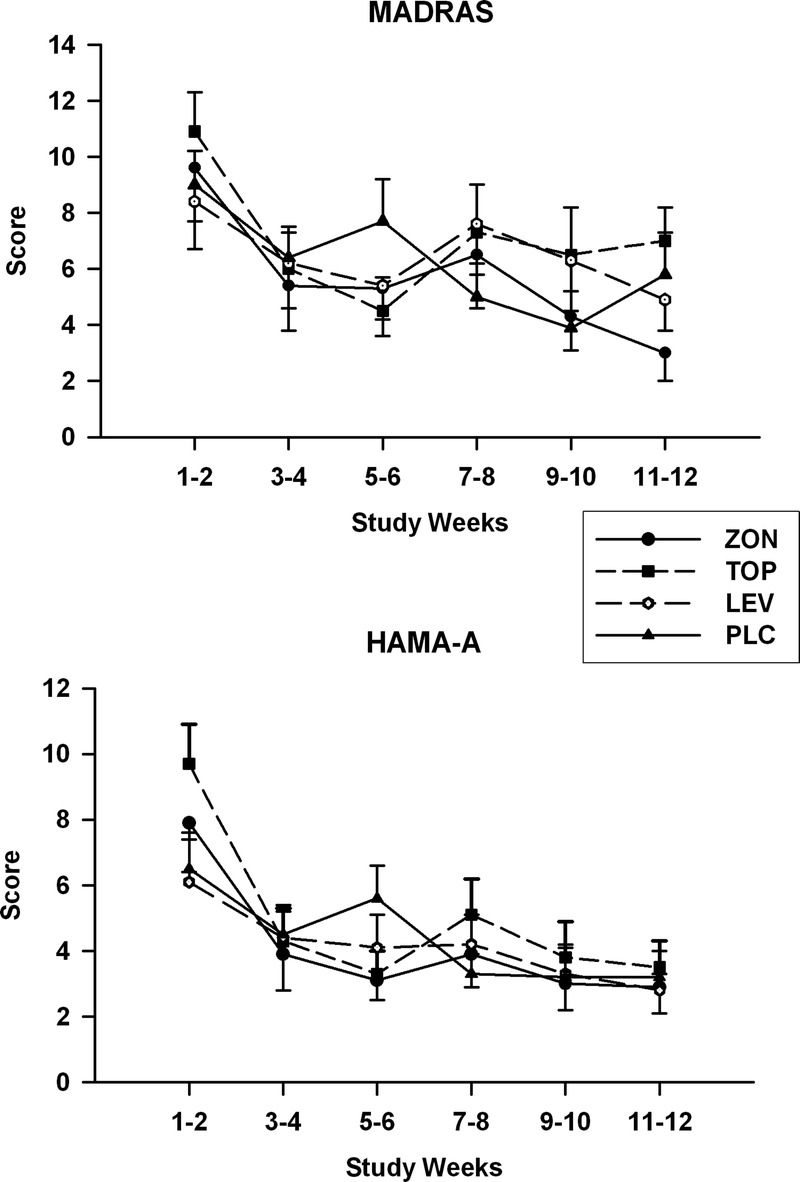
Mean ± SE MADRS (top) and HAM-A scale (bottom) scores for each treatment group obtained during the titration and maintenance phases of the study.
Mean ± SE total hours of sleep per night determined using the Sleep-MOS scales at baseline were 7.0 ± 0.3 for the levetiracetam group, 6.8 ± 0.3 for the placebo group, 6.9 ± 0.4 for the topiramate group, and 6.9 ± 0.2 for the zonisamide group. These baseline values did not differ significantly among the groups. Analysis of data for the total hours of sleep per night and the latency to the onset of sleep did not reveal any significant differences among the groups during the course of the treatment period.
For pairwise comparisons of topiramate versus placebo data for A-B Neurotoxicity Scales, the treatment effect (F1,39.2 = 8.0, P = 0.008) was significant for the mental slowing subscale, with the effect slices being significant only for the week 11 and 12 segment [P = 0.005) (see Supplemental Table 2, Supplemental Digital Content 3, http://links.lww.com/JCP/A258). For the total Neurotoxicity Scale scores, the α value for the treatment effect was 0.03, which is not significant after the correction for multiple comparisons, with an α value of 0.015 found for the week 11 and 12 segment. For the memory subscale of the Neurotoxicity Scale, none of the effects examined for the overall analysis were significant, but the tests of SLICE effects had an α value of 0.011 for the week 11 and 12 segment. Taken together, these results suggest that subjects on the topiramate group experienced greater neurotoxicity symptoms in the last 2 weeks of the maintenance phase of the study. A significant group-by-time interaction was found for the comparison between the zonisamide and placebo groups (F3,60.7 = 3.8, P = 0.015) for the Neurotoxicity memory subscale, with SLICE effects showing a significantly lower value for the zonisamide than for the placebo group in weeks 3 and 4 (ie, before the maintenance dose). The group-by-time interaction was also significant for pairwise comparisons between the levetiracetam and the placebo groups for data obtained on the Neurotoxicity memory subscale (F3,92.3 = 3.6, P = 0.017), with no SLICE effects showing a significant between-group difference during the treatment period.
The impairment of both verbal and visuospatial working memory in the topiramate and zonisamide groups was indicated by significant group-by-time interactions for comparisons between the placebo group and these medication groups for performance on the Forward portions of the Spatial and Digit Spans tests, with decreased scores being obtained for the 2 anticonvulsant groups whereas slight elevations were seen for scores of the placebo group (see Supplemental Table 2, in Supplemental Digital Content 3, http://links.lww.com/JCP/A258). Additional evidence for topiramate-induced deficits in working memory is indicated by significant group-by-time interactions for comparisons between the placebo group and this medication group for both age-adjusted and total scores of the Spatial Span and Digit Span tests. In addition, a significant interaction was found for the backward portion of the Digit Span test for the comparison of the topiramate and placebo, which is consistent with an impairment in verbal working memory having been produced by the administration of this anticonvulsant.
The interaction for comparisons for total scores for the Audio Visual Learning Test was significant, for both the topiramate and zonisamide groups, with reductions occurring in scores in both groups as compared with those obtained for the placebo group. These results indicate that these drugs produced impairment of verbal memory. A significant group-by-time interaction was also found for the topiramate-placebo comparison for the total recognition score for the Rey Complex Figure test, with scores being reduced only for the topiramate group. This suggests that topiramate may also adversely affect visual memory. Significant group-by-time interactions for comparison with the placebo group on both the Phonetic and Semantic portions of the COWAT seen for both the topiramate and zonisamide groups, with scores being decreased in each of these anticonvulsant groups, indicate that these 2 medications can impair verbal fluency. Similar results were found for both the intent-to-treat and sensitivity analyses of data obtained for the COWAT.
Performance on the Trail Making Test Part B was significantly impaired for the zonisamide group when compared with the placebo, with time to complete the trial being elevated in the zonisamide group. The group-by-time interaction for the Trails B Test approached significance when results were examined for the topiramate group (F1,18.1 = 8.0, P = 0.011). These results for the Trail Making Test are indicative of reductions in executive functioning, but results obtained for the Stroop Test and Wisconsin Card Sorting Test (WCST) suggest that either topiramate or zonisamide administration do not negatively influence many aspects of executive function.
No significant group-by-time interactions for comparisons between the placebo group and the levetiracetam group were found for any of the neuropsychological tests that were administered in this study (see Supplemental Table 3, in Supplemental Digital Content 4, http://links.lww.com/JCP/A259).
A serious adverse event involving a suicide attempt using the study medication occurred in 1 subject, who was in the zonisamide group. This serious adverse event, however, was rated as being only remotely related to the use of zonisamide. One subject in the topiramate group developed metabolic acidosis. Irritability occurred in a significantly larger proportion of subjects (24%) who were being treated with topiramate than the proportion for those being treated with placebo. Irritability was not reported by any of the subjects who were treated with zonisamide. Moreover, paresthesias occurred in 19% of the subjects who received topiramate, whereas none were found in subjects who were treated with zonisamide. Unexpectedly, 14% of the subjects in the topiramate group reported problems with erectile dysfunction.
DISCUSSION
The results of the present study demonstrate in subjects with AD that, compared with a treatment with placebo, the administration of either zonisamide or topiramate reduced ethanol intake on all of the 3 measures of alcohol consumption used, namely, percent days drinking per week, drinks consumed per day, and percent days heavy drinking per week. These findings are consistent with those of previous clinical trials indicating that zonisamide21 and topiramate1,2,55–57 decrease alcohol consumption in subjects with AUDS. Although previous placebo-controlled studies have failed to show that levetiracetam administration produces a decrease in alcohol consumption,32,33 in the present study, this anticonvulsant was found to significantly reduce the percent days heavy drinking. The factors that may account for the disparity in these findings remain to be determined.
In the present study, GGT levels were significantly lower at the end of maintenance phase for the topiramate as compared with the placebo group, and there was a significant group-by-time interaction for the pairwise comparison for this group, indicating that topiramate administration lowered GGT blood levels to a greater extent than did placebo. This finding is consistent with those previously reported for topiramate treatment of alcohol-dependent subjects.1 In so far as GGT levels can be considered to be a biomarker of alcohol consumption, they support the self-report data that topiramate significantly reduced alcohol consumption.
Significant differences were not found in comparisons of GGT concentrations obtained for either the zonisamide or the levetiracetam groups with those obtained for the placebo group. For the zonisamide group, in the present study, the value of using GGT as a biomarker for alcohol consumption is limited because mean concentrations of this enzyme, in contrast to those obtained for the other groups, were well within the normal range at screening.
The present study seems to be the first investigation in which the effects of either topiramate or zonisamide on cognitive function in individuals with AD were assessed using a full battery of neuropsychological tests. Treatment with either topiramate or zonisamide was associated with increased difficulty with verbal fluency and verbal working memory. In the present study, impairment of visual memory was detected in the topiramate but not the zonisamide group. Treatment with topiramate did not produce diminished executive functioning as assessed using the WCST or Stroop Test. There was, however, a trend for toward decreased performance on the Trail Making Test Part B, which may also assess aspects of executive function.58 Zonisamide administration did produce a decrement in the performance on the Trail Making Test Part B, suggesting a possible negative impact on executive function. Executive functioning, however, as measured by the WCST and the interference and color-word portions of the Stroop test was not impaired by this drug.
The findings for the A-B Neurotoxicity Scale reveal possible differences in subjects’ self-reports concerning their experience of the neurotoxic effects of these drugs. Results for the mental slowing subscale indicated that mental slowing was found to be worsened only by the administration of topiramate. Moreover, the SLICE effects analysis showed scores elevated above placebo group levels only for the topiramate group for both the memory and total scale scores for the Neurotoxicity Scale in the final 2 weeks of the maintenance therapy phase of the study. These results suggest that topiramate may have adverse neuropsychological effects in subjects with AUDS that are not detected by the cognitive tests used in the present study.
In contrast to treatment with either zonisamide or topiramate, levetiracetam administration did not produce any decrements in the performance on the neuropsychological tests used in this study. This result is consistent with other studies that have shown that levetiracetam treatment is not associated with cognitive impairment in patients with seizure disorder.29,30 The lack of the effects of levetiracetam on cognitive functioning may result from its comparatively selective actions on the brain, which most importantly may involve binding to the synaptic vesicle protein S2A.59,60
In the present study, total OCDS scores for the topiramate group became lower during the treatment period than for the placebo group. This is consistent with other findings that topiramate administration may significantly reduce craving for alcohol as measured by the OCDS in subjects with AD,2,57 although this was not found in a study in which this anticonvulsant was received for only 4 weeks.61 In contrast to topiramate, neither zonisamide nor levetiracetam administration resulted in the significant reduction of total OCDS scores to below control levels. Nevertheless, as can be seen in Figure 3, mean values for total OCDS scores for subjects in the zonisamide and levetiracetam groups seem to decline below levels reached by the subjects in the placebo group. Failure to detect significant differences in scores obtained for the OCDS between placebo and either the zonisamide or the levetiracetam group, therefore, might be related to the small size of these groups in the present study.
The neuronal mechanisms through which topiramate and zonisamide act to produce reductions in alcohol consumption in AD remain to be fully elucidated. One possible mechanism that these 2 drugs may share to modulate drinking behavior is to counteract the enhanced excitability that may result from the selective elevation in AMPA, N-methyl-d-aspartate, and/or kainate receptor subunits seen in the hippocampus, the orbital frontal cortex, and anterior cingulate cortex of individuals with AD.62,63 Topiramate may suppress alcohol-induced brain excitability through both positive modulatory interactions with GABAA receptors containing β1 or β3 subunits64 and antagonism of kainate receptors containing the GluK1 subunits.65,66 Indeed, a recent report indicates that sensitivity to topiramate-induced reductions in heavy drinking is associated with the presence of a specific polymorphism of the kainate receptor GluK1 subunit gene.67 Excitatory glutamatergic receptor activity may be reduced by zonisamide by the inhibition of the stimulated release of glutamate68 and by decreasing excitatory postsynaptic potentials through a postsynaptic mechanism that may involve a diminution in α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) receptor activity.69 Zonisamide may also reduce brain excitability by enhancing the activity of γ-aminobutyric acid (GABA) receptor systems by down-regulating GABA transporter proteins.70
The sample sizes used in the present study, although sufficient for comparisons between active medication and placebo, were too small to allow for meaningful between active medication group comparisons of the proportion of subjects in these groups who experienced a specific adverse event. Metabolic acidosis occurred only in 1 patient in the topiramate group, and 19% of the subjects in this group experienced paresthesias. In contrast, none of the subjects in the zonisamide reported having symptoms of paresthesia. These results are consistent with previous studies indicating that problems related to the inhibition of carbonic anhydrase are more likely to occur in individuals treated with topiramate than those who have received zonisamide.8
The primary limitation of this study is the small number of subjects included in each treatment group, which allows for only efficacy comparisons between active drugs and placebo but is not powered to detect efficacy differences between the study drugs. Another limitation of this study is that we did not enroll individuals with the most severe forms of AUDs, namely, those with advanced liver disease, severe neurological impairment, and/or an inability to maintain abstinence for even a short period, and consequently, the value of using the drugs evaluated in the present study in severe forms of AUDs needs further study.
The target maintenance doses of zonisamide and topiramate were chosen based on previous findings.2,19,31 In one previous clinical trial, a higher 500-mg daily dose of zonisamide was chosen for use. This dose also had efficacy in reducing alcohol consumption in subjects with AD. Although a 300-mg maintenance of topiramate was administered in the present study, other investigators report efficacy with 75- and 200-mg daily doses of this drug.56,57,67 It is possible that doses of zonisamide lower than 400 mg daily may also have efficacy in the treatment of AD in association with less cognitive impairment. At present, there has been, however, no systematic comparison of different doses of either zonisamide or topiramate on alcohol consumption or on cognitive functioning in subjects with AD.
The results of this study provide further support that zonisamide has efficacy as a medication that can facilitate reduced drinking in individuals with AD. This study has provided an initial characterization of precise areas of cognitive functioning that may be impaired by the administration of either topiramate or zonisamide in AD. Both agents seem to have the potential to produce modest deficits in cognitive function in the areas of verbal fluency and working memory. Evaluation of patients with the A-B Neurotoxicity Scale indicate that subjects with AUDS experience overall less impairment of cognition when treated with zonisamide than with topiramate, with the latter drug having more pronounced effects on mental slowing. The findings of this study leave unresolved the question of whether zonisamide produces fewer adverse effects related to the inhibition of carbonic anhydrase than does topiramate. They do, however, point to the value of further investigation of the many compounds that have been synthesized that are structurally related to these 2 drugs.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr Michael P. LaValley for his very helpful advice pertaining to the statistical analyses used in this article and for his assistance with the randomization of subjects in this study.
AUTHOR DISCLOSURE INFORMATION
This BU Clinical Studies Unit headed by Domenic A. Ciraulo, MD, has received previous funding from UCB PHARMA for the study of the effects of levetiracetam in AD.
The other authors declare no conflicts of interest.
Footnotes
This research was supported by National Institute of Health grant RO1 AA 015923 to Domenic A. Ciraulo, MD.
Clinical trials registry: clinicaltrials.gov, registration number NCT00862563.
Supplemental digital contents are available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the Journal’s Web site (www.psychopharmacology.com).
REFERENCES
- 1. Johnson BA, Rosenthal N, Capece JA, et al. Topiramate for treating alcohol dependence: a randomized controlled trial. JAMA. 2007; 298: 1641– 1651. [DOI] [PubMed] [Google Scholar]
- 2. Johnson BA, Ait-Daoud N, Bowden CL, et al. Oral topiramate for treatment of alcohol dependence: a randomised controlled trial. Lancet. 2003; 361: 1677– 1685. [DOI] [PubMed] [Google Scholar]
- 3. Johnson BA, Rosenthal N, Capece JA, et al. Improvement of physical health and quality of life of alcohol-dependent individuals with topiramate treatment: US multisite randomized controlled trial. Arch Intern Med. 2008; 168: 1188– 1199. [DOI] [PubMed] [Google Scholar]
- 4. Witt JA, Elger CE, Helmstarder C. Impaired verbal fluency under topiramate—evidence for synergistic negative effects of epilepsy, topiramate, and polytherapy. Eur J Neurol. 2013; 30: 130– 137. [DOI] [PubMed] [Google Scholar]
- 5. Sommer BR, Mitchell EL, Wroolie TE. Topiramate: effects on cognition in patients with epilepsy, migraine headache and obesity. Ther Adv Neurol Disord. 2013; 6: 211– 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meador KJ, Loring DW, Hulihan JF, et al. Differential cognitive and behavioral effects of topiramate and valproate. Neurology. 2003; 60: 1483– 1488. [DOI] [PubMed] [Google Scholar]
- 7. Ray Al, Miranda R, MacKillop J, et al. A preliminary pharmacogenetic investigation of adverse events from topiramate in heavy drinkers. Exp Clin Psychopharmacol. 2009; 17: 122– 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mirza NS, Alfirevic A, Jorgensen A, et al. Metabolic acidosis with topiramate and zonisamide: an assessment of its severity and predictors. Pharmacogenet Genomics. 2011; 21: 297– 302. [DOI] [PubMed] [Google Scholar]
- 9. Masuda Y, Utsui Y, Shiraishi Y, et al. Relationships between plasma concentrations of diphenylhydantoin, phenobarbital, carbamazepine, and 3-sulfamoylmethyl-1,2-benzisoxazole (AD-810), a new anticonvulsant agent, and their anticonvulsant or neurotoxic effects in experimental animals. Epilepsia. 1979; 20: 623– 633. [DOI] [PubMed] [Google Scholar]
- 10. McComsey DF, Smith-Swintosky VL, Parker MH, et al. Novel, broad-spectrum anticonvulsants containing a sulfamide group: pharmacological properties of (S)-N-[(6-chloro-2,3-dihydrobenzo[1,4]dioxin-2-yl)methyl]sulfamide (JNJ-26489112). J Med Chem. 2013; 56: 9019– 9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maryanoff BE, Costanzo MJ, Nortey SO, et al. Structure-activity studies on anticonvulsant sugar sulfamates related to topiramate. Enhanced potency with cyclic sulfate derivatives. J Med Chem. 1998; 41: 1315– 1343. [DOI] [PubMed] [Google Scholar]
- 12. Parker MH, Smith-Swintosky VL, McComsey DF, et al. Novel, broad-spectrum anticonvulsants containing a sulfamide group: advancement of N-((benzo[b]thien-3-yl)methyl)sulfamide (JNJ-26990990) into human clinical studies. J Med Chem. 2009; 52: 7528– 7536. [DOI] [PubMed] [Google Scholar]
- 13. Brodie MJ, Ben-Menachem E, Chouette I, et al. Zonisamide: its pharmacology, efficacy and safety in clinical trials. Acta Neurol Scand Suppl. 2012; 126 (suppl 194): 19– 28. [DOI] [PubMed] [Google Scholar]
- 14. Ashkenazi A, Benlifer A, Korenblit J, et al. Zonisamide for migraine prophylaxis in refractory patients. Cephalalgia. 2006; 26: 1199– 1202. [DOI] [PubMed] [Google Scholar]
- 15. Silberstein S, Diener HC, Lipton R, et al. Epidemiology, risk factors, and treatment of chronic migraine: a focus on topiramate. Headache. 2008; 48: 1087– 1095. [DOI] [PubMed] [Google Scholar]
- 16. McElroy SL, Kotwal R, Guerdjikova AI, et al. Zonisamide in the treatment of binge eating disorder with obesity: a randomized controlled trial. J Clin Psychiatry. 2006; 67: 1897– 1906. [DOI] [PubMed] [Google Scholar]
- 17. Knapp CM, Mercado M, Markley TL, et al. Zonisamide decreases ethanol intake in rats and mice. Pharmacol Biochem Behav. 2007; 87: 65– 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sarid-Segal O, Knapp CM, Burch W, et al. The anticonvulsant zonisamide reduces ethanol self-administration by risky drinkers. Am J Drug Alcohol Abuse. 2009: 35: 316– 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Knapp CM, Sarid-Segal O, Richardson MA, et al. Open label trial of the tolerability and efficacy of zonisamide in the treatment of alcohol dependence. Am J Drug Alcohol Abuse. 2010; 36: 102– 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rubio G, Lopez-Munoz F, Ferre F, et al. Effects of zonisamide in the treatment of alcohol dependence. Clin Neuropharmacol. 2010; 33: 250– 253. [DOI] [PubMed] [Google Scholar]
- 21. Arias AJ, Feinn R, Oncken C, et al. Placebo-controlled trial of zonisamide for the treatment of alcohol dependence. J Clin Psychopharmacol. 2010; 30: 318– 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ramsay E, Faught E, Krumholz A, et al. Efficacy, tolerability, and safety of rapid initiation of topiramate versus phenytoin in patients with new-onset epilepsy: a randomized double-blind clinical trial. Epilepsia. 2010; 51: 1970– 1977. [DOI] [PubMed] [Google Scholar]
- 23. Sankar R, Ramsay E, McKay A, et al. A multicenter, outpatient, open-label study to evaluate the dosing, effectiveness, and safety of topiramate as monotherapy in the treatment of epilepsy in clinical practice. Epilepsy Behav. 2009; 15: 506– 512. [DOI] [PubMed] [Google Scholar]
- 24. Ben-Menachem E, Sander JW, Stefan H, et al. Topiramate monotherapy in the treatment of newly or recently diagnosed epilepsy. Clin Ther. 2008; 30: 1180– 1195. [DOI] [PubMed] [Google Scholar]
- 25. Faught E. Review of United States and European clinical trials of zonisamide in the treament of refractory partial-onset seizures. Seizure. 2004; 13S: S59– S65. [DOI] [PubMed] [Google Scholar]
- 26. Park SP, Kim SY, Hwang YH, et al. Long-term efficacy and safety of zonisamide monotherapy in epilepsy patients. J Clin Neurol. 2007; 3: 175– 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berent S, Sackellares JC, Giordani B, et al. Zonisamide (CI-912) and cognition: results from preliminary study. Epilepsia. 1987; 28: 61– 67. [DOI] [PubMed] [Google Scholar]
- 28. Knapp CM, Sarid-Segal O, Richardson MA, et al. The effects of topiramate on alcohol use and cognitive function in alcohol dependence. Alcohol Clin Exp Res. 2006; 30: 107A. [Google Scholar]
- 29. Cho JR, Koo DL, Joo EY, et al. Effect of levetiracetam monotherapy on background EEG activity and cognition in drug-naive epilepsy patients. Clin Neurophysiol. 2012; 123: 883– 891. [DOI] [PubMed] [Google Scholar]
- 30. Gomer B, Wagner K, Frings L, et al. The influence of antiepileptic drugs on cognition: a comparison of levetiracetam with topiramate. Epilepsy Behav. 2007; 10: 486– 494. [DOI] [PubMed] [Google Scholar]
- 31. Sarid-Segal O, Piechniczek-Buczek J, Knapp C, et al. The effects of levetiracetam on alcohol consumption in alcohol-dependent subjects: an open label study. Am J Drug Alcohol Abuse. 2008; 34: 441– 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fertig JB, Ryan ML, Falk DE, et al. A double-blind, placebo-controlled trial assessing the efficacy of levetiracetam extended-release in very heavy drinking alcohol-dependent patients. Alcohol Clin Exp Res. 2012; 36: 1421– 1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Richter C, Effenberger S, Bschor T, et al. Efficacy and safety of levetiracetam for the prevention of alcohol relapse in recently detoxified alcohol-dependent patients: a randomized trial. J Clin Psychopharmacol. 2012; 32: 558– 562. [DOI] [PubMed] [Google Scholar]
- 34. Babor T, Higgins-Biddle JC, Saunders JB, et al. AUDIT—The Alcohol Use Disorders Identification Test: Guidelines for Use in Primary Care. Geneva, Switzerland: World Health Organization, Department of Mental Health and Substance Dependence; 2001. [Google Scholar]
- 35. Sullivan JT, Sykora K, Schneiderman J, et al. Assessment of alcohol withdrawal: the revised clinical institute withdrawal assessment for alcohol scale (CIWA-Ar). Br J Addict. 1989; 84: 1353– 1357. [DOI] [PubMed] [Google Scholar]
- 36. Johnson BA, DiClemente CC, Ait-Daoud N, et al. Brief Behavioral Compliance Enhancement Treatment (BBCET) manual. In: Johnson BA, Ruiz P, Galanter M, eds. Handbook of Clinical Alcholism Treatment. Baltimore, MD: Lippencott Williams & Wilkins; 2003: 282– 301. [Google Scholar]
- 37. Sobell LC, Sobell MB. Timeline follow-back: a technique for assessing self-reported alcohol consumption. In: Litten RZ, Allen JP, eds. Measuring Alcohol Consumption: Psychosocial and Biochemical Methods. Towota, NJ: Humana Press; 1992. [Google Scholar]
- 38. Litten RZ, Allen JP, Fertig JB. Gamma-glutamyltranspeptidase and carbohydrate deficient transferrin: alternative measures of excessive alcohol consumption. Alcohol Clin Exp Res. 1995; 19: 1541– 1546. [DOI] [PubMed] [Google Scholar]
- 39. Anton RF, Moak DH, Latham PK. The Obsessive Compulsive Drinking Scale: a new method of assessing outcome in alcoholism treatment studies. Arch Gen Psychiatry. 1996; 53: 225– 231. [DOI] [PubMed] [Google Scholar]
- 40. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979; 134: 382– 389. [DOI] [PubMed] [Google Scholar]
- 41. Hamilton M. The assessment of anxiety states by rating. Br J Med Psychol. 1959; 32: 50– 55. [DOI] [PubMed] [Google Scholar]
- 42. Hays R, Stewart A. Sleep measures. In: Stewart A, Ware J, eds. Measuring Functioning and Wellbeing: The Outcomes Study Approach. Durnam, NC: Duke University Press; 1992: 235– 259. [Google Scholar]
- 43. Aldenkamp AP, Baker G, Pieters MS, et al. The Neurotoxicity Scale: the validity of a patient-based scale, assessing neurotoxicity. Epilepsy Res. 1995; 20: 229– 239. [DOI] [PubMed] [Google Scholar]
- 44. Wechsler D. Wechsler Abbreviated Intelligence Scales. San Antonio, TX: The Psychological Corporation; 1999. [Google Scholar]
- 45. Wechsler D. Wechsler Memory Scales. 3rd ed San Antonio, TX: The Psychological Corporation; 1997. [Google Scholar]
- 46. Rey A. L’examen clinique en psychologie. Paris, France: Presses Universitaires de France; 1964. [Google Scholar]
- 47. Meyers J, Meyers K. The Rey Complex Figure and the Recognition Trial under four different administration procedures. Clin Neuropsychol. 1995; 9: 65– 67. [Google Scholar]
- 48. Benton AL, Hamsher KD. Multilingual Aphasia Examination. Iowa City, IA: AJA Associates; 1989. [Google Scholar]
- 49. Golden CJ, Freshwater SM. The Stroop Color and Word Test: A Manual for Clinical and Experimental Uses. Chicago, IL: Stoelting Co; 2002. [Google Scholar]
- 50. Reitan RM. Validity of the Trail Making Test as an indicator of organic brain damages. Percept Mot Skills. 1958; 19: 271– 276. [Google Scholar]
- 51. Heaton RK, Chelune GJ, Talley JL, et al. Wisconsin Card Sorting Test (WCST) Manual Revised and Expanded. Odessa, Ukraine: Psychological Assessment Resources Inc; 1993. [Google Scholar]
- 52. Smith A. Symbol Digit Modalities Test (SDMT): Manual (Revised). Los Angeles, CA: Western Psychological Services; 1982. [Google Scholar]
- 53. Klove H. Clinical Neuropsychology. Med Clin North Am. 1963; 47: 1647– 1658. [PubMed] [Google Scholar]
- 54. Shayh J, Shellenberger K, Canfax DM. Chemistry Biotransfromation and Pharmacokinetics in Antiepileptic Drugs. 5th ed Levy RH, Mattson RH, Meldrum BS, et al. eds. Philadelphia, PA: Lippincott Williams & Wilkins; 2002: 873– 879. [Google Scholar]
- 55. Baltieri DA, Daro FR, Ribeiro PL, et al. Comparing topiramate with naltrexone in the treatment of alcohol dependence. Addiction. 2008; 103: 2035– 2044. [DOI] [PubMed] [Google Scholar]
- 56. Paparrigopoulos T, Tzavellas E, Karaiskos D, et al. Treatment of alcohol dependence with low-dose topiramate: an open-label controlled study. BMC Psychiatry. 2011; 11: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Florez G, Garcia-Portilla P, Alvarez S, et al. Using topiramate or naltrexone for the treatment of alcohol-dependent patients. Alcohol Clin Exp Res. 2008; 32: 1251– 1259. [DOI] [PubMed] [Google Scholar]
- 58. Arbuthnott K, Frank J. Trail making test, part B as a measure of executive control: validation using a set-switching paradigm. J Clin Exp Neuropsychol. 2000; 22: 518– 528. [DOI] [PubMed] [Google Scholar]
- 59. Lynch BA, Lambeng N, Nocka K, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci U S A. 2004; 101: 9861– 9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Meehan AL, Yang X, McAdams BD, et al. A new mechanism for antiepileptic drug action: vesicular entry may mediate the effects of levetiracetam. J Neurophysiol. 2011; 106: 1227– 1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Miranda R, Jr, MacKillop J, Monti PM, et al. Effects of topiramate on urge to drink and the subjective effects of alcohol: a preliminary laboratory study. Alcohol Clin Exp Res. 2008; 32: 489– 497. [DOI] [PubMed] [Google Scholar]
- 62. Jin Z, Bhandage AK, Bazov I, et al. Selective increases of AMPA, NMDA, and kainate receptor subunit mRNAs in the hippocampus and orbitofrontal cortex but not in prefrontal cortex of human alcoholics. Front Cell Neurosci. 2014; 8: 1– 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kärkkäinen O, Kupila J, Häkkinen M, et al. AMPA receptors in post-mortem brains of Cloninger type 1 and 2 alcoholics: a whole-hemisphereautoradiography study. Psychiatry Res. 2013; 214: 429– 434. [DOI] [PubMed] [Google Scholar]
- 64. Simeone TA, Wilcox KS, White HS. Topiramate modulation of beta(1)- and beta(3)-homomeric GABA(A) receptors. Pharmacol Res. 2011; 64: 44– 52. [DOI] [PubMed] [Google Scholar]
- 65. Braga MF, Aroniadou-Anderjaska V, Li H, et al. Topiramate reduces excitability in the basolateral amygdala by selectively inhibiting GluK1 (GluR5) kainate receptors on interneurons and positively modulating GABAA receptors on principal neurons. J Pharmacol Exp Ther. 2009; 330: 558– 566. [DOI] [PubMed] [Google Scholar]
- 66. Gryder DS, Rogawski MA. Selective antagonism of GluR5 kainate-receptor-mediated synaptic currents by topiramate in rat basolateral amygdala neurons. J Neurosci. 2003; 23: 7069– 7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kranzler HR, Covault J, Feinn R, et al. Topiramate treatment for heavy drinkers: moderation by a GRIK1 polymorphism. Am J Psychiatry. 2014; 171: 445– 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Okada M, Kawata Y, Mizuno K, et al. Interaction between Ca2+, K+, carbamazepine and zonisamide on hippocampal extracellular glutamate monitored with a microdialysis electrode. Br J Pharmacol. 1998; 124: 1277– 1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Huang CW, Ueno S, Okada M, et al. Zonisamide at clinically relevant concentrations inhibits field EPSP but not presynaptic fiber volley in rat frontal cortex. Epilepsy Res. 2005; 67: 51– 60. [DOI] [PubMed] [Google Scholar]
- 70. Ueda Y, Doi T, Takumaru J, et al. Effect of zonisamide on molecular regulation of glutamate and GABA transporter proteins during epileptogenesis in rats with hippocampal seizures. Mol Brain Res. 2003; 116: 1– 6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.