

Abstract
Dichloroacetate (DCA) is biotransformed by glutathione transferase zeta 1 (GSTZ1), a bifunctional enzyme that, as maleylacetoacetate isomerase (MAAI), catalyzes the penultimate step in tyrosine catabolism. DCA inhibits GSTZ1/MAAI, leading to delayed plasma drug clearance and to accumulation of potentially toxic tyrosine intermediates. Haplotype variability in GSTZ1 influences short-term DCA kinetics in healthy adults, but the impact of genotype in children treated chronically with DCA is unknown. Drug kinetics was studied in 17 children and adolescents with congenital mitochondrial diseases administered 1,2-13C-DCA. Plasma drug half-life and trough levels varied 3–6 -fold, depending on GSTZ1/MAAI haplotype and correlated directly with urinary maleylacetone, a substrate for MAAI. However, chronic DCA exposure did not lead to progressive accumulation of plasma drug concentration; instead, kinetics parameters plateaued, consistent with the hypothesis that equipoise is established between the inhibitory effect of DCA on GSTZ1/MAAI and new enzyme synthesis.
Conclusion
GSTZ1/MAAI haplotype variability affects DCA kinetics and biotransformation. However, these differences appear to be stable in most individuals and are not associated with DCA plasma accumulation or drug-associated toxicity in young children.
Keywords: Pharmacokinetics, Pharmacogenetics, Congenital Lactic Acidosis, Dichloroacetate, Glutathione Transferase Zeta 1, Maleylacetoacetate Isomerase, Mitochondrial Disease, Tyrosine
Introduction
The mitochondrial pyruvate dehydrogenase complex (PDC) irreversibly decarboxylates pyruvate to acetyl CoA. Consequently, it serves to oxidatively remove lactate, which is in equilibrium with pyruvate, and to link glycolysis in the cytoplasm to the tricarboxylic acid cycle in mitochondria (Fig. 1A). PDC is regulated mainly by reversible phosphorylation, in which pyruvate dehydrogenase kinase (PDK) phosphorylates, and inhibits, PDC while pyruvate dehydrogenase phosphatase (PDP) dephosphorylates PDC and restores catalytic activity (1, 2).
Fig. 1.
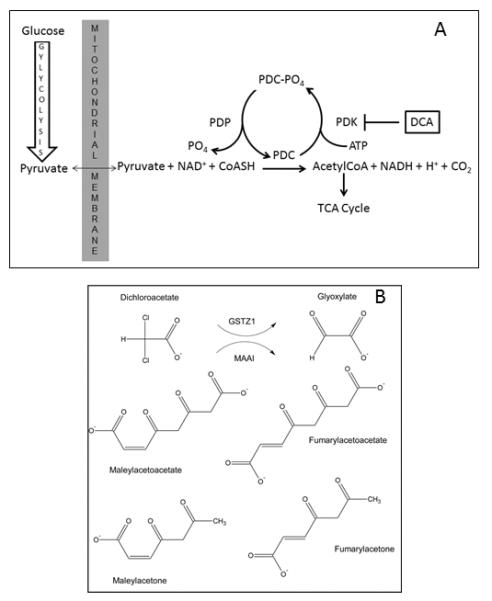
Site of action and biotransformation of DCA. Panel A: The pyruvate dehydrogenase complex (PDC) irreversibly oxidizes pyruvate to acetyl CoA, thereby linking cytoplasmic glycolysis to the mitochondrial tricarboxylic acid (TCA) cycle and oxidative phosphorylation. Pyruvate dehydrogenase kinase (PDK) reversibly inhibits PDC activity by phosphorylation, whereas, pyruvate dehydrogenase phosphatase (PDP) restores PDC activity via dephosphorylation. DCA inhibits PDK, thereby maintaining PDC in its catalytically active form. Panel B: DCA is dehalogenated by GSTZ1 to glyoxylate, upon which the xenobiotic enters the general carbon pool of the host. The same enzyme, as MAAI, catalyzes the penultimate step in the phenylalanine/tyrosine catabolic pathway, isomerizing maleylacetoacetate and maleylacetone to fumarylacetoacetate and fumarylacetone, respectively.
The PDC-PDK axis has been the focus of considerable multidisciplinary research, not only because of its critical role in cellular bioenergetics, but also because of its emerging presence as a therapeutic target for many congenital and acquired disorders in which cellular fuel metabolism is perturbed (3). Dichloroacetate (DCA), a prototypic inhibitor of PDK, maintains the PDC in its unphosphorylated active form, thereby stimulating mitochondrial oxidative metabolism. This mechanism provides the primary rationale for the use of DCA as an investigational drug for both primary and secondary mitochondrial diseases (reviewed in 4). Chronic DCA administration may cause reversible peripheral neuropathy in adults (5), but is generally well tolerated and appears safe in children and adolescents with primary mitochondrial diseases (6).
DCA is metabolized to glyoxylate by the bifunctional enzyme glutathione transferase zeta 1/maleylacetoacetate isomerase (GSTZ1/MAAI), which also catalyzes the penultimate step in the phenylalanine/tyrosine catabolic pathway (Fig. 1B) (7). The human GSTZ1/MAAI has three non-synonymous single nucleotide polymorphisms (SNPs): G94 > A (rs3177427) Glu→Lys at position 32; G124 > A (rs7972) Gly→Arg at position 42; and G245 > T (rs1046428) Thr → Met at position 92; there is also a promoter SNP-1002 G > A (rs7160195). The five reported haplotypes and their frequencies in the general population are: EGT (~50%); KGT (~30%); EGM (~10–20%); KRT (~5–10%); and KGM (<5%)(8). Carriers of at least one EGT (wild-type) allele show more rapid biotransformation and plasma clearance of DCA (“fast” metabolizers) than non-carriers of this allele (“slow” metabolizers)(8). Thus, haplotype variability in GSTZ1/MAAI directly influences DCA biotransformation and could lead to accumulation of DCA and potentially toxic tyrosine intermediates.
We previously determined in a sub-acute study that healthy adults who possess at least one EGT allele metabolized DCA more rapidly and accumulated lower levels of urinary maleylacetone (MA) than those lacking this allele (8). Inhibition of MAAI by DCA also shunts tyrosine carbon formation of succinylacetone, which inhibits a proximal step in heme synthesis, leading to accumulation of the reactive molecule δ-ALA (9).
It is uncertain the extent to which GSTZ1/MAAI haplotype variability influences DCA and tyrosine metabolism in children with primary mitochondrial disease who are chronically treated with the drug. Therefore, we investigated the impact of GSTZ1/MAAI haplotype on drug kinetics and tyrosine metabolism in children with inborn errors of mitochondrial metabolism who were chronically administered DCA.
Methods
This study was approved by the University of Florida Institutional Review Board and the Scientific Advisory Committee of the Clinical Research Center. Seventeen children (aged 1.2 – 19 yrs at entry) participated in a randomized, double-blind, controlled trial, the clinical outcome of which was previously reported (10, 11). Caucasians (n=12), African-Americans (n=1), Hispanics (n=1) and mixed race subjects (1 Asian/Caucasian; 2 Hispanic/Caucasian) comprised this cohort.
Patient safety was assessed at each visit by physical examination, measurement of fasting whole blood levels of glucose and lactate (YSI Glucose/Lactate Analyzer, Yellow Springs, OH), complete blood count and serum aspartate aminotransferase and alanine aminotransferase. Upper and lower extremity nerve conduction velocities were also obtained every six months, as reported previously (10). Tolerability to and compliance with administered DCA was determined by parental and patient interviews and by noting the amount of residual drug returned at each visit.
Patients received either 12.5 mg/kg/12h oral DCA or placebo for 6 months, after which subjects in the placebo arm were switched over to the DCA arm and all patients were followed for an indefinite period. At the start of DCA treatment, and at subsequent 6 month intervals, patients received an oral dose of 12.5 mg/kg of 100% 1, 2-13C-DCA to investigate DCA kinetics during chronic administration. Concentrations of plasma and urinary DCA and urinary MA and δ-ALA were analyzed by gas or liquid chromatography (GC-MS or LC-MS) (12, 13). The plasma concentration-time curve for all DCA measurements was fitted to a noncompartmental pharmacokinetic model for each patient using WinNonlin, version 5.01 software (Pharsight, Mountain View, CA), obtained through the academic license program. Through the WinNonlin software, we calculated the area under the plasma concentration-time curve from 0 to 1440 minutes (24 hours) for 13C-DCA (AUC 0–1440 min) using the linear-trapezoidal method. At least three sampling points were used by the modeling software to estimate the first-order elimination rate constant (λz) for each time-concentration curve. The mean, standard deviation and statistical significance of the data were determined using Excel software (Microsoft, Redmond, WA). A 2-sided Student t test was used to analyze kinetic and metabolic data between groups. In all cases, a P value of ≤ .05 was considered statistically significant.
DNA was isolated from peripheral blood and was genotyped for three non-synonymous single nucleotide polymorphisms (SNPs), rs7975 (E32K), rs7972 (G42R) and rs1046428 (T82M) by pyrosequencing as reported (11). In humans, these SNPs give rise to five GSTZ1/MAAI haplotypes, named here as EGT, KGT, EGM, KRT and KGM. Haplotypes were inferred by computational methods using the PHASE (version 2.1) Bayesian haplotype reconstruction program (14).
Results and Discussion
Tolerability to and compliance with DCA administration were excellent throughout the study. As reported previously (6), there were no significant changes in the patients' levels of blood glucose, hemogram or serum transaminases. Blood lactate levels remained normal throughout the trial. Serial nerve electrical measurements occasionally demonstrated clinically asymptomatic worsening of lower extremity nerve conduction velocity. These changes led to a temporary 50 percent dose reduction of DCA in patients 5 (24 months) and 14 (14 months)(6).
Table 1 shows the patient demographics and GSTZ1/MAAI haplotype distribution. There were 11 EGT carriers and 6 EGT non-carriers in the group. The allelic distribution was consistent with haplotype frequencies reported in the general population, which identified the EGT as the commonest haplotype, followed by the KGT allele (8). In this small patient sample, non-EGT carriers were predominantly male and younger than EGT carriers (4.3 yrs vs. 6.8 yrs).
Table 1.
Patient demographics and GSTZ1/MAAI haplotype distribution.
| Age at Entry (years) | Gender | Diagnosis | GSTZ1/MAAI Diplotype | |
|---|---|---|---|---|
| 1 | 3.7 | F | PDHE1α | (EGT/EGT) |
| 2 | 3.7 | F | PDHE1α | (EGT/EGT) |
| 3 | 9.6 | F | Complex I | (EGT/KGT) |
| 4 | 1.5 | M | Complex II | (EGT/KGT) |
| 5 | 1.7 | F | PDHE1α | (EGT/KGT) |
| 6 | 4.8 | M | PDHE1α | (EGT/KGT) |
| 7 | 9.7 | M | COX | (EGT/KGT) |
| 8 | 13.1 | M | MELAS | (EGT/KGT) |
| 9 | 19.1 | F | MELAS | (EGT/KGT) |
| 10 | 1.8 | F | Complex I | (EGT/KRT) |
| 11 | 6 | M | MELAS | (EGT/EGT) |
| 12 | 2.7 | M | PDHE1α | (KGT/KGM) |
| 13 | 1.2 | M | OXPHOS | (EGM/EGM) |
| 14 | 7.1 | M | Complex I and IV | (KGT/KGT) |
| 15 | 2.3 | M | Complex II, III, IV | (KGT/EGM) |
| 16 | 6.6 | M | PDHE1α | (EGM/EGM) |
| 17 | 5.8 | F | PDHE1α | (KGT/KGT) |
Abbreviations: PDHE1α, pyruvate dehydrogenase E1alpha subunit; OXPHOS, generalized oxidative phosphorylation defect; Complex I, II, III, V, respiratory chain complexes; MELAS, mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes. DCA slow metabolizers (EGT non-carriers) are in red.
13C-DCA kinetic parameters and urinary MA and δ-ALA levels are summarized in Table 2. As predicted, EGT carriers exhibited a more rapid plasma clearance of DCA (p = 0.01), with a shorter plasma elimination half-life (t ½; p = 0.03), compared to non-EGT carriers. In contrast, the area under the plasma concentration curve (AUC) was quite variable and did not differ significantly. (p = 0.16). Although peak plasma DCA concentration (Cmax) was unrelated to genotype, plasma trough drug levels were higher in non-EGT carriers (p = 0.02). Urinary MA (p = 0.02) and δ-ALA (p = 0.03) concentrations were higher in non-EGT carriers vs. EGT carriers. DCA plasma trough levels (Cmin) and t ½ correlated with GSTZ1/MAAI genotype and were directly associated with urinary MA or δ-ALA concentrations after 6 months and, particularly, after 30 months of drug exposure (Supplemental Figs. 1 and 2). Nevertheless, chronic DCA exposure did not lead to progressive plasma drug or urinary MA or δ-ALA accumulation. Rather, kinetic parameters tended to stabilize over time, resulting in a rough plateauing of DCA trough concentrations, regardless of genotype. This finding is most clearly illustrated in Fig. 2, which relates trough levels to duration of drug exposure in 6 patients in whom 13C-DCA kinetics were determined every 6 months for 30 consecutive months. Plasma DCA trough concentrations varied greatly, ranging from 2.9 μg/ml to 182 μg/ml. The highest trough levels occurred in patients 13 and 14, who were non-EGT carriers, consistent with prior data (8). 13 (Table 1).
Table 2.
13C-DCA kinetic parameters and urinary MA and δ-ALA levels.
| Subject Class | No. pts. | C max (μg/ml) | t ½ (hr) | Trough (μg/ml) | Plasma Cl (ml/hr) | AUC (μg/ml*hr) | Urinary MA (μg/g Cr) | Urinary δ-ALA (μg/g Cr) |
|---|---|---|---|---|---|---|---|---|
| EGT carriers (6 months) | 11 | 34.2 ± 10 | 6.4 ± 6.2 | 23 ± 43 | 114 ± 68 | 378 ± 474 | 1.2 ± 0.9 | 1.4 ± 0.9 |
| EGT non-carriers (6 months) | 6 | 47 ± 16 | 17.7 ± 9 * | 128 ± 80 * | 31.5 ± 21 * | 1362 ± 1099 | 6.9 ± 2.6 * | 4.2 ± 1.1 * |
| EGT carriers (30 months) | 6 | 35.5 ± 7.6 | 4.0 ± 2.5 | 12.5 ± 12 | 125 ± 66 | 236 ± 122 | 1.9 ± 1.1 | 1.2 ± 0.3 |
| EGT non-carriers (30 months) | 3 | 49.7 ± 11 | 13.5 ± 7 | 107 ± 55 | 40.3 ± 27 * | 747 ± 417 | 5.5 ± 1.2 * | 3.7 ± 0.9 * |
p ≤ 0.03. See text for details.
C max- maximum concentration (0–12 hrs)
t ½- plasma elimination half-life
Cl- clearance
AUC- Area under the time-concentration curve (0–12 hrs)
Fig. 2.

1, 2-13C-DCA trough levels as a function of duration of drug exposure in 6 selected subjects with differing GSTZ1/MAAI haplotypes. See Table 1 for further patient characteristics.
These data are consistent with the postulate that the response of the host chronically exposed to DCA is to establish a new dynamic equilibrium, in which the DCA-induced inhibition of the GSTZ1/MAAI enzyme is balanced by new protein synthesis. Although this possibility has not been examined directly, preliminary data indicate that haplotype variations in GSTZ1/MAAI do influence the expression of this gene in humans (15) and, presumably, also affect protein turnover. In addition, inhibition of GSTZ1/MAAI turnover in rats by DCA (15, 16) allows some product formation, even though enzyme protein synthesis is drastically reduced (17) and full GSTZ1 enzyme activity recovers slowly after DCA administration is withdrawn (18). Variability in enzyme inhibition is likely reflected by the higher drug trough levels in subjects who lack an EGT allele in adults, as well as in the pediatric subjects reported here. Such “slow metabolizers” would be predicted to achieve similar steady-state plasma DCA levels at lower doses than subjects who possess at least one EGT allele and are thus able to clear the drug more rapidly. Such genomics-based dosing was achieved in the safe, chronic administration of DCA to adults with recurrent malignant brain tumors in a phase 1 clinical trial (18 and unpublished observations).
MAAI catalyzes the penultimate step in the phenylalanine/tyrosine catabolic pathway. Loss-of-function mutations in the terminal enzyme fumarylacetoacetate hydrolase (FAH) lead to hereditary tyrosinemia type 1 and to severe neurological and hepatocellular toxicity, if untreated (9). It is thought that the adverse effects of FAH deficiency are due to accumulation of reactive intermediates of tyrosine metabolism, such as those depicted in Fig. 1B, and of δ-ALA, resulting from inhibition of heme metabolism by succinylacetone (SA). High doses of DCA are hepatotoxic and neurotoxic in inbred rodent strains and can lead to mild, asymptomatic and reversible elevation of serum transaminases and to reversible peripheral neuropathy in human's (4). We compared our current measurements of urinary MA and SA to those we previously reported (12) for 23 patients with untreated hereditary tyrosinemia type I, and to levels measured in MAAI knockout mice by expressing the results uniformly as μg/ml, because urinary creatinine was not determined in the prior studies. As shown in Table 3, urinary MA concentrations in the children with mitochondrial diseases after 6 and 30 months of DCA exposure were higher than those in children with tyrosinemia, but were vastly lower than the levels found in the knockout mice. In contrast, urinary SA was undetectable in the DCA-exposed patients but was present in both HT I subjects and in MAAI knockout mice. It is noteworthy that these mice showed no clinical or other biochemical evidence of toxicity from genetic ablation of MAAI. These data suggest that the inhibitory effect of chronic DCA on GSTZ1/MAAI leads to buildup of the MAAI substrates MA and maleylacetoacetate but little or no accumulation of SA. Nevertheless, urinary δ-ALA increases in children with chronic DCA (ref. 6 and Table 3), implying that either sufficient SA accumulates to inhibit δ-ALA metabolism and/or DCA itself may perturb heme synthesis. This latter postulate is inconsistent with the lack of changes in hemoglobin or hematocrit measured serially in children chronically exposed to DCA and this reported here and elsewhere (6). Regardless of the precise mechanism(s), it is possible that DCA's inhibitory effect on MAAI could lead to accumulation of toxic levels of MA, δ-ALA and other tyrosine intermediates, causing hepatocellular and neurological dysfunction in chronically exposed humans, particularly those who metabolize the drug slowly. However, despite the difference we found in MA and δ-ALA levels between EGT carriers and non-carriers, chronic DCA was well tolerated in both patient groups. Nevertheless, at the present time, we advise that any future clinical trials of DCA base dosing on GSTZ1/MAAI haplotype and prospectively monitor urinary MA levels and clinical signs of peripheral neuropathy. Incorporation of formal nerveconductiontesting should also be considered in subjects at higher risk of developing DCA-associated neuropathy, such as adult treated chronically with the drug.. Therefore, based on these observations, and on our prior studies in knockout mice (12), it appears likely that tissue concentrations of these intermediates in our chronically DCA-treated patients are not sufficiently high to be of major clinical significance, regardless of GSTZ1/MAAI haplotype.
Table 3.
Comparison of Urinary Maleylacetone and Succinylacetone Concentrations in DCA-Treated Children, Children with Untreated Hereditary Tyrosinemia Type I and Mice with Genetic Ablation of Maleylacetoacetate Isomerase.
| Population* | Urinary MA (mcg/ml) | Urinary SA (mcg/ml) |
|---|---|---|
|
| ||
| EGT carriers (6 months) | 0.81 ± 0.6 | <10 |
| EGT non-carriers (6 months) | 4.3 ± 2.1 | <10 |
| EGT carriers (30 months) | 1.2 ± 0.9 | <10 |
| EGT non-carriers (30 months) | 3.4 ± 1.2 | <10 |
| HT type I patients | 0.77 ± 0.63† | 57 ± 29† |
| MAAIKO mice | 1200 ± 500 | 350 ± 170 |
Data for EGT carriers and non-carriers converted from those presented in Table 2 for DCA-treated subjects. Samples from patients with HT type I were de-identified, so ages and sexes are unknown. All values are means ± S.D.
Values are from untreated patients. Samples taken from HT patients treated with NTBC had unmeasurable urinary MA levels (data not shown).
Abbreviations: HT, hereditary tyrosinemia; MAAI, maleylacetoacetate isomerase; KO, knockout.
Conclusion
Although GSTZ1/MAAI haplotype variability markedly affects DCA kinetics and tyrosine metabolism in chronically treated children, the differences appear to be stable and probably reflect attainment of a new equilibrium in which DCA induced enzyme inhibition is balanced by an increase in de novo enzyme synthesis. This postulate is consistent with experimental studies in rodents and with the chronic safety profile of DCA in children and adults, whose dosage is adjusted, based on GSTZ1 haplotype. (19).
Supplementary Material
Acknowledgments
Funding source: This study was supported by grants from the Muscular Dystrophy Association (MDA/92–95), the Orphan Products Division of the Food and Drug Administration (FD-R-001500), and the National Institutes of Health (R01 ES07355 and P42 ES07375, and UL1 TR000062 (CTSA).
Clinical Trial Registry: Clinicaltrials.gov, if clinical trial: N/A
Peter W. Stacpoole wrote the first draft of the manuscript and no honorarium, grant or other form of payment was given to anyone to produce the manuscript.
Abbreviations (nonstandard)
- DCA
dichloroacetate
- GSTZ1
glutathione transferase zeta 1
- MAAI
maleylacetoacetate isomerase
- δ-ALA
delta-aminolevulinate
- MA
maleylacetone
Footnotes
Financial Disclosure Statement: None
Conflict of Interest: None
References
- 1.Patel MS, Korotchkina LG. Regulation of mammalian pyruvate dehydrogenase complex by phosphorylation: complexity of multiple phosphorylation sites and kinases. Exp Mol Med. 2001;33(4):191–7. doi: 10.1038/emm.2001.32. [DOI] [PubMed] [Google Scholar]
- 2.Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998;329:191–196. doi: 10.1042/bj3290191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, Gottlieb E, Peeper DS. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013;498(7452):109–12. doi: 10.1038/nature12154. [DOI] [PubMed] [Google Scholar]
- 4.Stacpoole PW. The dichloroacetate dilemma: environmental hazard versus therapeutic goldmine--both or neither? Environ Health Perspect. 2011;119(2):155–158. doi: 10.1289/ehp.1002554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006;66(3):324–30. doi: 10.1212/01.wnl.0000196641.05913.27. [DOI] [PubMed] [Google Scholar]
- 6.Abdelmalak M, Lew A, Ramezani R, Shroads AL, Coats BS, Langaee T, et al. Long-term safety of dichloroacetate in congenital lactic acidosis. Mol Genet Metab. 2013;109(2):139–43. doi: 10.1016/j.ymgme.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stacpoole PW, Kurtz TL, Han Z, Langaee TY. Mitochondrial Medicine and Mitochondrion-Based Therapeutics; published in Advanced Drug Delivery Reviews. Vol. 60. Elsevier; Philadelphia: 2008. Role of dichloroacetate in the treatment of genetic mitochondrial diseases; pp. 1478–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shroads AL, Langaee T, Coats BS, Kurtz TL, Bullock JR, Weithorn D, et al. Human polymorphisms in the glutathione transferase zeta 1/maleylacetoacetate isomerase gene influence the toxicokinetics of dichloroacetate. J Clin Pharmacol. 2012;52:837–849. doi: 10.1177/0091270011405664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanguay RM, Lambert M, Grompe M, Mitchell GA. Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2004. Hypertyrosinemia; pp. 1777–1805. [Google Scholar]
- 10.Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, et al. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006;117:1519–31. doi: 10.1542/peds.2005-1226. [DOI] [PubMed] [Google Scholar]
- 11.Stacpoole PW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, Theriaque DW, et al. Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics. 2008;121:e1223–8. doi: 10.1542/peds.2007-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shroads AL, Henderson GN, Cheung J, James MO, Stacpoole PW. Unified gas chromatographic-mass spectrometric method for quantitating tyrosine metabolites in urine and plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;808:153–61. doi: 10.1016/j.jchromb.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 13.Felitsyn NM, Henderson GN, James MO, Stacpoole PW. Liquid chromatography-tandem mass spectrometry method for the simultaneous determination of delta-ALA, tyrosine and creatinine in biological fluids. Clin Chim Acta. 2004;350:219–30. doi: 10.1016/j.cccn.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 14.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson WB, Board PG, Gargano B, Anders MW. Inactivation of glutathione transferase zeta by dichloroacetic acid and other fluorine-lacking alpha-haloalkanoic acids. Chem Res Toxicol. 1999;12(12):1144–9. doi: 10.1021/tx990085l. [DOI] [PubMed] [Google Scholar]
- 16.James MO, Cornett R, Yan Z, Henderson GN, Stacpoole PW. Glutathione-dependent conversion to glyoxylate, a major pathway of dichloroacetate biotransformation in hepatic cytosol from humans and rats, is reduced in dichloroacetate-treated rats. Drug Metab Dispos. 1997;25(11):1223–7. [PubMed] [Google Scholar]
- 17.Ammini CV, Fernandez-Canon J, Shroads AL, Cornett R, Cheung J, James MO, et al. Pharmacologic or genetic ablation of maleylacetoacetate isomerase increases levels of toxic tyrosine catabolites in rodents. Biochem Pharmacol. 2003;66(10):2029–38. doi: 10.1016/j.bcp.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 18.Guo X, Dixit V, Liu H, Shroads AL, Henderson GN, James MO, et al. Inhibition and recovery of rat hepatic glutathione S-transferase zeta and alteration of tyrosine metabolism following dichloroacetate exposure and withdrawal. Drug Metab Dispos. 2006;34(1):36–42. doi: 10.1124/dmd.105.003996. [DOI] [PubMed] [Google Scholar]
- 19.Dunbar EM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, Shuster JJ, Wagner DA, Stacpoole Phase 1 trial of fichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest. New Drugs. 2014 doi: 10.1007/s10637-013-0047-4. DOI 10.1007/s10637-013-0047-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.