

Abstract
Previous studies have shown that activation of mouse MrgC11, a G-protein coupled receptor, by its peptide ligand BAM8-22 can inhibit chronic pain. A large scale screen has been carried out to isolate small molecule allosteric agonists of MrgX1, the human homologue of MrgC11. The goal of this study is to improve the efficacy and potency of the positive allosteric modulators with therapeutic implications of anti-chronic pain. Here, we report an iterative parallel synthesis effort and a structure-activity relationship of a series of arylsulfonamides, which led to the discovery of the first positive allosteric modulator (PAM) of MrgX1, ML382.
Keywords: MrgX1, ML382, chronic pain, positive allosteric modulator, MLPCN, molecular probes
Chronic pain is difficult to treat and has become a major health and economic burden worldwide reaching nearly epidemic levels with 20–25% of the population affected. Chronic pain is often refractory to current therapies and the many of the major analgesics (e.g., opioids) carry with them dose-limiting adverse events and serious risk of addiction and abuse. These risks of addiction and abuse present substantial barriers to their clinical use and have become a serious health threat. Thus the discovery of novel therapies to treat pain without the addiction potential is a major goal.
Mrgs (also named Mrgprs/SNSRs) are a family of G protein-coupled receptors (GPCRs) consisting of more than 50 members in the mouse genome.[1] Strikingly, the expression of many Mrgs, including MrgC11, is restricted to subsets of small-diameter sensory neurons in DRG and trigeminal ganglia as these genes have not been detected in the central nervous system or in the rest of the body.[2] Similarly, some of human MrgprXs such as MrgX1 are also selectively expressed in DRG neurons.[1b] BAM8-22, a 15 amino acid endogenous peptide, is a specific agonist for mouse MrgprC11 and human MrgX1.[1a, 2] Based on the expression pattern, ligand specificity, and similarity in signal pathways, MrgprC11 and MrgprX1 are functional orthologues.
Several complementary lines of evidence suggest that MrgprC11 functions as an endogenous regulator of persistent pain.[3] Particularly, spinal cord application of BAM8-22 and other MrgC11 agonists in mice significantly attenuates inflammatory hyperalgesia and neuropathic pain in an Mrgpr-dependent manner.[3a, 4] It has been shown recently that the activation of MrgC11 leads to inhibition high-voltage activating Ca2+ channels which play essential role in pain signal transmission.[5] Because of the specific expression, agonists and PAMs for MrgX1 may represent a new class of anti-hyperalgesics for chronic pain without side effects in the central nervous system.[6],[7]
The project commenced with a screen of the >300000 NIH Molecular Library Small Molecule Repository (MLSMR) compound collection using a triple addition protocol using BAM-22 as the as the ligand (AID: 588675) (Figure 1).[8] Using a HEK293 cell line that stably expresses the MrgX1 protein, the first addition is the drug itself (agonist mode), the second addition is an EC10 to EC30 of BAM-22 for identification of positive allosteric modulators, and the third addition uses an EC90 to Emax of BAM-22 for identification of antagonists/negative allosteric modulators. From the initial screen, ~1900 compound were identified as hits and after a medicinal chemistry triage, ~1100 compound were retested and then counter screened against HEK293 parental cells. There were ~150 compounds that showed no activity against the parental cells and these were then evaluated in a 5-point concentration response curve (CRC) leaving 29 active compounds. These compounds were then tested in 10-point CRC format revealing 19 confirmed MrgX1 positive allosteric modulators.
Figure 1.
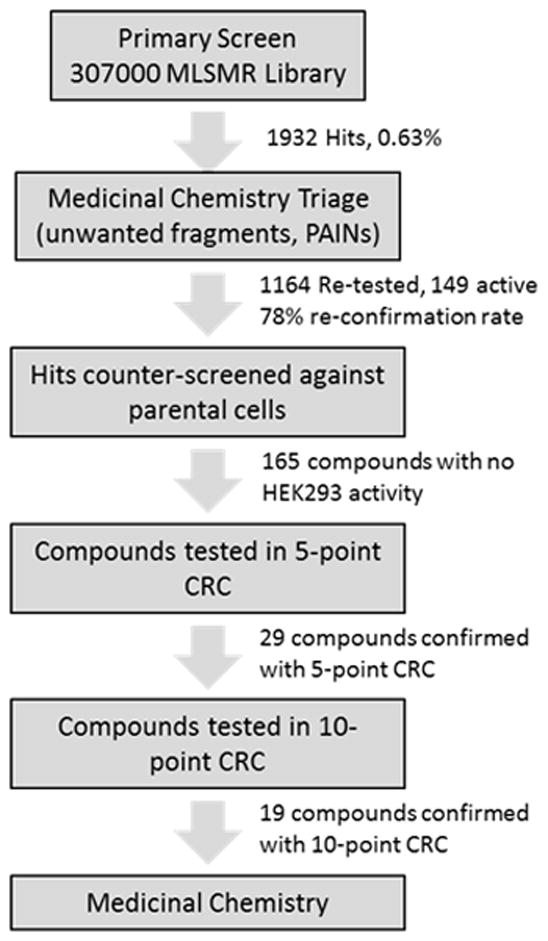
Flow chart of screening paradigm for MrgX1 positive allosteric modulators
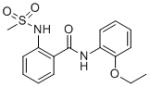
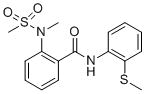
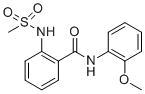
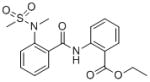
The results of the HTS screening campaign are summarized in Table 1. From the ~39 identified “hits”, there were four compounds from a sulfonamide benzamide series. Combining the activity in the MrgX1 assay and excellent selectivity profile as seen in the PubChem Assay and PubChem Hit rate[9], this series was chosen for further development towards a novel MrgX1 PAM. The initial SAR revealed a couple structural variants worth noting. First, the 2-ester analog was significantly less potent (3.85 μM), and second, the 2-methyl thioether was more potent than the 2-methoxy (~2-fold). The SAR chemical optimization strategy is highlighted in Table 1 wherein we evaluated multiple sites of the ligand in parallel through iterative synthesis.
Table 1.
Lead scaffolds from the HTS screen.
| SID | 4258711 | 4262381 | 3665540 | 3607659 |
|---|---|---|---|---|
|
|
|
|
|
| MrgX1 EC50 (μM) | 0.096 | 0.329 | 0.691 | 3.85 |
| PubChem Assays | 758 | 761 | 765 | 754 |
| PubChem Hits | 3 | 3 | 3 | 4 |
|
| ||||
|
| ||||
The synthesis of the MrgX1 compounds (generically shown as 4) is outlined in Scheme 1. The substituted anthranilic acid, 1, was converted to the acid chloride (SOCl2, DMF) and then coupled with aniline, 2, (pyridine, Et2O) to yield the amide, 3. The sulfonamide compounds, 4, were made by coupling, 3, with an appropriate sulfonyl chloride (RSO2Cl, pyridine, DCM). This synthesis represented a modular approach to the final compounds allowing for easy modification of R, R1, R2 and R3, which allowed for the synthesis of numerous analogs for this project.
Scheme 1.
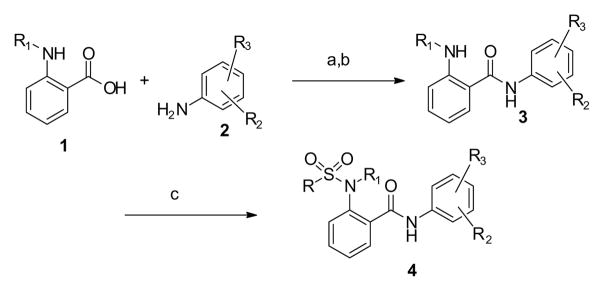
Synthesis of MrgX1 allosteric agonists, 4. Reagents and conditions: a) SOCl2, DMF, Et2O, 0 °C→ 70 °C, 2 h; b) pyridine, Et2O, 0 °C→rt, 2 h; c) RSO2Cl, pyridine, DCM, rt, 16h.
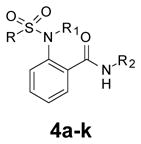
The SAR evaluation started with the re-synthesis and re-confirmation of the four initial hits that were identified in the HTS campaign (Table 2). These compounds, 4a, 4b, 4g, and 4h were all re-confirmed within ~2–5-fold of the initial HTS values (EC50), except for 4h which was inactive (AID: 743016). Those compounds with an Emax>50% were considered “active” and were progressed to an EC50 determination. From the SAR in Table 2, it was shown that ester groups on the R2 phenyl group were not tolerated (4d and 4h). In addition, phenyl sulphonamides were not tolerated (4j and 4k). Simple methyl sulphonamides coupled with either ether or thioether substituents on the R2 phenyl were well tolerated with many of the compounds showing nanomolar EC50’s (e.g., 4a, EC50 = 531 nM; 4c, EC50 = 419 nM; 4g, EC50 = 500 nM and 4i, EC50 = 551 nM). The 2-methoxy is less active than the 2-ethoxy or 2-methylthio groups. Also, substitution of the sulphonamide nitrogen was potency neutral compared to the unsubstituted NH sulfonamide (e.g., 4a vs 4i).
Table 2.
Evaluation of the right-hand phenyl group.
| |||||
|---|---|---|---|---|---|
| Cmpd | R | R1 | R2 | MrgX1 EC50, μMa | Emax (%)a |
| 4a | Me | H |
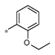
|
0.531 ± 0.16 | 238 ± 63 |
| 4b | Me | H |
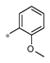
|
3.06± 1.50 | 188 ± 7 |
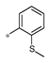
| 4c | Me | H |
|
0.42 ± 0.33 | 130 ± 28 |
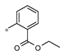
| 4d | Me | H |
|
Inactive | ND |
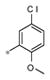
| 4e | Me | H |
|
1.01± 0.19 | 180 ± 9 |
| 4f | Me | H |
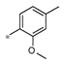
|
2.34± 1.30 | 241 ± 25 |
| 4g | Me | Me |
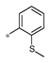
|
0.50± 0.12 | 184 ± 14 |
| 4h | Me | Me |
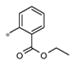
|
Inactive | ND |
| 4i | Me | Me |
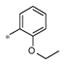
|
0.55±0.17 | 149 ± 19 |
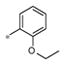
| 4j | Ph | H |
|
Inactive | ND |
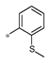
| 4k | Ph | H |
|
Inactive | ND |
Assay was run in the presence of 10 nM of BAM8-22; n = 2, N.D.: not determined since Emax is too low.
The final round of SAR utilized the 2-ethoxyphenyl group as the right-hand group and evaluated a variety of small, alkyl sulfonamides as these were identified from the previous round of SAR as the optimal substituents (Table 3). This round of SAR was far more productive than the previous round and showed a distinct SAR trend. Cyclopropyl sulfonamide, 4l was the most potent (EC50 = 190 nM; Figure 2) and efficacious (Emax = 195%). For this round of SAR, the Emax values were normalized to the positive control (4i, 100%). There were several compounds that showed >100% Emax. Another sub-micromolar compound was the 2-chloroethyl sulfonamide, 4m; however, as this compound possessed the reactive alkyl halo group, it was not progressed further. The isopropyl sulfonamide, 4o (EC50 = 1.82 μM; Emax = 255%) and the cyclopropyl methyl, 4p (EC50 = 4.47 μM, 267%) were significantly less potent than 4l. Based on the superior potency and efficacy, 4l has been declared the MLPCN probe compound for the MrgX1 allosteric agonist project, ML382.
Table 3.
Further SAR of the alkylsulfonamides.
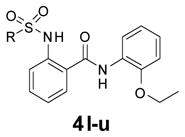
| |||
|---|---|---|---|
| Cmpd | R | MrgX1 EC50, μM | Emax a |
| 4l ML382 |
|
0.19 ± 0.06 | 195 ± 8 |
| 4m |
|
0.38 ± 0.05 | 204 ± 15 |
| 4n | Et | 0.91 ± 0.41 | 204 ± 10 |
| 4o |
|
1.82 ± 1.59 | 255 ± 53 |
| 4p |
|
4.47 ± 3.08 | 267 ± 101 |
| 4q |
|
N.D. | 46.1 |
| 4r |
|
N.D. | 29.6 |
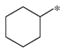
| 4s |
|
N.D. | 3.78 |
| 4t |
|
N.D. | 3.6 |
| 4u |
|
N.D. | 0.0 |
Emax as % of standard compound (4i)
N.D.: not determined since Emax<50%.
Figure 2.

Pharmacological profile of ML382 on MrgX1-expressing HEK293 cells. Dose response curve of ML382 in presence (red circles) and absence of BAM8-22 (black squares) determined by by Ca2+ imaging assay. EC50 of ML382 in presence of submaximal amount of BAM8-22 (10 nM) is 190 nM. In the absence of BAM8-22, ML382 itself does not activate MrgX1. RFU: relative fluorescence units. The fitting was dose response fitting. The experiments were repeated three times. Error bars represent as Mean±SEM.[11]
The dose responses studies show that ML382 enhances the potency of BAM8-22 on MrgX1 for >7-fold (i.e. EC50 of BAM8-22 from 18.7nM to 2.9nM; Supplemental Figure 1). However, ML382 does not affect the EMax of BAM8-22 when the maximum concentration of BAM8-22 was used (Supplemental Figure 1). Furthermore, in the absence of BAM8-22, ML382 does not activate MrgX1 suggesting ML382 itself does not has agonistic activity on MrgX1 (Figure 2). Pharmacological and selectivity profiling of ML382 were performed against the closely related MrgX2 (Supplemental Figure 2), and against a larger panel of receptors. ML382 was evaluated in HEK293 cells expressing MrgX2 as a selectivity screen. As can be seen in Supplemental Figure 2, ML382 showed no significant effect on activation of MrgX2 in the presence of the specific agonist peptide, PAMP (both EC50 and EMax). Also, ML382 had no effect on MrgprC11, the mouse homolog of MrgprX1 which indicates the compound is species selective (results not shown). In addition, ML382 was evaluated using EuroFin’s Lead Profiling Screen which is a binding assay panel of 68 GPCR’s, ion channels and transporters screened at 10 μM.[10] ML382 did not inhibit 67 of the 68 targets assayed (inhibition of radio ligand binding >50% at 10 μM); with the only target that was considered active was the serotonin (5-hydroxytryptamine), 5HT2B which showed 63% inhibition at 10 μM. Overall, ML382 displayed a very favourable selectivity profile.
Lastly, ML382 was further profiled in a battery of Tier 1 in vitro DMPK assays (Table 4). The intrinsic clearance was assessed in hepaticmicrosomes (rat/human) and ML382 was shown to be unstable to oxidative metabolism and predicted to display high clearance in both species (65.7 and 15.7 mL/min/kg, respectively). In addition, using an equilibrium dialysis approach, the protein binding of ML382 was evaluated and it was shown to have moderate free fraction in rat and low free fraction in human (1.7% in rat and 0.4% in human).
Table 4.
In Vitro Drug metabolism and pharmacokinetics (DMPK) profile of ML382.
| ML382 | |
|---|---|
| In Vitro PK Properties
| |
| Microsome Predicted Hepatic Clearance (mL/min/kg)
| |
| Rat CLHEP | 65.7 |
| Human CLHEP | 15.7 |
|
Percent Free Compound in Plasma | |
| Human PPB | 0.4 |
| Rat PPB | 1.7 |
In summary, we have developed 4l (also known as ML382 or VU0485891) as a potent and selective positive allosteric modulator (PAM) of MrgX1. This is the first report of a PAM for MrgX1. In the presence of the known ligand, BAM8-22, ML382 has an EC50 = 190 nM with an Emax = 148% and is inactive against the closely related MrgX2, and against a panel of selected receptors from the EuroFins panel. In addition, ML382 was profiled in our Tier 1 DMPK assay and possesses favourable free fraction in rat plasma protein binding; however, instability in rat liver microsomes indicates the utility of ML382 as an in vivo probe will be limited to non-oral dosing regimens (e.g., intraperitoneal (IP), subcutaneous (SC), or intrathecal). Further use of ML382 in an in vivo animal study will be reported in due course.
Experimental Section
Experimental procedures for the medicinal chemistry, pharmacology and drug metabolism studies, as well as compound characterization data are provided in the Supporting Information, available at http://dx.doi.org/XXXX.
Supplementary Material
Acknowledgments
This work was generously supported by the US National Institutes of Health/Molecular Libraries Probe Centers Network (MLPCN) grant U54 MH084659 (to C.W.L.) and U54 MH084691 (to M. L), and grant R03 DA033176 (to X.D.). M. W acknowledges additional funding supports from NIH (R03MH090837-01, R03 MH090849-01 and R03DA031670-01).
Contributor Information
Dr. Yan Wang, Email: lwwylwwy@hotmail.com.
Prof. Xinzhong Dong, Email: xdong2@jhmi.edu.
Prof. Corey R. Hopkins, Email: corey.r.hopkins@vanderbilt.edu.
References
- 1.a) Dong X, Han S-k, Zylka MJ, Simon MI, Anderson DJ. Cell. 2001;106:619–632. doi: 10.1016/s0092-8674(01)00483-4. [DOI] [PubMed] [Google Scholar]; b) Lembo PMC, Grazzini E, Groblewski T, O’Donnell D, Roy MO, Zhang J, Hoffert C, Cao J, Schmidt R, Pelletier M, Labarre M, Gosselin M, Fortin Y, Banville D, Shen SH, Ström P, Payza K, Dray A, Walker P, Ahmad S. Nature Neurosci. 2002;5:201–209. doi: 10.1038/nn815. [DOI] [PubMed] [Google Scholar]
- 2.Han S-k, Dong X, Hwang J-I, Zylka MJ, Anderson DJ, Simon MI. Proc Nat Acad Sci. 2002;99:14740–14745. doi: 10.1073/pnas.192565799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Guan Y, Liu Q, Tang Z, Raja SN, Anderson DJ, Dong X. Proc Nat Acad Sci. 2010;107:15933–15938. doi: 10.1073/pnas.1011221107. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) He SQ, Han L, Li Z, Xu Q, Tiwari V, Yang F, Guan X, Wang Y, Raja SN, Dong X, Guan Y. Neurosci. 2014;261:43–51. doi: 10.1016/j.neuroscience.2013.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He SQ, Li Z, Chu YX, Han L, Xu Q, Li M, Yang F, Liu Q, Tang Z, Wang Y, Hin N, Tsukamoto T, Slusher B, Tiwari V, Shechter R, Wei F, Raja SN, Dong X, Guan Y. Pain. 2014;155:534–544. doi: 10.1016/j.pain.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Z, He SQ, Xu Q, Yang F, Tiwari V, Liu Q, Tang Z, Han L, Chu YX, Wang Y, Hin N, Tsukamoto T, Slusher B, Guan X, Wei F, Raja SN, Dong X, Guan Y. Pain. 2014;155:1613–1621. doi: 10.1016/j.pain.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malik L, Kelly NM, Ma JN, Currier EA, Burstein ES, Olsson R. Bioorg Med Chem Lett. 2009;19:1729–1732. doi: 10.1016/j.bmcl.2009.01.085. [DOI] [PubMed] [Google Scholar]
- 7.Wroblowski B, Wigglesworth MJ, Szekeres PG, Smith GD, Rahman SS, Nicholson NH, Muir AI, Hall A, Heer JP, Garland SL, Coates WJ. J Med Chem. 2009;52:818–825. doi: 10.1021/jm800962k. [DOI] [PubMed] [Google Scholar]
- 8.Weaver CD. Curr Protoc Chem Biol. 2011;3:119–140. doi: 10.1002/9780470559277.ch110060. [DOI] [PubMed] [Google Scholar]
- 9.Assay ID’s (AIDs) can be viewed via the PubChem website.
- 10.http://www.eurofins.com/en.aspx
- 11.Kamohara M, Matsuo A, Takasaki J, Kohda M, Matsumoto M, Matsumoto S, Soga T, Hiyama H, Kobori M, Katou M. Biochem Biophys Res Commun. 2005;330:1146–1152. doi: 10.1016/j.bbrc.2005.03.088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.