

Abstract
Flexor tendon injuries caused by deep lacerations to the hands are a challenging problem as they often result in debilitating adhesions that prevent the movement of the afflicted fingers. Evidence exists that tendon adhesions as well as scarring throughout the body are largely precipitated by the pleiotropic growth factor, TGF-β1, but the effects of TGF-β1 are poorly understood in tendon healing. Using an in vitro model of tendon healing, we previously found that TGF-β1 causes gene expression changes in tenocytes that are consistent with scar tissue and adhesion formation, including upregulation of the anti-fibrinolytic protein, PAI-1. Therefore, we hypothesized that TGF-β1 contributes to scarring and adhesions by reducing the activity of proteases responsible for ECM degradation and remodeling, such as plasmin and MMPs, via upregulation of PAI-1. To test our hypothesis, we examined the effects of TGF-β1 on the protease activity of tendon cells. We found that flexor tendon tenocytes treated with TGF-β1 had significantly reduced levels of active MMP-2 and plasmin. Interestingly, the effects of TGF-β1 on protease activity were completely abolished in tendon cells from homozygous PAI-1 KO mice, which are unable to express PAI-1. Our findings support the hypothesis that TGF-β1 induces PAI-1, which suppresses plasmin and plasmin-mediated MMP activity, and provide evidence that PAI-1 may be a novel therapeutic target for preventing adhesions and promoting a scarless, regenerative repair of flexor tendon injuries.
Keywords: TGF-β1, PAI-1, Tendon, Adhesions, MMP, Tenocyte, Plasmin
Introduction
Flexor tendon injuries caused by deep lacerations to the hands are a challenging problem for surgeons. Such injuries are prone to the development of debilitating adhesions, scar tissue that prevents the normal gliding motion of the flexor tendons through their lubricating synovial sheaths, preventing the afflicted fingers from flexion (Silva et al., 2002; Zhao et al., 2004). Despite modern surgical and physical rehabilitation techniques, the formation of adhesions, which impair hand function continues to occur in as many as 30–60% of hand injuries (Caulfield et al., 2008). As there are presently no pharmacologic or biologic agents available for the prevention of tendon adhesions (Khanna et al., 2009), there is a definite need for a mechanistic, translational understanding of the factors which result in scarring and adhesion formation in flexor tendons, as well as other tissues prone to scarring and fibrosis.
Evidence exists that tendon adhesions (Chang et al., 2000; Jørgensen et al., 2005; Katzel et al., 2011) as well as scarring throughout the body (Gorvy et al., 2005; Shah et al., 1994; Shah et al., 1995) are largely mediated by the pleiotropic growth factor, Transforming Growth Factor Beta 1 (TGF-β1), the effects of which are not well-understood in tendon. Using an in vitro model of tendon healing, we previously found that TGF-β1 causes gene-expression changes in tenocytes that are consistent with scar tissue and adhesion formation (Farhat et al., 2012). Especially intriguing was the finding that TGF-β1 upregulated the expression of extracellular matrix (ECM) genes and downregulated gene expression of matrix metalloproteinases (MMPs). Moreover, TGF-β1 upregulated the expression of the important protease-suppressor, plasminogen activator inhibitor 1 (PAI-1), in flexor tendon tenocytes (Farhat et al., 2012).
MMPs are a family of proteases that play an important role in ECM remodeling and degradation as well as the regulation of signaling molecules, such as growth factors during development and wound healing (Clark et al., 2008; Gill and Parks, 2008). Defects in MMP activity and ECM remodeling are thought to contribute to fibrosis. As a potent inhibitor of both tissue- and urokinase-type Plasminogen Activators (tPA and uPA, respectively), PAI-1 acts as a master regulator of plasmin activity, thereby inhibiting fibrin degradation as well as plasmin-mediated activation of MMPs. As a repressor of both plasmin and MMP activity, it is little surprise that increased PAI-1 has been associated with a number of fibrotic pathologies, including skin, lung and liver fibroses (Ghosh and Vaughan, 2012).
As TGF-β1-mediated upregulation of PAI-1 expression has been implicated in numerous tissue fibroses, we hypothesize that upregulation of PAI-1 by TGF-β1 in tendon cells suppresses important proteases involved with ECM turnover and degradation, namely plasmin and MMPs. To test our hypothesis, we first evaluated whether TGF-β1 would suppress plasmin and MMP activity in a defined culture system that included critical components of the fibrinolytic pathway, namely plasminogen and tPA. We then determined whether PAI-1 was a critical mediator of TGF-β1’s effects on plasmin and MMP activity by comparing TGF-β1’s effects on tendon cells harvested from wild type versus PAI-1 KO mice.
Materials and Methods
Ethics statement
All of the mice (C57BL/6 and homozygous PAI-1 KO mice on a C57BL/6J background, Jackson Laboratory) used in this study were cared for in accordance with an animal use and care protocol approved by the University Committee on Animal Research of the University of Rochester. Mice were sacrificed in approved CO2 euthanasia chambers and death was verified using cervical dislocation prior to harvesting flexor digitorum longus (FDL) tendons from the hind paws.
Experimental Design
Murine flexor tendon tenocytes from wild type or PAI-1 KO mice were treated with various combinations of TGF-β1, plasminogen, and tPA for up to 48 h. For the initial experiments on wild type cells alone, two groups of samples were treated with control media (containing 1% serum) ± 10 ng/mL of TGF-β1, two groups with 20 μg/mL of plasminogen added to the system ±TGF-β1, and two more groups with both plasminogen and 50 ng/mL of tPA ±TGF-β1. For the subsequent wild type versus PAI-1 KO tendon cell experiment, samples from each cell type were treated with control media containing identical amounts of plasminogen and tPA shown above ± 10 ng/mL of TGF-β1.
In each experiment, the tendon cells were cultured in 6-well plates coated with rat tail collagen type I, to simulate the extracellular matrix (ECM) present in native tendon (Riley, 2004). Cells were incubated with up to 10 ng/mL of recombinant TGF-β1 for 48 h, because we previously found this dose and period of incubation sufficient to elicit significant changes in gene expression within 6–48 h in a similar culture model (Farhat et al., 2012). Although the dose of plasminogen (20 μg/mL) selected is only 15% of that found in normal plasma (Cederholm-Williams, 1981), this concentration was previously found to be sufficient for increasing plasmin-mediated MMP activity under analogous culture conditions (Sabeh et al., 2009). To activate the plasminogen, 50 ng/mL of tPA was added to the system, a concentration that is about 5-fold higher than that found in plasma under normal conditions (Wanhainen et al., 2007).
Tissue harvest & cell culture
FDL tendons were obtained from the hind limbs of five freshly sacrificed, 7 month old C57BL/6 mice and two 1.5 month old PAI-1 KO mice. They were processed for tendon cell (i.e. tenocyte) culture as described previously (Farhat et al., 2012). Briefly, specimens were stripped of surrounding tissue, washed in DPBS with 1% Pen Strep (Gibco), minced into 1 mm pieces, and trypsinized (Gibco, #25200) for 1 hour at room temperature under sterile conditions. The tendon fragments were then cultured in MEM-α (Gibco, #12561) supplemented with 20% FBS (Sigma-Aldrich, #F6178), 1% Pen Strep, and 6.5 μL/L of 2-Mercaptoethanol (Sigma-Aldrich, #M7522). The wild type cells that emerged were serially passaged five times, and then cryopreserved at −80°C in 50% MEM-α, 40% FBS and 10% DMSO (Sigma-Aldrich, #D2650). The tendon cells were later thawed, plated, expanded and used at passage 7 for each of the experiments. The PAI-1 KO cells were passaged 4 times and then immediately used for the experiment.
Preparation of collagen-coated culture plates
6-well tissue culture plates were pre-coated with 50 μg/cm2 of rat tail tendon collagen I (BD Biosciences, #354236) for the experiment. First, rat tail collagen I stock was diluted to 300 μg/mL, made isotonic and brought to a pH of 7.4 using a combination of 1X DPBS (Gibco, #14190), 10X PBS (Gibco, #14190), and 0.1 M NaOH under sterile conditions. After verifying the neutrality of a sample of the collagen solution using pH strips, 1.58 mL of the collagen solution (50 μg collagen/cm2 of culture surface) was added to each well. The collagen solution was incubated overnight at 37°C to allow for gelation, and the next day the plates were carefully transferred to a culture hood where they were allowed to dry overnight under sterile conditions with their lids removed. The next day, the collagen coated plates were washed three times with 3 mL of sterile distilled water for 30 minutes, being careful not to damage the fragile collagen coating throughout the procedure. After the final wash was removed, the plates were allowed to dry for 1 hour under the culture hood as before. They were then covered and stored until the day of the experiment.
Seeding and treatment of cells for the experiment
The day before each experiment, near-confluent tenocytes were trypsinized for 10 minutes in 0.25% Trypsin-EDTA in a humidified incubator (5% CO2, 37°C). Cells were then washed in culture media, centrifuged, strained to ensure a single cell suspension, and counted using a hemocytometer. The cells were then pelleted, suspended in control media (MEM α supplemented with 1% FBS and 1% Pen Strep), seeded onto the collagen-coated plates (70,000 cells/2 mL media/well) and incubated overnight in control media (MEM α and 1% FBS). The next day, time 0 samples were collected, and the remaining samples were treated with fresh control media with or without 10 ng/mL of TGF-β1 (R&D Systems, #240-B-010), 20 μg/mL of human glu-plasminogen (Haematologic Technologies, HCPG-0130), 50 ng/mL of tPA (Technoclone, #TC41072), 5 μg/mL of tranexamic acid (Sigma-Aldrich, #857653) or their respective vehicles (specified by the manufacturers). After 48 h, cells and media samples were collected and immediately stored at −80°C for later assessment of gene, protein, and protease activity levels. Four separate experiments (n=3 per experiment) were performed, but the number of samples used for each assay varied and is indicated where appropriate.
Gene, protein, and protease activity assays
RNA purification was performed using a combination of TRIzol (Invitrogen, #15596-026) and mini-spin columns (Epoch Life Science, #1660050), and gene expression was evaluated with Real-Time Polymerase Chain Reaction (RT-PCR) and normalized to a house keeping gene (Eef1a1). Validated primer sequences for all of the genes analyzed (Table 1) were obtained from PrimerBank (Spandidos et al., 2010) and validated with NCBI’s Primer-BLAST for specificity to the desired genes, as described previously (Farhat et al., 2012).
Table 1.
Primers used for gene expression analysis with RT-PCR
| Gene | Forward Primer (5′-3′) | Reverse Primer (5′-3′) | Amplicon Length |
|---|---|---|---|
| Mmp2 (Matrix Metalloproteinase-2) | CAAGTTCCCCGGCGATGTC | TTCTGGTCAAGGTCACCTGTC | 171 |
| Serpine1 ( Plasminogen Activator Inhibitor 1) | TTCAGCCCTTGCTTGCCTC | ACACTTTTACTCCGAAGTCGGT | 116 |
| Plat (tPa) | AACGCAGACAACTTACCAACA | GTTCGCTGCAACTTCGGAC | 131 |
| Plau (uPa) | GCGCCTTGGTGGTGAAAAAC | TTGTAGGACACGCATACACCT | 100 |
| Eef1a1 (Eukaryotic translation elongation factor 1 alpha 1) | TACGCCTGGGTCTTAGACAAA | TCCACAGGGAGATGTCAATAGT | 70 |
Commercial ELISA kits were used to assess the protein levels of total PAI-1 (Molecular Innovations, #MPAIKT-TOT) and glu-plasminogen (Technoclone, TC12040) in media samples diluted 100- and 200-fold in assay buffer, respectively. The tPA and plasmin activity of undiluted media samples was assessed after incubating samples for 30 minutes at room temperature with their respective fluorogenic substrates (AnaSpec, #72160 and #72125, respectively), according to the manufacturer’s instructions. Similarly, MMP activity was assessed with MMP FRET Substrate IX (Anaspec, 60576-01). To do so, 0.1 mg of solid substrate was dissolved in DMSO at 1 mM. The stock solution was then diluted to 100 μM in assay buffer (such as 10 mM HEPES, pH 7.4, 150 mM NaCl, 0.005% Tween 20) aliquoted and stored at −20°C. On the day of the assay, 100 μM substrate was then further diluted 70-fold in assay buffer, and 50 μL of diluted substrate was combined with 50 μL of freshly collected culture media into a black, flat-bottomed 96-well plate. Fluorescence was measured in a plate reader (Ex/Em 490/520 nm) and was recorded every 5 min for 1 hour, and 1 hour measurements were used for MMP activity quantification.
Pro- and active MMP-2 in culture media was assessed using gelatin zymography. Media samples were first concentrated about 10-fold using centrifugal devices with a 10-kDa molecular weight cut-off (Pall Corporation, #OD010C34) as directed by the manufacturer. Samples were then assessed with gelatin zymography as previously described (Hu and Beeton, 2010) using commercial reagents (Invitrogen) with one notable modification to their technique. In the final stages of staining the zymogram, larger volumes of stain solution and longer incubation times were used in order to improve the signal-to-noise ratio of the zymograms. Specifically, best results were achieved when the zymograms were incubated overnight in 40 mL of SimplyBlue SafeStain (Invitrogen, #LC6060) and then immediately scanned without any consequent destaining. The molecular weight of MMPs detected in the zymograms was assessed using commercial protein standards (Invitrogen, #LC5800) and 10 ng/well of purified, recombinant MMP-2 (Calbiochem, #PF037).
Data collection and statistical analysis
Gene expression data was analyzed using previously described methods (Farhat et al., 2012). Protein standards provided with the commercial ELISA kits were used to interpolate samples protein concentrations with Prism 4 (GraphPad Software). Bands of MMP activity observed in the gelatin zymograms were normalized to 10 ng/well of APMA-activated MMP-2 standard and quantified in ImageJ (available at http://rsb.info.nih.gov/ij; developed by Wayne Rasband, National Institutes of Health, Bethesda, MD). Significant differences (p<0.05) for all data were determined using 1-way ANOVAs and Bonferroni post-tests in Prism 4, unless otherwise indicated in the figure legends.
Results
TGF-β1 upregulates PAI-1 gene and protein levels in wild type tendon cells
Serpine1 (PAI-1 gene) expression was evaluated using real time RT-PCR after 0 and 48 hours of treating wild type flexor tendon cells with different combinations of TGF-β1, plasminogen, and tPA (Figure 1A). Expression of Serpine1 did not change significantly over the course of 48 h when no factors were added to the media. TGF-β1 increased gene expression 6-fold over control media (p<0.0001), and 2-fold in media that was supplemented with plasminogen (p<0.01). In the presence of tPA and plasminogen, TGF-β1 increased gene expression by as much as 70%, but this was not significantly different from untreated controls. Neither plasminogen nor tPA supplementation caused significant changes in the expression of Serpine1 in the absence of TGF- β1.
Figure 1. TGF-β1 significantly upregulated PAI-1 gene and protein levels.

(A) Relative Serpine1 (PAI-1) gene expression increased 2–6 fold with TGF-β1 treatment in all groups except those supplemented with tPA (n=5–9 from 2 separate experiments). In the presence of tPA, TGF-β1 increased Serpine1 expression by as much as 70%, but was not significantly different than untreated controls. (B) PAI-1 protein levels in culture media, on the other hand, were significantly elevated by 12- to 25-fold in all groups treated with TGF-β1, including those supplemented with plasminogen and tPA. n=12 from 4 separate experiments.
A commercial sandwich ELISA kit was used to measure the amount of total PAI-1 (including active, latent, and tPA-bound PAI-1) in culture media at the conclusion of the 48 h experiment (Figure 1B). TGF-β1 increased the level of PAI-1 by 12- to 25-fold in the presence and absence of plasminogen and tPA (p<0.0001). In the absence of TGF- β1, neither plasminogen nor tPA supplementation caused significant changes in the amount of PAI-1 detected.
TGF-β1 reduces tPA, but not uPA gene expression in wild type tendon cells
Plat (tPA gene) and Plau (uPA gene) expression were evaluated using real time RT-PCR at 0 and 48 hours after treating wild type tendon cells with TGF-β1, plasminogen, and tPA (Figure 2). Expression of Plat increased by about 1.9-fold over the course of 48 h when no factors were added to the media (p<0.001, Figure 2A). Treatment with TGF-β1 reduced the expression of Plat by 81% compared with control media (p<0.0001). Although TGF-β1 reduced the expression of Plat by about 70% in the presence of plasminogen ± tPA, these differences were not statistically significant.
Figure 2. Effects of TGF-β1 on Plat (tPA) and Plau (uPA) expression.

(A) Gene expression analysis with RT-PCR showed that Plat (tPA) was significantly downregulated 81% by TGF-β1 in the absence of plasminogen and tPA, but this effect was reduced in the presence of those two factors and did not reach significance. (B) Plau (uPA) was not significantly affected by TGF-β1 alone, but different combinations of TGF-β1, plasminogen, and tPA significantly reduced Plau expression by 47–55%. n=5–9 samples per treatment per time point from two separate experiments.
Expression of Plau (uPA) did not change significantly over the course of 48 h when no factors were added to the media (Figure 2B). While Plau expression did not appear to be affected by treatment with TGF-β1 alone, the combination of TGF-β1 and plasminogen, plasminogen and tPA, and all three factors combined reduced Plau expression by about 50% compared with control media (p<0.05).
TGF-β1 reduces plasminogen conversion to active plasmin in wild type tendon cells
The amount of plasminogen in culture media samples remaining in the inactive, pro-form at the conclusion of the 48 h treatment was assessed using a commercial ELISA kit (Figure 3A). Plasminogen was barely detectable in control and TGF-β1-treated groups that were not supplemented with plasminogen. As should be expected, addition of plasminogen caused a large increase in the amount of non-activated plasminogen detected, and treatment with TGF-β1 appeared to preserve plasminogen to a minor extent. The addition of tPA to the system in the absence of TGF- β1 caused a significant, 89% decrease in plasminogen when compared to non-tPA supplemented groups, but only 56% of Plasminogen was depleted when TGF-β1 was added.
Figure 3. TGF-β1 reduced Plasminogen conversion to Plasmin.

(A) Plasminogen in culture media was assessed by ELISA, and showed that tPA supplementation caused a depletion of 85% of plasminogen when compared to non-tPA supplemented control media. TGF-β1 reduced plasminogen depletion to only 40% of controls. (B) Plasmin levels were assessed with a fluorogenic substrate assay. Plasmin activity was minimal in groups that were not treated with exogenous Plasminogen. Addition of Plasminogen caused a 5-fold increase in Plasmin activity (not significant), but this increase was completely reversed by treatment with TGF-β1. Addition of tPA caused a 150-fold increase in plasmin activity over plasminogen -and tPA-free media, but TGF-β1 reduced plasmin activity by 84% in the presence of plasminogen and tPA. n=9 samples from three separate experiments.
The level of active plasmin in culture media at the conclusion of the 48 h treatment was assessed with a fluorescence resonance energy transfer (FRET)-based assay (Figure 3B). Plasmin activity was minimal in groups that were not treated with exogenous plasminogen. Addition of plasminogen caused a 5-fold increase in plasmin activity (not significant), but this increase was completely reversed by treatment with TGF-β1. The addition of both plasminogen and tPA caused a 150-fold increase in plasmin activity over baseline levels (p<0.001), which was reduced as much as 84% by treatment with TGF-β1 (p<0.01). Together, these data provide evidence that TGF-β1 is suppressing the activation of plasminogen to plasmin by tPA.
TGF-β1 suppresses plasmin-mediated MMP-2 activation in wild type tendon cells
The gene expression of Mmp2 was evaluated after 0 and 48 hours of treatment using RT-PCR (Figure 4A). Expression of Mmp2 did not change significantly over the course of 48 h when no factors were added to the media, or with any of the treatments administered.
Figure 4. Plasmin-mediated MMP-2 activity was suppressed by TGF-β1.

(A) Gene expression analysis with RT-PCR showed that Mmp2 was not significantly regulated by TGF-β1 in the presence or absence of plasminogen and tPA. (B) Representative gelatin zymogram that was used to assess pro- and active MMP-2 in media after treating samples for 48 h as indicated below panel D. (C) Pro-MMP-2 was not significantly different among any of the treatments. (D) TGF-β1 reduced active MMP-2 by 90% in the presence of plasminogen alone, however this was not significant. In the presence of plasminogen and tPA, TGF-β1 reduced active MMP-2 by 71% (p<0.0001). n=8 samples from three separate experiments. (E) Representative zymogram from samples treated with plasminogen and tPA, as well as combinations of TGF-β1 and tranexamic acid (plasmin inhibitor). (F) Pro-MMP-2 was lower in groups treated with tranexamic acid. (G) Tranexamic acid reduced active MMP-2 by 87%, which was a reduction similar to the 78% reduction seen with TGF-β1 treatment. n=3 samples from one experiment.
Gelatin zymography was used to assess pro- and active MMP-2 in culture media after the 48 h treatment (Figure 4B). The higher-molecular weight bands (measured at ~65 kDa) corresponded with bands produced by commercial pro-MMP-2 standards (data not shown), while the lower molecular weight bands (~55 kDa) represent active MMP-2 (Hussain et al., 2010),(Cotrim et al., 2002). Pro-MMP-2 levels were not significantly different among any of the treatment groups (Figure 4C). Active MMP-2 was barely detectable in the absence of plasminogen (Figure 4D). Upon addition of plasminogen, MMP-2 activity increased and TGF-β1 reduced active MMP-2 by as much as 90%, but this was not statistically significant. When plasminogen and tPA were added, MMP-2 activity increased 3-fold above samples treated with plasminogen alone, and TGF-β1 reduced the level of active MMP-2 in plasminogen- and tPA-treated samples by about 71% (p<0.0001).
The small molecule plasmin inhibitor, tranexamic acid (TA), was used to determine whether the increase in MMP-2 activity observed with plasminogen and tPA supplementation was the result of plasmin-mediated MMP-2 activation. Gelatin zymography (Figure 4E) performed on media samples showed a decrease in Pro-MMP-2 levels (Figure 4F) in groups treated with tranexamic acid (p<0.01). On the other hand, tranexamic acid completely abolished 87% of active MMP-2 compared with control samples (Figure 4G), a level similar to that of TGF-β1 treatment, which showed a 78% decrease (p<0.001). Collectively, these results demonstrate that TGF-β1 reduces plasmin-mediated MMP-2 activation.
TGF-β1’s suppression of tPA, plasmin and MMP activity are abolished in tendon cells from PAI-1 KO mice
Flexor tendon cells from wild type and PAI-1 KO mice were treated with and without TGF-β1 in the presence of supplemented plasminogen and tPA. After 48 h, the levels of active tPA, plasmin and MMPs in the culture media were assessed with fluorescence resonance energy transfer FRET substrates (Figure 5).
Figure 5. TGF-β1 suppresses tPA, plasmin and MMP activity in wild type, but not PAI-1 KO tendon cells.

(A) tPA activity in culture media collected from the samples after 48 hours of treatment with or without TGF-β1 was measured with a fluorescent substrate (FRET) assay. TGF-β1 suppressed tPA activity by 81% in wild type cells (p<0.05), but did not suppress tPA activity in tendon cells null for PAI-1. (B) Plasmin activity measured with a FRET substrate also showed that TGF-β1 suppressed plasmin activity by 96% in wild type cells (p<0.05), but not in tendon cells null for PAI-1. (C) MMP activity measured with a FRET substrate showed that TGF-β1 suppressed MMP activity by 59% in media collected from wild type cells (p<0.0001), but did not suppress MMP activity in media from tendon cells null for PAI-1. n=6 samples from two separate experiments. Significance for tPA and plasmin activity was determined using Dunn’s post test to correct for multiple comparisons.
tPA activity was suppressed 81% by TGF-β1 in wild type cells (p<0.05), but not in PAI-1 KO (Figure 5A). Plasmin activity was similarly suppressed 96% by TGF-β1 in wild type cells (p<0.05), but was not suppressed in PAI-1 KO cells (Figure 5B). MMP activity assessed with a FRET substrate was suppressed 59% by TGF-β1 (p<0.0001), but not in PAI-1 KO cells (Figure 5C). These data provide evidence that TGF-β1 is suppressing the activity of tPA, plasmin and MMP activity via upregulation of PAI-1.
Discussion
Adult tissues, including tendon, heal with fibrotic scar formation. This can be a particularly challenging problem when the flexor tendons of the hand are injured, as scar formation can lead to adhesions that severely impair tendon gliding and limit flexion or extension of the fingers. The conventional paradigm in scar etiology points to an imbalance between ECM synthesis and remodeling (Farhat et al., 2012). ECM synthesis is important for reestablishing tissue continuity and structural integrity, but excessive synthesis coupled with deficits in protease-mediated ECM remodeling are thought to cause numerous fibrotic conditions throughout the body, including flexor tendon adhesions.
While it is recognized that plasmin and MMPs are important mediators of ECM remodeling throughout the body (Mercer and Chambers, 2013), the extent of their importance in the context of flexor tendon adhesions has yet to be elucidated. MMP activity is partially dependent on the activity of plasmin, and plasmin activity is regulated primarily by PAI-1, a potent suppressor of the plasminogen activators, tPA and uPA (Figure 6). Therefore, PAI-1 is likely to play an important regulatory role in both plasmin and plasmin-mediated MMP activity during tendon healing as it does in other tissues (Ghosh and Vaughan, 2012; Iwaki et al., 2012). Here, we used various combinations of TGF-β1, plasminogen, and tPA in order to test the hypothesis that TGF-β1 suppresses the plasmin activity and plasmin-mediated MMP-2 activity of tendon cells.
Figure 6. Proposed interactions of TGF-β1 with the plasminogen activation system and MMPs.
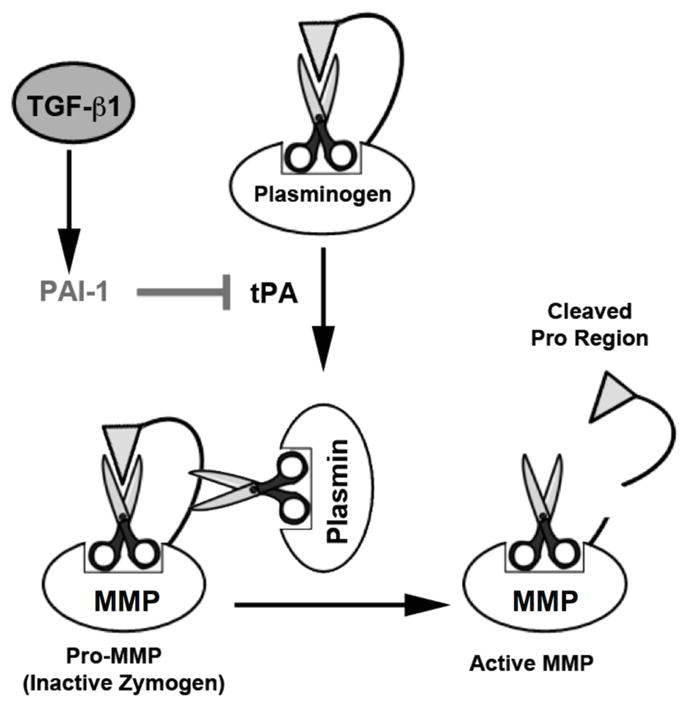
MMPs may be activated from their “Pro” state by cleavage of the inhibitory pro-peptide by plasmin. However, plasmin must itself first be activated from its pro-form, “plasminogen,” via either tissue- or urokinase-type Plasminogen Activators (tPA and uPA, respectively). Both tPA and uPA are inhibited by PAI-1, which is upregulated by TGF-β1 in a number of tissues and diseases. Thus, TGF-β1 may inhibit both plasmin and MMP activity through PAI-1.
As a first step toward identifying whether PAI-1 could be an important regulator of adhesion formation during flexor tendon healing, we tested whether TGF-β1 significantly upregulates the expression of PAI-1 gene and protein expression in flexor tendon tenocytes. We found that the PAI-1 gene, Serpine1, (PAI-1 gene) was significantly upregulated 2 to 6-fold by TGF-β1 under almost all the conditions tested, while PAI-1 protein levels were significantly elevated 12 to 25-fold by TGF-β1. The apparent discrepancy between the extent of gene and protein upregulation at 48 h may be the result of more profound early increases in PAI-1 gene expression (i.e. at 6 h) that were not measured in this experiment, with gene expression changes diminishing at the later time point of 48 h. This explanation is consistent with the temporal expression pattern that we have reported earlier from studying tenocyte-seeded collagen gels that were also treated with TGF-β1 (Farhat et al., 2012).
The plasminogen activation system is crucial for the degradation of the fibrin clot that initially forms in an injury as well as the regulation of other proteases that play a role throughout the healing process, such as MMPs (Toriseva and Kahari, 2009). Nonetheless, the interaction between the plasminogen activation system and tendon cells throughout the healing process is largely unknown. Therefore, we evaluated the effects of TGF-β1, plasminogen and tPA on the activation of plasminogen to plasmin. In the absence of supplemented tPA, there was very little conversion of plasminogen to plasmin. This suggests that the basal levels of tPA and uPA in this system were too low to activate a considerable amount of plasminogen over 48 h. Upon addition of exogenous tPA, there was an almost-total depletion of the available plasminogen by the end of the experiment, and a 150-fold increase in plasmin activity. Our finding that tPA supplementation was necessary for plasminogen activation is consistent with reports that tPA is largely produced by vascular endothelial cells in vivo (Lijnen and Collen, 1997), the likes of which were not present in this system, which necessitated the addition of exogenous tPA in our in vitro system. Addition of TGF-β1 to the system reduced plasminogen activation significantly, as evidenced by an increase in the amount of preserved plasminogen and an 84% decrease in plasmin activity after 48 h. This data strongly suggests that TGF-β1 suppressed plasminogen activation by neutralizing the effects of tPA, and a reasonable explanation for this finding is that TGF-β1 inhibited tPA activity via upregulation of PAI-1.
Reduced MMP-2 activity has been associated with increased fibrosis in models of liver (Onozuka et al., 2011) and renal fibrosis (Wang and Hirschberg, 2003), and TGF-β1 is thought to be a major contributor to those disease processes (Wynn, 2008). Interestingly, MMP-2 plays an important role in tendon development (Jung et al., 2009) and is also expressed during tendon healing (Loiselle et al., 2009). However, it remains unknown whether TGF-β1 contributes to flexor tendon adhesions through suppression of MMP-2 activity during tendon healing. To address this question, we examined the interaction between TGF-β1, plasminogen and tPA on the MMP-2 activity of tendon cells using gelatin zymography. We found that active MMP-2 was barely detectable in the absence of supplemented plasminogen, and increased considerably when plasminogen was added. However, addition of TGF-β1 almost completely reversed the effects of adding plasminogen to the system on the activation of MMP-2. Addition of tPA to the system caused an even greater increase in MMP-2 activity, providing more evidence that MMP-2 activation in this system is largely plasmin-dependent, and addition of the plasmin inhibitor, tranexamic acid, completely eliminated this increase in MMP-2 activity. This is consistent with the results of others (Sabeh et al., 2009), which showed under similar culture conditions that the activity of secreted MMPs (e.g. MMP-2) is highly dependent on plasminogen being present in the system. Therefore, the reduction of plasmin-mediated MMP activity by TGF-β1 in wild type cells but not in PAI-1 KO cells support the role of PAI-1 as an important regulator of MMP activity (Figure 6).
After confirming that TGF-β1 suppressed plasmin and MMP-2 activity in the culture media collected from tendon cells, we compared the effects of TGF-β1 on wild type versus PAI-1 null tendon cells in order to evaluate whether upregulation of PAI-1 was essential for TGF-β1’s pro-fibrotic effects. In doing so, we found that TGF-β1’s suppression of tPA, plasmin and MMP activity was completely abolished in the absence of PAI-1, implicating PAI-1 as a major contributor to TGF-β1’s pro-fibrotic effects in tendon. Our finding that PAI-1 mediates TGF-β1’s suppression of protease activity in tendon cells is significant, because it implies that targeting PAI-1 could be a novel way to reduce the formation of tendon adhesions. As evidence of this, one study reported that neutralizing antibodies against PAI-1 reduced abdominal adhesions in mice (Falk et al., 2001). Another study found that a small molecule PAI-1 inhibitor, TM5275, reversed the pathologic effects of constitutively expressed TGF-β1 in a mouse model of lung fibrosis (Huang et al., 2012). In addition, PAI-1 knockout (KO) mice were protected from the collagen accumulation associated with renal (reviewed in (Iwaki et al., 2012)) and lung (Eitzman et al., 1996) fibrosis, while PAI-1 overexpressing mice had more severe fibrosis in both cases. PAI-1 KO mice have also been reported to have accelerated skin wound closure (John et al., 2010). These findings support the importance of PAI-1 as an inhibitor of the matrix remodeling associated with wound healing. However, the complete absence of PAI-1 in KO mice has also been associated with poor outcomes in models of myocardial infarction (reviewed in (Iwaki et al., 2012)) and glomerulonephritis (Hertig et al., 2003), indicating that complete removal of PAI-1 could have detrimental effects in the context of certain diseases and organ systems. To our knowledge, role of PAI-1 on tendon healing and adhesion formation has yet to be explored beyond the experiments described in this work.
MMPs are secreted as inactive zymogens, and there are at least 3 physiological mechanisms that have been identified as responsible for their activation, all of which involve proteolytic cleavage of the pro-domain (Murphy et al., 1999). In addition to plasmin, membrane-bound MMPs are known to play a critical role in the proteolytic activation of secreted MMPs, including MMP-2 (Jung et al., 2009) as well as in cell-mediated collagen degradation (Sabeh et al., 2009). Indeed, we found that TGF-β1 caused significant reductions in the expression of genes coding for membrane-bound MMPs (Mmp14 and Mmp16; data not shown). This is consistent with our previous finding in tenocytes cultured in collagen gels (Farhat et al., 2012). Therefore, downregulation of membrane-bound MMPs by TGF-β1 may constitute a PAI-1-independent pathway through which TGF-β1 could reduce MMP activity during tendon healing, which merits further investigation. Moreover, it has also been reported that the activation of secreted pro-MMPs (including MMP-2) can be partially or entirely autoproteolytic (Morgunova et al., 1999), but this mechanism remains poorly understood. Tissue inhibitors of matrix metalloproteinases (TIMPs) are also involved in inhibiting MMPs once they are activated (Choi et al., 2002). While our previous findings suggest that TGF-β1 has no effects on TIMP-2 in tenocytes cultured in collagen gels (Farhat et al., 2012), the possibility of the involvement of this mechanism in the context of flexor tendon healing cannot be excluded and merits an independent investigation.
In conclusion, the reduced activity of proteases such as plasmin and MMP-2 is thought to play an important role in the accumulation of scar tissue throughout the body (Ghosh and Vaughan, 2012). The data presented here provides evidence that TGF-β1 suppresses the plasmin and plasmin-dependent MMP activity of flexor tendon cells cultured in vitro via upregulation of PAI-1. With that said, it is possible that TGF-β1 also suppresses MMP and plasmin activity in other ways, such as through the downregulation of tPA, uPA and/or membrane-bound MMP expression in tenocytes or other cell types not represented in our simplified and controlled in vitro system, but which play an active role in the healing process, such as synovial fibroblasts and vascular endothelial cells. More work is required to decipher the relative roles of each of these potential mechanisms and cell types in contributing to adhesion formation. However, the identification of PAI-1 as a precipitating factor in the inhibitory effects of TGF-β1 on plasmin-mediated MMP activation in our in vitro system might have significant therapeutic value towards the biological modification of adhesions following flexor tendon injury and repair. It remains to be determined whether antagonizing PAI-1’s effects on protease (plasmin and MMP) activity in vivo will have therapeutic effects for the scarless healing of tendon injuries.
Acknowledgments
Contract Grant Sponsors: National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) and the National Institutes of Health (NIH),
Contract Grant Numbers: R01AR056696, P30AR061307, T32 GM07356, T32 AR053459
The study was supported by grant numbers R01 AR056696 and P30 AR061307 from NIAMS/NIH. Youssef Farhat is a former trainee in the Medical Scientist Training Program funded by NIH T32 GM07356, and a current trainee of the Training In Orthopaedic Research Program funded by NIH T32 AR053459. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or NIH.
Literature Cited
- Caulfield RH, Maleki-Tabrizi A, Patel H, Coldham F, Mee S, Nanchahal J. Comparison of zones 1 to 4 flexor tendon repairs using absorbable and unabsorbable four-strand core sutures. The Journal of Hand Surgery. 2008;33(4):412–417. doi: 10.1177/1753193408090758. European Volume. [DOI] [PubMed] [Google Scholar]
- Cederholm-Williams SA. Concentration of plasminogen and antiplasmin in plasma and serum. Journal of clinical pathology. 1981;34(9):979–981. doi: 10.1136/jcp.34.9.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Thunder R, Most D, Longaker MT, Lineaweaver WC. Studies in flexor tendon wound healing: neutralizing antibody to TGF-beta1 increases postoperative range of motion. Plast Reconstr Surg. 2000;105(1):148–155. doi: 10.1097/00006534-200001000-00025. [DOI] [PubMed] [Google Scholar]
- Choi HR, Kondo S, Hirose K, Ishiguro N, Hasegawa Y, Iwata H. Expression and enzymatic activity of MMP-2 during healing process of the acute supraspinatus tendon tear in rabbits. J Orthop Res. 2002;20(5):927–933. doi: 10.1016/S0736-0266(02)00016-5. [DOI] [PubMed] [Google Scholar]
- Clark IM, Swingler TE, Sampieri CL, Edwards DR. The regulation of matrix metalloproteinases and their inhibitors. The International Journal of Biochemistry & Cell Biology. 2008;40(6–7):1362–1378. doi: 10.1016/j.biocel.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Cotrim P, de Andrade CR, Line S, de Almeida OP, Coletta RD. Expression and activity of matrix metalloproteinase-2 (MMP-2) in the development of rat first molar tooth germ. Brazilian dental journal. 2002;13(2):97–102. doi: 10.1590/s0103-64402002000200004. [DOI] [PubMed] [Google Scholar]
- Eitzman DT, McCoy RD, Zheng X, Fay WP, Shen T, Ginsburg D, Simon RH. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. The Journal of clinical investigation. 1996;97(1):232–237. doi: 10.1172/JCI118396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk K, Bjorquist P, Stromqvist M, Holmdahl L. Reduction of experimental adhesion formation by inhibition of plasminogen activator inhibitor type 1. The British journal of surgery. 2001;88(2):286–289. doi: 10.1046/j.1365-2168.2001.01647.x. [DOI] [PubMed] [Google Scholar]
- Farhat YM, Al-Maliki AA, Chen T, Juneja SC, Schwarz EM, O’Keefe RJ, Awad HA. Gene Expression Analysis of the Pleiotropic Effects of TGF-beta1 in an In Vitro Model of Flexor Tendon Healing. PloS one. 2012;7(12):e51411. doi: 10.1371/journal.pone.0051411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. Journal of cellular physiology. 2012;227(2):493–507. doi: 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SE, Parks WC. Metalloproteinases and their inhibitors: regulators of wound healing. The International Journal of Biochemistry & Cell Biology. 2008;40(6–7):1334–1347. doi: 10.1016/j.biocel.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorvy DA, Herrick SE, Shah M, Ferguson MWJ. Experimental manipulation of transforming growth factor-beta isoforms significantly affects adhesion formation in a murine surgical model. The American Journal of Pathology. 2005;167(4):1005–1019. doi: 10.1016/s0002-9440(10)61190-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertig A, Berrou J, Allory Y, Breton L, Commo F, Costa De Beauregard MA, Carmeliet P, Rondeau E. Type 1 plasminogen activator inhibitor deficiency aggravates the course of experimental glomerulonephritis through overactivation of transforming growth factor beta. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2003;17(13):1904–1906. doi: 10.1096/fj.03-0084fje. [DOI] [PubMed] [Google Scholar]
- Hu X, Beeton C. Detection of functional matrix metalloproteinases by zymography. Journal of visualized experiments : JoVE. 2010;(45) doi: 10.3791/2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WT, Vayalil PK, Miyata T, Hagood J, Liu RM. Therapeutic value of small molecule inhibitor to plasminogen activator inhibitor-1 for lung fibrosis. American journal of respiratory cell and molecular biology. 2012;46(1):87–95. doi: 10.1165/rcmb.2011-0139OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain AA, Lee Y, Marshall J. High molecular-weight gelatinase species of human Bruch’s membrane: compositional analyses and age-related changes. Investigative ophthalmology & visual science. 2010;51(5):2363–2371. doi: 10.1167/iovs.09-4259. [DOI] [PubMed] [Google Scholar]
- Iwaki T, Urano T, Umemura K. PAI-1, progress in understanding the clinical problem and its aetiology. British journal of haematology. 2012;157(3):291–298. doi: 10.1111/j.1365-2141.2012.09074.x. [DOI] [PubMed] [Google Scholar]
- John J, Quinlan AT, Silvestri C, Billiar K. Boundary stiffness regulates fibroblast behavior in collagen gels. Ann Biomed Eng. 2010;38(3):658–673. doi: 10.1007/s10439-009-9856-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen HG, McLellan SD, Crossan JF, Curtis ASG. Neutralisation of TGF beta or binding of VLA-4 to fibronectin prevents rat tendon adhesion following transection. Cytokine. 2005;30(4):195–202. doi: 10.1016/j.cyto.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Jung JC, Wang PX, Zhang G, Ezura Y, Fini ME, Birk DE. Collagen fibril growth during chicken tendon development: matrix metalloproteinase-2 and its activation. Cell Tissue Res. 2009;336(1):79–89. doi: 10.1007/s00441-009-0755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzel EB, Wolenski M, Loiselle AE, Basile P, Flick LM, Langstein HN, Hilton MJ, Awad HA, Hammert WC, O’Keefe RJ. Impact of Smad3 loss of function on scarring and adhesion formation during tendon healing. J Orthop Res. 2011;29(5):684–693. doi: 10.1002/jor.21235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna A, Friel M, Gougoulias N, Longo UG, Maffulli N. Prevention of adhesions in surgery of the flexor tendons of the hand: what is the evidence? British Medical Bulletin. 2009;90:85–109. doi: 10.1093/bmb/ldp013. [DOI] [PubMed] [Google Scholar]
- Lijnen HR, Collen D. Endothelium in hemostasis and thrombosis. Progress in cardiovascular diseases. 1997;39(4):343–350. doi: 10.1016/s0033-0620(97)80032-1. [DOI] [PubMed] [Google Scholar]
- Loiselle AE, Bragdon GA, Jacobson JA, Hasslund S, Cortes ZE, Schwarz EM, Mitten DJ, Awad HA, O’Keefe RJ. Remodeling of murine intrasynovial tendon adhesions following injury: MMP and neotendon gene expression. J Orthop Res. 2009;27(6):833–840. doi: 10.1002/jor.20769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer PF, Chambers RC. Coagulation and coagulation signalling in fibrosis. Biochimica et biophysica acta. 2013;1832(7):1018–1027. doi: 10.1016/j.bbadis.2012.12.013. [DOI] [PubMed] [Google Scholar]
- Morgunova E, Tuuttila A, Bergmann U, Isupov M, Lindqvist Y, Schneider G, Tryggvason K. Structure of human pro-matrix metalloproteinase-2: activation mechanism revealed. Science. 1999;284(5420):1667–1670. doi: 10.1126/science.284.5420.1667. [DOI] [PubMed] [Google Scholar]
- Murphy G, Stanton H, Cowell S, Butler G, Knauper V, Atkinson S, Gavrilovic J. Mechanisms for pro matrix metalloproteinase activation. APMIS : acta pathologica, microbiologica, et immunologica Scandinavica. 1999;107(1):38–44. doi: 10.1111/j.1699-0463.1999.tb01524.x. [DOI] [PubMed] [Google Scholar]
- Onozuka I, Kakinuma S, Kamiya A, Miyoshi M, Sakamoto N, Kiyohashi K, Watanabe T, Funaoka Y, Ueyama M, Nakagawa M, Koshikawa N, Seiki M, Nakauchi H, Watanabe M. Cholestatic liver fibrosis and toxin-induced fibrosis are exacerbated in matrix metalloproteinase-2 deficient mice. Biochemical and biophysical research communications. 2011;406(1):134–140. doi: 10.1016/j.bbrc.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Riley G. The pathogenesis of tendinopathy. A molecular perspective. Rheumatology (Oxford, England) 2004;43(2):131–142. doi: 10.1093/rheumatology/keg448. [DOI] [PubMed] [Google Scholar]
- Sabeh F, Li XY, Saunders TL, Rowe RG, Weiss SJ. Secreted versus membrane-anchored collagenases: relative roles in fibroblast-dependent collagenolysis and invasion. The Journal of biological chemistry. 2009;284(34):23001–23011. doi: 10.1074/jbc.M109.002808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah M, Foreman DM, Ferguson MW. Neutralising antibody to TGF-beta 1,2 reduces cutaneous scarring in adult rodents. Journal of Cell Science. 1994;107 ( Pt 5):1137–1157. doi: 10.1242/jcs.107.5.1137. [DOI] [PubMed] [Google Scholar]
- Shah M, Foreman DM, Ferguson MW. Neutralisation of TGF-beta 1 and TGF-beta 2 or exogenous addition of TGF-beta 3 to cutaneous rat wounds reduces scarring. Journal of Cell Science. 1995;108 ( Pt 3):985–1002. doi: 10.1242/jcs.108.3.985. [DOI] [PubMed] [Google Scholar]
- Silva MJ, Boyer MI, Gelberman RH. Recent progress in flexor tendon healing. J Orthop Sci. 2002;7(4):508–514. doi: 10.1007/s007760200090. [DOI] [PubMed] [Google Scholar]
- Spandidos A, Wang X, Wang H, Seed B. PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 2010;38(Database issue):D792–799. doi: 10.1093/nar/gkp1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toriseva M, Kahari VM. Proteinases in cutaneous wound healing. Cellular and molecular life sciences : CMLS. 2009;66(2):203–224. doi: 10.1007/s00018-008-8388-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Hirschberg R. BMP7 antagonizes TGF-beta -dependent fibrogenesis in mesangial cells. Am J Physiol Renal Physiol. 2003;284(5):F1006–1013. doi: 10.1152/ajprenal.00382.2002. [DOI] [PubMed] [Google Scholar]
- Wanhainen A, Nilsson TK, Bergqvist D, Boman K, Bjorck M. Elevated tissue plasminogen activator in patients with screening-detected abdominal aortic aneurysm. Journal of vascular surgery. 2007;45(6):1109–1113. doi: 10.1016/j.jvs.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Cellular and molecular mechanisms of fibrosis. The Journal of Pathology. 2008;214(2):199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Amadio PC, Paillard P, Tanaka T, Zobitz ME, Larson DR, An KN. Digital resistance and tendon strength during the first week after flexor digitorum profundus tendon repair in a canine model in vivo. J Bone Joint Surg Am. 2004;86-A(2):320–327. doi: 10.2106/00004623-200402000-00015. [DOI] [PubMed] [Google Scholar]