

Abstract
Here we report that targeting casein kinase 1 (CSNK1α1) is a potential novel treatment strategy in multiple myeloma (MM) therapy distinct from proteasome inhibition. CSNK1α1 is expressed in all the tested MM cell lines and patient MM cells, and is not altered during bortezomib-triggered cytotoxicity. Inhibition of CSNK1α1 kinase activity in MM cells with targeted therapy D4476 or shRNAs triggers cell G0/G1 phase arrest, prolonged G2/M phase, and apoptosis. D4476 also induced cytotoxicity in bortezomib-resistant MM cells and enhances bortezomib-triggered cytotoxicity. CSNK1α1 signaling pathways include CDKN1B, P53 and FADD; gene signatures involved included interferon alpha (IFNα), tumor necrosis factor alpha (TNFα), and LIN9. Additionally, reduction of Csnk1α1 prevents cMYC/KRAS12V transformation of BaF3 cells independent of IL3. Impartially, reducing Csnk1α1 prevented development of cMYC/KRAS12V-induced plasmacytomas (PCTs) in mice, suggesting that CSNK1α1 may be involved in MM initiation and progression. Our data suggest that targeting CSNK1α1, alone or combined with bortezomib, is a potential novel therapeutic strategy in MM. Moreover, inhibition of CSNK1α1 may prevent the progression of monoclonal gammopathy of undetermined significance (MGUS) to MM.
Keywords: Multiple Myeloma (MM), CSNK1α1, D4476, Bortezomib, cMYC/KRAS12V, plasmacytomas (PCTs)
Introduction
Multiple myeloma (MM) is a plasma cell neoplasm characterized by the accumulation of monoclonal plasma cells in bone marrow and monoclonal protein in serum and urine, associated with bone fractures, anemia, kidney dysfunction, hypercalcemia, and immunodeficiency. 1 Currently, all newly diagnosed patients receive novel immune-modulating drugs (Thalidomide/Lenalidomide) and/or proteasome inhibitors (bortezomib). 2, 3 In spite of these therapies, development of resistance and relapsed remains a major treatment challenge 4, and novel targeted MM therapies are urgently needed.
Pathologic activation of MYC family members plays a critical role in MM pathogenesis and relapse. 5 Amplification of MYC associated with progression from MGUS to MM for MYC 6 and the high frequency of MYC deregulation family members in MM, suggest that MYC is an attractive therapeutic target. Moreover, previous studies already demonstrated that targeting MYC significantly inhibited MM cell survival both in vitro and in vivo. 7 However, MYC family members play essential functions in proliferative tissues, 8 and inhibiting MYC activity for long period could cause toxicity. An alternative strategy to target MYC signaling is to inhibit its partner functions. For example, previous studies indicated that interaction between MYC and CSNK1 family members, and that targeting CSNK1 family member can halt MYC-driven cancer growth. 9, 10
CSNK1 kinase family members are serine/threonine specific protein kinases including seven mammalian isoforms (α, β, γ1, γ2, γ3, δ, and ε) and their various splice variants. 11 CSNK1 family members are involved in multiple cellular processes including gene transcription, DNA repair, cell division, nuclear localization, and membrane transport. 11 Analysis of gene profiling shows increased expression of CSNK1α1 in most newly diagnosed MM compared with MGUS. 12, 13 CSNK1α1 has also been shown to be involved in other signaling pathways, such as Wnt-signaling. Specifically, ablation of Csnk1α1 stabilizes β-catenin and increases transcription of Wnt target genes. 9 More increasing evidences support that Csnk1α1 plays positive roles in tumor pathogenesis and is a potential target for tumor therapy 14. Csnk1α1 negatively regulates tumor suppressor genes by directly or indirectly interacting with these proteins. For example, Csnk1α1 physically interacts with murine double minute chromosome 2 (Mdm2) oncogene resulting in degradation of p53. 15–17 CSNK1α1 also negatively regulate apoptosis; CSNK1α1 phosphorylates p75 tumor necrosis factor and (FADD) thereby impeding their apoptotic activities. 18, 19 CSNK1α1 regulates Fas-mediated apoptosis through phosphorylation of Bid, which prevents caspase 8-dependant cleavage of Bid and inhibits Fas response. 20 Finally, CSNK1α1 interacts and/or phosphorylates RXR to modulate RXR agonist-mediated apoptosis, which prevents cytochrome C releasing from the mitochondria. 21
To date, the functions of CSNK1α1 in MM pathogenesis and disease progression are not defined. Here we report that CSNK1α1 is critical for MM cell survival and represent a novel therapeutic target.
Results
Inhibiting CSNK1α1 activity impaired MM cell survival
Previous studies have reported that CSNk1α1 is highly expressed in many kinds of cancers and a potential target for cancer chemotherapy. 14 In MM studies, gene expression profiling revealed that CSNk1α1 was highly expressed in MM and plasma cell leukemia patients compared to MGUS and normal plasma cells. 12, 13, 22 Consistent with gene expression results, we confirmed that CSNk1α1 protein was presented in all examined MM cell lines and CD138+ MM patient cells (Fig. 1A, B).
Fig. 1. CSNK1α1 is not involved in bortezomib-triggered cytotoxicity.

(A), (B) Immunoblotting analysis shows that CSNK1α1 is expressed in MM (MM lines, n=8; MM patient cells, n=5). (C) MM1S cells were treated with bortezomib at the concentrations indicated for 12h (left) or with 20nM bortezomib for time internal, as indicated (right). Whole cells lysates were analyzed with immunoblotting against ubiquitin, CSNK1α1, and GAPDH. GAPDH or Tubulin served as loading controls.
To evaluate whether CSNK1α1 represents a novel target for MM therapy, we next treated MM lines with D4476, a CSNK1α1 inhibitor. 23, 24 We found that D4476 induced cytotoxicity in ANBL6, INA6 and RPMI8226 lines; MM1S and U266 lines were less sensitive; and OPM1 line was totally resistant. High concentrations (50μM) D4476 induced toxicity in all MM lines (Fig. 1C). Inhibition of CSNK1α1 activity reduced cell entering S phase and prolonged G2/M phase, as well as induced MM cell apoptosis (Fig. 1D and E).
Previous studies showed that D4476 selectively inhibits CSNK1α1, as well as inhibits TGF-β type-I receptor (ALK5) activity. 23 To verify the function of CSNK1α1 in MM cells, we used shRNA to specifically knock down CSNK1α1. The biological efficiency of shRNAs in reducing CSNK1α1 protein was tested in RPMI8226 cells: relative to control vector and other shRNAs, shRNA3 and 5 showed most efficient CSNK1α1 targeting (Fig. 2A). Importantly, down-regulation of CSNK1α1 with shRNA3 or 5 significantly decreased MM cell survival and proliferation (Fig. 2B). As with D4476, down-regulation of CSNK1α1 with shRNAs reduced cell entering S phase arrest and prolonged G2/M phase, as well as induced apoptosis (Fig. 2C and D). These results suggest that CSNK1α1 plays a critical role in MM cell survival.
Fig. 2. D4476-induced cytotoxicity in MM lines.

(A) Six MM lines were treated with D4476 (0–50μM) for 72h, or with 20μM D4476 for 0–72h. Viable cells were measured with MTT. (B) D4476 induced MM cell apoptosis. Annexin/PI staining and FACS analysis for apoptosis were performed on RPMI8226 cells treated with D4476 (0–30μM) for 72h. Viable cells are represented in Q4 (lower left). Increasing percentages of apoptosis cells are represented in Q2 (upper right) and Q3 (Lower right). (C) D4476 affected cell cycle progression. Cell cycle was examined with Flow Cytometry in RPMI8226 cells stained with PI and exposed to D4476 (0–30 μM) for 72h.
Inhibition of CSNK1α1 causes upregulation of TP53, P21, and FADD. 9, 18 After MM cells were exposed to D4476, protein levels of TP53, P27, and FADD were increased (Supplemental data, Fig. S1A and B), associated with cell cycle progression and induction of apoptosis. TP53 is mutant in both RPMI8226 and ANBL6 lines, therefore TP53 signaling is unlikely to mainly contribute to the cytotoxicity.
To further delineate the role of CSNK1α1 in MM cells, we compared gene transcription changes in RPMI8226 cells transfected with control vector, versus shRNA3 and 5. CSNK1α1 mRNAs were also evaluated with RT-PCR. Consistent with immunoblotting results, both shRNA3 and 5 significantly reduced CSNK1α1 mRNA level (Fig. 3A). In this analysis, gene expression profiling of shRNA3 and 5 was evaluated with Gene Set Enrichment Analysis (GSEA). GSEA demonstrated that gene, upregulated by interferon alpha (IFNα) in ovarian cancer progenitor cells 25 were significantly upregulated and enriched by shRNA3 and 5 in RPMI8226 cells (Fig. 3B), suggesting that targeting CSNK1α1 may induce anti-tumor effects similar as does IFNα. We selected six genes (DDX60, IFIH1, IFI44L, IFIT1, MX1, and SAMD9L), which are ranked with high score in GSEA analysis gene list, to represent the gene signature. Consistent with microarray data, all six genes were upregulated in RT-PCR analysis (Fig. 3C). To further confirm the mechanism whereby inhibition of CSNK1α1 induced cytotoxicity, we treated RPMI8226 and ANBL6 cells with D4476 (30μM for 24h) and examined gene transcription pattern changes by RT-PCR in both RPMI8226 and ANBL6 cells (Fig. 3D, E). These results show that inhibition of CSNK1α1 activity with either shRNAs or D4476 induced cytotoxicity via a mechanism similar with IFNα.
Fig. 3. Reduction of CSNK1α1 protein decrease MM cell survival and inhibit cell cycle progression.

RPMI8226 cells were transduced with control vector or CSNK1α1 shRNAs as indicated. (A) CSNK1α1 was examined with immunoblotting of whole-cell lysates of Ctrl or shRNAs-transfected RPMI8226 cells. GAPDH served as a loading control. (B) Viability was assayed with MTT. Data represent triplicate cultures. (C) Cells were harvested at 72h post viral transduction and stained with PI. Cell cycle was analyzed with Flow Cytometry. (D) Annexin/PI staining and FACS analysis for apoptosis were performed on RPMI8226 cells transduced with control vector, sh3 or 5. Viable cells are represented in Q4 (lower left). Increasing percentages of apoptotic cells are represented in Q2 (upper right) and Q3 (Lower right).
Several other CSNK1α1 gene signatures were also identified. For example, genes in IRF4 signature were significantly enriched and downregulated after CSNK1α1 inhibition by shRNA3 and 5 (Supplemental data, Fig. S2A). 26 Genes in LIN9 signaling regulating cell mitosis were also enriched and downregulated, consistent with the observation that inhibition of CSNK1α1 activity blocks cell mitosis (Supplemental data, Fig. S2B). 27 We also found that TNFα 28 and STAT3 29 gene sets were significantly upregulated and enriched after CSNK1α1 knock-down by shRNA3 and 5 (Supplemental data, Fig. S2C and D).
Factors impeding the cytotoxicity effects of targeting CSNK1α1 in MM cells
OPM1 cells were resistant to D4476-triggered cytotoxicity, whereas MM1S and U266 cells were less sensitive. To delineate a potential the mechanisms of resistance, MM1S and OPM1 lines were treated with either D4476 (30μM) or equal concentrations of DMSO for 24h. Total RNA was extracted and mRNA changes of the six selected genes were examined with RT-PCR. Compared with RPMI8226 sensitive cell line, four of six genes were slightly upregulated and two genes were downregulated in MM1S cells. In OPM1 line, three genes were down-regulated, two genes were slightly upregulated, and one gene showed no changes (Fig. 4A) (Supplemental data, Table. S1). OPM1 line was established from a patient with t(4;14) translocation MM, in which fibroblast growth-factor receptor 3 (FGFR3) gene is involved in pathogenesis. 30, 31 To examine whether FGFR3 confers OPM1 line resistance, we ectopically expressed FGFR3 in RPMI8226 cells and established a FGFR3 stable transfection cell line, named with RPMI-FGFR3. Importantly, RPMI-FGFR3, it remains sensitive to D4476-induced cytotoxicity (Fig. 4B), suggesting that OPM1 resistance to D4476 is not due to FGFR3.
Fig. 4. Gene expression signatures in MM cells with inhibition of CSNK1α1 activity.

(A) RT-PCR shows CSNK1α1 mRNA level changes in RPMI8226 cells transduced with sh3 or 5 relative to control cells (Ctrl). All data represent mean of triplicate experiments. (B) GSEA showed genes in IFNα signature were significantly upregulated and enriched in RPMI8226 cells transduced with shRNA3 and 5. (C) RT-PCR confirmed mRNA level changes of six selected genes in RPMI8226 cells relative to control cells (Ctrl). (D), (E) RT-PCR shows mRNA level changes of selected genes in RPMI82226 or ANBL6 cells exposed to D4476 (30μM for 24h) relative to mRNA level in DMSO only treated cells (Ctrl).
Targeting CSNK1α1 overcame bortezomib resistance and enhanced bortezomib-induced cytotoxicity
To test whether targeting CSNK1α1 could overcome bortezomib resistance, a bortezomib resistant line, ANBL6-VR5, was treated with D4476. D4476 significantly impaired ANBL6-VR5 cell survival (Fig. 4C), suggesting that targeting CSNK1α1 can overcome bortezomib resistance. This result also indicates that combination of D4476 and bortezomib may trigger synergistic cytotoxicity effects on MM cells. To verify the possibility, we first treated MM cells with bortezomib (0–20 nM) for 12h or with bortezomib (20 nM) for 0–8h. Although proteasome activity was significantly inhibited, CSNk1α1 protein was not affected (Fig. 4D), suggesting that CSNk1α1 is worthy to be targeted in bortezomib-triggered cytotoxicity. Then, we treated RPMI8226 cells with both D4476 and bortezomib and found that the combination triggered a synergistic cytotoxicity in MM cells (Fig. 4E).
CSNK1α1 is required for cMYC/KRAS12V-induced plasamacytomas (PCTs) in vivo
Our previous studies have shown that transfection of cMYC with KRAS12V can induce PCTs in BABL/c mice. 32 Based on previous reports, Ras increases CSNK1α1 activity via MEKK1. 33, 34 To determine whether Csnk1α1 plays a role in cMYC/KRAS12V-induced PCT genesis, we expressed cMYC/KRAS12V and inhibited Csnk1α1 in targeted cells using the same vector. Specifically, we generated two this kind of constructs (MKCsnk1α1R1 and MKCsnk1α1R2) for these studies (Fig. 5A). To examine whether the shRNA cassette impacts its upstream oncogene expression, we transiently transfected 293T cells with these constructs and examined gene expression with immunoblotting. The shRNA cassette does not affect KRAS12V, cMYC, and GFP expression (Fig. 5B) (GFP expression was observed by microscopy). To examine biological sequelea, MK, MKCsnk1α1R1, and MKCsnk1α1R2 were retrovirally transfected into BaF3 cells. GFP+ cells were sorted at 48h post viral transduction, then cultured for 48h, pass to harvesting and immunoblotting against Csnk1α1. Csnk1α1 level was obviously decreased in MKCsnk1α1R1 transduced cells, but not MK or MKCsnk1α1R2 transfected BaF3 cells (Fig. 5C); and MKCsnk1α1R1 could not drive BaF3 cell growth independent of IL3, as did MK and MKCsnk1α1R2 (Fig. 5D). This data indicated that Csnk1α1 is required for cMYC/KRAS12V driven BaF3 cell growth independent of IL3.
Fig. 5. Targeting CSNK1α1 induced cytotoxicity in bortezomib-resistant multiple myeloma cells.


(A) Expression of selected genes was tested with RT-PCR in MM1S (left) or OPM1 (right) cells exposed to D4476 (30μM for 24h). All data represent mean of triplicate experiments. (B) FGFR3 was ectopically expressed in RPMI8826 cells. Compared with control vector transfected cells, cells with high expression of FGFR3 remained sensitive to D4476. (C) RPMI8226 cells were treated for 72h with DMSO, bortezomib (2.5nM), D4476 (20μM), or bortezomib (2.5nM) and D4476 (20μM). (D) ANBL6-WT and ANBL6-VR5 cells were treated with D4476 (20μM for 72h). Cell viability was measured with MTT.
To determine whether MKCsnk1α1R1 can transform BALB/cJ T2 B cells and induce PCTs in vivo, we transplanted cells transfected with MK or MKCsnk1α1R1 into lethally irradiated syngeneic recipient mice. Recipients receiving MK-transduced cells developed lethal PCTs by 39d post transplantation. Recipients receiving MKCsnk1α1R1-transfected cells remained healthy at 20 weeks post transplantation (Fig. 6A). Consistent with previous data, 32 mice receiving MK-transduced cells died with lethal PCTs characterized by peritoneal tumor, as cites, and enlarged spleen as in previous studies. 32 Bone marrow (BM) and spleen cells were isolated from MK and MKCsnk1α1R1 group mice and analyzed for the expression of GFP and CD138 by flow cytometry. Importantly, there were no tumor cells (GFP+CD138+) detectable in MKCsnk1α1R1 group samples (Fig. 6B). Consistent with FACS results, PCT cells only developed in MK, but not in MKCsnk1α1R1 group mice (Fig. 6C). These results suggest that downregulation of Csnk1α1 can prevent MYC/KRAS12V-induced PCTs in BALB/c mice.
Fig. 6. cMYC/KRAS12V cannot transform BaF3 cells independent of IL3 with low level of Csnk1α1.

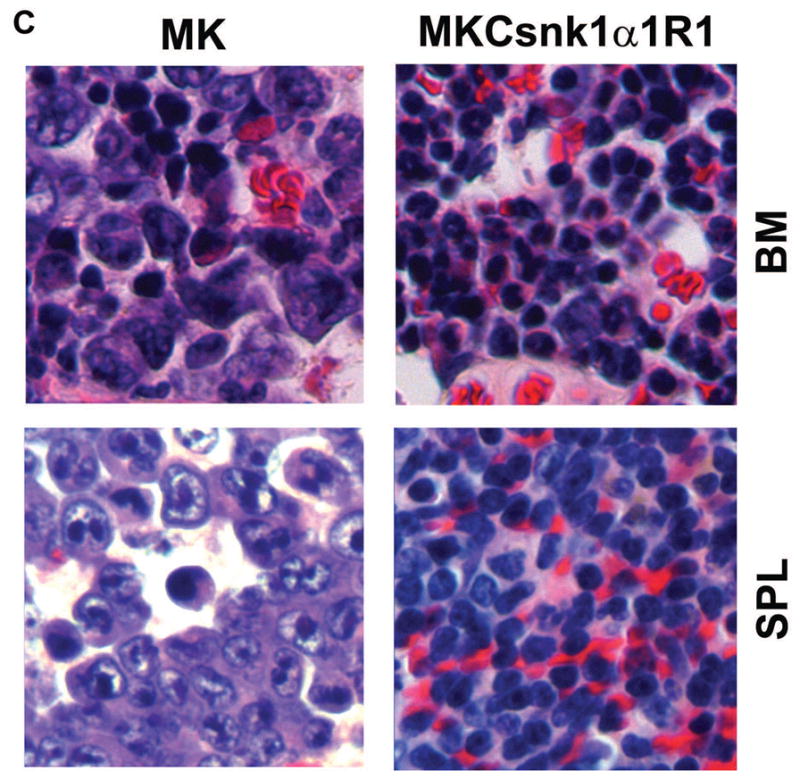
(A) Schematic diagram of MSCV-based retroviral vectors: MK, MKCsnk1α1R1 and MKCsnk1α1R2. (B) Immunoblotting analysis shows expression of cMYC and KRAS12V in 293T cells transfected with the MK and MKCsnk1α1Rs vectors. GAPDH served as a loading control. (C) Immunoblotting shows Csnk1α1 expression in BaF3 cells transfected with MK and MKCsnk1α1Rs vectors. GAPDH served as a loading control. (D) Photographs of BaF3 cells transduced with MK, MKCsnk1α1R1, or MKCsnk1α1R2 vectors. MKCsnk1α1R1 transfection cannot drive BaF3 cell growth independent of IL3 in vitro (middle).
Discussion
In this study, we demonstrated that CSNK1α1 is present in patient MM cells and MM lines. Inhibition of CSNK1α1 activity, by either inhibitor (D4476) or shRNAs, causes G0/G1 phase arrest, prolongs G2/M phase, and induces apoptosis in some MM cell lines. Targeting CSNK1α1 signaling affects several classic cell proliferation and apoptosis pathways including CDKN1B, P53, FADD, IFNα, TNF, and LIN9, in MM cells. Specifically, CSNK1α1 suppresses P53 activity by altering its phosphorylation status, as well as increases MDM2 activity. 17 However, our results suggest that P53 does not play a critical role in CSNK1α1-triggered cytotoxicity, since RPMI8226 cell with P53 temperature sensitive mutation (E285K) 35 are more sensitive to D4476 or CSNK1α1 shRNA than MM1S cells with wild type P53.
To further reveal the potential pathways involved in CSNK1α1 signaling, we compared gene expression profiling of RPMI8226 cells transfected with either control vector or with CSNK1α1 shRNA3 and 5. Genes modulated by IFNα and TNFα are significantly upregulated and enriched by CSNK1α1 knockdown. The anti-proliferative effects of IFNα are associated G0/G1 phase blocked and S-phase prolonged, 36 as is observed in MM cells treated with D44476 or transfected with CSNK1α1 shRNAs. In previous reports of IFNα clinical studies, 5–10% of MM patients obtained a significant prolongation in event-free survival and overall survival. 37 Our in vitro studies also show that only certain MM cell lines are sensitive to CSNK1α1 inhibitor or shRNAs. To date, however, there is no biomarker to identify those patients who are likely to benefit from IFNα treatment. 37 Based on our studies, we hypothesized that expression of genes in the IFNα signature may identify these patients 28. To test this hypothesis, we compared the expression of these genes among RPMR8226, MM1S and OPM2 MM cell lines (GSE22759 at GEO database). As expected, most IFNα signature genes are expressed highly in RPMR8226 cells, moderately in MM1S cells, and weakly in OPM2 cells (Supplemental data, Fig. S3). These data suggest that patients with MM cells which highly express these genes may well respond to either IFNα or inhibition of CSNK1α1 treatment.
Before investigating the roles of CSNK1α1 in MM, we analyzed its expression in MM patients using Oncomine database system (https://www.oncomine.org/). CSNK1α1 was highly expressed in smoldering myeloma versus normal plasma cells, 13 and its expression was significantly increased in MM or plasma cell leukemia patients compared to MGUS (Supplemental data, Fig. S4). 12, 13, 22 These data suggest a role of CSNK1α1 in the progression of MGUS to MM. To examine this possibility, we used our retroviral transfection/transplantation PCT mouse model. 32 Specifically, we used cMYC and KRAS12V transfection to drive PCT formation, since previous studies indicate that dysregulation or mutants of cMYC and RAS play major roles in progression from MGUS to MM and MM relapse. 38 Moreover, CSNK1α1 is not only involved in MYC signaling, but also RAS signaling via MEKK1. 33, 34 Our in vitro data show that cMYC/KRAS12V without Csnk1α1 could not promote BaF3 growth independent of IL3, suggesting that Csnk1α1 plays an important role in cell transformation by MYC and RAS. Furthermore, our in vivo results demonstrate that inhibiting Csnk1α1 prevents development of cMYC/KRAS12V-induced PCTs in mice. These results indicate that Csnk1α1 is required for cMYC/KRAS12V-induced development of PCTs in mice, and suggests a role of CSNK1α1 in progression from MGUS to MM, especially, in MM with cMYC or KRAS dysregulation. Our studies therefore delineate the role of CSNK1α1 in MM pathogenesis, and provide the framework for clinical evaluation of CSNK1α1 inhibitors to treat MM, as well as to prevent progression from MGUS to MM.
Materials and Experimental methods
Cell lines and MM patient cells
Human MM cell lines including MM1S, RPMI8226, U266, MM1R, OPM1, OPM2, INA6, ANBL6, ANBL-6VR, LR5 and RPMI-DOX40 (DOX40) are maintained and cultured in RPMI1640 medium supplemented with 10% fetal bovine serum and 100 units/ml penicillin/streptomycin. INA6, ANBL6, and ANBL-6VR also require 2ng/ml IL6; and ANBL-6VR line is cultured with 2.5nM bortezomib. CD138+ tumor cells were purified from bone marrow (BM) aspirates of MM patients. BaF3 cells are cultured in RPMI1640 medium supplemented with 10% fetal bovine serum 100 units/ml penicillin/streptomycin, 50μM β-mercaptoethanol, and 10% WEHI3 cell culture supernatant.
Immunoblotting
Whole cells were lysed with RIPA buffer plus protease and phosphatase inhibitors. Proteins were separated with 4–15% PAGE gel electrophoresis, transferred onto membranes, and immunoblotted with primary antibodies; followed by incubation with horseradish peroxidase-conjugated secondary antibodies. Final signal is detected with enhanced chemiluminescence (ECL).
Cell biological assays
In all cell assays, final cell concentration was 1x105 cells/ml. bortezomib and D4476 were dissolved in dimethyl sulfoxide (DMSO) and stored at −20°C. Cell viability assay: MM cells were seeded in triplicate into 96-well plates in 100μL culture media. D4476 was added to each well at concentrations of 0, 5, 10, 20, 30, 40, and 50μM in another 100μL culture media. Cell viability was measured with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) at the 72h drug exposure. Absorbance was measured at 570nm with spectrophotometer.
MM cells were exposed to DMSO or D4476 at 10, 20, and 30μM. For cell apoptosis analysis, cells were harvested at 72h and stained with FITC conjugated annexin V and propidium iodide (PI) for Flow Cytometric analysis. For cell cycle analysis, cells were harvested at 24h and permeabilized with 5ml 70% ethanol on ice for 30mins, resuspended the cells with 1ml staining solution (PBS containing 0.1% Triton X-100, 50μg/mL PI and 20 units/mL RNase-A), and incubated at 37°C for 30mins. Cell cycle was analyzed with Flow Cytometry using Watson model.
Lentiviral stocks and cell transfection
Lentiviral-based small hairpin RNA (shRNA) vectors include one control vector (shRNA sequence targeting Lactose gene) and five shRNAs designed specifically against CSNK1α1. To produce virus, constructs were co-transfected with packaging vectors (psPAX2 and MD2.G) into 293T cells. Viral supernatant was collected at 48h. For each sample, three million cells were re-suspended with viral transfection medium (2ml cell culture medium, 2ml viral stocks, 40μL PH7.4 HEPES, 2μg/ml polybrene), and centrifuged at 1000g for 90mins in 6-well-plates. After another 3h culture, cells were transferred to 25cm2 flasks with 10ml fresh medium. All processes were performed in a BL2l enti room.
Gene expression profiling
Total RNA was extracted with RNA mini kit, and gene expression profiling was performed with the Affymetrix Human Gene 2.0 ST chips. Microarray data was analyzed with Bioconductor oligo and OncchannelGUI packages in R workplace. Differential expression was determined using LIMMA model. Gene signatures were analyzed with Gene Set Enrichment Analysis (GSEA) in Molecular Signatures Database (MSigDB).
DNA constructs
The retroviral vectors (MIGm, MIKMG) were generated as previously descripted 32. The MIGmI vector was created by inserting a specific sequence (GATCGTTTAAAGGGAGGTAGTGAGTCGACCAGTAGATCTCTCGAGGATATCTAGACCCAGCTTTCTTGTACAAAGTGGTTGATCTAGAGGGCCCGCGGTTCGCTGATGTCG A) into MIGm vector at BglII/XhoI sites. The RNAi sequences specifically targeting mouse Csnk1α1 (GCAGAATTTGCCATGTACTTA and GCTGTACGAGAGCAAACTGTA) were cloned into pcDNA6.2-GW/EmGFP-miR vector, and then were cut off with BamHI/XhoI and inserted into MIGmI vector to obtain MIGmICsnk1α1Rs. Oncogenic cassette (cMYC-2a-eGFP-IRES-KRAS12V) was cut from MIKMG with restriction enzymes SpeI and NotI, and then inserted into MIGmICsnk1α1Rs at same enzyme digesting sites to obtain MKCsnk1α1R1 and MKCsnk1α1R2. Knockdown efficiency was assessed following retroviral transduction into BaF3 cells, as previously described 32.
Mouse model
The procedures for retroviral transfection and transplantation in the PCT mouse model, as well as phenotypic analysis were referred as in previous studies 32.
Statistics
All in vitro experiments were performed in triplicate and repeated at least 3 times. Statistical significance was determined with t tests, with minimal significance level P < 0.05 using GraphPad Prism software.
Supplementary Material
Fig. S1. P53, CDKN1B (P27), and FADD are involved in D4476 triggered cytotoxicity. (A) RPMI8226 and ANBL6 cells were treated with D4476 at the concentrations as indicated for 72h. Whole cells lysates were analyzed with immunoblotting against P53, CDKN1B (P27), and GAPDH. (B) RPMI8226 and ANBL6 cells were treated with D4476 at the concentrations as indicated for 72h. Whole cell lysates were analyzed with immunoblotting against FADD and GAPDH. GAPDH served as a loading control.
Fig. S2 Gene expression signatures in MM cells with inhibition of CSNK1α1 activity. (A) GSEA showed genes in IRF4 signature were significantly downregulated and enriched in RPMI8226 cells transduced with shRNAs. (B) GSEA showed genes in LIN9 signature were significantly downregulated and enriched in RPMI8226 cells transduced with shRNAs. (C) GSEA showed genes in TNFα signature were significantly upregulated and enriched in RPMI8226 cells transduced with shRNAs. (D) GSEA showed genes in STAT3 signature were significantly upregulated and enriched in RPMI8226 cells transduced with shRNAs. (E) Table of the gene signatures related to reductions of CSNK1α1 in RPMI8226 cells.
Fig. S3 Expression density of genes in IFNα signature in OPM2, MM1S, and RPMI8226 lines. Gene expression dataset GSE22759 was downloaded from GEO database and analyzed with OneChannelGUI packages at R workspace. Expression values of genes in IFNα signature were exported to GraphPad Prism 6 and graphed. Signal density is shown as indicated.
Fig. S4 CSNK1α1 expression patterns in MM patients. The expression of CSNK1α1 in MM patients was analyzed in Oncomine database. All probe values were exported.
Table S1 RPMI8226, MM1S and OPM1 cells were exposed to D4476 at 30μM for 24h, and expression change of gene including DDX60, IFIH1, IFIT1, IFI44L, MX1, and SAM9L, was tested with RT-PCR. All data represent mean fold change of triplicate experiments.
Significance.
Case in kinase 1 (CSNK1) family members are attractive targets for cancer therapies. In this study, we identified that Casein kinase 1 alpha 1 (CSNK1α1) was a novel target for multiple myeloma (MM) treatment. Targeting CSNK1α1 was shown to be effective in TP53 mutation or proteasome inhibitor resistance MM cells. Moreover, the combination of D4476 (a CSNK1α1 inhibitor) and bortezomib (proteasome inhibitor) triggered synergistic cytotoxicity. Based on the studies of our rodent tumor model, cMYC and KRAS12V without Csnk1α1 could not induce plasmacytomas, which indicates that targeting CSNK1α1 can prevent progression from monoclonal gammopathy of undetermined significance (MUGS, a non-diseased condition) to MM.
Acknowledgments
We would like to thank Mei Zheng for performing the histological sections; Reddy Gali, who is part of Harvard Catalyst grant member, for his conduction in microarray analysis; the Microarray and the Flow Cytometry Core Facilities at Dana-Farber Cancer Institute for technical support. This study was supported in part by National Institutes of Health Grants P50-100707, P01-78378 and RO1-50947. K.C.A. is an American Cancer Society Clinical Research Professor.
Footnotes
Conflict-of-interest disclosure: The authors have no conflicting financial interests.
Supplementary information is available at the journal’s website.
References
- 1.Palumbo A, Anderson K. Multiple myeloma. The New England journal of medicine. 2011 Mar 17;364(11):1046–1060. doi: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- 2.Anderson KC. New insights into therapeutic targets in myeloma. Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program. 2011;2011:184–190. doi: 10.1182/asheducation-2011.1.184. [DOI] [PubMed] [Google Scholar]
- 3.Kane RC, Bross PF, Farrell AT, Pazdur R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. The oncologist. 2003;8(6):508–513. doi: 10.1634/theoncologist.8-6-508. [DOI] [PubMed] [Google Scholar]
- 4.Field-Smith A, Morgan GJ, Davies FE. Bortezomib (Velcadetrade mark) in the Treatment of Multiple Myeloma. Therapeutics and clinical risk management. 2006 Sep;2(3):271–279. doi: 10.2147/tcrm.2006.2.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple myeloma and its premalignant precursor. The Journal of clinical investigation. 2012 Oct 1;122(10):3456–3463. doi: 10.1172/JCI61188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chng WJ, Huang GF, Chung TH, Ng SB, Gonzalez-Paz N, Troska-Price T, et al. Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed multiple myeloma. Leukemia. 2011 Jun;25(6):1026–1035. doi: 10.1038/leu.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011 Sep 16;146(6):904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trumpp A, Refaeli Y, Oskarsson T, Gasser S, Murphy M, Martin GR, et al. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature. 2001 Dec 13;414(6865):768–773. doi: 10.1038/414768a. [DOI] [PubMed] [Google Scholar]
- 9.Elyada E, Pribluda A, Goldstein RE, Morgenstern Y, Brachya G, Cojocaru G, et al. CKIalpha ablation highlights a critical role for p53 in invasiveness control. Nature. 2011 Feb 17;470(7334):409–413. doi: 10.1038/nature09673. [DOI] [PubMed] [Google Scholar]
- 10.Toyoshima M, Howie HL, Imakura M, Walsh RM, Annis JE, Chang AN, et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proceedings of the National Academy of Sciences of the United States of America. 2012 Jun 12;109(24):9545–9550. doi: 10.1073/pnas.1121119109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, Stoter M. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal. 2005 Jun;17(6):675–689. doi: 10.1016/j.cellsig.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 12.Dickens NJ, Walker BA, Leone PE, Johnson DC, Brito JL, Zeisig A, et al. Homozygous deletion mapping in myeloma samples identifies genes and an expression signature relevant to pathogenesis and outcome. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010 Mar 15;16(6):1856–1864. doi: 10.1158/1078-0432.CCR-09-2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhan F, Barlogie B, Arzoumanian V, Huang Y, Williams DR, Hollmig K, et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood. 2007 Feb 15;109(4):1692–1700. doi: 10.1182/blood-2006-07-037077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knippschild U, Wolff S, Giamas G, Brockschmidt C, Wittau M, Wurl PU, et al. The role of the casein kinase 1 (CK1) family in different signaling pathways linked to cancer development. Onkologie. 2005 Oct;28(10):508–514. doi: 10.1159/000087137. [DOI] [PubMed] [Google Scholar]
- 15.Huart AS, MacLaine NJ, Meek DW, Hupp TR. CK1alpha plays a central role in mediating MDM2 control of p53 and E2F-1 protein stability. J Biol Chem. 2009 Nov 20;284(47):32384–32394. doi: 10.1074/jbc.M109.052647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huart AS, MacLaine NJ, Narayan V, Hupp TR. Exploiting the MDM2-CK1alpha protein-protein interface to develop novel biologics that induce UBL-kinase-modification and inhibit cell growth. PloS one. 2012;7(8):e43391. doi: 10.1371/journal.pone.0043391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meek DW, Knippschild U. Posttranslational modification of MDM2. Mol Cancer Res. 2003 Dec;1(14):1017–1026. [PubMed] [Google Scholar]
- 18.Alappat EC, Feig C, Boyerinas B, Volkland J, Samuels M, Murmann AE, et al. Phosphorylation of FADD at serine 194 by CKIalpha regulates its nonapoptotic activities. Molecular cell. 2005 Aug 5;19(3):321–332. doi: 10.1016/j.molcel.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 19.Beyaert R, Vanhaesebroeck B, Declercq W, Van Lint J, Vandenabele P, Agostinis P, et al. Casein kinase-1 phosphorylates the p75 tumor necrosis factor receptor and negatively regulates tumor necrosis factor signaling for apoptosis. J Biol Chem. 1995 Oct 6;270(40):23293–23299. doi: 10.1074/jbc.270.40.23293. [DOI] [PubMed] [Google Scholar]
- 20.Desagher S, Osen-Sand A, Montessuit S, Magnenat E, Vilbois F, Hochmann A, et al. Phosphorylation of bid by casein kinases I and II regulates its cleavage by caspase 8. Molecular cell. 2001 Sep;8(3):601–611. doi: 10.1016/s1097-2765(01)00335-5. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Y, Qin S, Atangan LI, Molina Y, Okawa Y, Arpawong HT, et al. Casein kinase 1alpha interacts with retinoid X receptor and interferes with agonist-induced apoptosis. J Biol Chem. 2004 Jul 16;279(29):30844–30849. doi: 10.1074/jbc.M404651200. [DOI] [PubMed] [Google Scholar]
- 22.Agnelli L, Mosca L, Fabris S, Lionetti M, Andronache A, Kwee I, et al. A SNP microarray and FISH-based procedure to detect allelic imbalances in multiple myeloma: an integrated genomics approach reveals a wide gene dosage effect. Genes, chromosomes & cancer. 2009 Jul;48(7):603–614. doi: 10.1002/gcc.20668. [DOI] [PubMed] [Google Scholar]
- 23.Rena G, Bain J, Elliott M, Cohen P. D4476, a cell-permeant inhibitor of CK1, suppresses the site-specific phosphorylation and nuclear exclusion of FOXO1a. EMBO Rep. 2004 Jan;5(1):60–65. doi: 10.1038/sj.embor.7400048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tillement V, Lajoie-Mazenc I, Casanova A, Froment C, Penary M, Tovar D, et al. Phosphorylation of RhoB by CK1 impedes actin stress fiber organization and epidermal growth factor receptor stabilization. Exp Cell Res. 2008 Sep 10;314(15):2811–2821. doi: 10.1016/j.yexcr.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 25.Moserle L, Indraccolo S, Ghisi M, Frasson C, Fortunato E, Canevari S, et al. The side population of ovarian cancer cells is a primary target of IFN-alpha antitumor effects. Cancer Res. 2008 Jul 15;68( 14):5658–5668. doi: 10.1158/0008-5472.CAN-07-6341. [DOI] [PubMed] [Google Scholar]
- 26.Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, Xiao W, et al. IRF4 addiction in multiple myeloma. Nature. 2008 Jul 10;454(7201):226–231. doi: 10.1038/nature07064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reichert N, Wurster S, Ulrich T, Schmitt K, Hauser S, Probst L, et al. Lin9, a subunit of the mammalian DREAM complex, is essential for embryonic development, for survival of adult mice, and for tumor suppression. Molecular and cellular biology. 2010 Jun;30(12):2896–2908. doi: 10.1128/MCB.00028-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sana TR, Janatpour MJ, Sathe M, McEvoy LM, McClanahan TK. Microarray analysis of primary endothelial cells challenged with different inflammatory and immune cytokines. Cytokine. 2005 Mar 21;29(6):256–269. doi: 10.1016/j.cyto.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 29.Dauer DJ, Ferraro B, Song L, Yu B, Mora L, Buettner R, et al. Stat3 regulates genes common to both wound healing and cancer. Oncogene. 2005 May 12;24(21):3397–3408. doi: 10.1038/sj.onc.1208469. [DOI] [PubMed] [Google Scholar]
- 30.Chesi M, Nardini E, Brents LA, Schrock E, Ried T, Kuehl WM, et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nature genetics. 1997 Jul;16(3):260–264. doi: 10.1038/ng0797-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richelda R, Ronchetti D, Baldini L, Cro L, Viggiano L, Marzella R, et al. A novel chromosomal translocation t(4; 14)(p16.3; q32) in multiple myeloma involves the fibroblast growth-factor receptor 3 gene. Blood. 1997 Nov 15;90(10):4062–4070. [PubMed] [Google Scholar]
- 32.Hu Y, Zheng M, Gali R, Tian Z, Topal Gorgun G, Munshi NC, et al. A novel rapid-onset high-penetrance plasmacytoma mouse model driven by deregulation of cMYC cooperating with KRAS12V in BALB/c mice. Blood cancer journal. 2013;3:e156. doi: 10.1038/bcj.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu J, Shibasaki F, Price R, Guillemot JC, Yano T, Dotsch V, et al. Intramolecular masking of nuclear import signal on NF-AT4 by casein kinase I and MEKK1. Cell. 1998 May 29;93(5):851–861. doi: 10.1016/s0092-8674(00)81445-2. [DOI] [PubMed] [Google Scholar]
- 34.Russell M, Lange-Carter CA, Johnson GL. Direct interaction between Ras and the kinase domain of mitogen-activated protein kinase kinase kinase (MEKK1) J Biol Chem. 1995 May 19;270(20):11757–11760. doi: 10.1074/jbc.270.20.11757. [DOI] [PubMed] [Google Scholar]
- 35.Teoh G, Tai YT, Urashima M, Shirahama S, Matsuzaki M, Chauhan D, et al. CD40 activation mediates p53-dependent cell cycle regulation in human multiple myeloma cell lines. Blood. 2000 Feb 1;95(3):1039–1046. [PubMed] [Google Scholar]
- 36.Oberg K. The action of interferon alpha on human carcinoid tumours. Seminars in cancer biology. 1992 Feb;3(1):35–41. [PubMed] [Google Scholar]
- 37.Blade J, Esteve J. Viewpoint on the impact of interferon in the treatment of multiple myeloma: benefit for a small proportion of patients? Medical oncology. 2000 May;17(2):77–84. doi: 10.1007/BF02796202. [DOI] [PubMed] [Google Scholar]
- 38.Bergsagel PL, Kuehl WM. Molecular pathogenesis and a consequent classification of multiple myeloma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005 Sep 10;23(26):6333–6338. doi: 10.1200/JCO.2005.05.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. P53, CDKN1B (P27), and FADD are involved in D4476 triggered cytotoxicity. (A) RPMI8226 and ANBL6 cells were treated with D4476 at the concentrations as indicated for 72h. Whole cells lysates were analyzed with immunoblotting against P53, CDKN1B (P27), and GAPDH. (B) RPMI8226 and ANBL6 cells were treated with D4476 at the concentrations as indicated for 72h. Whole cell lysates were analyzed with immunoblotting against FADD and GAPDH. GAPDH served as a loading control.
Fig. S2 Gene expression signatures in MM cells with inhibition of CSNK1α1 activity. (A) GSEA showed genes in IRF4 signature were significantly downregulated and enriched in RPMI8226 cells transduced with shRNAs. (B) GSEA showed genes in LIN9 signature were significantly downregulated and enriched in RPMI8226 cells transduced with shRNAs. (C) GSEA showed genes in TNFα signature were significantly upregulated and enriched in RPMI8226 cells transduced with shRNAs. (D) GSEA showed genes in STAT3 signature were significantly upregulated and enriched in RPMI8226 cells transduced with shRNAs. (E) Table of the gene signatures related to reductions of CSNK1α1 in RPMI8226 cells.
Fig. S3 Expression density of genes in IFNα signature in OPM2, MM1S, and RPMI8226 lines. Gene expression dataset GSE22759 was downloaded from GEO database and analyzed with OneChannelGUI packages at R workspace. Expression values of genes in IFNα signature were exported to GraphPad Prism 6 and graphed. Signal density is shown as indicated.
Fig. S4 CSNK1α1 expression patterns in MM patients. The expression of CSNK1α1 in MM patients was analyzed in Oncomine database. All probe values were exported.
Table S1 RPMI8226, MM1S and OPM1 cells were exposed to D4476 at 30μM for 24h, and expression change of gene including DDX60, IFIH1, IFIT1, IFI44L, MX1, and SAM9L, was tested with RT-PCR. All data represent mean fold change of triplicate experiments.