

Abstract
Multiple system atrophy (MSA) is a rare, yet fatal neurodegenerative disease that presents clinically with autonomic failure in combination with parkinsonism or cerebellar ataxia. MSA impacts on the autonomic nervous system affecting blood pressure, heart rate and bladder function, and the motor system affecting balance and muscle movement. The cause of MSA is unknown, no definitive risk factors have been identified, and there is no cure or effective treatment. The definitive pathology of MSA is the presence of α-synuclein aggregates in the brain and therefore MSA is classified as an α-synucleinopathy, together with Parkinson's disease and dementia with Lewy bodies. Although the molecular mechanisms of misfolding, fibrillation and aggregation of α-synuclein partly overlap with other α-synucleinopathies, the pathological pathway of MSA is unique in that the principal site for α-synuclein deposition is in the oligodendrocytes rather than the neurons. The sequence of pathological events of MSA is now recognized as abnormal protein redistributions in oligodendrocytes first, followed by myelin dysfunction and then neurodegeneration. Oligodendrocytes are responsible for the production and maintenance of myelin, the specialized lipid membrane that encases the axons of all neurons in the brain. Myelin is composed of lipids and two prominent proteins, myelin basic protein and proteolipid protein. In vitro studies suggest that aberration in protein distribution and lipid transport may lead to myelin dysfunction in MSA. The purpose of this perspective is to bring together available evidence to explore the potential role of α-synuclein, myelin protein dysfunction, lipid dyshomeostasis and ABCA8 in MSA pathogenesis.
Keywords: Multiple system atrophy, oligodendrocyte, α-synuclein, myelin, lipid dyshomeostasis
INTRODUCTION
Multiple system atrophy (MSA) is a sporadic neurodegenerative disease characterized by a combination of parkinsonism, cerebellar ataxia and autonomic dysfunction [1]. The distribution of pathology classically encompasses three functional systems in the central nervous system - the striatonigral system, olivopontocerebellar system and autonomic system - impacting on movement, muscle control, blood pressure, heart rate and bladder function [2,3]. MSA affects equally both men and women primarily in their 50s, although it can strike as early as 30s. The progression of disease is rapid and patients are confined to bed within 5 years of onset of symptoms and death results within ~9 years [4]. MSA shares some similarities with Parkinson's disease (PD) with overlapping clinical presentation of motor impairments, and as such, MSA is commonly misdiagnosed as PD [1,5]. However, in comparison to PD, MSA is relatively rare, with a prevalence rate of 3~4 per 100,000 [6,7,8]. The aetiology of MSA is largely unknown, although studies point to a possible genetic component [9,10], as well as environmental factors capable of increasing susceptibility [5,11]. Based on current information the sequence of pathological events of MSA is now recognized as abnormal protein redistributions in oligodendrocytes first, followed by myelin dysfunction and then neurodegeneration and loss of neurons (Fig. 1).
Fig. 1.

A putative pathogenic pathway of multiple system atrophy.
NEUROPATHOLOGY OF MSA
Current understanding of MSA neuropathology is that both grey and white matter pathology occur in the form of neurodegeneration, gliosis, myelin loss and axonal degeneration [12]. These changes also typically occur in specific anatomical locations that include subcortical regions within the olivopontocerebellar pathway (e.g. inferior olives, pons, cerebellum), striatonigral pathway (e.g. striatum and substantia nigra), and in autonomic nuclei within the spinal cord and brainstem [13,14]. Furthermore, although earlier studies reported the cerebral cortex was spared in MSA, later studies have reported decreased neuronal density in the primary and supplementary motor cortex of MSA patients [14,15], as well as atrophy occurring in regions within the frontal lobe [7]. Therefore, as exemplified by these studies, neuropathological changes occur widely throughout various subcortical and cortical regions in MSA brains.
However, while these neuropathological changes are commonly observed in post-mortem brains of MSA patients [13], the most consistent pathological hallmark of MSA is glial cytoplasmic inclusions (GCIs) (Fig. 2). These inclusions are variably shaped, filamentous protein aggregates that form in the cytoplasm of oligodendrocytes, which are thought to play a primary role in the pathogenesis of MSA [12], as their anatomical distribution correlates with regions where neurodegeneration occurs [16,17]. In terms of their constituents, GCIs are composed of a multitude of proteins including ubiquitin, the heat shock protein αβ-crystallin, and the microtubule proteins, α- and β-tubulin [18]. However, the predominant constituent is the α-synuclein protein [19,20]. Normally, α-synuclein is mainly localized to the presynaptic terminals of neurons as a non-phosphorylated, soluble and unfolded monomer [8,21]. α-Synuclein is putatively involved in regulating synaptic plasticity [22] and presynaptic events [23,24], although gaps in our understanding of its normal physiological role still remain. Nonetheless, the presence of these α-synuclein aggregates thus places MSA in a category of diseases known as α-synucleinopathies alongside PD and Lewy body dementia, which are similarly characterized by abnormal α-synuclein aggregates.
Fig. 2.
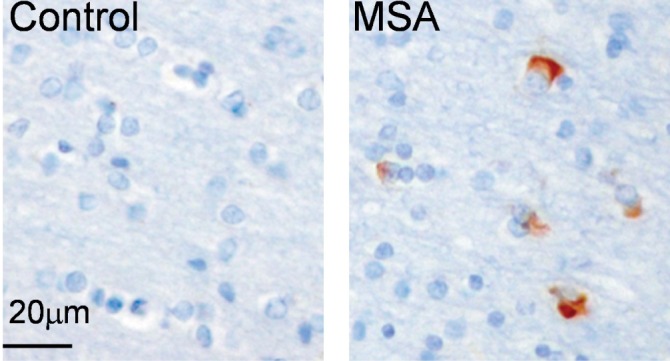
The pathological hallmark of MSA is the presence of glial cytoplasmic inclusions (GCIs) in the brain.
α-SYNUCLEIN PATHOLOGY AND NEURODEGENERATION
In contrast to the neuronal localization and normal structure of α-synuclein in healthy brains [8,21], during the MSA disease process, the localization and structure of α-synuclein is vastly altered. As suggested by the location of GCIs in MSA, α-synuclein becomes aberrantly translocated to the cytoplasm of oligodendrocytes. However, the mechanism by which this occurs remains unknown, although it is possible neurons secrete α-synuclein into the extracellular environment, which is subsequently taken-up by surrounding oligodendrocytes [12,25,26,27]. Nonetheless, α-synuclein also undergoes multiple structural modifications including phosphorylation at serine residue 129, while also developing an ordered β-sheet-rich secondary structure, with the latter being facilitated by an increase in surrounding lipid concentrations [28,29,30,31]. Together, these structural changes are thought to promote the self-aggregation of α-synuclein into intermediate species (e.g. oligomers and protofibrils) that precede the formation of mature fibrils [32,33].
The formation of GCIs and intermediate species of α-synuclein also cause alterations in α-synuclein function in MSA. That is, rather than contributing to the regulation of synaptic plasticity and presynaptic events [22,23,24], α-synuclein appears to contribute towards neurodegeneration, albeit the precise mechanism(s) that induces this remains unclear. Potential mechanisms suggested by in vitro studies of other α-synucleinopathies include oxidative stress and neurotoxicity evoked by mitochondrial and lysosomal damage [34,35], as well as impaired transport of crucial presynaptic proteins (e.g. synapsin-1), which cause synaptic dysfunction to contribute towards neuronal death [36]. Furthermore, another hypothesis posits that annular shaped intermediate species form abnormal membranous pore-like channels that are capable of altering membrane permeability and reducing the integrity of presynaptic vesicles [37]. This is then subsequently thought to promote dysregulated ion homeostasis and dopamine release respectively, to cause the excessive release of dopamine and ions such as calcium into the extracellular space, to ultimately result in neurodegeneration induced by neurotoxicity.
The downstream consequences of these α-synuclein induced mechanisms of neurodegeneration have also been illustrated through transgenic mice overexpressing human α-synuclein (e.g. under the control of the murine myelin basic protein promoter). More specifically, these mice demonstrated selective accumulation and aggregation of α-synuclein in oligodendrocytes that preceded neurodegeneration, as well as gliosis, myelin loss and axonal abnormalities [38,39,40]. The severity of these neuropathological features also correlated with the degree of α-synuclein overexpression, whereby mice expressing higher amounts of α-synuclein exhibited more severe neuropathological changes and vice versa, ultimately providing further evidence for a causative relationship between α-synuclein and downstream neuropathological changes. Furthermore, these consequences from α-synuclein accumulation and aggregation also appeared sufficient to produce motor impairments and induce death, which draws similarities with the clinical profile of MSA patients [1,38,39].
Therefore, given the apparent ability of α-synuclein to cause a variety of neuropathological abnormalities that are similarly observed in MSA patients, alterations in its localization and function have been incorporated into the current working hypothesis of MSA pathogenesis. This working hypothesis suggests that the initiating culprit of MSA pathogenesis is the uptake of α-synuclein into oligodendrocytes, and its subsequent aggregation into GCIs. The latter is facilitated by p25α, which is normally involved in myelination and stabilization of microtubules [41], but re-localized from myelin to cell soma during the early stages of MSA, where it acts as potent stimulator of α-synuclein aggregation to promote GCI formation [42,43]. The relocation of p25α and the formation of GCIs then leads to oligodendrocyte dysfunction, with the retraction of myelinating processes. Consequently this demyelination causes myelin loss-induced axonal and neuronal degeneration that subsequently account for the onset of clinical symptoms [12].
MYELIN PROTEIN FUNCTION
Another important, yet under-studied aspect of MSA pathogenesis is myelin dysfunction. Myelin is a large modified membrane produced by oligodendrocytes that encases the axons of all neurons. It provides the insulation required to facilitate rapid signal transmission between neurons [44]. Critical to myelin function is membrane-associated proteins. These include proteolipid protein (PLP), which spans the myelin membrane bilayer, and myelin basic protein (MBP), which is located on the cytoplasmic surface of myelin membranes [45,46]. Together PLP and MBP constitute the majority of the total myelin proteins (85%). Furthermore, they are both located in the compact portion of myelin, which is where adjacent myelin layers become fused in forming the central segment of the myelin sheath, as opposed to the non-compact portion where layers do not fuse and form the periphery of the myelin sheath [47]. Here, PLP and MBP function to ensure the proper compaction of myelin layers and thus stabilize the ultrastructure of compact myelin [44]. More specifically, PLP stabilizes the intraperiod line (IPL) to maintain a separation between myelin layers, whereas MBP stabilizes the major dense line (MDL) to facilitate the adhesion of these layers [48,49].
Maintaining the necessary levels of MBP and PLP for the proper compaction of myelin is integral for normal myelination. This has been demonstrated through previous studies investigating the myelination capabilities of PLP and MBP mutant mice, whereby mice exhibited defects in myelin compaction corresponding to the specific ultrastructure they are known to stabilize. That is, in both MBP and PLP mutant mice that have undetectable levels of the proteins, a complete absence of the MDL and IPL was observed respectively [50,51]. The detrimental effects resulting from the absence of these ultrastructures was subsequently exemplified by myelin instability in the form of dysmyelination (i.e. myelin is formed, but in an aberrant pattern) and hypomyelination (i.e. inability to form adequate amounts of myelin) in both PLP and MBP mutant mice [50,52]. Notably however, these abnormalities were more pronounced in MBP deleted mice [46,50], thus suggesting either MBP holds a more important role in myelination, or that other unknown molecules may compensate for the functional and structural consequences associated with the loss of PLP to consequently reduce the severity of myelin abnormalities [44]. Nonetheless, in conjunction with the trend of increasing myelination throughout life [53] and the requirement of continual myelin turnover to maintain neuronal networks even at older ages [54], it is clear retaining adequate levels of PLP and MBP is essential for both the maintenance and formation of myelin throughout life. Thus, reductions in the amount of these proteins in myelin could account for myelin dysfunction observed in MSA brains.
As suggested by the aforementioned studies, genetic mutations are one way in which the levels of MBP and PLP can be reduced. However, given the sporadic origin of MSA, this may be unlikely in MSA and hence, a more relevant mechanism could be through the disruption of their synthesis and subsequent transport from oligodendrocyte cell body to its myelin. The appropriate synthesis and subsequent transport of myelin constituents is of utmost importance, as different compartments of myelin (e.g. compact, non-compact) have different compositions required for executing their functions [55]. Furthermore, similar to their contrasting functions in myelination, the synthesis and transport pathways of MBP and PLP are also considerably different. That is, MBP mRNA is transcribed in the nucleus of oligodendrocytes and directly transported as mRNA granules towards myelin [56,57]. The protein synthesis of MBP then occurs de novo in myelin to prevent non-specific interaction during its transport to myelin, due to the highly adhesive properties of MBP in protein form [58,59]. In contrast, PLP protein is synthesized in the ER before being packaged into vesicles for transport to the Golgi apparatus [55,60]. From here, PLP indirectly reaches the myelin membrane by associating with a lipid raft domain of the oligodendrocyte membrane [61,62].
PROTEIN-LIPID INTERACTION IN MYELIN
Originally implicated in the transport of apical epithelial cell membrane constituents [61], lipid rafts are involved in the sorting of myelin constituents, as observed with PLP [60]. However, this is with an exception of MBP, as it does not appear to interact with lipid rafts for transport to myelin [55,59,63], although the MBP transcriptional regulator, fyn, is localized in lipid rafts [64,65]. Hence instead of transport, this suggests lipid rafts may indirectly influence the production of MBP by acting upstream of the protein itself. Nonetheless, the transport of PLP to myelin is assisted by lipid rafts being enriched in particular lipids, namely sphingomyelin and cholesterol, as studies have indicated protein-lipid interactions are essential for proper PLP transport. This includes the study by Kramer-Albers and colleagues [66], who used mice with missense mutations in the PLP gene that subsequently interfered with the ability of PLP to interact with cholesterol. Interestingly, this lack of a PLP-cholesterol association in mice consequently impeded their ability to transport PLP to lipid rafts [66]. Thus, since lipid rafts are required for the delivery of PLP to myelin, these findings were thought to disturb the delivery of PLP to myelin, to ultimately prevent the downstream compaction of myelin layers necessary for myelination. Furthermore, in vitro studies have also demonstrated the depletion of cholesterol is capable of abolishing PLP-lipid raft associations, with the inhibition of sphingomyelin synthesis exerting similar consequences [67]. Hence, disruption of these interactions, whether it is through alterations in myelin proteins or disrupted brain lipid homeostasis, could initiate myelin impairment in MSA. Therefore, with regards to the latter, given that members of the ATP-binding cassette (ABC) transporter family regulate brain lipid homeostasis, aberrant function in ABCA members could potentially contribute to myelin impairment and loss in MSA.
ATP-BINDING CASSETTE (ABC) TRANSPORTERS
ABC transporters are a large superfamily of transmembrane proteins involved in the active translocation of various substrates (e.g. lipids, ions, sugars and peptides) across membranes, by binding and utilizing ATP hydrolysis for energy [68]. Thus far, 48 transcriptionally active human ATP transporter genes have been identified, and are divided into the seven subfamilies designated ABCA to ABCG based on their sequence homology, gene structure and transmembrane domain [68,69]. ABCA transporters hold a specialized role in maintaining lipid homeostasis by transporting lipids across cellular membranes [69]. For example in the periphery, ABCA1 mediates cholesterol efflux from peripheral tissues to the lipid acceptor apolipoprotein A1 for subsequent liver metabolism [70], whereas ABCA3 transports phospholipids to lamellar bodies in lungs for the synthesis of pulmonary surfactants [71,72]. Furthermore, as genetic mutations in these ABCA transporters result in disorders such as Tangier's disease and respiratory distress syndrome, respectively, this clearly exemplifies the importance of these transporters for the maintenance of lipid homeostasis [73,74]. These deleterious effects following dysfunctional lipid transport from ABCA gene mutations affects the central nervous system similarly.
ABCA8 IN THE EARLY STAGES OF MSA PATHOGENESIS
These findings suggestive of ABCA transporters being involved in the regulation of lipid homeostasis have recently been extended to the relatively unknown member ABCA8 [75], another putative brain lipid transporter of the ABCA subfamily [76]. Initial evidence suggestive of its role in regulating brain lipid homeostasis arose from its expression in the choroid plexus of adult mice [77] and in the human brain [75], along with its ability to transport the lipophilic substrate, leukotriene C4, across Xenopus laevis oocyte membranes [77]. A recent study by Kim and colleagues [75] has also provided the first in-depth functional study of ABCA8. Here, it was revealed ABCA8 specifically regulates sphingomyelin production in oligodendrocytes. Thus, given the necessity of sphingomyelin for the formation of lipid rafts to properly transport myelin constituents necessary for myelination [61], this suggested ABCA8 was involved in myelination. This was also consistent with the finding that ABCA8 expression correlated with the normal pattern of myelination throughout life [75], whereby it increases from neonatal periods to adulthood [53]. Additionally, the expression of ABCA8 was also highly elevated in the myelin-enriched white matter of the superior frontal lobe, in comparison to its grey matter counterpart, which also contains myelin, albeit to a much lesser degree [75]. Taken together, it was suggested ABCA8 appears to play a role in myelination through the regulation of sphingomyelin homeostasis. Therefore, in conjunction with the finding that ABCA8 expression is increased seven-fold in the brains of MSA patients in comparison to control brains [78], it is possible the dysregulation of lipid homeostasis could contribute to myelin dysfunction in the early stages of MSA. However, it remains to be determined whether ABCA8 is also capable of influencing other key myelin constituents such as MBP and PLP in carrying out this role, and whether their levels are altered by ABCA8 during MSA pathogenesis.
Besides myelination, recent evidence has also alluded to the involvement of ABCA8 in α-synuclein related pathogenic processes. This was suggested through the stimulation of the α-synuclein production upon overexpression of ABCA8 in cultured oligodendrocytes [78], thus suggesting a potential relationship between ABCA8 and α-synuclein production. If there is local aberrant α-synuclein synthesis in MSA oligodendrocytes following increased ABCA8 expression, the increased p25α expression that has been observed early in MSA [43] may stimulate the α-synuclein aggregation into GCIs [42]. Overall, these lines of evidence suggest ABCA8 is upregulated during the earlier stages of MSA, and may contribute to aberrant α-synuclein production and aggregation through dysregulated lipid homeostasis.
ACKNOWLEDGEMENTS
This work was supported by a National Health and Medical Research Council of Australia (NHMRC) project grants (#1022325). GMH is a NHMRC Senior Principal Research Fellow (#630434). Tissues were received from the Sydney Brain Bank at Neuroscience Research Australia and the New South Wales Tissue Resource Centre at the University of Sydney which are supported by the NHMRC, University of New South Wales, Neuroscience Research Australia, Schizophrenia Research Institute and National Institute of Alcohol Abuse and Alcoholism (NIH (NIAAA) R24AA012725).
References
- 1.Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, Wood NW, Colosimo C, Dürr A, Fowler CJ, Kaufmann H, Klockgether T, Lees A, Poewe W, Quinn N, Revesz T, Robertson D, Sandroni P, Seppi K, Vidailhet M. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–676. doi: 10.1212/01.wnl.0000324625.00404.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wenning GK, Tison F, Ben Shlomo Y, Daniel SE, Quinn NP. Multiple system atrophy: a review of 203 pathologically proven cases. Mov Disord. 1997;12:133–147. doi: 10.1002/mds.870120203. [DOI] [PubMed] [Google Scholar]
- 3.Ozawa T, Okuizumi K, Ikeuchi T, Wakabayashi K, Takahashi H, Tsuji S. Analysis of the expression level of alpha-synuclein mRNA using postmortem brain samples from pathologically confirmed cases of multiple system atrophy. Acta Neuropathol. 2001;102:188–190. doi: 10.1007/s004010100367. [DOI] [PubMed] [Google Scholar]
- 4.Wüllner U, Schmitz-Hübsch T, Abele M, Antony G, Bauer P, Eggert K. Features of probable multiple system atrophy patients identified among 4770 patients with parkinsonism enrolled in the multicentre registry of the German Competence Network on Parkinson's disease. J Neural Transm. 2007;114:1161–1165. doi: 10.1007/s00702-007-0746-0. [DOI] [PubMed] [Google Scholar]
- 5.Litvan I, Goetz CG, Jankovic J, Wenning GK, Booth V, Bartko JJ, McKee A, Jellinger K, Lai EC, Brandel JP, Verny M, Chaudhuri KR, Pearce RK, Agid Y. What is the accuracy of the clinical diagnosis of multiple system atrophy? A clinicopathologic study. Arch Neurol. 1997;54:937–944. doi: 10.1001/archneur.1997.00550200007003. [DOI] [PubMed] [Google Scholar]
- 6.Schrag A, Ben-Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross-sectional study. Lancet. 1999;354:1771–1775. doi: 10.1016/s0140-6736(99)04137-9. [DOI] [PubMed] [Google Scholar]
- 7.Konagaya M, Konagaya Y, Sakai M, Matsuoka Y, Hashizume Y. Progressive cerebral atrophy in multiple system atrophy. J Neurol Sci. 2002;195:123–127. doi: 10.1016/s0022-510x(01)00692-x. [DOI] [PubMed] [Google Scholar]
- 8.Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T. The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- 9.Hara K, Momose Y, Tokiguchi S, Shimohata M, Terajima K, Onodera O, Kakita A, Yamada M, Takahashi H, Hirasawa M, Mizuno Y, Ogata K, Goto J, Kanazawa I, Nishizawa M, Tsuji S. Multiplex families with multiple system atrophy. Arch Neurol. 2007;64:545–551. doi: 10.1001/archneur.64.4.545. [DOI] [PubMed] [Google Scholar]
- 10.Soma H, Yabe I, Takei A, Fujiki N, Yanagihara T, Sasaki H. Heredity in multiple system atrophy. J Neurol Sci. 2006;240:107–110. doi: 10.1016/j.jns.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Nee LE, Gomez MR, Dambrosia J, Bale S, Eldridge R, Polinsky RJ. Environmental-occupational risk factors and familial associations in multiple system atrophy: a preliminary investigation. Clin Auton Res. 1991;1:9–13. doi: 10.1007/BF01826052. [DOI] [PubMed] [Google Scholar]
- 12.Wenning GK, Stefanova N, Jellinger KA, Poewe W, Schlossmacher MG. Multiple system atrophy: a primary oligodendrogliopathy. Ann Neurol. 2008;64:239–246. doi: 10.1002/ana.21465. [DOI] [PubMed] [Google Scholar]
- 13.Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome) J Neurol Sci. 1989;94:79–100. doi: 10.1016/0022-510x(89)90219-0. [DOI] [PubMed] [Google Scholar]
- 14.Papp MI, Lantos PL. The distribution of oligodendroglial inclusions in multiple system atrophy and its relevance to clinical symptomatology. Brain. 1994;117:235–243. doi: 10.1093/brain/117.2.235. [DOI] [PubMed] [Google Scholar]
- 15.Su M, Yoshida Y, Hirata Y, Watahiki Y, Nagata K. Primary involvement of the motor area in association with the nigrostriatal pathway in multiple system atrophy: neuropathological and morphometric evaluations. Acta Neuropathol. 2001;101:57–64. doi: 10.1007/s004010000273. [DOI] [PubMed] [Google Scholar]
- 16.Ozawa T, Paviour D, Quinn NP, Josephs KA, Sangha H, Kilford L, Healy DG, Wood NW, Lees AJ, Holton JL, Revesz T. The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations. Brain. 2004;127:2657–2671. doi: 10.1093/brain/awh303. [DOI] [PubMed] [Google Scholar]
- 17.Halliday GM, Holton JL, Revesz T, Dickson DW. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol. 2011;122:187–204. doi: 10.1007/s00401-011-0852-9. [DOI] [PubMed] [Google Scholar]
- 18.Burn DJ, Jaros E. Multiple system atrophy: cellular and molecular pathology. Mol Pathol. 2001;54:419–426. [PMC free article] [PubMed] [Google Scholar]
- 19.Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson's disease and dementia with Lewy bodies. Neurosci Lett. 1998;251:205–208. doi: 10.1016/s0304-3940(98)00504-7. [DOI] [PubMed] [Google Scholar]
- 20.Gai WP, Pountney DL, Power JH, Li QX, Culvenor JG, McLean CA, Jensen PH, Blumbergs PC. alpha-Synuclein fibrils constitute the central core of oligodendroglial inclusion filaments in multiple system atrophy. Exp Neurol. 2003;181:68–78. doi: 10.1016/s0014-4886(03)00004-9. [DOI] [PubMed] [Google Scholar]
- 21.Eliezer D, Kutluay E, Bussell R, Jr, Browne G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 22.Cheng F, Vivacqua G, Yu S. The role of α-synuclein in neurotransmission and synaptic plasticity. J Chem Neuroanat. 2011;42:242–248. doi: 10.1016/j.jchemneu.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 23.Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaugler MN, Genc O, Bobela W, Mohanna S, Ardah MT, El-Agnaf OM, Cantoni M, Bensadoun JC, Schneggenburger R, Knott GW, Aebischer P, Schneider BL. Nigrostriatal overabundance of α-synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta Neuropathol. 2012;123:653–669. doi: 10.1007/s00401-012-0963-y. [DOI] [PubMed] [Google Scholar]
- 25.Kisos H, Pukaß K, Ben-Hur T, Richter-Landsberg C, Sharon R. Increased neuronal α-synuclein pathology associates with its accumulation in oligodendrocytes in mice modeling alpha-synucleinopathies. PLoS One. 2012;7:e46817. doi: 10.1371/journal.pone.0046817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reyes JF, Rey NL, Bousset L, Melki R, Brundin P, Angot E. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia. 2014;62:387–398. doi: 10.1002/glia.22611. [DOI] [PubMed] [Google Scholar]
- 28.Dickson DW, Liu W, Hardy J, Farrer M, Mehta N, Uitti R, Mark M, Zimmerman T, Golbe L, Sage J, Sima A, D'Amato C, Albin R, Gilman S, Yen SH. Widespread alterations of alpha-synuclein in multiple system atrophy. Am J Pathol. 1999;155:1241–1251. doi: 10.1016/s0002-9440(10)65226-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campbell BC, McLean CA, Culvenor JG, Gai WP, Blumbergs PC, Jäkälä P, Beyreuther K, Masters CL, Li QX. The solubility of alpha-synuclein in multiple system atrophy differs from that of dementia with Lewy bodies and Parkinsons disease. J Neurochem. 2001;76:87–96. doi: 10.1046/j.1471-4159.2001.00021.x. [DOI] [PubMed] [Google Scholar]
- 30.Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 31.Dikiy I, Eliezer D. Folding and misfolding of alpha-synuclein on membranes. Biochim Biophys Acta. 2012;1818:1013–1018. doi: 10.1016/j.bbamem.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marques O, Outeiro TF. Alpha-synuclein: from secretion to dysfunction and death. Cell Death Dis. 2012;3:e350. doi: 10.1038/cddis.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hashimoto M, Kawahara K, Bar-On P, Rockenstein E, Crews L, Masliah E. The Role of alpha-synuclein assembly and metabolism in the pathogenesis of Lewy body disease. J Mol Neurosci. 2004;24:343–352. doi: 10.1385/JMN:24:3:343. [DOI] [PubMed] [Google Scholar]
- 36.Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J Neurosci. 2010;30:8083–8095. doi: 10.1523/JNEUROSCI.1091-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27:9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shults CW, Rockenstein E, Crews L, Adame A, Mante M, Larrea G, Hashimoto M, Song D, Iwatsubo T, Tsuboi K, Masliah E. Neurological and neurodegenerative alterations in a transgenic mouse model expressing human alpha-synuclein under oligodendrocyte promoter: implications for multiple system atrophy. J Neurosci. 2005;25:10689–10699. doi: 10.1523/JNEUROSCI.3527-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yazawa I, Giasson BI, Sasaki R, Zhang B, Joyce S, Uryu K, Trojanowski JQ, Lee VM. Mouse model of multiple system atrophy alpha-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron. 2005;45:847–859. doi: 10.1016/j.neuron.2005.01.032. [DOI] [PubMed] [Google Scholar]
- 40.Ubhi K, Rockenstein E, Mante M, Inglis C, Adame A, Patrick C, Whitney K, Masliah E. Neurodegeneration in a transgenic mouse model of multiple system atrophy is associated with altered expression of oligodendroglial-derived neurotrophic factors. J Neurosci. 2010;30:6236–6246. doi: 10.1523/JNEUROSCI.0567-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hlavanda E, Kovács J, Oláh J, Orosz F, Medzihradszky KF, Ovádi J. Brain-specific p25 protein binds to tubulin and microtubules and induces aberrant microtubule assemblies at substoichiometric concentrations. Biochemistry. 2002;41:8657–8664. doi: 10.1021/bi020140g. [DOI] [PubMed] [Google Scholar]
- 42.Lindersson E, Lundvig D, Petersen C, Madsen P, Nyengaard JR, Højrup P, Moos T, Otzen D, Gai WP, Blumbergs PC, Jensen PH. p25alpha Stimulates alpha-synuclein aggregation and is co-localized with aggregated alpha-synuclein in alpha-synucleinopathies. J Biol Chem. 2005;280:5703–5715. doi: 10.1074/jbc.M410409200. [DOI] [PubMed] [Google Scholar]
- 43.Song YJ, Lundvig DM, Huang Y, Gai WP, Blumbergs PC, Højrup P, Otzen D, Halliday GM, Jensen PH. p25alpha relocalizes in oligodendroglia from myelin to cytoplasmic inclusions in multiple system atrophy. Am J Pathol. 2007;171:1291–1303. doi: 10.2353/ajpath.2007.070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aggarwal S, Yurlova L, Simons M. Central nervous system myelin: structure, synthesis and assembly. Trends Cell Biol. 2011;21:585–593. doi: 10.1016/j.tcb.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 45.Morell P, Quarles RH. Myelin formation, structure and biochemistry. In: Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD, editors. Basic neurochemistry: molecular, cellular and medical aspects. 6th ed. Philadelphia, PA: Lippincott-Raven Publishers; 1999. pp. 69–94. [Google Scholar]
- 46.Jahn O, Tenzer S, Werner HB. Myelin proteomics: molecular anatomy of an insulating sheath. Mol Neurobiol. 2009;40:55–72. doi: 10.1007/s12035-009-8071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paz Soldán MM, Pirko I. Biogenesis and significance of central nervous system myelin. Semin Neurol. 2012;32:9–14. doi: 10.1055/s-0032-1306381. [DOI] [PubMed] [Google Scholar]
- 48.Greer JM, Lees MB. Myelin proteolipid protein--the first 50 years. Int J Biochem Cell Biol. 2002;34:211–215. doi: 10.1016/s1357-2725(01)00136-4. [DOI] [PubMed] [Google Scholar]
- 49.Harauz G, Boggs JM. Myelin management by the 18.5-kDa and 21.5-kDa classic myelin basic protein isoforms. J Neurochem. 2013;125:334–361. doi: 10.1111/jnc.12195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duncan ID, Hammang JP, Trapp BD. Abnormal compact myelin in the myelin-deficient rat: absence of proteolipid protein correlates with a defect in the intraperiod line. Proc Natl Acad Sci U S A. 1987;84:6287–6291. doi: 10.1073/pnas.84.17.6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boison D, Stoffel W. Disruption of the compacted myelin sheath of axons of the central nervous system in proteolipid protein-deficient mice. Proc Natl Acad Sci U S A. 1994;91:11709–11713. doi: 10.1073/pnas.91.24.11709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Privat A, Jacque C, Bourre JM, Dupouey P, Baumann N. Absence of the major dense line in myelin of the mutant mouse shiverer. Neurosci Lett. 1979;12:107–112. doi: 10.1016/0304-3940(79)91489-7. [DOI] [PubMed] [Google Scholar]
- 53.Durston S, Hulshoff Pol HE, Casey BJ, Giedd JN, Buitelaar JK, van Engeland H. Anatomical MRI of the developing human brain: what have we learned? J Am Acad Child Adolesc Psychiatry. 2001;40:1012–1020. doi: 10.1097/00004583-200109000-00009. [DOI] [PubMed] [Google Scholar]
- 54.Ando S, Tanaka Y, Toyoda Y, Kon K. Turnover of myelin lipids in aging brain. Neurochem Res. 2003;28:5–13. doi: 10.1023/a:1021635826032. [DOI] [PubMed] [Google Scholar]
- 55.Baron W, Hoekstra D. On the biogenesis of myelin membranes: sorting, trafficking and cell polarity. FEBS Lett. 2010;584:1760–1770. doi: 10.1016/j.febslet.2009.10.085. [DOI] [PubMed] [Google Scholar]
- 56.Barbarese E, Koppel DE, Deutscher MP, Smith CL, Ainger K, Morgan F, Carson JH. Protein translation components are colocalized in granules in oligodendrocytes. J Cell Sci. 1995;108:2781–2790. doi: 10.1242/jcs.108.8.2781. [DOI] [PubMed] [Google Scholar]
- 57.Müller C, Bauer NM, Schäfer I, White R. Making myelin basic protein -from mRNA transport to localized translation. Front Cell Neurosci. 2013;7:169. doi: 10.3389/fncel.2013.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barbarese E, Brumwell C, Kwon S, Cui H, Carson JH. RNA on the road to myelin. J Neurocytol. 1999;28:263–270. doi: 10.1023/a:1007097226688. [DOI] [PubMed] [Google Scholar]
- 59.Masaki T. Polarization and myelination in myelinating glia. ISRN Neurol. 2012;2012:769412. doi: 10.5402/2012/769412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maier O, Hoekstra D, Baron W. Polarity development in oligodendrocytes: sorting and trafficking of myelin components. J Mol Neurosci. 2008;35:35–53. doi: 10.1007/s12031-007-9024-8. [DOI] [PubMed] [Google Scholar]
- 61.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 62.Korade Z, Kenworthy AK. Lipid rafts, cholesterol, and the brain. Neuropharmacology. 2008;55:1265–1273. doi: 10.1016/j.neuropharm.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pasquini JM, Guarna MM, Besio-Moreno MA, Iturregui MT, Oteiza PI, Soto EF. Inhibition of the synthesis of glycosphingolipids affects the translocation of proteolipid protein to the myelin membrane. J Neurosci Res. 1989;22:289–296. doi: 10.1002/jnr.490220309. [DOI] [PubMed] [Google Scholar]
- 64.Umemori H, Kadowaki Y, Hirosawa K, Yoshida Y, Hironaka K, Okano H, Yamamoto T. Stimulation of myelin basic protein gene transcription by Fyn tyrosine kinase for myelination. J Neurosci. 1999;19:1393–1397. doi: 10.1523/JNEUROSCI.19-04-01393.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krämer EM, Koch T, Niehaus A, Trotter J. Oligodendrocytes direct glycosyl phosphatidylinositol-anchored proteins to the myelin sheath in glycosphingolipid-rich complexes. J Biol Chem. 1997;272:8937–8945. doi: 10.1074/jbc.272.14.8937. [DOI] [PubMed] [Google Scholar]
- 66.Krämer-Albers EM, Gehrig-Burger K, Thiele C, Trotter J, Nave KA. Perturbed interactions of mutant proteolipid protein/DM20 with cholesterol and lipid rafts in oligodendroglia: implications for dysmyelination in spastic paraplegia. J Neurosci. 2006;26:11743–11752. doi: 10.1523/JNEUROSCI.3581-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Simons M, Krämer EM, Thiele C, Stoffel W, Trotter J. Assembly of myelin by association of proteolipid protein with cholesterol- and galactosylceramide-rich membrane domains. J Cell Biol. 2000;151:143–154. doi: 10.1083/jcb.151.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res. 2001;42:1007–1017. [PubMed] [Google Scholar]
- 69.Kim WS, Weickert CS, Garner B. Role of ATP-binding cassette transporters in brain lipid transport and neurological disease. J Neurochem. 2008;104:1145–1166. doi: 10.1111/j.1471-4159.2007.05099.x. [DOI] [PubMed] [Google Scholar]
- 70.Kolovou GD, Mikhailidis DP, Anagnostopoulou KK, Daskalopoulou SS, Cokkinos DV. Tangier disease four decades of research: a reflection of the importance of HDL. Curr Med Chem. 2006;13:771–782. doi: 10.2174/092986706776055580. [DOI] [PubMed] [Google Scholar]
- 71.Veldhuizen R, Possmayer F. Phospholipid metabolism in lung surfactant. Subcell Biochem. 2004;37:359–388. doi: 10.1007/978-1-4757-5806-1_11. [DOI] [PubMed] [Google Scholar]
- 72.Kaminski WE, Piehler A, Wenzel JJ. ABC A-subfamily transporters: structure, function and disease. Biochim Biophys Acta. 2006;1762:510–524. doi: 10.1016/j.bbadis.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 73.Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HO, Loubser O, Ouelette BF, Fichter K, Ashbourne-Excoffon KJ, Sensen CW, Scherer S, Mott S, Denis M, Martindale D, Frohlich J, Morgan K, Koop B, Pimstone S, Kastelein JJ, Genest J, Jr, Hayden MR. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet. 1999;22:336–345. doi: 10.1038/11905. [DOI] [PubMed] [Google Scholar]
- 74.Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med. 2004;350:1296–1303. doi: 10.1056/NEJMoa032178. [DOI] [PubMed] [Google Scholar]
- 75.Kim WS, Hsiao JH, Bhatia S, Glaros EN, Don AS, Tsuruoka S, Shannon Weickert C, Halliday GM. ABCA8 stimulates sphingomyelin production in oligodendrocytes. Biochem J. 2013;452:401–410. doi: 10.1042/BJ20121764. [DOI] [PubMed] [Google Scholar]
- 76.Tsuruoka S, Ishibashi K, Yamamoto H, Wakaumi M, Suzuki M, Schwartz GJ, Imai M, Fujimura A. Functional analysis of ABCA8, a new drug transporter. Biochem Biophys Res Commun. 2002;298:41–45. doi: 10.1016/s0006-291x(02)02389-6. [DOI] [PubMed] [Google Scholar]
- 77.Matsumoto N, Kitayama H, Kitada M, Kimura K, Noda M, Ide C. Isolation of a set of genes expressed in the choroid plexus of the mouse using suppression subtractive hybridization. Neuroscience. 2003;117:405–415. doi: 10.1016/s0306-4522(02)00827-8. [DOI] [PubMed] [Google Scholar]
- 78.Bleasel JM, Hsiao JH, Halliday GM, Kim WS. Increased expression of ABCA8 in multiple system atrophy brain is associated with changes in pathogenic proteins. J Parkinsons Dis. 2013;3:331–339. doi: 10.3233/JPD-130203. [DOI] [PubMed] [Google Scholar]