

Abstract
Parkinson's disease is a multifactorial disorder with several genes linked to the familial types of the disease. ATP13A2 is one of those genes and encode for a transmembrane protein localized in lysosomes and late endosomes. Previous studies suggested the roles of this protein in lysosomal functions and cellular ion homeostasis. Here, we set out to investigate the role of ATP13A2 in lysosomal function and in metabolism of α-synuclein, another PD-linked protein whose accumulation is implicated in the pathogenesis. We generated non-sense mutations in both copies of ATP13A2 gene in SH-SY5Y human neuroblastoma cells. We examined lysosomal function of ATP13A2-/- cells by measuring the accumulation of lysosomal substrate proteins, such as p62 and polyubiquitinated proteins, induction of acidic compartments, and degradation of ectopically introduced dextran. None of these measures were altered by ATP13A2 deficiency. The steady-state levels of α-synuclein in cells or secretion of this protein were unaltered either in ATP13A2-/- compared to the normal cells. Therefore, the proposed roles of ATP13A2 in lysosomal functions may not be generalized and may depend on the cellular context. The ATP13A2-/- cells generated in the current study may provide a useful control for studies on the roles of PD genes in lysosomal functions.
Keywords: ATP13A2, PARK9, Lysosome, Parkinson's disease, alpha-synuclein, SH-SY5Y cell
INTRODUCTION
Parkinson's disease (PD) is the second most prevalent neurodegenerative disease, with clinical characteristics of resting tremor, muscle tone rigidity, bradykinesia, and postural instability [1]. However, patients with PD also suffer from wide-range of non-motor symptoms, including psychiatric, autonomic, sensory, and sleep abnormalities [2]. These symptoms become diverse and severe as the disease progresses, indicating broad spectrum pathology in the central nervous system as well as the peripheral nervous system [3,4,5,6]. Pathologically, PD is characterized by the loss of specific neurons, such as dopaminergic neurons in the substantia nigra pars compacta, and the occurrence of proteinacious inclusion bodies known as Lewy bodies and Lewy neurites [7]. These inclusion bodies are composed of numerous proteins and vesicles, among which amyloid fibril aggregates of a neuronal protein α-synuclein are the major constituents [8].
Progress in PD research has been driven by human genetic studies that identified about 20 genes that are associated with PD [9,10]. Among these genes, SNCA, which encodes α-synuclein, has been not only linked to several inherited forms of PD [11] but also identified as the most consistent and strongest genetic risk factor for sporadic PD from genome-wide association studies [12].
Mutations in ATP13A2 have been linked to different diseases including Kufor-Rakeb syndrome, PD, and Neuronal Ceroid Lipofuscinosis (NCL) [13,14,15,16,17,18,19,20,21,22,23,24,25]. ATP13A2 protein is localized in lysosomes and late endosomes [13] and belongs to the P-type ATPase pump. Consistent with the localization and the structural features, the normal function of ATP13A2 has been suggested in mitophagy [26,27,28], autophagic protein degradation [29,30,31], and metal/cation homeostasis [28,31,32,33,34,35,36,37,38,39,40,41].
ATP13A2 knockout mice display neuropathological phenotypes that resemble those of NCL and PD, such as hippocampal accumulation of α-synuclein and lipofuscinosis [29]. Systems biology analysis suggested that ATP13A2 and α-synuclein were integral components in the common interaction network [35].
Several genes linked to PD have been suggested to function in the lysosomal degradation pathway and in formation of α-synuclein pathology [42,43,44]. Here, we generated a human neuroblastoma cell line lacking ATP13A2 and investigated the role of this protein in the general lysosomal function and in α-synuclein metabolism.
MATERIALS AND METHODS
Materials
The following antibodies were used in this study: ATP13A2 polyclonal antibody (Abcam, ab135674, Cambridge, MA; 1:1,000), β-actin monoclonal antibody AC-15 (Sigma-Aldrich, A5441, St. Louis, MO; 1:10,000), p62 monoclonal antibody (BD Transduction Laboratories, c2384-0B, Swampscott, MA; 1:1,000), ubiquitin polyclonal antibodies (Dako, #z0458, Glostrup, Denmark, and Chemicon, Temecula, CA; 1:1,000), α-synuclein monoclonal antibody (BD Biosciences, #610787, San Diego, CA; 1:1,500), α-synuclein monoclonal antibody Ab274 (1:1,500), α-synuclein monoclonal antibody Ab62 (1:1,000), HRP-conjugated goat anti-mouse IgG (H+L) (Bio-Rad Laboratories, 172-1011, Hercules, CA; 1:3,000), and HRP-conjugated goat anti-rabbit IgG (H+L) (Bio-Rad Laboratories; 1:3,000).
Fluorescein-conjugated dextran (10,000 molecular weight; D-1821), TO-PRO-3 iodide (T3605), and LysoTracker Red DND-99 (L-7528) were purchased from Invitrogen (Carlsbad, CA).
Generation of ATP13A2 knockout cell lines
SH-SY5Y cells (ATCC, CRL-2266, Manassas, VA) were transfected with plasmids encoding zinc-finger nuclease and a magnetic reporter (ToolGen, Seoul, Korea) by using electroporation. After incubation for 48 h, cells were trypsinized and mixed with magnetic bead-conjugated antibody against H-2Kk (MACSelect Kk microbeads, Miltenyi Biotech, Germany). The mixture was applied to a MACS LS column (Miltenyi Biotech). A single cell isolated from eluates was maintained until the clonal colony was picked from the culture dish. Nonsense mutations in the ATP13A2 gene were confirmed by DNA sequencing.
Cell culture
SH-SY5Y human neuroblastoma cell lines were subcultured as described previously [43]. Cells were maintained every 2 days at 37℃ in humidified air with 5% CO2 in Dulbecco's modified eagle's medium (DMEM) (HyClone, SH30243.01, Logan, UT) containing 10% fetal bovine serum (HyClone, SH30396.03), 100 units/mL penicillin, and 100 units/mL streptomycin (Gibco, 15140-122, Grand Island, NY). To differentiate SH-SY5Y cells, cells were maintained in the presence of 50 µM all-trans-retinoic acid (Sigma-Aldrich, R2625) for 7 days. For overexpression of human α-synuclein, differentiated SH-SY5Y cells were infected with a recombinant adenoviral vector (serotype Ad5, CMV promoter) containing human α-synuclein cDNA at a multiplicity of infection of 33.3.
Preparation of cell extracts
After washing with ice-cold phosphate-buffered saline (PBS) twice, cells were lysed in extraction buffer (1% Triton X-100 and 1% (v/v) protease inhibitor cocktail (Sigma) in PBS). Cell lysates were incubated on ice for 10 min and centrifuged at 16,000 × g for 10 min. The Triton X-100 insoluble fraction was resuspended in 1× Laemmli sample buffer and sonicated briefly.
Western blotting
Western blotting was performed as previously described [43]. Images were obtained and quantified using a Luminescent Image Analyzer (LAS-3000) and MultiGauge version 3.0 software (Fujifilm, Tokyo, Japan).
Characterization of lysosomal dysfunction
To analyze the accumulation of acidic compartments, SH-SY5Y cells were incubated with 75 nM LysoTracker solution diluted in growth medium. After incubation for 1 h at 37℃ in a CO2 incubator, cells were washed with ice-cold PBS and fixed in a 4% paraformaldehyde (PFA) solution. To analyze the degradation ratio of internalized dextran, cells were incubated with 20 µg/mL of fluorescein isothiocyanate (FITC)-labeled dextran (Invitrogen) for 2 h. After washing with DMEM, cells were incubated with fresh growth medium for 30 min and fixed with a 4% PFA solution. The fluorescence intensity was measured using Olympus FV1000 software. The extent of degradation of internalized dextran-FITC was calculated using the equation (Ftime0-Ftime30)/Ftime0, where Ftime0 and Ftime30 are the integrated fluorescence intensities at 0 and 30 min, respectively.
Enzyme-linked immunosorbent assay (ELISA)
ELISA was performed as previously described [45]. 96-well ELISA plates (Nalgene Nunc International, Rochester, NY) were coated with 1 mg/mL capture antibody (Ab62) in 50 mM carbonate buffer (pH 9.6) at 4℃ overnight. After washing with PBS with 0.05% Tween 20 (PBST), the plate was incubated with SuperBlock T20 (PBS) Blocking Buffer (Thermo Scientific, Rockford, IL) at room temperature (RT) with shaking for 1 h, and washed five times in PBST. Samples and standards were incubated at RT for 2.5 h with shaking. After washing with PBST, 1 µg/mL biotinylated Ab62 in blocking buffer was added to each well. After 1.5 h incubation at RT with shaking, the plates were washed with PBST. Avidin-conjugated peroxidase (ExtrAvidin, Sigma) was incubated for 1 h at RT. After washing with PBST, 100 µL of 3,3',5,5'-tetramethylbenzidine solution (Sigma) was added to each well and plate was incubated for 15 min with shaking. To stop the reaction, 50 µL of 2N H2SO4 was added to each well. The absorbance was measured at 450 nm.
Statistical analysis
Values shown in the figures are means±S.E.M. To analyze the statistical significance, p values were calculated by means of paired, two-tailed Student's t tests by using InStat version 3.05 software (GraphPad Software, San Diego, CA).
RESULTS
To generate a human neuronal cell line deficient in ATP13A2 gene, we designed zinc finger nuclease (ZFN) constructs targeting the exon 2 of the gene (Fig. 1A), and SH-SY5Y cells were transfected with the ZFN vectors. Transfected cells were screened for nonsense mutations in the targeted gene as described in the Materials and Methods. We selected a cell line with nonsense mutations in both copies of ATP13A2 gene (ATP13A2-/-, Fig. 1A). Gene deficiency was confirmed by western blotting showing the lack of protein expression (Fig. 1B).
Fig. 1.
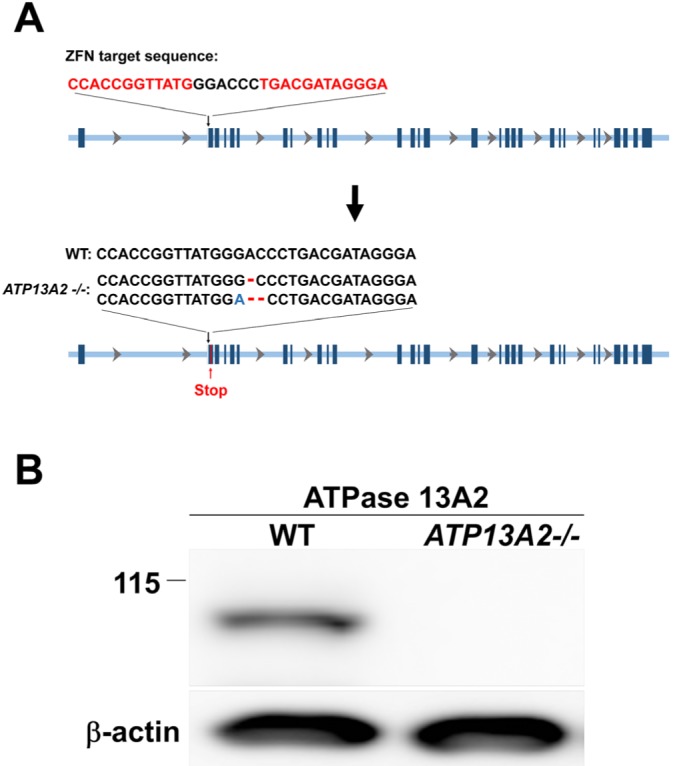
Generation of ATP13A2 deficient cell line by using zinc-finger nucleases. (A) To generate nonsense mutations in two alleles of the ATP13A2 gene, SH-SY5Y human neuroblastoma cells were transfected with zinc-finger nucleases (ZFNs) targeting exon 2. After clonal selection, the nonsense mutations in ATP13A2 exon were confirmed by DNA sequencing. (B) Western blot analysis of ATPase 13A2 in cell lysates. ATPase 13A2 protein was completely depleted in ATP13A2 -/- cell lines.
To examine the role of ATP13A2 in lysosomal functions, we performed the following analyses. First, we measured the steady state levels of lysosomal substrate proteins, p62 and polyubiquitinated proteins. The levels of these proteins were not altered significantly in the knockout cells (Fig. 2A, B). Second, we measured the amounts of cytoplasmic acidic compartments using lysotracker, a protonophilic fluorescent dye. It has been shown that production of acidic compartments were elevated in cells with lysosomal dysfunction, perhaps as a compensatory mechanism [43]. ATP13A2-/- cells contained the similar amounts of lysotracker-positive compartments as the control cells (Fig. 2C). Finally, we measured the rates of dextran degradation. Fluorescence-labeled dextran was fed to cells, and the fluorescence decay rates reflect lysosomal degradation activities. The normal cells and ATP13A2-/- cells showed similar rates of dextran degradation (Fig. 2D). These results, collectively, suggest that in SH-SY5Y neuroblastoma cells, ATP13A2 is not necessary for the normal lysosomal functions.
Fig. 2.
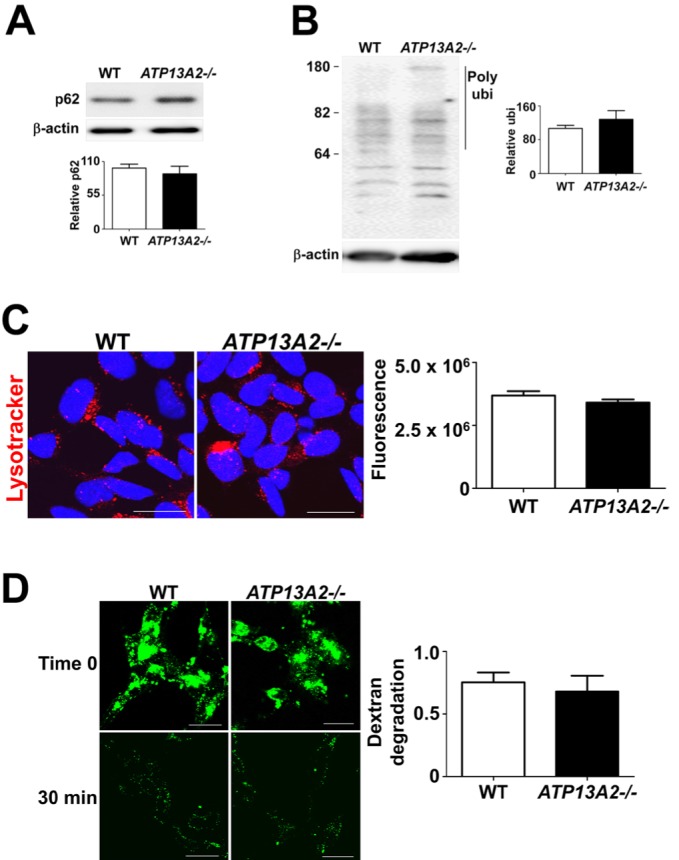
ATP13A2 depletion in SH-SY5Y cell line did not alter the lysosomal function. (A, B) The accumulation of p62 (A) and polyubiquitinated proteins (B) were analyzed in triton X-100 insoluble fraction. For quantification of polyubiquitinated proteins, the quantified size range is indicated by the line to the right of the blot. The ATP13A2 deficiency did not result in the accumulation of p62 and polyubiquitinated proteins in SH-SY5Y cells. n=5. (C) The level of acidic cellular compartment was measured by using LysoTracker. The number of LysoTracker-positive compartments did not increase in ATP13A2 deficient SH-SY5Y cell. n=5, Scale bars: 20 µm. (D) To test lysosomal degradation rate, degradation rate of internalized dextran-fluorescein isothiocyanate (FITC) was analyzed. ATP13A2 deficient SH-SY5Y cells did not show the decrease in the degradation rate. n=3, Scale bars: 20 µm.
Previous studies suggested that ATP13A2 and α-synuclein exert their pathogenic actions in the same pathway [35]. Lysosomal functions were postulated to play a role in the cooperation between these two proteins in PD pathogenesis. To examine the functional link between these proteins, we measured the steady state levels of α-synuclein in the control and ATP13A2-/- cells. The levels of α-synuclein in both Triton-soluble and Triton-insoluble fractions were not altered in ATP13A2 deficient cells (Fig. 3). We also measured the levels of secreted, extracellular α-synuclein, which might be a key player in progression of PD [46]. ELISA showed that the levels of secreted α-synuclein in the culture media were not changed in ATP13A2-/- cells compared with the control cells (Fig. 4).
Fig. 3.

α-synuclein did not accumulate in ATP13A2 deficient SH-SY5Y cells. (A-C) Westernblot analysis of α-synuclein. Human α-synuclein was overexpressed both in the WT and ATP13A2 -/- SH-SY5Y cells. On day 3 after infection, the levels of α-synuclein was measured in triton X-100 soluble (B) and insoluble fraction (C). ATP13A2 depletion in SH-SY5Y cell did not change the level of α-synuclein. n=3.
Fig. 4.

ATP13A2 depletion did not increase the secretion of α-synuclein aggregates. (A) To test the alteration of α-synuclein aggregate secretion, α-synuclein conditioned media was obtained from α-synuclein overexpressing WT and ATP13A2 -/- SH-SY5Y cells. On day 2 after infection with adenovirus, cells were incubated with serum free media for 18 h, after washing with DMEM three times. After incubation for 18 h at 37℃, the media was collected from cells. The level of secreted α-synuclein aggregates was analyzed by using α-synuclein aggregate specific ELISA. The level of secreted α-synuclein aggregates was not altered in ATP13A2 deficient SH-SY5Y cells. n=4. The level of α-synuclein aggregates was normalized with the level of secretogranin II (SGII) in culture media (B).
DISCUSSION
A large number of previous studies suggested that ATP13A2 deficiency caused lysosomal dysfunction and α-synuclein accumulation in both in vitro and in vivo models [29,30,33,34]. The relationship between ATP13A2 and α-synuclein seems to be conserved throughout the evolutionary stages from yeast through nematode to mammals [35]. The lack of effect of ATP13A2 deficiency on lysosomal functions and α-synuclein metabolism presented in the current study is therefore unexpected and could be interpreted in various ways. One possibility is that ATP13A2 is not critical for lysosomal functions at least in SH-SY5Y neuroblastoma cells. This does not exclude the possibility that ATP13A2 may play critical functions in lysosomes in different cell types. Another possibility is that the function of ATP13A2 is redundant. ATP13A2 is a member of P5-type ATPase pump (ATP13A1-5). Although functions of these family members have not been determined, it is possible that at least some of the functions are redundant. Therefore, expression of other P5-type ATPases may compensate for the lack of ATP13A2. A recent study concluded that lowering the levels of ATP13A2 did not alter Mn+ sensitivity in SH-SY5Y cells [33].
Although many previous studies showed the role of ATP13A2 in lysosomal functions and metal homeostasis, our study suggests that functional manifestation of ATP13A2 deficiency varies depending on the cell types. SH-SY5Y cells are commonly used in the field of neurological diseases, particularly frequently among PD researchers for their catecholaminergic phenotypes. Based on the results of the current study, we conclude that SH-SY5Y cells are not a suitable model system to study the loss-of-function effects of ATP13A2 on lysosomal functions, however, can be useful as a negative control to study lysosomal dysfunction caused by genetic modifications. We do not exclude the possibility that SH-SY5Y/ATP13A2-/- cells have phenotypes that are not related to lysosomal functions, such as ion homeostasis. Further characterization of the cells should clarify the issue.
ACKNOWLEDGMENTS
This work was supported by a National Research Foundation grant funded by the Korean Government (2010-0015188) and the Basic Science Research Program, Ministry of Education, Republic of Korea (2013R1A1A2062861).
References
- 1.Fahn S. Description of Parkinson's disease as a clinical syndrome. Ann N Y Acad Sci. 2003;991:1–14. doi: 10.1111/j.1749-6632.2003.tb07458.x. [DOI] [PubMed] [Google Scholar]
- 2.Chaudhuri KR, Healy DG, Schapira AH National Institute for Clinical Excellence. Non-motor symptoms of Parkinson's disease: diagnosis and management. Lancet Neurol. 2006;5:235–245. doi: 10.1016/S1474-4422(06)70373-8. [DOI] [PubMed] [Google Scholar]
- 3.Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 4.Lee SJ, Desplats P, Sigurdson C, Tsigelny I, Masliah E. Cell-to-cell transmission of non-prion protein aggregates. Nat Rev Neurol. 2010;6:702–706. doi: 10.1038/nrneurol.2010.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hyun M, Lee SJ. Topographical propagation of α-synuclein pathology in Parkinson's disease: phenomenology and hypothetical mechanism. Exp Neurobiol. 2009;18:19–25. [Google Scholar]
- 6.Lee HJ, Suk JE, Lee KW, Park SH, Blumbergs PC, Gai WP, Lee SJ. Transmission of synucleinopathies in the enteric nervous system of A53T alpha-synuclein transgenic mice. Exp Neurobiol. 2011;20:181–188. doi: 10.5607/en.2011.20.4.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forno LS. Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol. 1996;55:259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klein C, Westenberger A. Genetics of Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a008888. doi: 10.1101/cshperspect.a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klein C, Schlossmacher MG. The genetics of Parkinson disease: implications for neurological care. Nat Clin Pract Neurol. 2006;2:136–146. doi: 10.1038/ncpneuro0126. [DOI] [PubMed] [Google Scholar]
- 11.Thomas B, Beal MF. Parkinson's disease. Hum Mol Genet. 2007;2:R183–R194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- 12.Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat. 2010;31:763–780. doi: 10.1002/humu.21277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 14.Di Fonzo A, Chien HF, Socal M, Giraudo S, Tassorelli C, Iliceto G, Fabbrini G, Marconi R, Fincati E, Abbruzzese G, Marini P, Squitieri F, Horstink MW, Montagna P, Libera AD, Stocchi F, Goldwurm S, Ferreira JJ, Meco G, Martignoni E, Lopiano L, Jardim LB, Oostra BA, Barbosa ER;, Bonifati V Italian Parkinson Genetics Network, Bonifati V. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology. 2007;68:1557–1562. doi: 10.1212/01.wnl.0000260963.08711.08. [DOI] [PubMed] [Google Scholar]
- 15.Ning YP, Kanai K, Tomiyama H, Li Y, Funayama M, Yoshino H, Sato S, Asahina M, Kuwabara S, Takeda A, Hattori T, Mizuno Y, Hattori N. PARK9-linked parkinsonism in eastern Asia: mutation detection in ATP13A2 and clinical phenotype. Neurology. 2008;70:1491–1493. doi: 10.1212/01.wnl.0000310427.72236.68. [DOI] [PubMed] [Google Scholar]
- 16.Santoro L, Breedveld GJ, Manganelli F, Iodice R, Pisciotta C, Nolano M, Punzo F, Quarantelli M, Pappatà S, Di Fonzo A, Oostra BA, Bonifati V. Novel ATP13A2 (PARK9) homozygous mutation in a family with marked phenotype variability. Neurogenetics. 2011;12:33–39. doi: 10.1007/s10048-010-0259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin CH, Tan EK, Chen ML, Tan LC, Lim HQ, Chen GS, Wu RM. Novel ATP13A2 variant associated with Parkinson disease in Taiwan and Singapore. Neurology. 2008;71:1727–1732. doi: 10.1212/01.wnl.0000335167.72412.68. [DOI] [PubMed] [Google Scholar]
- 18.Paisàn-Ruiz C, Guevara R, Federoff M, Hanagasi H, Sina F, Elahi E, Schneider SA, Schwingenschuh P, Bajaj N, Emre M, Singleton AB, Hardy J, Bhatia KP, Brandner S, Lees AJ, Houlden H. Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord. 2010;25:1791–1800. doi: 10.1002/mds.23221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fong CY, Rolfs A, Schwarzbraun T, Klein C, O'Callaghan FJ. Juvenile parkinsonism associated with heterozygous frameshift ATP13A2 gene mutation. Eur J Paediatr Neurol. 2011;15:271–275. doi: 10.1016/j.ejpn.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 20.Malakouti-Nejad M, Shahidi GA, Rohani M, Shojaee SM, Hashemi M, Klotzle B, Fan JB, Elahi E. Identification of p.Gln858* in ATP13A2 in two EOPD patients and presentation of their clinical features. Neurosci Lett. 2014;577:106–111. doi: 10.1016/j.neulet.2014.06.023. [DOI] [PubMed] [Google Scholar]
- 21.Djarmati A, Hagenah J, Reetz K, Winkler S, Behrens MI, Pawlack H, Lohmann K, Ramirez A, Tadić V, Brügemann N, Berg D, Siebner HR, Lang AE, Pramstaller PP, Binkofski F, Kostić VS, Volkmann J, Gasser T, Klein C. ATP13A2 variants in early-onset Parkinson's disease patients and controls. Mov Disord. 2009;24:2104–2111. doi: 10.1002/mds.22728. [DOI] [PubMed] [Google Scholar]
- 22.Eiberg H, Hansen L, Korbo L, Nielsen IM, Svenstrup K, Bech S, Pinborg LH, Friberg L, Hjermind LE, Olsen OR, Nielsen JE. Novel mutation in ATP13A2 widens the spectrum of Kufor-Rakeb syndrome (PARK9) Clin Genet. 2012;82:256–263. doi: 10.1111/j.1399-0004.2011.01745.x. [DOI] [PubMed] [Google Scholar]
- 23.Bras J, Verloes A, Schneider SA, Mole SE, Guerreiro RJ. Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis. Hum Mol Genet. 2012;21:2646–2650. doi: 10.1093/hmg/dds089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farias FH, Zeng R, Johnson GS, Wininger FA, Taylor JF, Schnabel RD, McKay SD, Sanders DN, Lohi H, Seppälä EH, Wade CM, Lindblad-Toh K, O'Brien DP, Katz ML. A truncating mutation in ATP13A2 is responsible for adult-onset neuronal ceroid lipofuscinosis in Tibetan terriers. Neurobiol Dis. 2011;42:468–474. doi: 10.1016/j.nbd.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 25.Wöhlke A, Philipp U, Bock P, Beineke A, Lichtner P, Meitinger T, Distl O. A one base pair deletion in the canine ATP13A2 gene causes exon skipping and late-onset neuronal ceroid lipofuscinosis in the Tibetan terrier. PLoS Genet. 2011;7:e1002304. doi: 10.1371/journal.pgen.1002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grünewald A, Arns B, Seibler P, Rakovic A, Münchau A, Ramirez A, Sue CM, Klein C. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol Aging. 2012;33:1843.e1–1843.e7. doi: 10.1016/j.neurobiolaging.2011.12.035. [DOI] [PubMed] [Google Scholar]
- 27.Gusdon AM, Zhu J, Van Houten B, Chu CT. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol Dis. 2012;45:962–972. doi: 10.1016/j.nbd.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park JS, Koentjoro B, Veivers D, Mackay-Sim A, Sue CM. Parkinson's disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum Mol Genet. 2014;23:2802–2815. doi: 10.1093/hmg/ddt623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schultheis PJ, Fleming SM, Clippinger AK, Lewis J, Tsunemi T, Giasson B, Dickson DW, Mazzulli JR, Bardgett ME, Haik KL, Ekhator O, Chava AK, Howard J, Gannon M, Hoffman E, Chen Y, Prasad V, Linn SC, Tamargo RJ, Westbroek W, Sidransky E, Krainc D, Shull GE. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and age-dependent sensorimotor deficits. Hum Mol Genet. 2013;22:2067–2082. doi: 10.1093/hmg/ddt057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci. 2012;32:4240–4246. doi: 10.1523/JNEUROSCI.5575-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A. 2012;109:9611–9616. doi: 10.1073/pnas.1112368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramonet D, Podhajska A, Stafa K, Sonnay S, Trancikova A, Tsika E, Pletnikova O, Troncoso JC, Glauser L, Moore DJ. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum Mol Genet. 2012;21:1725–1743. doi: 10.1093/hmg/ddr606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kong SM, Chan BK, Park JS, Hill KJ, Aitken JB, Cottle L, Farghaian H, Cole AR, Lay PA, Sue CM, Cooper AA. Parkinson's disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes alpha-Synuclein externalization via exosomes. Hum Mol Genet. 2014;23:2816–2833. doi: 10.1093/hmg/ddu099. [DOI] [PubMed] [Google Scholar]
- 34.Tsunemi T, Krainc D. Zn(2)(+) dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum Mol Genet. 2014;23:2791–2801. doi: 10.1093/hmg/ddt572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, Caldwell KA, Caldwell GA, Cooper AA, Rochet JC, Lindquist S. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. 2009;41:308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lambie EJ, Tieu PJ, Lebedeva N, Church DL, Conradt B. CATP-6, a C. elegans ortholog of ATP13A2 PARK9, positively regulates GEM-1, an SLC16A transporter. PLoS One. 2013;8:e77202. doi: 10.1371/journal.pone.0077202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chesi A, Kilaru A, Fang X, Cooper AA, Gitler AD. The role of the Parkinson's disease gene PARK9 in essential cellular pathways and the manganese homeostasis network in yeast. PLoS One. 2012;7:e34178. doi: 10.1371/journal.pone.0034178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rentschler G, Covolo L, Haddad AA, Lucchini RG, Zoni S, Broberg K. ATP13A2 (PARK9) polymorphisms influence the neurotoxic effects of manganese. Neurotoxicology. 2012;33:697–702. doi: 10.1016/j.neuro.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan J, Zhang T, Jiang L, Chi J, Hu D, Pan Q, Wang D, Zhang Z. Regulation of intracellular manganese homeostasis by Kufor-Rakeb syndrome-associated ATP13A2 protein. J Biol Chem. 2011;286:29654–29662. doi: 10.1074/jbc.M111.233874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt K, Wolfe DM, Stiller B, Pearce DA. Cd2+, Mn2+, Ni2+ and Se2+ toxicity to Saccharomyces cerevisiae lacking YPK9p the orthologue of human ATP13A2. Biochem Biophys Res Commun. 2009;383:198–202. doi: 10.1016/j.bbrc.2009.03.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Covy JP, Waxman EA, Giasson BI. Characterization of cellular protective effects of ATP13A2/PARK9 expression and alterations resulting from pathogenic mutants. J Neurosci Res. 2012;90:2306–2316. doi: 10.1002/jnr.23112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hyun CH, Yoon CY, Lee HJ, Lee SJ. LRRK2 as a Potential Genetic Modifier of Synucleinopathies: Interlacing the Two Major Genetic Factors of Parkinson's Disease. Exp Neurobiol. 2013;22:249–257. doi: 10.5607/en.2013.22.4.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bae EJ, Yang NY, Song M, Lee CS, Lee JS, Jung BC, Lee HJ, Kim S, Masliah E, Sardi SP, Lee SJ. Glucocerebrosidase depletion enhances cell-to-cell transmission of α-synuclein. Nat Commun. 2014;5:4755. doi: 10.1038/ncomms5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang NY, Lee YN, Lee HJ, Kim YS, Lee SJ. Glucocerebrosidase, a new player changing the old rules in Lewy body diseases. Biol Chem. 2013;394:807–818. doi: 10.1515/hsz-2012-0322. [DOI] [PubMed] [Google Scholar]
- 45.Lee HJ, Bae EJ, Jang A, Ho DH, Cho ED, Suk JE, Yun YM, Lee SJ. Enzyme-linked immunosorbent assays for alpha-synuclein with species and multimeric state specificities. J Neurosci Methods. 2011;199:249–257. doi: 10.1016/j.jneumeth.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 46.Lee HJ, Bae EJ, Lee SJ. Extracellular alpha--synuclein-a novel and crucial factor in Lewy body diseases. Nat Rev Neurol. 2014;10:92–98. doi: 10.1038/nrneurol.2013.275. [DOI] [PubMed] [Google Scholar]