

Abstract
Mitochondrial Ca2+ is known to regulate diverse cellular functions, for example energy production and cell death, by modulating mitochondrial dehydrogenases, inducing production of reactive oxygen species, and opening mitochondrial permeability transition pores. In addition to the action of Ca2+ within mitochondria, Ca2+ released from mitochondria is also important in a variety of cellular functions. In the last 5 years, the molecules responsible for mitochondrial Ca2+ dynamics have been identified: a mitochondrial Ca2+ uniporter (MCU), a mitochondrial Na+–Ca2+ exchanger (NCLX), and a candidate for a mitochondrial H+–Ca2+ exchanger (Letm1). In this review, we focus on the mitochondrial Ca2+ release system, and discuss its physiological and pathophysiological significance. Accumulating evidence suggests that the mitochondrial Ca2+ release system is not only crucial in maintaining mitochondrial Ca2+ homeostasis but also participates in the Ca2+ crosstalk between mitochondria and the plasma membrane and between mitochondria and the endoplasmic/sarcoplasmic reticulum.
Keywords: Mitochondria, Ca2+ dynamics, NCLX, Letm1, Cellular function
Introduction
Mitochondria are crucial organelles in ATP production as well as in Ca2+ storage. They also serve as master switches determining cell fate on exposure to different stimuli [1–3]. Mechanisms of Ca2+ homeostasis in mitochondria have been extensively studied over the last half century, so the importance of mitochondrial Ca2+ in regulating mitochondrial functions is well recognized. Ca2+ enters mitochondria mainly via a mitochondrial Ca2+ uniporter, a protein known as MCU or CCDC109A [4, 5]. The characteristics and physiological and pathophysiological functions of this protein, and its associated proteins have been widely studied [6–8]. On the other hand, studies of the molecules responsible for Ca2+ release by mitochondria have just begun, although functional characterization of the release system started in the 1970s [9]. The mitochondrial Ca2+ release system mainly consists of an Na+–Ca2+ exchanger and an H+–Ca2+ exchanger. The molecule responsible for the former (NCLX) was identified in 2010 [10]. A possible molecular candidate for the latter (Letm1) was reported in 2009 [11], although this is still controversial.
It is now well understood that some mitochondria are in close contact with the plasma membrane and others with the endoplasmic reticulum (ER)/sarcoplasmic reticulum (SR). Although the molecular mechanisms of tethering of mitochondria to the plasma membrane are not well understood, several tethering protein complexes involved in interactions between mitochondria and the ER/SR have been identified, and details of the molecular mechanisms have been reviewed [12, 13]. It is believed that these interactions are important in modulating a variety of cellular functions.
In this paper we review recent progress in the study of mitochondrial Ca2+ release system, specifically, interactions between mitochondria and the ER/SR and interactions between mitochondria and the plasma membrane, and discuss their physiological and pathophysiological significance, focusing on the destiny of Ca2+ released by mitochondria.
Physiological roles of Ca2+ in mitochondria and released by mitochondria
Mitochondrial Ca2+ regulates diverse cellular functions
It is now well accepted that mitochondrial Ca2+ is important in regulation of diverse cellular functions (Fig. 1). For example, an increase of mitochondrial Ca2+ activates three dehydrogenases in the mitochondrial matrix: pyruvate dehydrogenase, oxoglutarate dehydrogenase, and isocitrate dehydrogenase. As a result, the mitochondrial NADH-to-NAD ratio increases and, hence, flow of electrons down the respiratory chain increases, adjusting ATP synthesis to the increased ATP needs of a cell [14–16]. Mitochondrial Ca2+-mediated NADH oxidase activation may result in increased production of mitochondrial reactive oxygen species (ROS). In fact, an increase of mitochondrial Ca2+ has been reported to increase production of ROS in the heart and in neurons, resulting in impaired respiration and in cytotoxicity [17, 18]. Several studies have indicated that Ca2+ activates F1-Fo ATP synthase, balancing ATP utilization increased by Ca2+, and that it is probably the intra-mitochondrial Ca2+ which regulates F1-Fo ATP synthase, although this is still controversial [19, 20]. A large increase of mitochondrial Ca2+ results in opening of a non-specific pore called the mitochondrial permeability transition pore (PTP). PTP opening is followed by inner mitochondrial membrane depolarization, uncoupling of oxidative phosphorylation, and massive mitochondrial swelling, resulting in necrosis [1, 21–23]. PTP is also involved in apoptosis. Apoptosis-promoting factors, for example cytochrome c and pro-caspases, are released to the cytosol through PTP [24–26]. Furthermore, cardiac mitochondrial nitric oxide (NO) synthase is activated by mitochondrial Ca2+, contributing to NO-mediated cardioprotection against PTP opening [27]. Accordingly, mitochondrial Ca2+ is crucial in tuning a variety of mitochondrial and, thus, cellular functions.
Fig. 1.
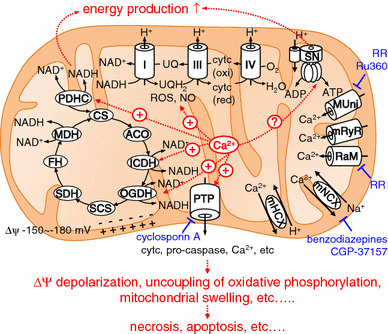
Regulation of mitochondrial functions by mitochondrial Ca2+. Mitochondrial Ca2+ activates three dehydrogenases in the mitochondrial matrix: pyruvate dehydrogenase (PDHC), oxoglutarate dehydrogenase (OGDH), and isocitrate dehydrogenase (ICDH). ROS production is stimulated by increased mitochondrial Ca2+, possibly via increased NADH production. F1-Fo ATP synthase is activated by mitochondrial Ca2+, although this is still controversial. This regulatory activity contributes to energy homeostasis. PTP opening is activated by a large increase of mitochondrial Ca2+, resulting in the release of a variety of compounds from mitochondria, for example cytochrome c (cytc), pro-caspase, and Ca2+, leading to apoptosis or necrosis. CS citrate synthase, ACO aconitase, SCS succinyl-CoA synthase, SDH succinate dehydrogenase, FH fumarate hydratase, MDH malate dehydrogenase, SN F1-Fo ATP synthase, RR ruthenium red, MUni mitochondrial Ca2+ uniporter, mRyR mitochondrial RyR, mNCX mitochondrial Na+–Ca2+ exchanger, mHCX mitochondrial H+–Ca2+ exchanger
What is the source of the mitochondrial Ca2+? The location of the mitochondria within the cell seems important for mitochondrial Ca2+ uptake. Some mitochondria are located in proximity to the plasma membrane or the ER/SR. Ca2+ flowing through the plasma membrane or released from the ER/SR predominantly enters neighbouring mitochondria. In other words, there is Ca2+ crosstalk between mitochondria and the plasma membrane and between mitochondria and the ER/SR (Fig. 2). A substantial amount of the Ca2+ flowing through the plasma membrane or released by the ER/SR binds to cytosolic soluble Ca2+-binding proteins, for example calmodulin, troponin C, and calbindin. Of these, calmodulin is a ubiquitous protein well conserved across eukaryotes and Ca2+-bound calmodulin regulates a variety of cellular functions by binding to calmodulin-dependent kinase, calcineurin, plasma membrane Ca2+ ATPase, and the ryanodine receptor, among others [28]. Further information about these Ca2+-buffering proteins is available in a detailed review [29].
Fig. 2.
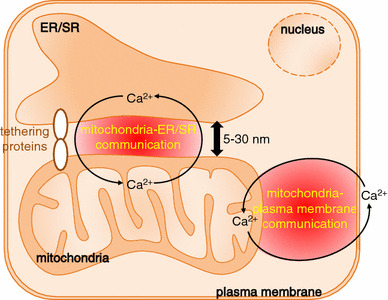
Ca2+ communication between mitochondria and the plasma membrane and between mitochondria and the ER/SR
Ca2+ crosstalk between mitochondria and the plasma membrane and between mitochondria and the ER/SR
Ca2+ enters cells via Ca2+ channels in the plasma membrane, for example the voltage-gated Ca2+ channel (VDCC) and the Ca2+ release-activated Ca2+ channel (CRAC) or store-operated Ca2+ channel. The Ca2+ which enters the cell is efficiently taken up by mitochondria located near the plasma membrane [30–33]. Lawrie et al. [30] found that after depletion of stored Ca2+, re-addition of extracellular Ca2+ evoked an increase in mitochondrial Ca2+ but not in cytoplasmic Ca2+ in human umbilical vein endothelial cell line ECV304, in which 14 % of mitochondria are located within 700 nm of the inner surface of the plasma membrane. They suggested that Ca2+ levels increase in microdomains beneath the plasma membrane, causing predominant Ca2+ uptake by mitochondria facing the microdomains. However, the contribution of the Ca2+ microdomains depends on cell type, because this phenomenon was not observed for a clone of a HeLa cell line, in which <6 % of mitochondria are located in the proximity of the plasma membrane. Park et al. [32] reported that store-operated Ca2+ influx via CRAC channels through the basolateral membrane led to predominant Ca2+ uptake by sub-plasmalemmal mitochondria in pancreatic acinar cells. These reports suggest Ca2+ communication, through plasma membrane Ca2+ channels, with mitochondria located in proximity to plasma membrane. Ca2+ communication in the opposite direction has also been suggested. Mitochondrial membrane depolarization suppressed store-operated Ca2+ entry in T lymphocytes and in rat basophilic leukaemia cells [34, 35]. Accordingly, bidirectional Ca2+ crosstalk between mitochondria and the plasma membrane is important in the regulation of cellular functions. As will be described below, the mitochondrial Na+-Ca2+ exchanger NCLX is involved in this Ca2+ crosstalk.
The ER/SR is major Ca2+ store within the cells. The ER/SR is located close to mitochondria, approximately 5–30 nm, and narrow interorganellar spaces exist between the organelles in a variety of cell types, including B lymphocytes and cardiomyocytes [36]. Figure 3 shows 3D-reconstructed confocal images of mitochondria and the ER/SR of cultured B lymphocytes and cardiomyocytes. The two organelles are located close to each other, although narrow interorganellar spaces are not visible. The close contact between mitochondria and the ER/SR is supported by tethering proteins, for example MIRO, DRP1, MFN2, and Mmm1/Mdm10/Mdm12/Mdm34 complex [12, 13]. Mitochondria–ER/SR communication is important for regulation of a variety of cellular processes, including lipid biosynthesis, mitochondrial division, and Ca2+ signalling [12, 37]. With regard to Ca2+, these narrow interorganellar spaces may serve to create Ca2+ level much higher than in the bulk cytosol, up to 9–50 μM [38, 39], enabling the mitochondrial Ca2+ uniporter, the Ca2+ affinity of which is relatively low, to transport enough Ca2+ into mitochondria. Several studies reported that an inositol 1,4,5-trisphosphate (IP3) receptor of the ER and a voltage-dependent anion channel of the outer mitochondrial membrane are located in the mitochondria–ER region, creating a Ca2+ pathway from the ER to the mitochondria. This may aid regulation of energy metabolism and apoptosis in such cells as HeLa cells, CHO cells, fibroblasts, and yeast [40–42]. Ca2+ released from the ryanodine receptor (RyR) on the SR also accumulates in mitochondria of rat ventricular myocytes, indicating preferential coupling of Ca2+ transport from the SR to mitochondria [38]. It has been suggested that reverse Ca2+ movement, from mitochondria to the ER/SR, is important for refilling ER/SR Ca2+ after ligand stimulation of such cells as HeLa cells, endothelial cell line, and vascular smooth muscle cells, to minimize cytosolic Ca2+ elevation and to prevent depletion of ER/SR Ca2+ [43–45]. The crucial importance of NCLX in the movement of Ca2+ from mitochondria to the ER/SR was clearly demonstrated in our recent studies [46–48], as described below. The bidirectional Ca2+ crosstalk between mitochondria and the ER/SR is also important for regulating cellular functions.
Fig. 3.
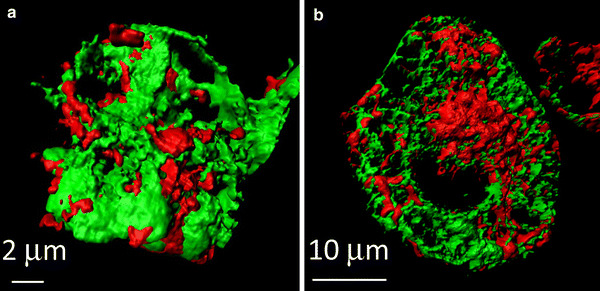
Close location of mitochondria and the ER/SR in A20 B lymphocytes (a) and in HL-1 cardomyocytes (b). Cells were co-transfected with mitochondria-targeted pTagRFP-mito (red) and ER/SR-targeted cameleon D1ER (green). Images were acquired by use of a laser-scanning confocal microscope (LSM710, Carl Zeiss) with a ×63 oil objective lens, and 3D images were reconstructed by use of Imaris (Bitplane) (color figure online)
Taken together, Ca2+ crosstalk between mitochondria and the plasma membrane and between mitochondria and the ER/SR is important in controlling cellular functions. Understanding the roles of each molecule participating in the Ca2+ crosstalk is particularly important for fully understanding cellular physiological and pathophysiological functions.
Ca2+-transporting systems in mitochondria
Studies of mitochondrial Ca2+ dynamics started more than half a century ago. The existence of a respiration-dependent pathway of Ca2+ into isolated rat kidney mitochondria was reported in the early 1960s [49, 50]. In the 1970s, pathways of Ca2+ out of isolated rat heart mitochondria (Na+-dependent) and out of isolated rat liver mitochondria (Na+-independent and H+-dependent) were discovered [9, 51]. Since then, characteristics of mitochondrial Ca2+ dynamics have been extensively studied [1, 52–54].
The major pathways of Ca2+ uptake into and efflux out of mitochondria are summarized in Fig. 1. Ca2+ uptake into mitochondria is mainly mediated by a mitochondrial Ca2+ uniporter driven by a highly negative membrane potential (mitochondrial membrane potential ΔΨ is −150 to −180 mV) [55]. Another uptake system is a rapid uptake mode (RaM) which might contribute to mitochondrial Ca2+ uptake from fast cytosolic Ca2+ transients. A mitochondrial ryanodine receptor is also reported to mediate Ca2+ uptake into rat heart mitochondria. Details are available in other reviews [1, 52]. It was not until the 2010s that the identities of the molecules responsible for the mitochondrial Ca2+ uniport were revealed. A regulator of the Ca2+ uniporter, MICU, was discovered in 2010 before cloning of the mitochondrial Ca2+ uniporter [56]. MCU (or CCDC109A) was then discovered as a gene coding the mitochondrial Ca2+ uniporter [4, 5]. Very recently, characteristics of MCU knockout have been reported. Although MCU knockout in Trypanosoma brucei resulted in marked dysregulation of mitochondrial bioenergetics, causing autophagy and cell death [57], relatively minor alteration of basal energetics was observed for MCU knockout mice [58]. MCU and its regulators have been reviewed in detail elsewhere [7, 8].
Ca2+ efflux from mitochondria is mainly mediated by two saturable pathways, an Na+-dependent (Na+–Ca2+ exchanger; benzodiazepines and CGP-37157-sensitive) pathway and an Na+-independent (H+–Ca2+ exchanger; ruthenium red-insensitive) pathway. Under pathophysiological conditions in which the PTP opens, PTP functions as a Ca2+ transporter from mitochondria. In the sections below we describe, in detail, the two physiological pathways of mitochondrial Ca2+ release.
Mitochondrial Na+–Ca2+ exchanger
Biophysical properties of the mitochondrial Na+–Ca2+ exchanger
The mitochondrial Na+–Ca2+ exchanger was first discovered by Carafoli et al. [9] in 1974 in isolated rat heart mitochondria. This Na+–Ca2+ exchange activity is found in a wide variety of tissues and is dominant in the heart, brain, skeletal muscle, parotid gland, adrenal cortex, and brown fat [52, 59]. The Na+–Ca2+ exchanger is also present in liver, kidney, and lung mitochondria, although its activity is weak [60]. In tissues in which mitochondrial Na+–Ca2+ exchange activity is low, H+–Ca2+ exchange activity is of dominant importance in the release of Ca2+ from mitochondria [61].
One interesting characteristic of the mitochondrial Na+–Ca2+ exchanger, which is distinct from the plasmalemmal Na+–Ca2+ exchanger (NCX), is that Li+ can substitute for Na+ [9]. This unique characteristic contributed to identification of NCLX, a gene responsible for the mitochondrial Na+–Ca2+ exchanger, as will be described in the next section. The stoichiometry (ion-exchange ratio) and the electrogenicity of the mitochondrial Na+–Ca2+ exchanger were controversial, but it was believed to be electroneutral [62, 63]. Our group clearly demonstrated, by use of permeabilized rat ventricular myocytes, that the mitochondrial Na+–Ca2+ exchanger is voltage-dependent and electrogenic, which suggests the stoichiometry is >3Na+ for one Ca2+ [64]. We also predicted by computer simulation that the voltage dependence of the mitochondrial Na+–Ca2+ exchanger changes, the affinity becoming lower with mitochondrial membrane depolarization [64]. Because of these features, the mitochondrial Na+–Ca2+ exchanger dynamically changes the exchange mode (forward or reverse) and modulates the mitochondrial Ca2+ concentration in a manner dependent on cytosolic Na+ concentration and mitochondrial membrane potential. Recently, several molecules which may modulate the mitochondrial Na+–Ca2+ exchanger have been identified. Gandhi et al. [18] reported that a deficiency of a PINK1, a 581-amino-acid protein consisting of a mitochondrial targeting motif and a serine/threonine kinase domain homologous with that of the Ca2+/calmodulin family, causes impairment of mitochondrial Ca2+ efflux via the mitochondrial Na+–Ca2+ exchanger. Mutations in the PINK1 gene are known to cause autosomal recessive Parkinson’s disease [18]. Da Cruz et al. [65] showed that a stomatin-like protein 2 (SLP-2), a novel member of the stomatin superfamily found in several types of human tumour, negatively modulates mitochondrial Na+–Ca2+ exchange activity in HeLa cells, regulating the capacity of mitochondria to store Ca2+. Although detailed mechanisms underlying the regulation of mitochondrial Na+–Ca2+ exchange activity by these proteins have not yet been clarified, recent identification of NCLX as a mitochondrial Na+–Ca2+ exchanger will surely accelerate understanding of the mechanisms.
Cloning, tissue distribution, and cellular localization of NCLX
In 2004, two independent research groups reported the cloning of a new transporter mediating Na+–Ca2+ exchange, which was subsequently identified as a mitochondrial Na+–Ca2+ exchanger [66, 67]. Cai and Lytton employed a bioinformatics-based search of the GenBank™ database using a conserved amino acid sequence of the α-repeat regions of the K+-dependent Na+–Ca2+ exchanger (NCKX) gene family 2 (NCKX2) [66]. The amino acid sequence of the identified clone was divergent from that of NCX and NCKX family members, but was slightly closer to that of NCKX. The clone was therefore named “NCKX6”. They also found an alternative spliced isoform of mouse NCKX6. Although the long isoform was retained in the ER fraction and was not functional when heterologously expressed in HEK293 cells, the short isoform was targeted in the plasma membrane and had K+-dependent Na+–Ca2+ exchange activity. Very soon after the publication by Cai and Lytton [66], Sekler’s group found the same clone during a search for the Na+–Zn2+ exchanger gene [67]. In contrast with the report by Cai and Lytton [66], both long and short clones had K+-independent Na+–Ca2+ exchange activity and did not have Na+–Zn2+ exchange activity when heterologously expressed in HEK293 cells. They also found that Li+ can substitute for Na+ to release Ca2+ from the cells, so they named the clone “NCLX”. The discrepant results obtained by the two groups might be a result of different experimental conditions, i.e. Cai and Lytton [66] used Li+ as substituent for Na+ to examine K+-dependent Na+–Ca2+ exchange activity. This might have affected the responsiveness of the clone. In addition, FLAG epitope might have interfered with the sorting system in the Cai and Lytton [66] experiments. Anyway, both groups found that NCKX6/NCLX had Na+–Ca2+ exchange activity and broad tissue distribution, for example heart, pancreas, skeletal muscle, stomach, spleen, and brain.
It took another 6 years, however, for Sekler’s group to discover that NCKX/NCLX is the long-sought mitochondrial Na+–Ca2+exchanger. In 2010, Sekler’s group beautifully demonstrated that NCLX was located in mitochondria and its characteristics corresponded well to those of the mitochondrial Na+–Ca2+ exchanger [10]. Direct measurements of mitochondrial Ca2+ by use of the mitochondria-targeted Ca2+ sensing protein Pericam-mito [68] clearly showed that Ca2+ efflux from mitochondria was accelerated by overexpressing NCLX in an Na+ and Li+-dependent manner and was decelerated by knocking down NCLX. The fact that Li+ can substitute for Na+ in releasing Ca2+ from mitochondria corresponds well to the unique characteristic of mitochondrial Na+–Ca2+ exchange. This NCLX-mediated Ca2+ efflux from mitochondria was inhibited by the mitochondrial Na+–Ca2+ exchange inhibitor CGP-37157. It was also shown that mitochondrial Na+, measured by use of the mitochondrial Na+-sensing dye CoroNa Red, increased during the mitochondrial Ca2+ efflux phase, and the rate became much faster with NCLX overexpression. These results confirm that NCLX is the gene responsible for the mitochondrial Na+–Ca2+ exchanger. Since then, information about the involvement of NCLX in the physiological and pathophysiological functions of different types of cell has accumulated. We will describe, in detail, recent findings on the roles of NCLX in pancreatic β-cells, astrocytes, B lymphocytes, and cardiomyocytes. Experimental results from NCLX knockout/knockdown in each type of cell are summarized in Table 1.
Table 1.
Effects of NCLX reduction on cellular functions
| Mitochondrial Ca2+ at rest | Mitochondrial Ca2+ transient | Cytosolic Ca2+ transient | SOCE | ATP production | Others | |
|---|---|---|---|---|---|---|
| Pancreatic β-cells ([70, 71]) | Increased | Faster rise slower decline | Smaller | N.D. | Unchanged at steady state. Slightly accelerated during stimulation | Reduced insulin secretion |
| Astrocytes ([77]) | Increased | Faster rise slower decline | Smaller | Smaller and slower | N.D. |
Reduced glutamate release Impaired wound healing Impaired proliferation |
| B lymphocytes ([44, 45, 84]) | N.D. | N.D. | Diminished cytosolic Ca2+ increase after BCR stimulation | Smaller and slower | N.D. |
Increased apoptosis Reduced chemotaxis |
| Cardiomyocytes ([46, 102]) | Increased | N.D. | Slower upstroke | N.D. | Unaffected | Cycle length prolongation |
N.D. not determined
NCLX in pancreatic β-cells (Fig. 4a)
Fig. 4.
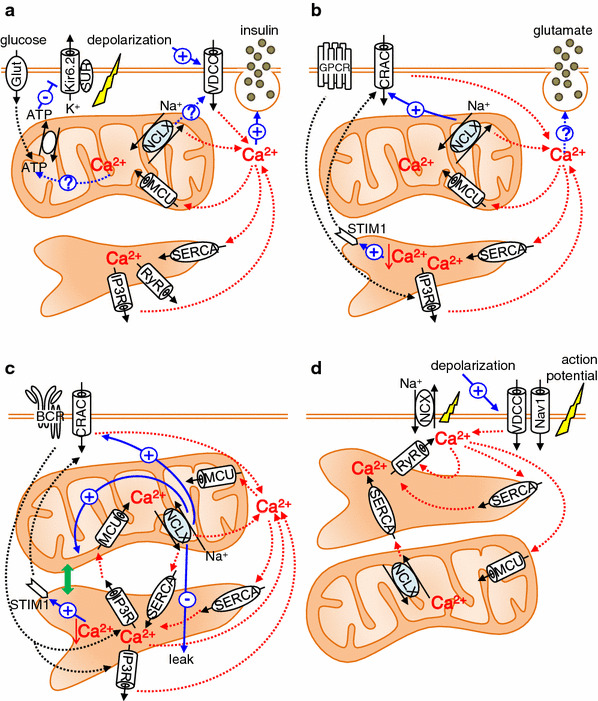
Roles of NCLX in pancreatic β-cells (a), astrocytes (b), B lymphocytes (c), and cardiomyocytes (d). See text for details
When the plasma glucose level increases, ATP production in pancreatic β-cells increases. ATP-dependent K+ channels consequently close, resulting in depolarization of the plasma membrane. Subsequently, voltage-gated Ca2+ channels open, causing Ca2+ influx into the cell, and exocytosis of insulin granules occurs [69]. In addition to the metabolic roles of mitochondria in the regulation of insulin secretion [70, 71], Tarasov et al. and Nita et al. [72, 73] demonstrated the contribution of NCLX to the function of pancreatic β-cells by use of the pancreatic β cell line MIN6 and mouse pancreatic primary β-cells. By use of pericam-mito to directly sense the mitochondrial Ca2+, Nita et al. [73] found that membrane depolarization-mediated Ca2+ influx, ATP-mediated ER Ca2+ depletion, and treatment with 20 mM glucose all caused a mitochondrial Ca2+ transient. In all cases, silencing of NCLX by use of siRNA or dominant negative mutant resulted in a more rapid increase in mitochondrial Ca2+ and slower mitochondrial Ca2+ decline. The resting mitochondrial Ca2+ level was higher in NCLX knockdown cells. These results not only suggest that NCLX functions as a mitochondrial Na+–Ca2+ exchanger, but also show that Ca2+ efflux via NCLX is already activated at the early phase of mitochondrial Ca2+ influx. Interestingly, silencing NCLX caused a smaller and slower cytosolic Ca2+ increase induced by membrane depolarization or by treatment with 20 mM glucose. Furthermore, NCLX knockdown was followed by a delay in glucose-dependent insulin secretion, whereas ATP production was little affected. These results indicate that NCLX is a critical component of the Ca2+ crosstalk between mitochondria and the plasma membrane in modulation of the insulin secretion pattern. Tarasov et al. obtained results essentially similar to those of Nita et al. [73] in repetitively depolarized MIN6 cells with regard to NCLX function. In addition, Tarasov et al. [72] found that glucose-induced mitochondrial Ca2+ response and subsequent ATP/ADP increase was impaired under the glucolipotoxic conditions often observed with type 2 diabetes. Because mRNA expression levels of MCU and NCLX were unaltered, it was suggested that changes in the intracellular distribution of mitochondria [74] were involved in this altered Ca2+ dysfunction and ATP production. In 2013, Proverbio et al. [75] used a multi-step screening strategy to identify genes related to congenital hyperinsulinism, characterized by severe hypoglycaemia as a result of inappropriate insulin secretion by pancreatic β-cells. They found a mutation in the NCLX-encoding SLC24A6 gene, which results in amino acid change at position 564 from tyrosine to histidine. Because this position resides in the middle of putative transmembrane domain 12 and is conserved among a variety of species, including chimpanzee, mouse, rat, cattle, chicken, and zebrafish, it is possible that the mutation causes a functional change of NCLX. Further analysis is required to clarify how NCLX is involved in the pathophysiological condition of pancreatic β-cells.
NCLX in astrocytes (Fig. 4b)
Brain is another tissue highly expressing NCLX. Among the different kinds of cell constituting the brain, astrocytes constitute approximately half the volume. They express a large number of G protein-coupled receptors (GPCRs), linked to a diverse array of intracellular cascades including elevation of intracellular Ca2+. It is also well known that astrocytes release neurotransmitters called gliotransmitters, for example glutamate, ATP and d-serine, which bind to neuronal receptors to modulate synaptic transmission and activity [76, 77]. Thus astrocytes not only interact with neuronal activity but also modulate this activity via gliotransmitters. Although still controversial, several reports suggest that release of gliotransmitters by astrocytes depends on elevation of cytosolic Ca2+ [77, 78]. The increase of cytosolic Ca2+ in astrocytes occurs via the phospholipase C (PLC)/IP3 pathway. That is, upon GPCR activation, PLC hydrolyses the membrane lipid phosphatidylinositol 4,5-bisphosphate to generate diacylglycerol and IP3, activating the IP3 receptor (IP3R) and releasing Ca2+ from the ER. Parnis et al. [79], by use of a mouse astrocyte culture, examined how NCLX is involved in Ca2+ dynamics and the release of gliotransmitters. They used pericam-mito to sense mitochondrial Ca2+ and found that the resting mitochondrial Ca2+ level increased, and that the rise and decline of mitochondrial Ca2+ transients caused by extracellular ATP application became faster and slower, respectively, on knocking down NCLX. These results were very similar to those observed for pancreatic β-cells [73]. Interestingly, NCLX knockdown reduced Ca2+ entry into the cytosol both from extracellular space and from the ER, but the effect on the former was stronger. Detailed analysis revealed that NCLX knockdown impairs store-operated Ca2+ entry (SOCE), indicative of strong Ca2+ crosstalk between mitochondria and the plasma membrane, as in pancreatic β-cells. NCLX knockdown also significantly reduced such processes as exocytotic glutamate release, in vitro wound closure, and proliferation, which may be regulated in a Ca2+-dependent manner [79].
NCLX in B lymphocytes (Fig. 4c)
Activation of B lymphocytes by antigens is followed by an increase in cytosolic Ca2+, resulting in rapid proliferation and differentiation or apoptosis, depending on the differentiation stage of the cells [80]. Upon binding of the antigen to the B cell surface receptor (BCR), IP3 increases and facilitates Ca2+ release from the IP3R on the ER membrane. Ca2+ release from the ER causes the initial cytosolic Ca2+ increase after receptor activation. Subsequent depletion of Ca2+ in the ER causes translocation of stromal interaction molecule 1 (STIM1) to the vicinity of plasmalemma, inducing a sustained and oscillatory cytosolic Ca2+ increase by activation of SOCE through CRAC channels encoded by ORAI1 [81, 82]. We discovered that NCLX is crucial in this antigen receptor -mediated Ca2+ signalling from a combined study of mathematical simulations and NCLX knockout/knockdown in DT40 and A20 B lymphocytes [46, 47]. NCLX reduction greatly reduced cytosolic Na+-dependent mitochondrial Ca2+ efflux in saponin-permeabilized cells loaded with the mitochondrial Ca2+-sensitive dye Rhod-2, confirming that NCLX is responsible for mitochondrial Na+–Ca2+ exchange in B lymphocytes. Interestingly, mitochondrial membrane potential, evaluated by JC-1 staining, was more depolarized and DNA fragmentation (sub G1) was increased in NCLX knockout cells, suggesting that the progress of apoptosis was accelerated in NCLX knockout cells. Mathematical model revealed that NCLX inhibition reduces basal ER Ca2+ content and suppresses BCR-mediated cytosolic Ca2+ rise. These predictions were validated by experiments. i.e., ER Ca2+ content decreased and the cytosolic increase of Ca2+ was diminished in NCLX knockout/knockdown cells after the BCR activation. Comparable with the results obtained by use of astrocytes, SOCE activity in B lymphocytes was also diminished by silencing NCLX, probably further contributing to impairment of the cytosolic Ca2+ increase after BCR activation [47]. The reduction in ER Ca2+ content was a result of impaired Ca2+ supply from mitochondria via NCLX, because ER Ca2+ uptake via the ER Ca2+ pump SERCA was decelerated by NCLX reduction when mitochondrial respiration was intact (Fig. 5a) whereas it was comparable when mitochondrial respiration was disturbed [47]. Interesting findings were that NCLX reduction resulted in impaired co-localization of mitochondria with ER and augmented ER Ca2+ leak. Although the cause remains unresolved, NCLX may be associated with the tethering proteins connecting ER and mitochondria [83, 84], and reduction of NCLX may weaken mitochondria–ER interactions. Alternatively, mitochondrial depolarization induced by NCLX knockout/knockdown might result in elimination of the impaired mitochondria by mitophagy [85]. Taken together, our results not only show that NCLX is involved in the crosstalk between mitochondria and the plasma membrane, as revealed by SOCE regulation, but also indicate that NCLX-mediated crosstalk between mitochondria and ER Ca2+ dynamics is crucial for physiological functioning of B lymphocytes; i.e. a response to BCR activation. Very recently we found that silencing NCLX reduced the chemotaxis of B lymphocytes triggered by chemokine CXCL12, although the mechanism is still unclear [86]. Although further analysis is needed, it is likely that NCLX is important in regulation of immune systems.
Fig. 5.
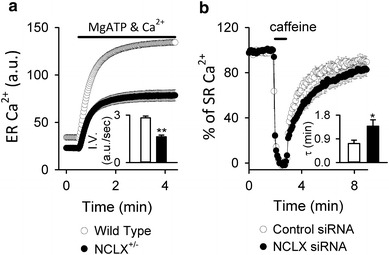
Modulation of SERCA activity by NCLX reduction in (a) DT40 B lymphocytes and (b) HL-1 cardiomyocytes. a ER Ca2+ uptake by wild type and NCLX+/− DT40 cells. ER Ca2+ uptake were activated by applying 0.1 mM MgATP and 100 nM Ca2+ in Mag Fluo-4-loaded and permeabilized cells under conditions in which mitochondrial respiration was intact. The bar graph depicts the initial velocity of the ER Ca2+ increase. Data are mean ± SEM of independent recordings. Modified from Kim et al. [44]. b SR Ca2+ reuptake by HL-1 cardiomyocytes. Plasmid harbouring Cameleon D1ER, an indicator of SR Ca2+, was co-transfected with control (white) or NCLX siRNA (black) into HL-1 cardiomyoyctes. After emptying Ca2+ in SR with 10 mM caffeine, recovery of SR Ca2+ was measured. The bar graph depicts the recovery time constant τ, showing that SR Ca2+ reuptake was slower in NCLX knockdown cells. **p < 0.01, *p < 0.05. Modified from Takeuchi et al. [46]
NCLX in cardiomyocytes (Fig. 4d)
The heart is one of the most studied organs for investigation of the characteristics of the mitochondrial Na+–Ca2+ exchanger, which serves as a major Ca2+ release system [87, 88]. The roles of the mitochondrial Na+–Ca2+ exchanger in cardiac energetics have been widely investigated. For example, activation of the mitochondrial Na+–Ca2+ exchanger by increasing cytosolic Na+ causes a decrease of mitochondrial Ca2+, resulting in an imbalance in energy demand and supply or in an increase of ROS production [89, 90]. In contrast, the contribution of the mitochondrial Na+–Ca2+ exchanger to cytosolic Ca2+ transients and to action potential generation has been regarded as negligible, because the contribution of mitochondrial Ca2+ uptake to Ca2+ release from cardiomyocytes is as low as 1–2 % [91]. However, we recently discovered that NCLX participates in modulation of action potential configuration and in regulation of the automaticity of HL-1, a spontaneously beating cardiac cell line originating from mouse atrial myocytes [48]. The expression patterns of ion channels and transporters in HL-1 cells are similar to those in adult atrial myocytes, except that HL-1 cells highly express the T-type Ca2+ channel (I CaT) and the hyperpolarization-activated cation channel (I ha), which are known to be involved in the automaticity of cardiac pacemaker sinoatrial (SA) node cells [92–95]. NCLX reduction using siRNA reduced NCLX protein expression by ~50 %, resulting in a ~50 % reduction of the rate of cytosolic Na+-dependent mitochondrial Ca2+ efflux, confirming that NCLX is responsible for mitochondrial Na+–Ca2+ exchange in HL-1 cells. Mitochondrial Ca2+ content, evaluated as the Ca2+ chelation-responsive fraction of the intensity of mitochondrial Ca2+ sensor pCase12-mito, was larger in NCLX knockdown cells. Although beat-to-beat change of mitochondrial Ca2+ was not observed in HL-1 cells, the result indicated that NCLX contributes to the steady state mitochondrial Ca2+ content in intact HL-1 cells. Cellular energetics seemed to be unaffected by NCLX knockdown, because there were no differences in cellular ATP content, mitochondrial membrane potential, or mitochondrial ROS between control cells and NCLX knockdown cells. This may be because protein expression of NCLX knockdown using siRNA was reduced by 50 % only.
An interesting finding was that NCLX knockdown caused marked prolongation of the cycle length of spontaneous action potentials and Ca2+ transients [48]. Kinetic analysis of electrophysiological data and Ca2+ transients obtained by line scanning cells loaded with cytosolic Ca2+ indicator Fluo-4 revealed that NCLX knockdown slowed the upstrokes of both action potentials and cytosolic Ca2+ transients. SR Ca2+ dynamics, which is known to contribute to the upstroke of cytosolic Ca2+ transients, was evaluated by use of ER/SR Ca2+ FRET protein cameleon D1ER [96]. As a result, SR Ca2+ content of NCLX knockdown cells was smaller and SR Ca2+ reuptake rate was slower (Fig. 5b), suggesting that the NCLX reduction-mediated prolongation of cycle length is related to compromised SR Ca2+ dynamics. The mechanisms underlying the NCLX reduction-mediated prolongation of cycle length was further studied with a newly constructed mathematical model of HL-1 cells [48]. The HL-1 cell model well reproduces the spontaneous generation of action potentials and Ca2+ transients, and the prolongation of the cycle length induced by knocking down NCLX. The model analysis indicated that automaticity of HL-1 cells is determined by spontaneous Ca2+ leak from the SR. Simulation of NCLX reduction showed that Ca2+ supply from the mitochondria to the SR decreased to slow down the rate of spontaneous Ca2+ leak from the SR. The timing of Ca2+-induced Ca2+ release (CICR), activation of the inward current of the plasma membrane NCX (I NCX), and thus the timing of activation of voltage-dependent Na+ current (I Na) and voltage-dependent T and L type Ca2+ channels (I CaT and I CaL) was thus delayed, resulting in prolongation of the cycle length. Taken together, our combined experiments and simulations indicated that NCLX regulates the rhythmicity of HL-1 cells via crosstalk between mitochondria and SR Ca2+ dynamics. Considering that NCLX reduction resulted in modification of plasma membrane NCX activity, NCLX may also be indirectly involved in mitochondria–plasma membrane Ca2+ crosstalk in HL-1 cells. Interestingly, Opuni and Reeves reported the functional coupling of mitochondrial function, possibly mitochondrial Na+–Ca2+ exchange activity, and plasma membrane NCX activity in Chinese hamster ovary cells stably transfected with bovine cardiac NCX [97]. Further analysis is necessary to elucidate the interaction of mitochondrial NCLX and plasma membrane NCX. Because HL-1 cells are derived from atrial myocytes, which have no automaticity under physiological conditions, NCLX may be involved in the abnormal automaticity of atria, for example atrial flutter or atrial ectopic tachycardia. It may also be possible that occurrence of arrhythmia in patients with mitochondrial disease [98, 99] is caused by abnormal NCLX function. Further analysis is required to clarify the involvement of NCLX in these arrhythmias.
Whether NCLX participates in the automaticity of normal pacemaker cells, sinoatrial (SA) node cells, is a big issue. Recently, Yaniv et al. [100] reported that mitochondrial Na+–Ca2+ exchange inhibitor CGP-37157 slowed the generation of automaticity of rabbit SA node cells, suggesting that mitochondrial Na+–Ca2+ exchange is involved in generation of the automaticity of SA node cells. However, because CGP-37157 also blocks I CaL, which is related to generation of the automaticity of SA node cells, a nonspecific effect of CGP-37157 on I CaL cannot be ignored. In addition, the automaticity of the SA node cells they used significantly depends on spontaneous and local subsarcolemmal Ca2+ releases from SR, the so called “Ca2+ clock” mechanism [101]. However, this automaticity mechanism has been controversial. It has been accepted for a long time that I ha and a variety of other inward membrane currents determine the automaticity [102–104]. This mechanism is called the “membrane clock”. Whether NCLX contributes to the automaticity of all types of SA node cells, including the cells driven by “membrane clock”, must be studied.
We examined the contribution of NCLX using mathematical models of SA node cells [105]. In the original SA node model developed by Yaniv et al. [100], complete reduction of mitochondrial Na+–Ca2+ exchange resulted in prolongation of cycle length by 2.7 % only. The larger effect reported by Yaniv et al. [100] is probably because of their simultaneous reduction of the amplitude factor of SERCA. To further test the contribution of NCLX to generation of the automaticity of SA node cell models, we incorporated our model of mitochondrial Ca2+ dynamics into two representative SA node cell models: a membrane clock model by Himeno et al. [102, 103] and a Ca2+ clock model by Maltsev and Lakatta [101]. In both models, NCLX reduction reduced the SR Ca2+ content, supporting the idea of Ca2+ communication between mitochondria and the SR in SA node cells. However, the effect on the automaticity was different between the models. The cycle length was prolonged in the Maltsev and Lakatta model whereas it was shortened in the Himeno model [105]. Furthermore, model analysis revealed that cytosolic Na+ and Na+-permeable inward current (sustained inward current I st) [106] are crucial factors distinguishing the effect of NCLX on pace-making in the two models. In the Himeno model, NCLX reduction reduced cytosolic Ca2+, which decreased the inward I NCX thus reducing the cytosolic Na+ concentration. This increased the amplitude of the inward I st and overcame the decrease of inward I NCX, accelerating the firing rate. In contrast, the amplitude of I st is set smaller in the Maltsev and Lakatta model so that NCLX reduction only reduces inward I NCX and slows diastolic depolarization, decelerating the firing rate [105]. Accordingly, it is likely that the Ca2+ communication between mitochondria and the SR via NCLX functions also in SA node cells. However, it is necessary to investigate quantitatively how much the ‘‘Ca2+ clock’’ mechanism, cytoplasmic Na+, and I st contribute to SA node automaticity.
Mitochondrial H+–Ca2+ exchanger
H+–Ca2+ exchange has a dominant effect on release of Ca2+ from mitochondria in tissues in which mitochondrial Na+–Ca2+ exchange activity is low, for example the liver, kidney, lung, and smooth muscle [61]. The H+–Ca2+ exchanger also occurs in the heart, though the activity is weak [61, 107].
The stoichiometry of the H+–Ca2+ exchanger is regarded as 2H+ for 1Ca2+, being electroneutral. However, because the rate of efflux via the H+–Ca2+ exchanger decreases with increasing pH gradient in rat isolated liver mitochondria, it is suggested that the mechanism is not a passive Ca2+ for 2H+ exchanger, but an active Ca2+ for 2H+ exchanger [108].
The molecular identity of the mitochondrial H+–Ca2+ exchanger is still controversial. The candidate is the Letm1 (leucine–zipper–EF hand-containing transmembrane region). Jiang et al. [11] conducted high-throughput RNA interference screening of Drosophila genes and identified a gene affecting mitochondrial Ca2+ and H+, the human homolog of which is Letm1. By using digitonin-permeabilized S2 or 293 cells expressing mitochondrial Ca2+ sensor protein pericam, or by using purified Letm1 reconstituted in liposomes, they found that Letm1 mediates H+–Ca2+ exchange. However, because a drastic reduction of mitochondrial Ca2+ uptake was observed when Letm1 protein expression was suppressed, and because H+–Ca2+ exchange via Letm1 was sensitive to an inhibitor of mitochondrial Ca2+ influx, ruthenium red, Letm1 is regarded as mediating Ca2+ influx into mitochondria, at least at low cytosolic Ca2+ level. This idea was confirmed by subsequent work by Jiang et al. [109] in which Letm1 knockdown in 293 cells resulted in dramatically reduced mitochondrial Ca2+ content. In the same work they produced Letm1 knockout mice and found that Letm1 homozygous knockout mice were embryonic lethal and so were half of the heterozygous knockout mice. The surviving mice had altered glucose metabolism, impaired control of brain ATP levels, and increased seizure activity, suggesting involvement of Letm1 in the pathology of Wolf–Hirschhorn syndrome, in which Letm1 is known to be one of the genes deleted [110]. Nowikovsky et al. [111] analysed theoretically the direction of Ca2+ flux through the mitochondrial membrane in energized mitochondria with different H+:Ca2+ stoichiometry. They proposed that Ca2+ enters mitochondria with 1H+:1Ca2+ whereas Ca2+ leaves mitochondria with 2H+:1Ca2+ or 3H+:1Ca2+ stoichiometry under physiological respiration conditions. Tsai et al. [112] reported that Letm1 mediates the electroneutral 2H+:1Ca2+ antiport and is insensitive to ruthenium red, by using Letm1 reconstituted proteoliposome. These results combined with the theoretical analysis by Nowikovsky et al. [111] strongly suggest that Letm1 is the long-awaited molecular identity of the mitochondrial H+–Ca2+ exchanger. There is, currently, no reasonable explanation of the discrepancy of the sensitivity of Letm1 to ruthenium red. We independently found that a substantial part of the Ca2+ efflux from mitochondria was independent of cytosolic Na+ when mitochondria were loaded with a lower concentration of Ca2+ than when examining Na+–Ca2+ exchange activity using A20 B lymphocytes [46]. This fraction, which probably represents the H+–Ca2+ exchange system, was sensitive to ruthenium red. Further analysis is necessary to determine whether Letm1 is, indeed, the H+–Ca2+ exchanger mediating Ca2+ extrusion from mitochondria.
Summary
Ca2+, not only inside mitochondria but also released from mitochondria, is crucially important in regulating a variety of cellular physiological functions. Identification of the molecules responsible for the pathways of Ca2+ release enables us to isolate the contributions of these molecules. For example, NCLX has been shown to participate in insulin secretion in pancreatic β-cells, glutamate release in astrocytes, Ca2+ responsiveness to BCR stimulation in B lymphocytes, and generation of the spontaneous rhythmicity of cadiomyocytes, via Ca2+ crosstalk between mitochondria and the plasma membrane and/or between mitochondria and the ER/SR (Fig. 4). However, many issues remain unanswered. One is the question of whether mitochondrial Ca2+ release proteins are indeed located in the tethering spots between mitochondria and the ER/SR or between mitochondria and the plasma membrane. Another is the contribution of the mitochondrial Ca2+ release system to mitochondrial energetics. Distinct phenotypes of NCLX knockdown cells related to mitochondrial energetics in cardiomyocytes and in pancreatic β-cells have not been observed, while Letm1 knockout mice had impaired mitochondrial energetics. Complete knockout of NCLX in mice or cells will clarify the matter.
Acknowledgments
This work was supported by JSPS Kakenhi grant numbers 25136707, 23689011, and 23390042 (A.T. and S.M.).
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 2.Murgia M, Giorgi C, Pinton P, Rizzuto R. Controlling metabolism and cell death: at the heart of mitochondrial calcium signalling. J Mol Cell Cardiol. 2009;46:781–788. doi: 10.1016/j.yjmcc.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Celsi F, Pizzo P, Brini M, Leo S, Fotino C, Pinton P, Rizzuto R. Mitochondria, calcium and cell death: a deadly triad in neurodegeneration. Biochim Biophys Acta. 2009;1787:335–344. doi: 10.1016/j.bbabio.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Niescier RF, Chang KT, Min KT. Miro, MCU, and calcium: bridging our understanding of mitochondrial movement in axons. Front Cell Neurosci. 2013;7:148. doi: 10.3389/fncel.2013.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patron M, Raffaello A, Granatiero V, Tosatto A, Merli G, De Stefani D, Wright L, Pallafacchina G, Terrin A, Mammucari C, Rizzuto R. The mitochondrial calcium uniporter (MCU): molecular identity and physiological roles. J Biol Chem. 2013;288:10750–10758. doi: 10.1074/jbc.R112.420752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marchi S, Pinton P. The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J Physiol. 2014;592:829–839. doi: 10.1113/jphysiol.2013.268235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carafoli E, Tiozzo R, Lugli G, Crovetti F, Kratzing C. The release of calcium from heart mitochondria by sodium. J Mol Cell Cardiol. 1974;6:361–371. doi: 10.1016/0022-2828(74)90077-7. [DOI] [PubMed] [Google Scholar]
- 10.Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A. 2010;107:436–441. doi: 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science. 2009;326:144–147. doi: 10.1126/science.1175145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol. 2012;13:607–625. doi: 10.1038/nrm3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Vliet AR, Verfaillie T, Agostinis P (2014) New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta (in press). doi:10.1016/j.bbamcr.2014.03.009 [DOI] [PubMed]
- 14.McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- 15.Jo H, Noma A, Matsuoka S. Calcium-mediated coupling between mitochondrial substrate dehydrogenation and cardiac workload in single guinea-pig ventricular myocytes. J Mol Cell Cardiol. 2006;40:394–404. doi: 10.1016/j.yjmcc.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 16.Satrústegui J, Pardo B, Del Arco A. Mitochondrial transporters as novel targets for intracellular calcium signalin. Physiol Rev. 2007;87:29–67. doi: 10.1152/physrev.00005.2006. [DOI] [PubMed] [Google Scholar]
- 17.Chacon E, Acosta D. Mitochondrial regulation of superoxide by Ca2+: an alternate mechanism for the cardiotoxicity of doxorubicin. Toxicol Appl Pharmacol. 1991;107:117–128. doi: 10.1016/0041-008x(91)90336-d. [DOI] [PubMed] [Google Scholar]
- 18.Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, Duchen MR, Abramov AY. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009;33:627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris DA. Regulation of the mitochondrial ATP synthase in rat heart. Biochem Soc Trans. 1993;21:778–781. doi: 10.1042/bst0210778. [DOI] [PubMed] [Google Scholar]
- 20.Scholz TD, Balaban RS. Mitochondrial F1-ATPase activity of canine myocardium: effects of hypoxia and stimulation. Am J Physiol. 1994;266:H2396–H2403. doi: 10.1152/ajpheart.1994.266.6.H2396. [DOI] [PubMed] [Google Scholar]
- 21.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- 22.Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly-Dyson E, Di Lisa F, Forte MA. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273:2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- 23.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 24.Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–2933. [PubMed] [Google Scholar]
- 25.Hajnóczky G, Davies E, Madesh M. Calcium signaling and apoptosis. Biochem Biophys Res Commun. 2003;304:445–454. doi: 10.1016/s0006-291x(03)00616-8. [DOI] [PubMed] [Google Scholar]
- 26.Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta. 2009;1787:1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dedkova EN, Blatter LA. Characteristics and function of cardiac mitochondrial nitric oxide synthase. J Physiol. 2009;587:851–872. doi: 10.1113/jphysiol.2008.165423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tidow H, Nissen P. Structural diversity of calmodulin binding to its target sites. FEBS J. 2013;280:5551–5565. doi: 10.1111/febs.12296. [DOI] [PubMed] [Google Scholar]
- 29.Gilabert JA. Cytoplasmic calcium buffering. Adv Exp Med Biol. 2012;740:483–498. doi: 10.1007/978-94-007-2888-2_20. [DOI] [PubMed] [Google Scholar]
- 30.Lawrie AM, Rizzuto R, Pozzan T, Simpson AW. A role for calcium influx in the regulation of mitochondrial calcium in endothelial cells. J Biol Chem. 1996;271:10753–10759. doi: 10.1074/jbc.271.18.10753. [DOI] [PubMed] [Google Scholar]
- 31.Babcock DF, Herrington J, Goodwin PC, Park YB, Hille B. Mitochondrial participation in the intracellular Ca2+ network. J Cell Biol. 1997;136:833–844. doi: 10.1083/jcb.136.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park MK, Ashby MC, Erdemli G, Petersen OH, Tepikin AV. Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J. 2001;20:1863–1874. doi: 10.1093/emboj/20.8.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, Pozzan T. Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol Cell. 2010;38:280–290. doi: 10.1016/j.molcel.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 34.Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J Cell Biol. 1997;137:633–648. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glitsch MD, Bakowski D, Parekh AB. Store-operated Ca2+ entry depends on mitochondrial Ca2+ uptake. EMBO J. 2002;21:6744–6754. doi: 10.1093/emboj/cdf675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Csordás G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnóczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006;174:915–921. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma VK, Ramesh V, Franzini-Armstrong C, Sheu SS. Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J Bioenerg Biomembr. 2000;32:97–104. doi: 10.1023/a:1005520714221. [DOI] [PubMed] [Google Scholar]
- 39.Csordás G, Várnai P, Golenár T, Roy S, Purkins G, Schneider TG, Balla T, Hajnóczky G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell. 2010;39:121–132. doi: 10.1016/j.molcel.2010.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rizzuto R. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- 41.Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, Rodrigues MA, Gomez MV, Nathanson MH, Leite MF. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem. 2005;280:40892–40900. doi: 10.1074/jbc.M506623200. [DOI] [PubMed] [Google Scholar]
- 42.Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arnaudeau S, Kelley WL, Walsh JV, Jr, Demaurex N. Mitochondria recycle Ca2+ to the endoplasmic reticulum and prevent the depletion of neighboring endoplasmic reticulum regions. J Biol Chem. 2001;276:29430–29439. doi: 10.1074/jbc.M103274200. [DOI] [PubMed] [Google Scholar]
- 44.Malli R, Frieden M, Trenker M, Graier WF. The role of mitochondria for Ca2+ refilling of the endoplasmic reticulum. J Biol Chem. 2005;280:12114–12122. doi: 10.1074/jbc.M409353200. [DOI] [PubMed] [Google Scholar]
- 45.Poburko D, Liao CH, van Breemen C, Demaurex N. Mitochondrial regulation of sarcoplasmic reticulum Ca2+ content in vascular smooth muscle cells. Circ Res. 2009;104:104–112. doi: 10.1161/CIRCRESAHA.108.180612. [DOI] [PubMed] [Google Scholar]
- 46.Kim B, Takeuchi A, Koga O, Hikida M, Matsuoka S. Pivotal role of mitochondrial Na+–Ca2+ exchange in antigen receptor mediated Ca2+ signalling in DT40 and A20 B lymphocytes. J Physiol. 2012;590:459–474. doi: 10.1113/jphysiol.2011.222927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim B, Takeuchi A, Koga O, Hikida M, Matsuoka S. Mitochondria Na+–Ca2+ exchange in cardiomyocytes and lymphocytes. Adv Exp Med Biol. 2013;961:193–201. doi: 10.1007/978-1-4614-4756-6_16. [DOI] [PubMed] [Google Scholar]
- 48.Takeuchi A, Kim B, Matsuoka S. The mitochondrial Na+–Ca2+ exchanger, NCLX, regulates automaticity of HL-1 cardiomyocytes. Sci Rep. 2013;3:2766. doi: 10.1038/srep02766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deluca HF, Engstrom GW. Calcium uptake by rat kidney mitochondria. Proc Natl Acad Sci U S A. 1961;47:1744–1750. doi: 10.1073/pnas.47.11.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vasington FD, Murphy JV. Ca++ uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J Biol Chem. 1962;237:2670–2677. [PubMed] [Google Scholar]
- 51.Fiskum G, Lehninger AL. Regulated release of Ca2+ from respiring mitochondria by Ca2+/2H+ antiport. J Biol Chem. 1979;254:6236–6239. [PubMed] [Google Scholar]
- 52.Gunter TE, Yule DI, Gunter KK, Eliseev RA, Salter JD. Calcium and mitochondria. FEBS Lett. 2004;567:96–102. doi: 10.1016/j.febslet.2004.03.071. [DOI] [PubMed] [Google Scholar]
- 53.Carafoli E. The fateful encounter of mitochondria with calcium: how did it happen? Biochim Biophys Acta. 2010;1797:595–606. doi: 10.1016/j.bbabio.2010.03.024. [DOI] [PubMed] [Google Scholar]
- 54.Malli R, Graier WF. Mitochondrial Ca2+ channels: great unknowns with important functions. FEBS Lett. 2010;584:1942–1947. doi: 10.1016/j.febslet.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 56.Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature. 2010;467:291–296. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang G, Vercesi AE, Docampo R. Essential regulation of cell bioenergetics in Trypanosoma brucei by the mitochondrial calcium uniporter. Nat Commun. 2013;4:2865. doi: 10.1038/ncomms3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–1472. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Crompton M, Moser R, Lüdi H, Carafoli E. The interrelations between the transport of sodium and calcium in mitochondria of various mammalian tissues. Eur J Biochem. 1978;82:25–31. doi: 10.1111/j.1432-1033.1978.tb11993.x. [DOI] [PubMed] [Google Scholar]
- 60.Haworth RA, Hunter DR, Berkoff HA. Na+ releases Ca2+ from liver, kidney and lung mitochondria. FEBS Lett. 1980;110:210–218. doi: 10.1016/0014-5793(80)80076-7. [DOI] [PubMed] [Google Scholar]
- 61.Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- 62.Affolter H, Carafoli E. The Ca2+–Na+ antiporter of heart mitochondria operates electroneutrally. Biochem Biophys Res Commun. 1980;95:193–196. doi: 10.1016/0006-291x(80)90723-8. [DOI] [PubMed] [Google Scholar]
- 63.Wingrove DE, Gunter TE. Kinetics of mitochondrial calcium transport. II. A kinetic description of the sodium-dependent calcium efflux mechanism of liver mitochondria and inhibition by ruthenium red and by tetraphenylphosphonium. J Biol Chem. 1986;261:15166–15171. [PubMed] [Google Scholar]
- 64.Kim B, Matsuoka S. Cytoplasmic Na+-dependent modulation of mitochondrial Ca2+ via electrogenic mitochondrial Na+–Ca2+ exchange. J Physiol. 2008;586:1683–1697. doi: 10.1113/jphysiol.2007.148726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Da Cruz S, De Marchi U, Frieden M, Parone PA, Martinou JC, Demaurex N. SLP-2 negatively modulates mitochondrial sodium-calcium exchange. Cell Calcium. 2010;47:11–18. doi: 10.1016/j.ceca.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 66.Cai X, Lytton J. Molecular cloning of a sixth member of the K+-dependent Na+/Ca2+ exchanger gene family, NCKX6. J Biol Chem. 2004;279:5867–5876. doi: 10.1074/jbc.M310908200. [DOI] [PubMed] [Google Scholar]
- 67.Palty R, Ohana E, Hershfinkel M, Volokita M, Elgazar V, Beharier O, Silverman WF, Argaman M, Sekler I. Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J Biol Chem. 2004;279:25234–25240. doi: 10.1074/jbc.M401229200. [DOI] [PubMed] [Google Scholar]
- 68.Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+ . Proc Natl Acad Sci U S A. 2001;98:3197–3202. doi: 10.1073/pnas.051636098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ashcroft FM. ATP-sensitive K+ channels and disease: from molecule to malady. Am J Physiol Endocrinol Metab. 2007;293:E880–E889. doi: 10.1152/ajpendo.00348.2007. [DOI] [PubMed] [Google Scholar]
- 70.Ashcroft SJ, Weerasinghe LC, Randle PJ. Interrelationship of islet metabolism, adenosine triphosphate content and insulin release. Biochem J. 1973;132:223–231. doi: 10.1042/bj1320223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kennedy HJ. Glucose generates sub-plasma membrane ATP microdomains in single islet β-cells. Potential role for strategically located mitochondria. J Biol Chem. 1999;274:13281–13291. doi: 10.1074/jbc.274.19.13281. [DOI] [PubMed] [Google Scholar]
- 72.Tarasov AI, Semplici F, Ravier MA, Bellomo EA, Pullen TJ, Gilon P, Sekler I, Rizzuto R, Rutter GA. The mitochondrial Ca2+ uniporter MCU is essential for glucose-induced ATP increases in pancreatic β-cells. PLoS ONE. 2012;7:e39722. doi: 10.1371/journal.pone.0039722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nita II, Hershfinkel M, Fishman D, Ozeri E, Rutter GA, Sensi SL, Khananshvili D, Lewis EC, Sekler I. The mitochondrial Na+/Ca2+ exchanger upregulates glucose dependent Ca2+ signalling linked to insulin secretion. PLoS ONE. 2012;7:e46649. doi: 10.1371/journal.pone.0046649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Molina AJ, Wikstrom JD, Stiles L, Las G, Mohamed H, Elorza A, Walzer G, Twig G, Katz S, Corkey BE, Shirihai OS. Mitochondrial networking protects β-cells from nutrient-induced apoptosis. Diabetes. 2009;58:2303–2315. doi: 10.2337/db07-1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Proverbio MC, Mangano E, Gessi A, Bordoni R, Spinelli R, Asselta R, Valin PS, Di Candia S, Zamproni I, Diceglie C, Mora S, Caruso-Nicoletti M, Salvatoni A, De Bellis G, Battaglia C. Whole genome SNP genotyping and exome sequencing reveal novel genetic variants and putative causative genes in congenital hyperinsulinism. PLoS ONE. 2013;8:e68740. doi: 10.1371/journal.pone.0068740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Agulhon C, Petravicz J, McMullen AB, Sweger EJ, Minton SK, Taves SR, Casper KB, Fiacco TA, McCarthy KD. What is the role of astrocyte calcium in neurophysiology? Neuron. 2008;59:932–946. doi: 10.1016/j.neuron.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Navarrete M, Araque A. Endocannabinoids mediate neuron-astrocyte communication. Neuron. 2008;57:883–893. doi: 10.1016/j.neuron.2008.01.029. [DOI] [PubMed] [Google Scholar]
- 78.Serrano A, Haddjeri N, Lacaille JC, Robitaille R. GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J Neurosci. 2006;26:5370–5382. doi: 10.1523/JNEUROSCI.5255-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Parnis J, Montana V, Delgado-Martinez I, Matyash V, Parpura V, Kettenmann H, Sekler I, Nolte C. Mitochondrial exchanger NCLX plays a major role in the intracellular Ca2+ signaling, gliotransmission, and proliferation of astrocytes. J Neurosci. 2013;33:7206–7219. doi: 10.1523/JNEUROSCI.5721-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Scharenberg AM, Humphries LA, Rawlings DJ. Calcium signalling and cell-fate choice in B cells. Nat Rev Immunol. 2007;7:778–789. doi: 10.1038/nri2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7:690–702. doi: 10.1038/nri2152. [DOI] [PubMed] [Google Scholar]
- 82.Vig M, Kinet JP. Calcium signaling in immune cells. Nat Immunol. 2009;10:21–27. doi: 10.1038/ni.f.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27:6407–6418. doi: 10.1038/onc.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Csordás G, Hajnóczky G. SR/ER-mitochondrial local communication: calcium and ROS. Biochim Biophys Acta. 2009;1787:1352–1362. doi: 10.1016/j.bbabio.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim B, Takeuchi A, Matsuoka S (2013) Mitochondrial NCX controls directional migration of B lymphocyte. J Physiol Sci 63:S136
- 87.Crompton M, Künzi M, Carafoli E. The calcium-induced and sodium-induced effluxes of calcium from heart mitochondria. Evidence for a sodium-calcium carrier. Eur J Biochem. 1977;79:549–558. doi: 10.1111/j.1432-1033.1977.tb11839.x. [DOI] [PubMed] [Google Scholar]
- 88.Wei AC, Liu T, Cortassa S, Winslow RL, O’Rourke B. Mitochondrial Ca2+ influx and efflux rates in guinea pig cardiac mitochondria: low and high affinity effects of cyclosporine A. Biochim Biophys Acta. 2011;1813:1373–1381. doi: 10.1016/j.bbamcr.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu T, O’Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103:279–288. doi: 10.1161/CIRCRESAHA.108.175919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Bohm M, O’Rourke B, Maack C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–1613. doi: 10.1161/CIRCULATIONAHA.109.914911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bassani RA, Bassani JW, Bers DM. Relaxation in ferret ventricular myocytes: unusual interplay among calcium transport systems. J Physiol. 1994;476:295–308. doi: 10.1113/jphysiol.1994.sp020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Claycomb WC, Lanson NAJ, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJJ. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.White SM, Constantin PE, Claycomb WC. Cardiac physiology at the cellular level: use of cultured HL-1 cardiomyocytes for studies of cardiac muscle cell structure and function. Am J Physiol Heart Circ Physiol. 2004;286:H823–H829. doi: 10.1152/ajpheart.00986.2003. [DOI] [PubMed] [Google Scholar]
- 94.Yang Z, Murray KT. Ionic mechanisms of pacemaker activity in spontaneously contracting atrial HL-1 cells. J Cardiovasc Pharmacol. 2011;57:28–36. doi: 10.1097/FJC.0b013e3181fda7c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Adachi T, Shibata S, Okamoto Y, Sato S, Fujisawa S, Ohba T, Ono K. The mechanism of increased postnatal heart rate and sinoatrial node pacemaker activity in mice. J Physiol Sci. 2013;63:133–146. doi: 10.1007/s12576-012-0248-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Palmer AE, Jin C, Reed JC, Tsien RY. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci U S A. 2004;101:17404–17409. doi: 10.1073/pnas.0408030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Opuni K, Reeves JP. Feedback inhibition of sodium/calcium exchange by mitochondrial calcium accumulation. J Biol Chem. 2000;275:21549–21554. doi: 10.1074/jbc.M003158200. [DOI] [PubMed] [Google Scholar]
- 98.Khatami M, Houshmand M, Sadeghizadeh M, Eftekharzadeh M, Heidari MM, Saber S, Banihashemi K, Scheiber-Mojdehkar B. Accumulation of mitochondrial genome variations in Persian LQTS patients: a possible risk factor? Cardiovasc Pathol. 2010;19:e21–e27. doi: 10.1016/j.carpath.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 99.Koopman WJ, Willems PH, Smeitink JA. Monogenic mitochondrial disorders. N Engl J Med. 2012;366:1132–1141. doi: 10.1056/NEJMra1012478. [DOI] [PubMed] [Google Scholar]
- 100.Yaniv Y, Spurgeon HA, Lyashkov AE, Yang D, Ziman BD, Maltsev VA, Lakatta EG. Crosstalk between mitochondrial and sarcoplasmic reticulum Ca2+ cycling modulates cardiac pacemaker cell automaticity. PLoS ONE. 2012;7:e37582. doi: 10.1371/journal.pone.0037582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Maltsev VA, Lakatta EG. Synergism of coupled subsarcolemmal Ca2+ clocks and sarcolemmal voltage clocks confers robust and flexible pacemaker function in a novel pacemaker cell model. Am J Physiol Heart Circ Physiol. 2009;296:H594–H615. doi: 10.1152/ajpheart.01118.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Himeno Y, Sarai N, Matsuoka S, Noma A. Ionic mechanisms underlying the positive chronotropy induced by beta1-adrenergic stimulation in guinea pig sinoatrial node cells: a simulation study. J Physiol Sci. 2008;58:53–65. doi: 10.2170/physiolsci.RP015207. [DOI] [PubMed] [Google Scholar]
- 103.Himeno Y, Toyoda F, Satoh H, Amano A, Cha CY, Matsuura H, Noma A. Minor contribution of cytosolic Ca2+ transients to the pacemaker rhythm in guinea pig sinoatrial node cells. Am J Physiol Heart Circ Physiol. 2011;300:H251–H261. doi: 10.1152/ajpheart.00764.2010. [DOI] [PubMed] [Google Scholar]
- 104.Severi S, Fantini M, Charawi LA, DiFrancesco D. An updated computational model of rabbit sinoatrial action potential to investigate the mechanisms of heart rate modulation. J Physiol. 2012;590:4483–4499. doi: 10.1113/jphysiol.2012.229435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Takeuchi A, Matsuoka S. Mitochondrial Na–Ca exchanger NCLX-mediated mitochondria-sarcoplasmic reticulum Ca crosstalk and cardiomyocyte automaticity. J Physiol Sci. 2014;64:S68. [Google Scholar]
- 106.Guo J, Mitsuiye T, Noma A. The sustained inward current in sino-atrial node cells of guinea-pig heart. Pflügers Arch. 1997;433:390–396. doi: 10.1007/s004240050293. [DOI] [PubMed] [Google Scholar]
- 107.Jurkowitz MS, Brierley GP. H+-dependent efflux of Ca2+ from heart mitochondria. J Bioenerg Biomembr. 1982;14:435–449. doi: 10.1007/BF00743069. [DOI] [PubMed] [Google Scholar]
- 108.Gunter KK, Zuscik MJ, Gunter TE. The Na+-independent Ca2+ efflux mechanism of liver mitochondria is not a passive Ca2+/2H+ exchanger. J Biol Chem. 1991;266:21640–21648. [PubMed] [Google Scholar]
- 109.Jiang D, Zhao L, Clish CB, Clapham DE. Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc Natl Acad Sci U S A. 2013;110:E2249–E2254. doi: 10.1073/pnas.1308558110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bergemann AD, Cole F, Hirschhorn K. The etiology of Wolf-Hirschhorn syndrome. Trends Genet. 2005;21:188–195. doi: 10.1016/j.tig.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 111.Nowikovsky K, Pozzan T, Rizzuto R, Scorrano L, Bernardi P. Perspectives on: SGP symposium on mitochondrial physiology and medicine: the pathophysiology of LETM1. J Gen Physiol. 2012;139:445–454. doi: 10.1085/jgp.201110757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tsai MF, Jiang D, Zhao L, Clapham D, Miller C. Functional reconstitution of the mitochondrial Ca2+/H+ antiporter Letm1. J Gen Physiol. 2014;143:67–73. doi: 10.1085/jgp.201311096. [DOI] [PMC free article] [PubMed] [Google Scholar]