

Background: A Gs-biased agonist for the β2-adrenergic receptor (β2AR) has yet to be reported.
Results: A screen of β2AR pepducins identified receptor-dependent and receptor-independent pepducins that selectively activate Gs.
Conclusion: Receptor-dependent pepducins promote a Gs-biased conformation of the β2AR, whereas receptor-independent pepducins directly activate Gs.
Significance: Gs-biased pepducins provide a valuable tool for the continued study of β2AR function and may prove useful as next-generation asthma therapeutics.
Keywords: Adrenergic Receptor, Asthma, Drug Discovery, G Protein-coupled Receptor (GPCR), Peptides, Signal Transduction, β2-Adrenergic Receptor, Pepducin, Biased Agonist, Functional Selectivity
Abstract
The β2-adrenergic receptor (β2AR) is a prototypical G protein-coupled receptor that mediates many hormonal responses, including cardiovascular and pulmonary function. β-Agonists used to combat hypercontractility in airway smooth muscle stimulate β2AR-dependent cAMP production that ultimately promotes airway relaxation. Chronic stimulation of the β2AR by long acting β-agonists used in the treatment of asthma can promote attenuated responsiveness to agonists and an increased frequency of fatal asthmatic attacks. β2AR desensitization to β-agonists is primarily mediated by G protein-coupled receptor kinases and β-arrestins that attenuate receptor-Gs coupling and promote β2AR internalization and degradation. A biased agonist that can selectively stimulate Gs signaling without promoting receptor interaction with G protein-coupled receptor kinases and β-arrestins should serve as an advantageous asthma therapeutic. To identify such molecules, we screened ∼50 lipidated peptides derived from the intracellular loops of the β2AR, known as pepducins. This screen revealed two classes of Gs-biased pepducins, receptor-independent and receptor-dependent, as well as several β-arrestin-biased pepducins. The receptor-independent Gs-biased pepducins operate by directly stimulating G protein activation. In contrast, receptor-dependent Gs-biased pepducins appear to stabilize a Gs-biased conformation of the β2AR that couples to Gs but does not undergo G protein-coupled receptor kinase-mediated phosphorylation or β-arrestin-mediated internalization. Functional studies in primary human airway smooth muscle cells demonstrate that Gs-biased pepducins are not subject to conventional desensitization and thus may be good candidates for the development of next generation asthma therapeutics. Our study reports the first Gs-biased activator of the β2AR and provides valuable tools for the study of β2AR function.
Introduction
The β2-adrenergic receptor (β2AR)3 is a G protein-coupled receptor (GPCR) responsible for hormonal signal transduction in functions such as cardiac muscle contraction, airway smooth muscle relaxation, and blood vessel dilation. The β2AR has served as a prototypical model for understanding GPCR signaling and regulation (1). Crystallographic and biophysical characterization has provided insight into the structure of the basal state of the receptor as well as the conformational changes associated with agonist-stimulated receptor activation and G protein binding (2–8). A diverse set of ligands for the β2AR have also been developed that are now mainstays in the clinic (9–13). β-Antagonists have been used extensively in the treatment of hypertension, and some inverse agonists such as carvedilol have been used in the treatment of congestive heart failure (11, 13). β2AR agonists, including salbutamol and formoterol, are commonly prescribed drugs for the treatment of asthma and chronic obstructive pulmonary disease (9, 12).
Asthma is a chronic condition by which airway inflammation and bronchoconstriction promote peak airflow restriction. Bronchotone, determined by the contractile state of airway smooth muscle (ASM), is the product of differential signaling through a number of GPCRs. These include the β2AR, which is a critical regulator of airway smooth muscle relaxation. β-Agonists stimulate Gs activation leading to an increase in intracellular cAMP and ASM relaxation (9). Iterative β2AR signaling is regulated by GPCR kinase (GRK)-mediated phosphorylation and subsequent β-arrestin recruitment that promotes receptor desensitization, internalization, and degradation (1, 14). Current therapeutic strategies to combat airway constriction include the use of both short acting and long acting β-agonists (15). Treatment with long acting β-agonists has been linked to an increased incidence of an asthmatic episode that results in fatality (16). Prolonged β2AR stimulation promotes recalcitrance to β-agonists by increasing receptor desensitization and degradation. The recruitment of β-arrestins may play an important role in the mechanism behind the severe side effect of long acting β-agonist use, as a murine model demonstrated the ability of β-arrestin2 to attenuate β-agonist-stimulated cAMP production, whereas a β-arrestin knockdown in human ASM partially reduced β2AR desensitization (17).
Traditional β-agonists operate through the extracellular ligand-binding pocket to propagate intracellular signaling (1). One strategy to potentially modulate β2AR interaction with G proteins, GRKs, and β-arrestins is to target the intracellular surface of the receptor using pepducins. Pepducins are cell-penetrating palmitoylated peptides derived from the intracellular loops of a GPCR (18). Pepducins have been generated from many GPCRs, including PAR1, PAR2, PAR4, sphingosine 1-phosphate receptor-3, formyl peptide receptor 2, melanocortin-4 receptor, Smoothened, CXCR1, CXCR2, and CXCR4, and have been shown to function as allosteric agonists or antagonists of their cognate receptor (19–25). A recent study also found that pepducins might function in a biased manner as the CXCR4 pepducin ATI-2341 selectively promoted interaction with Gi over G13, GRKs and β-arrestins that are typically associated with CXCR4 stimulation (26). Although the mechanism of action is unclear, pepducins are proposed to directly interact with a receptor and allosterically modulate receptor signaling (18).
In this study, we focused on determining whether pepducins derived from the β2AR could function as biased modulators. We identified multiple Gs-biased pepducins that stimulated cAMP production without the recruitment of β-arrestins to the β2AR as well as several β-arrestin-biased pepducins. The Gs-biased pepducins did not promote β2AR desensitization, GRK-mediated phosphorylation, or β-arrestin-mediated internalization over an extended time course. These pepducins fell into two classes with receptor-independent pepducins promoting cAMP production by direct activation of Gs, whereas receptor-dependent pepducins induced a β2AR conformation that selectively activated Gs. These pepducins are the first reported Gs-biased molecules operating through the β2AR and show promise in the development of next generation asthma therapeutics.
EXPERIMENTAL PROCEDURES
Pepducin Synthesis
A pepducin library was generated from sequences derived from intracellular loops 1–3 (ICL1–3) of the human β2AR. Pepducin synthesis was performed by a standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) solid-phase protocol with an N-terminal palmitoylation and C-terminal amidation on each peptide. >98% purity was accomplished by C18 reverse-phase chromatography (JPT Peptide Technologies, Peptide 2.0).
cAMP Measurement
HEK 293 cells were grown to confluency in 24-well plates at 37 °C in Dulbecco's modified Eagle's medium (DMEM, Cellgro) supplemented with 10% fetal bovine serum (FBS). Cells were stimulated with 1 μm isoproterenol, 5 μm salbutamol, or 10 μm pepducin for various times at 37 °C in the presence of 0.5 mm 3-isobutyl-1-methylxanthine (IBMX). For the initial screen of all ICL1 and ICL3-1 to ICL3-11 pepducins, cells were lysed by adding 270 μl of 0.1 m HCl followed by 20 min at room temperature on an orbital shaker. Lysates were cleared by centrifugation at 1000 × g for 10 min. cAMP levels were measured using the cyclic AMP EIA kit following the manufacturer's instructions (Cayman Chemical). In all other cAMP measurements, stimulation was stopped on ice by aspirating the media, adding 500 μl of ice-cold ethanol, and incubating for 2 h at room temperature on an orbital shaker. Samples were lyophilized until dry and resuspended in 300 μl of assay buffer (50 mm sodium acetate, pH 6.2). cAMP was measured by radioimmunoassay using an anti-cAMP antibody (a generous gift from Dr. Mario Ascoli, University of Iowa) and 125I-labeled cAMP tracer (Biomedical Technologies, Inc., and PerkinElmer Life Sciences) as described (27).
Analysis of β-Arrestin2 Binding to the β2AR Using Bioluminescence Resonance Energy Transfer (BRET)
β-Arrestin2 recruitment was monitored following the protocol of Hamdan et al. (28). HEK 293 cells were grown in 6-well plates to 80% confluence in DMEM with 10% FBS. Cells were co-transfected with pcDNA3-β-arrestin2-GFP10 (energy acceptor) and pcDNA3-β2AR-RLucII (energy donor) using Lipofectamine 2000 (Invitrogen) for 4 h in serum-free OptiMEM (Invitrogen). Cells were allowed to recover overnight in growth media and then replated in poly-l-ornithine (Sigma)-coated opaque 96-well plates (Optiplate, PerkinElmer Life Sciences) at a density of 100,000 cells per well. After overnight incubation at 37 °C in DMEM with high glucose (Invitrogen), cells were washed three times with PBS plus glucose (Invitrogen) and incubated with PBS plus glucose. Coelenterazine 400a was added to 2.5 μm final concentration and incubated at 37 °C for 2 min. BRET was measured at 510 nm following addition of β-agonist or pepducin using a Tecan Infinite F500 microplate reader. BRET ratios were calculated as the light emitted by the GFP10 acceptor (510 nm) divided by the total light emitted by the donor RLucII (400 nm). ΔBRET was calculated by subtracting the BRET ratio of the unstimulated trials from the stimulated trials.
Detection of β2AR Phosphorylation Using Phosphospecific Antibodies
HEK 293 cells stably overexpressing FLAG-β2AR (a generous gift from Dr. Mark von Zastrow, University of California, San Francisco) were grown to confluency in 10-cm dishes at 37 °C in DMEM supplemented with 10% FBS and 500 μg/ml G418 sulfate (Cellgro). Cells were stimulated with 1 μm isoproterenol, 5 μm salbutamol, or 10 μm pepducin for given time points at 37 °C. Media were removed, and cells were washed on ice three times with PBS (Cellgro). Cells were lysed on ice by the addition of 500 μl of lysis buffer (20 mm Tris-HCl, pH 7.5, 100 mm NaCl, 2 mm EDTA, 1% Triton X-100, 1 Complete mini protease inhibitor tablet, and 1 PhosSTOP phosphatase inhibitor tablet (Roche Applied Science)). Cells were scraped, briefly sonicated, and cleared by centrifugation at 1000 × g for 10 min. Equal protein concentrations were immunoprecipitated using rabbit polyclonal anti-FLAG (Sigma) and protein A-agarose beads (Roche Applied Science) for the detection of PKA phosphorylation. For detection of GRK phosphorylation, cell lysates were immunoprecipitated using mouse monoclonal M2 anti-FLAG (Sigma) and protein G-agarose PLUS beads (Santa Cruz Biotechnology). Samples were incubated overnight at 4 °C and briefly centrifuged to pellet beads from immunodepleted lysate. Pelleted beads were washed with lysis buffer three times, and the washed pellets were resuspended in 60 μl of 2× Laemmli buffer. Immunoprecipitated proteins were separated by SDS-PAGE on a 10% polyacrylamide gel and receptor phosphorylation was analyzed by Western blotting. GRK phosphorylation was detected using a phosphospecific antibody (1:500) against β2AR phosphoserines 355 and 356 (Ser(P)355/6) (Santa Cruz Biotechnology). Equal receptor loading was confirmed by blotting using rabbit polyclonal anti-FLAG (Sigma), at 1:1000 in Tris-buffered saline with Tween 20 (TBST, 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, and 0.1% Tween 20) plus 5% BSA, and rabbit anti-human β2AR (Santa Cruz Biotechnology) at 1:500 (in TBST with 5% BSA). PKA phosphorylation was detected by blotting using a phosphospecific antibody, 2G3, against phosphoserine 262 (a generous gift from Dr. Richard Clark, University of Texas at Houston) at a 1:1000 dilution (29). Equal receptor loading was confirmed by blotting using mouse monoclonal M2 anti-FLAG (Sigma) at 1:1000. Chemiluminescence was measured using Pico chemiluminescent substrate (Thermo Scientific).
Receptor Internalization
HEK 293 cells stably overexpressing FLAG-β2AR were seeded into 24-well plates precoated with poly-l-lysine (Sigma) at a density of 150,000 cells per well and grown at 37 °C in DMEM supplemented with 10% FBS and 500 μg/ml G418 sulfate (Cellgro). At confluency, cells were washed once with warm DMEM and then treated with 1 μm isoproterenol, 5 μm salbutamol, or 10 μm pepducin in complete media for given time points at 37 °C. The media were removed, and cells were fixed on ice with 3.7% paraformaldehyde in Tris-buffered saline (TBS) for 10 min. Cells were washed twice with TBS and blocked for 45 min with blocking buffer (TBS, 1% BSA, and 1 mm CaCl2) at room temperature. Cell surface FLAG-β2AR was detected by ELISA as described previously (30). Briefly, cells were incubated with rabbit polyclonal anti-FLAG (Sigma) for 1 h at room temperature, anti-rabbit HRP secondary antibody (Vector Laboratories) for 1 h at room temperature, and then washed twice with cold blocking buffer, developed by adding a one-step 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) substrate (Thermo Scientific), and incubated at room temperature for 25 min. 100 μl of the developed solution was transferred to a 96-well plate, and the absorbance was measured on a plate reader (Bio-Rad) at 405 nm.
Functional Desensitization
Primary human airway smooth muscle cells were isolated from donors with no chronic illness or medication use. ASM cell cultivation and characterization were described previously (31, 32). Passages 4–7 ASM cells were maintained in Ham's F-12 medium supplemented with 10% FBS. Use of human ASM cells does not constitute research of human subjects because all donor tissue was harvested anonymously and de-identified. For assays measuring total cAMP, primary human ASM cells were seeded into 24-well plates precoated with poly-l-lysine in DMEM and 10% FBS and grown to confluence at 37 °C. Media were removed, and wells were washed twice with Dulbecco's phosphate-buffered saline (DPBS, Cellgro), and 500 μl of DPBS with calcium and magnesium was added to each well. Cells were then treated with 1 μm isoproterenol, 5 μm salbutamol, or 10 μm pepducin for given time points. Stimulation was stopped on ice by adding 750 μl of ice-cold ethanol and incubating for 2 h at room temperature on an orbital shaker. cAMP was measured using the 125I-labeled cAMP radioimmunoassay protocol described above. For assays measuring intracellular cAMP levels, the same procedure was performed as above except the DPBS was removed before the addition of ice-cold ethanol.
Pepducin Specificity
CHO-K1 cells were seeded into 6-well plates and grown to 60% confluency at 37 °C in Ham's F-12 media supplemented with 10% FBS. CHO-K1 cells were transfected with pcDNA3-FLAG-β1AR (a generous gift from Dr. Robert Lefkowitz, Duke University), pcDNA3-FLAG-β2AR and pcDNA3-FLAG-EP2R (a generous gift from Dr. Raymond Penn, Thomas Jefferson University), or pcDNA3 for 4 h with Lipofectamine 2000 (Invitrogen). The cells were allowed to recover for 48 h and then treated with 1 μm isoproterenol, 10 μm PGE2, or 10 μm pepducin for 10 min in the presence of IBMX. Stimulation was stopped on ice by aspirating the media, adding 500 μl of ice-cold ethanol, and incubating for 2 h at room temperature on an orbital shaker. cAMP was measured by the radioimmunoassay described above.
Expression and Purification of Gs Heterotrimer
Bovine Gαs short, His6-rat Gβ1, and bovine Gγ2 were expressed in High Five insect cells (Expression Systems Inc.) grown in ESF921 media (Expression Systems Inc.). Cultures were grown to a density of 3 million cells/ml and then infected with two separate viruses containing the Gαs and Gβγ cDNAs at a 1:1 multiplicity of infection. After 48 h of incubation, the infected cells were harvested by centrifugation and resuspended in 200 ml of lysis buffer (20 mm HEPES, pH 7.5, 100 μm MgCl2, 5 mm β-mercaptoethanol, 10 μm GDP, 2.5 μg/ml leupeptin, and 160 μg/ml benzamidine) per liter of cell culture for 30 min. Lysates were centrifuged for 10 min at 18,000 × g and then resuspended in 100 ml of solubilization buffer (20 mm HEPES, pH 7.5, 100 mm NaCl, 1% sodium cholate, 0.05% dodecylmaltoside (DDM), 5 mm MgCl2, 5 μl of calf intestinal alkaline phosphatase (Sigma), 5 mm β-mercaptoethanol, 10 μm GDP, 5 mm imidazole, 2.5 μg/ml leupeptin, and 160 μg/ml benzamidine). Samples were Dounce-homogenized for 20 strokes and stirred for 1 h at 4 °C followed by centrifugation at 18,000 × g for 30 min. 2 ml of pre-equilibrated nickel-nitrilotriacetic acid resin per liter of cell culture was added to the solubilized supernatant and stirred for 1.5 h at 4 °C. Bound Gs was collected by centrifugation at 4000 × g for 10 min and washed with solubilization buffer three times in a 50-ml conical tube. The Gs-bound resin was washed for 30 min with 50% E1 buffer (solubilization buffer plus 15 mm imidazole) plus 50% E2 buffer (20 mm HEPES, pH 7.5, 50 mm NaCl, 0.1% DDM, 1 mm MgCl2, 5 mm β-mercaptoethanol, 20 μm GDP, 20 mm imidazole) followed by washes with 25% E1/75% E2, 10% E1/90% E2, and 5% E1/95% E2. Gs was eluted with E2 buffer supplemented with 200 mm imidazole. The eluate was passed through a 0.2-μm filter and applied to ion exchange chromatography as described previously (6) to separate Gs heterotrimer from free Gβγ complex. The fractions containing pure Gs heterotrimer were pooled and dialyzed against 20 mm HEPES, pH 7.5, 100 mm NaCl, 0.1% DDM, 1 mm MgCl2, 100 μm tris(2-carboxyethyl)phosphine (TCEP), and 20 μm GDP. The protein was concentrated to ∼15 mg/ml using a 100-kDa cutoff concentrator (Millipore). and glycerol was added to 15%. The final sample was aliquoted, flash-frozen, and stored at −80 °C.
Expression and Purification of β2AR from Baculovirus-infected Sf9 Cells
Recombinant baculovirus was prepared using Bestbac expression system (Expression Systems Inc.) with pVL1392 as vector. The full-length β2AR (termed “PN1”) was expressed by infecting Sf9 cells at a density of 4.5 million cells/ml with second passage baculovirus stock using 20 ml of virus stock per liter of cell culture. 1 μm alprenolol was added to stabilize the receptor during expression. The infected cells were harvested after 48 h of incubation at 27 °C. Cell pellets were lysed by stirring in lysis buffer for 20 min (20 mm HEPES, pH 7.5, 5 mm EDTA, 1 μm alprenolol, 2.5 μg/ml leupeptin, 160 μg/ml benzamidine; 10 ml of buffer/g of cell pellet). The receptor was then extracted from the membrane using Dounce homogenization in solubilization buffer (20 mm HEPES, pH 7.5, 100 mm NaCl, 1% DDM, 1 μm alprenolol, 2.5 μg/ml leupeptin, 160 μg/ml benzamidine) for 1 h at room temperature. 10 ml of solubilization buffer was added per g of cell pellet. After addition of 2 mm CaCl2, the solubilized receptor was clarified by high speed centrifugation at 18,000 × g for 30 min. The N-terminal FLAG-tagged receptor was then captured by M1 antibody affinity chromatography (Sigma). The column was extensively washed with HMS-CHS buffer (20 mm HEPES, pH 7.5, 350 mm NaCl, 0.1% DDM, 0.01% cholesterol hemisuccinate) plus 2 mm CaCl2 to remove impurities and alprenolol. The receptor was then eluted with HMS-CHS buffer supplemented with 5 mm EDTA and 200 μg/ml free FLAG peptide.
Analysis of Gs Activation by [35S]GTPγS Binding
Purified Gs was diluted to 3 μm in 10 mm HEPES, pH 8, 1 mm EDTA, and 0.1% Lubrol and then diluted 2-fold in 50 mm HEPES, pH 8, 1 mm EDTA, 125 mm MgCl2, and 200 mm NaCl. Pepducins or 0.5% DMSO were incubated with Gs for 15 min on ice, and the binding reaction was initiated by addition of 10 μm GTPγS (cold plus hot, ∼1300 cpm/fmol). Samples were incubated for 15 min at 4 °C and then quenched by addition of cold GTPγS wash buffer (20 mm Tris-HCl, pH 8, 25 mm MgCl2, 100 mm NaCl) followed by rapid filtration through BA85 filters (Millipore). The filters were washed four times with 4 ml of cold GTPγS wash buffer, and [35S]GTPγS binding was quantified by liquid scintillation counting.
We also evaluated Gs activation in lipid bicelles. Purified Gs (18 μm) in 2% 3:1 dimyristoyl phosphatidylcholine (DOPC)/CHAPSO bicelles with 1.13 mm CHS, 20 mm HEPES, pH 7.5, and 100 mm NaCl was incubated in the presence or absence of β2AR (1.26 μm) for 2 h on ice to allow protein incorporation into the lipid bicelles. 2 μl of reconstituted β2AR-Gs or reconstituted Gs alone was diluted 200-fold in 20 mm HEPES, pH 7.5, 150 mm NaCl, 1 mm MgCl2, and 38.5 nm [35S]GTPγS (PerkinElmer Life Sciences). 20-μl reactions were initiated by the addition of 1 μm isoproterenol or 10 μm pepducin and incubated for the indicated times at room temperature. Non-pepducin trials included 0.05% DMSO. Bound [35S]GTPγS was collected by rapid filtration on GF/B filters (Whatman), washed four times with 4 ml of cold GTPγS wash buffer, and analyzed by liquid scintillation counting.
Analysis of Gαs Engagement to the β2AR Using BRET
Gαs interaction with the β2AR was assayed by BRET using a β2AR construct tagged with GFP10 at the C terminus of the receptor and Gαs constructs tagged with RLucII either at the N terminus (RLucII-Gαs) or at residue 67 of Gαs (Gαs67-RLucII). For the Gαs67-RLucII studies, HEK 293T cells were cultured in DMEM supplemented with 10% FBS, 0.2 units/ml penicillin, 100 μg/ml streptomycin (Wisent Inc.) and were seeded in 6-well plates at 600,000 cells/well 24 h before transfection. Transient transfections with β2AR-GFP10 and Gαs67-RLucII in the presence of untagged Gβ1 and Gγ2 were performed using linear polyethyleneimine, 25-kDa (Polysciences, Inc.), as transfecting agent at a 3:1 ratio of polyethyleneimine/DNA. Two h after transfection, culture medium was replaced with fresh media, and the cells were then maintained in culture for 48 h before BRET experiments. The expression level of the acceptor was determined as total fluorescence, using a FlexStationII fluorometer (Molecular Devices) with 400-nm excitation and 510-nm emission filters. The expression level of the donor was measured as total luminescence, using a Mithras LB940 Multimode Microplate Reader (Berthold Technologies), following the addition of 2.5 μm coelenterazine 400a. Cells were washed once with Hanks' balanced salt solution (Invitrogen) containing 20 mm HEPES (HBSS) and detached in HBSS supplemented with 0.1% BSA (HBSS/BSA) (Sigma) at room temperature. 100,000 cells/well were then distributed in a white 96-well microplate (Greiner). Cells were then treated with or without different concentrations of ligand, and BRET values were collected using the Mithras LB940 Reader equipped with BRET400-GFP10 filter set (acceptor, 515 ± 20-nm, and donor, 400 ± 70-nm filters), following the addition of coelenterazine 400a. BRET signals were determined as the ratio of the light emitted by the acceptor over donor. The specific BRET signal (net BRET) was determined by subtracting the background signal detected in cells transfected with the luciferase donor alone from the BRET obtained in cells expressing both energy donor and acceptor. The ligand-promoted BRET signal (ΔBRET) was calculated by subtracting the BRET values obtained in the vehicle condition from the one measured in the presence of ligand.
For the RLucII-Gαs studies, HEK 293 cells were grown in 6-well plates to 80% confluence in DMEM with 10% FBS. Cells were co-transfected with pcDNA3.1-RLucII-Gαs (donor) and pGFP-β2AR-GFP10 (acceptor) using Lipofectamine 2000 (Invitrogen) for 4 h in serum-free OptiMEM (Invitrogen). Cells were allowed to recover overnight in growth media and then replated in poly-l-ornithine (Sigma)-coated opaque 96-well plates (Optiplate, PerkinElmer Life Sciences) at a density of 100,000 cells per well. After overnight incubation at 37 °C in DMEM with high glucose (Invitrogen), cells were washed three times with PBS plus glucose (Invitrogen) and incubated with PBS plus glucose. Coelenterazine 400a was added to 2.5 μm final concentration and incubated at 37 °C for 2 min. BRET was measured at 510 nm following addition of 1 μm isoproterenol or 10 μm pepducin using a Tecan Infinite F500 microplate reader. BRET ratios were calculated as the light emitted by the GFP10 acceptor (510 nm) divided by the total light emitted by the RLucII donor (400 nm). ΔBRET was calculated by subtracting the BRET ratio of the unstimulated trials from the stimulated trials.
[125I]Iodocyanopindolol Binding
HEK 293 cells stably expressing FLAG-β2AR were isolated and washed three times with assay buffer (HBSS with calcium and magnesium, 0.1% BSA, pH 7.4), diluted to 50,000 cells/ml, and incubated with 1 nm [125I]iodocyanopindolol in the presence or absence of pepducin or propranolol for 1 h at 25 °C. Incubations were terminated by the addition of 4 ml of cold assay buffer and rapid filtration on GF/B filters. Filters were washed four times with 4 ml of cold assay buffer, and [125I]iodocyanopindolol binding was quantitated by gamma emission counting.
Monobromobimane Labeling of β2AR
Purified FLAG-β2AR and 20 μm monobromobimane (Invitrogen) were incubated for 1 h on ice for labeling. The monobromobimane-labeled receptor was then purified by affinity chromatography using alprenolol-Sepharose as described previously to select functional receptors (4). 300 μm alprenolol was used to elute the receptor to a tandemly linked M1 FLAG column. The column was washed with HMS-CHS buffer for removal of alprenolol to prepare unliganded receptor. The receptor was then eluted from M1 resin with HMS-CHS buffer supplemented with 5 mm EDTA, 200 μg/ml free FLAG peptide. Size-exclusion chromatography on a Superdex-200 column (GE Healthcare) equilibrated in HMS-CHS buffer was used to increase the purity. The receptor was concentrated to 125 μm with purity greater than 95% as assessed by SDS-PAGE.
Analysis of Monobromobimane-β2AR Fluorescence
Monobromobimane-labeled β2AR (mBB-β2AR) was incorporated into 2% DOPC/CHAPSO (3:1) with 1.13 mm CHS lipid bicelles by incubating for 30 min on ice. Lipid bicelles containing 50 nm mBB-β2AR were incubated for 15 min at 25 °C in 20 mm HEPES, pH 7.5, 100 mm NaCl with isoproterenol or pepducin. Isoproterenol samples also contained 0.1 or 0.5% DMSO to account for the pepducin solvent. In experiments using Gs, 200 nm Gs was incubated for 20 min at 25 °C alone or post-agonist addition depending on the experimental setup. mBB-β2AR fluorescence was measured by excitation at 370 nm and recording emission from 430 to 490 nm at 1-nm increments with 1 nm s−1 integration on a Spex FluoroMax-3 spectrofluorometer (Jobin Yvon Inc.) in photon counting mode set at a 4-nm emission bandwidth pass. Background fluorescence contributed by the assay buffer and ligand was subtracted from the experimental spectra.
RESULTS
Characterization of a Library of β2AR Pepducins
Recent studies have shown that a pepducin from the first intracellular loop (ICL) of CXCR4 can effectively activate Gi without promoting appreciable coupling to G13, GRKs, or β-arrestins (26). In an effort to identify Gs-biased pepducins, we synthesized a library of 51 pepducins corresponding to sequences derived from ICL1, ICL2, and ICL3 of the human β2AR, a GPCR that primarily couples to Gs (Fig. 1). These pepducins were then screened for their ability to promote cAMP production in HEK 293 cells. This screen yielded multiple pepducins that promote cAMP production with four demonstrating efficacy comparable with the partial agonist salbutamol (Fig. 2A). The majority of these pepducins were from the proximal portion of ICL3 (ICL3-2, ICL3-7,and ICL3-8), and others were from the central region of ICL3 (ICL3-9) or from ICL1 (ICL1-15).
FIGURE 1.

Amino acid sequences of the β2AR pepducin library. Residues in red are located in the transmembrane domains, and residues in black are located in the intracellular loops. Dashed line denotes skipped amino acids from the β2AR sequence. All pepducins were synthesized with an N-terminal palmitate and C-terminal amide.
FIGURE 2.

Analysis of β2AR pepducins for cAMP production and β-arrestin binding. A, cAMP assay was performed in HEK 293 cells. Cells were stimulated with 10 μm pepducin or 5 μm salbutamol in DMEM with 10% FBS in the presence of 500 μm IBMX. cAMP was measured at 10 min by ELISA. Data are represented by the mean of three independent experiments ± S.D. B, schematic of β-arrestin recruitment analysis by BRET. Upon β-agonist stimulation, GFP10-β-arrestin2 is recruited to β2AR-RLucII. Upon β-arrestin2 binding to the β2AR, GFP10 will be within the BRET radius of RLucII allowing GFP10 emission readout to be indicative of β-arrestin recruitment. C, HEK 293 cells co-transfected with GFP10-β-arrestin2 and β2AR-RLucII were preincubated with coelenterazine 400a for 2 min and stimulated with 10 μm pepducin, 1 μm isoproterenol, or 5 μm salbutamol for the indicated times. BRET was monitored at the indicated times post-addition. Data are expressed as ΔBRET as the background BRET has been subtracted. The data are represented by the means of four independent experiments ± S.D. D, cAMP output (10 min) as a function of β-arrestin recruitment reveals multiple Gs-biased and β-arrestin-biased pepducins. Balanced agonists, such as isoproterenol, can effectively promote both cAMP production and β-arrestin recruitment with similar efficacies. An agonist that promotes cAMP production more effectively than β-arrestin recruitment is a Gs-biased agonist (i.e. ICL3-8 and ICL3-9), whereas agonists that couple β-arrestins more effectively than stimulating cAMP production are considered β-arrestin-biased. E, HEK 293 cells were stimulated with various concentrations of isoproterenol, ICL3-8, or ICL3-9 for 10 min in the presence of 500 μm IBMX. ICL3-8 has an EC50 of 577 ± 14 nm, whereas ICL3-9 has an EC50 of 4.7 ± 0.1 μm. cAMP production is represented as percentage normalized to maximal isoproterenol stimulation. The data are represented by the mean of three independent experiments ± S.D.
To gain a more complete understanding of the diverse signaling profiles from the β2AR pepducin library, all pepducins were also analyzed for their ability to promote β-arrestin recruitment to the β2AR using BRET. This assay involved treating HEK 293 cells co-expressing a β2AR-Renilla reniformis luciferase II fusion (β2AR-RLucII) and GFP10-tagged β-arrestin2 with the various pepducins (Fig. 2B) (28). Several ICL1-derived pepducins effectively promoted β-arrestin2 interaction with the β2AR, whereas ICL2 and ICL3 pepducins had no effect (Fig. 2C and data not shown). Thus, multiple pepducins derived from ICL3 activate cAMP accumulation without promoting β-arrestin binding to the β2AR and therefore appear to function as Gs-biased allosteric agonists, whereas several pepducins from ICL1 (ICL1-4, ICL1-11, and ICL120) appear to be β-arrestin-biased allosteric agonists as they effectively stimulate β-arrestin2 engagement with the β2AR without promoting any cAMP production (Fig. 2D).
Because the primary goal of this study was to develop Gs-biased agonists, further characterization was limited to two of the candidate Gs-biased pepducins, ICL3-8 and ICL3-9. ICL3-8 is representative of a family of sequence-related pepducins from the proximal portion of ICL3, whereas ICL3-9 is primarily from the central portion of ICL3 (Fig. 1). Both pepducins promoted ∼40% cAMP production compared with the full agonist isoproterenol with ICL3-8 having an EC50 of 577 ± 14 nm and ICL3-9 an EC50 of 4.7 ± 0.1 μm (Fig. 2E).
Gs-biased Pepducins Do Not Induce Receptor Desensitization
Agonist-specific desensitization of the β2AR is primarily mediated by GRK phosphorylation of the receptor, which promotes high affinity binding of β-arrestins and attenuates G protein coupling (1). Phosphorylation of Ser355 and Ser356 (Ser355/6) on the C-terminal tail of the β2AR has been attributed to GRK5/6 and is partially responsible for β-arrestin recruitment and receptor internalization (Fig. 3A) (29, 33, 34). Ligand-promoted phosphorylation of the β2AR was monitored using phosphospecific antibodies targeting Ser355/6. Stimulation with either isoproterenol or salbutamol induced rapid and robust phosphorylation at Ser355/6, whereas treatment with either ICL3-8 or ICL3-9 did not induce appreciable receptor phosphorylation at this site (Fig. 3B).
FIGURE 3.

Gs-biased pepducins do not promote GRK-mediated β2AR phosphorylation. A, schematic diagram of kinase-specific phosphorylation sites on the β2AR upon β-agonist stimulation. GRK5/6 phosphorylates Ser355/6 on the β2AR C-terminal tail, whereas PKA can phosphorylate Ser261/2 in ICL3. B and C, HEK 293 cells stably overexpressing FLAG-β2AR were stimulated with 1 μm isoproterenol, 5 μm salbutamol, or 10 μm pepducin in DMEM with 10% FBS. 0.05% DMSO was included in non-pepducin-stimulated cells. B, GRK5/6 phosphorylation at Ser355/6 was monitored on immunoprecipitated FLAG-β2AR at the indicated time points by Western blotting using α-Ser(P)-355/Ser356. C, PKA phosphorylation at Ser261/2 was monitored on immunoprecipitated FLAG-β2AR at the indicated time points by Western blotting using α-Ser(P)262.
Distinct phosphorylation sites for the cAMP-dependent protein kinase (PKA) have been identified at Ser261 and Ser262 in ICL3 of the β2AR (Fig. 3A) as well as in the C-terminal tail (Ser345 and Ser346). To monitor ligand-promoted phosphorylation by PKA, we used a phosphospecific antibody against Ser262 (29). Isoproterenol and salbutamol promoted an increase in phosphorylation that was observed at 5–10 min after stimulation (Fig. 3C). ICL3-8 and ICL3-9 also promoted effective phosphorylation of Ser262 with kinetics and efficacy similar to that observed with isoproterenol (Fig. 3C). This result correlates well with the ability of ICL3-8 and ICL3-9 to promote cAMP production and thereby activate PKA.
β-Arrestin recruitment couples the β2AR to the internalization machinery leading to a loss of cell surface receptors and further propagating receptor desensitization (35). Agonist-promoted β2AR internalization was analyzed post-stimulation with isoproterenol, salbutamol, ICL3-8, and ICL3-9 in HEK 293 cells stably overexpressing FLAG-β2AR by cell surface ELISA. Although both isoproterenol and salbutamol induced rapid internalization of the receptor, the pepducins did not induce any internalization over a 1-h period (Fig. 4A).
FIGURE 4.

Gs-biased pepducins do not promote β2AR internalization or desensitization. A, β2AR internalization was monitored by cell surface ELISA in HEK 293 cells stably overexpressing FLAG-β2AR post-stimulation with 1 μm isoproterenol, 5 μm salbutamol, or 10 μm pepducin in DMEM with 10% FBS. 0.05% DMSO was included in non-pepducin-stimulated cells. The data are represented by the mean of three independent experiments ± S.D. B, total cAMP was measured post-stimulation with 1 μm isoproterenol, 5 μm salbutamol, or 10 μm pepducin in DPBS at the indicated time points in human ASM cells. C, intracellular cAMP was measured post-stimulation with 1 μm isoproterenol, 5 μm salbutamol, or 10 μm pepducin in DPBS at the indicated time points in human ASM cells. 0.05% DMSO was included in non-pepducin-stimulated cells. The data are represented by the mean of three independent experiments ± S.D.
The long term use of β-agonists in the treatment of asthma has been implicated in chronic airway desensitization (15, 16, 36–39). To evaluate whether the Gs-biased pepducins induce functional desensitization of the β2AR, we studied cAMP production in primary human airway smooth muscle cells. Receptor desensitization was observed post-β-agonist stimulation as noted by the decreasing rate of total cAMP production over a 2-h time course (Fig. 4B). In contrast, the pepducins promoted a steady rate of total cAMP production, suggesting that signaling through the pepducins is not subject to the conventional desensitization mechanisms (Fig. 4B). The root of the linearity was further characterized by monitoring intracellular cAMP production over time. Isoproterenol and salbutamol stimulated a rapid peak in cAMP levels that decreased over time as desensitized receptors were likely unable to maintain Gs activation (Fig. 4C). In contrast, the pepducins slowly achieved a steady state level of cAMP production (Fig. 4C). Taken together, these results reveal that the ICL3-8 and ICL3-9 induce less desensitization compared with isoproterenol and salbutamol.
Mechanism of Pepducin-mediated Activation of Gs
Stimulation of the β2AR by β-agonists promotes rapid engagement of Gs to the receptor which, in turn, promotes GDP dissociation, GTP binding, and G protein activation. To assess whether the pepducins promote Gs binding to the β2AR, we used BRET to measure association of Gαs67-RLucII and β2AR-GFP10 (Fig. 5A) (40). Upon isoproterenol treatment, Gs was rapidly engaged by the β2AR as indicated by the change in BRET ratio (Fig. 5B). ICL3-9 also promoted Gs interaction with the β2AR, although this occurred on a slower time scale (Fig. 5B). The EC50 value for ICL3-9-promoted β2AR-Gs interaction was 3.3 ± 0.4 μm (Fig. 5C), which is comparable with the EC50 value observed for cAMP production (Fig. 2E). In contrast, ICL3-8 was unable to stimulate β2AR-Gs coupling and therefore may be activating Gs in a manner independent of receptor-mediated nucleotide exchange (Fig. 5B).
FIGURE 5.

Gs-biased pepducins can operate in a receptor-dependent or receptor-independent manner. A, schematic of β2AR-Gs engagement analysis by BRET. Upon β-agonist stimulation, Gαs67-RLucII engages the β2AR-GFP10 causing a change in the distance between RLucII and GFP10 and a subsequent change in GFP10 emission. B, HEK 293 cells co-transfected with Gαs67-RLucII and β2AR-GFP10 were preincubated with coelenterazine 400a for 5 min and stimulated with 10 μm pepducin or 10 μm isoproterenol. Changes in BRET were monitored over the indicated time points. The data are represented by the mean of three independent experiments ± S.E. C, Gαs67-RLucII engagement was measured over increasing concentrations of ICL3-9 or isoproterenol at 15 min. The ICL3-9 EC50 for Gαs67-RLucII recruitment was 3.3 ± 0.4 μm. The data are represented by the mean of four independent experiments ± S.E. D, receptor-dependent mechanism of action suggests the pepducin promotes a productive interaction between the G protein and β2AR. A receptor-independent pepducin might directly stimulate the G protein. E, lipid bicelles (0.02% DOPC/CHS with 11.3 μm CHS) containing reconstituted 12.6 nm β2AR, 180 nm Gs, or 180 nm Gs alone were stimulated with 1 μm isoproterenol or 10 μm pepducin in the presence of 38.5 nm [35S]GTPγS in assay buffer (20 mm HEPES, 7.5, 150 mm NaCl, 1 mm MgCl2). Samples were isolated by rapid filtration on BA85 filters. 0.05% DMSO was included in non-pepducin-stimulated samples. The data are represented by the mean of four independent experiments ± S.D. F, 1.5 μm purified Gs in 0.05% lubrol was preincubated for 15 min with 100 μm pepducin or 0.5% DMSO at 4 °C, and GTPγS exchange was initiated by the addition of 10 μm GTPγS. The data are represented by the mean of three independent experiments ± S.D.
From the two-dimensional screen, it is possible for pepducins to produce a Gs-biased profile in either a receptor-dependent or a receptor-independent manner. A receptor-dependent pepducin would stimulate G protein activation by promoting receptor-G protein coupling, whereas a receptor-independent pepducin might directly activate the G protein (Fig. 5D). Receptor dependence was initially assessed in lipid bicelles by monitoring activation of purified Gs by GTPγS exchange in the presence or absence of purified β2AR. Both isoproterenol and ICL3-9 were able to promote G protein activation only when the β2AR was included in the assay (Fig. 5E). In contrast, ICL3-8 promoted effective GTPγS binding to Gs independent of whether the β2AR was present (Fig. 5E). To confirm ICL3-8 as a bona fide direct activator of Gs, we also evaluated GTPγS exchange on purified detergent-solubilized Gs. ICL3-8 was found to rapidly and robustly stimulate GTPγS binding, whereas ICL3-9 had no effect (Fig. 5F). Higher concentrations of pepducin (100 μm) were necessary to observe maximal efficacy as the N-terminal lipidation of the pepducin was unable to contribute to its potency in the detergent-solubilized assay. Overall, these studies demonstrate that ICL3-8 directly activates Gs, whereas ICL3-9 activates Gs in a β2AR-dependent manner.
Receptor Specificity of ICL3-9
The ability of an agonist to promote receptor-dependent activation of downstream signaling is critical in drug targeting and predicting off-target effects. As multiple GPCRs can couple to Gs, it is important to define the receptor specificity of ICL3-9. To evaluate this, we transfected CHO-K1 cells with the Gs-coupled β1AR, β2AR, or prostaglandin E2 receptor (EP2R). Control-transfected CHO-K1 cells lack endogenous expression of these receptors as an agonist-induced increase in cAMP was only observed in the cells transfected with the specific receptor (Fig. 6). As expected, ICL3-8 activated cAMP production similarly in all of the cell lines further corroborating its receptor-independent activity. In contrast, ICL3-9 effectively stimulated cAMP production in cells expressing either the β1AR or β2AR but had no effect in EP2R- or control-transfected cells (Fig. 6). Thus, ICL3-9 is able to utilize both the β1AR and β2AR to activate cAMP production suggesting that it can function on closely related family members. ICL1, ICL2, and the proximal and distal portions of ICL3 are highly conserved in β1AR and β2AR, although the central portion of the β1AR ICL3 contains a proline-rich insert that interrupts the partially conserved ICL3-9 sequence. The significant homology between the β1AR and β2AR along with the same G protein coupling profile may help to explain the dual-specificity of ICL3-9.
FIGURE 6.

ICL3-9 demonstrates distinct receptor specificity. CHO-K1 cells were transfected with pcDNA3.1, FLAG-β1AR, FLAG-β2AR, or FLAG-EP2R and stimulated with 1 μm isoproterenol, 10 μm PGE2, or 10 μm pepducin in the presence of 500 μm IBMX for 10 min in Ham's F-12 with 10% FBS. 0.05% DMSO was included in non-pepducin-stimulated cells. The data are represented by the mean of three independent experiments ± S.D.
Mutagenesis of ICL3-9
To understand the residues critical for ICL3-9 function, truncation and triple-alanine substitution variants were synthesized and assessed for their ability to stimulate cAMP production (Fig. 7A). As expected, the peptide palmitoylation and amidation were essential for ICL3-9 activity as removing these modifications markedly reduced functionality (Fig. 7B). This is likely due to the inability of the peptides to access the inner leaflet of the cell membrane. Similarly, both the N and C termini also seem critical in ICL3-9 activity as any truncation at these locations fully abrogated its ability to stimulate cAMP accumulation (Fig. 7B).
FIGURE 7.

ICL3-9 truncations and mutations modulate pepducin efficacy and potency. A, sequences of ICL3-9 truncation and substitution variants. B, HEK 293 cells were stimulated with 10 μm ICL3-9 truncation variants in DMEM with 10% FBS in the presence of 500 μm IBMX. 0.05% DMSO was added to cells not stimulated by pepducin. cAMP was measured at 10 min by radioimmunoassay. Data are represented by the mean of three independent experiments ± S.D. C, HEK 293 cells were stimulated with 10 μm ICL3-9 mutations in DMEM with 10% FBS in the presence of 500 μm IBMX. 0.05% DMSO was added to cells not stimulated by pepducin. cAMP was measured at 10 min by radioimmunoassay. Data are represented by the mean of three independent experiments ± S.D. D, cAMP was measured in HEK 293 cells stimulated with ICL3-9 substitution variants for 10 min in the presence of 500 μm IBMX. 0.05% DMSO was added to cells not stimulated by pepducin. cAMP production is normalized to isoproterenol stimulation. The EC50 of ICL3-9A1 is ∼1.5 ± 0.8 μm and the EC50 of ICL3-9A2 is ∼0.18 ± 0.24 μm. E, lipid bicelles containing reconstituted 12.6 nm β2AR/180 nm Gs or 180 nm Gs alone were preincubated with 5 μm of the ICL3-9 substitution variants for 10 min at room temperature and stimulated with 5 μm ICL3-9 in the presence of 38.5 nm [35S]GTPγS in assay buffer (20 mm HEPES, 7.5, 150 mm NaCl, 1 mm MgCl2). Samples were isolated by rapid filtration on BA85 filters. The data are represented by the mean of three independent experiments ± S.D.
Triple-alanine substitutions through the central portion of ICL3-9 appeared to have different effects on ICL3-9 functionality. Substitutions in the N-terminal half of ICL3-9, as represented by ICL3-9A1 and ICL3-9A2, reduced the efficacy by ∼70% (Fig. 7C). Interestingly, the ICL3-9A2 substitution displayed an ∼25-fold increase in potency despite the reduced efficacy (Fig. 7D). Alanine substitutions in the C-terminal half of ICL3-9 yielded activity-null variants. The loss in efficacy could be attributed to the exchange of critical residues necessary for the pepducin functionality or residues that are participating in the interaction with the β2AR. Pepducins that lack residues critical for activity might still have the ability to interact with the receptor and act as an antagonist in competition with ICL3-9, whereas binding-defective mutants would lack the ability to compete with ICL3-9. ICL3-9A3, ICL3-9A4, and ICL3-9A5 were unable to modulate ICL3-9-promoted GTPγS binding in a β2AR-Gs binding assay, although ICL3-9A1 and ICL3-9A2 partially reduced G protein activation (Fig. 7E). It is unknown whether our results are assessing the necessity of the substituted residues or whether modulating the secondary structure of the pepducin contributes to the change in efficacy and potency.
Mechanism of Receptor-dependent Gs Bias of ICL3-9
The traditional definition of an orthosteric receptor agonist is a ligand that binds within the ligand-binding pocket of the receptor and elicits a biological response that is subject to inhibition by a receptor antagonist (41, 42). To assess whether ICL3-8 or ICL3-9 interact with the ligand-binding pocket of the β2AR, we tested their ability to compete for binding of the β2AR antagonist [I125]iodocyanopindolol. Although [I125]iodocyanopindolol binding to the β2AR was effectively inhibited by propranolol, there was no effect of ICL3-8 or ICL3-9 (Fig. 8A). Thus, ICL3-9 does not appear to interact with the orthosteric ligand-binding pocket of the β2AR.
FIGURE 8.
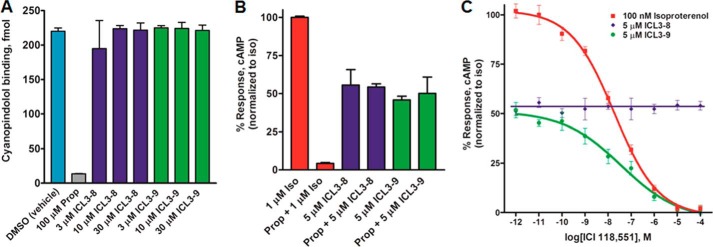
Gs-biased pepducins do not interact with the orthosteric binding site of the β2AR antagonist but ICL3-9 is sensitive to the inverse agonist ICI118,551. A, HEK 293 cells stably expressing FLAG-β2AR were incubated with 1 nm [125I]cyanopindolol for 1 h at room temperature in HBSS with calcium and magnesium and 0.1% BSA in the presence or absence of 3–30 μm pepducin or 100 μm propranolol. 0.05% DMSO was included in non-pepducin-stimulated cells. The data are represented by the mean of three independent experiments ± S.D. B, HEK 293 cells were stimulated with 1 μm isoproterenol or 5 μm pepducin in the presence of 500 μm IBMX for 10 min at 37 °C with or without a 10-min preincubation with 100 μm propranolol. 0.05% DMSO was included in non-pepducin-stimulated cells. The data are represented by the mean of three independent experiments ± S.D. C, HEK 293 cells were preincubated with ICI118,551 for 10 min and stimulated with 100 nm isoproterenol or 5 μm pepducin in the presence of 500 μm IBMX for 10 min in DMEM with 10% FBS. 0.05% DMSO was included in non-pepducin-stimulated cells. The data are represented by the mean of three independent experiments ± S.D.
Inverse agonists have the ability to occupy the ligand-binding pocket and, unlike receptor antagonists, attenuate spontaneous signal activation from a receptor (42, 43). For the β2AR, inverse agonists are proposed to constrict the receptor conformational plasticity needed for spontaneous receptor signaling (44). The Gs-biased pepducins were evaluated for sensitivity to two different β2AR inverse agonists. The weak inverse agonist propranolol effectively blocked isoproterenol-promoted cAMP production in HEK 293 cells but had no effect on ICL3-8- or ICL3-9-stimulated cAMP production (Fig. 8B). In contrast, the potent inverse agonist ICI118,551 effectively suppressed both isoproterenol- and ICL3-9-promoted cAMP production with an IC50 of ∼10 nm, whereas the responsiveness to ICL3-8 was unaffected (Fig. 8C). As ICL3-9 is not sensitive to a weak inverse agonist and is not operating through the ligand-binding pocket, its sensitivity to ICI118,551 likely stems from a competition between an ICL3-9-promoted Gs-biased conformation and an ICI118,551-promoted inactive conformation of the β2AR.
ICL3-9 Antagonizes β-Agonist-promoted β2AR Internalization
As both isoproterenol and ICL3-9 stimulate Gs through the β2AR, it is not possible to detect a functional difference between the ICL3-9- and isoproterenol-induced active states through monitoring cAMP production. Because isoproterenol promotes β-arrestin binding to the receptor, although ICL3-9 does not, if ICL3-9 stabilizes a conformation distinct from that promoted by isoproterenol, it might be able to modulate the efficacy of isoproterenol to promote receptor internalization. Consistent with this notion, ICL3-9 was able to inhibit isoproterenol-promoted β2AR internalization in a dose-dependent manner (Fig. 9A). The ability of ICL3-9 to inhibit isoproterenol-stimulated internalization can be attributed to reduced β-arrestin binding as increasing concentrations of ICL3-9 also attenuate β-arrestin2 recruitment to the β2AR as monitored by BRET (Fig. 9B). These results suggest that the β2AR conformation induced by ICL3-9 appears to be different from that promoted by the β-agonist isoproterenol. Alternatively, it is possible that ICL3-9 binding to the β2AR might sterically hinder receptor interaction with GRKs and β-arrestin.
FIGURE 9.
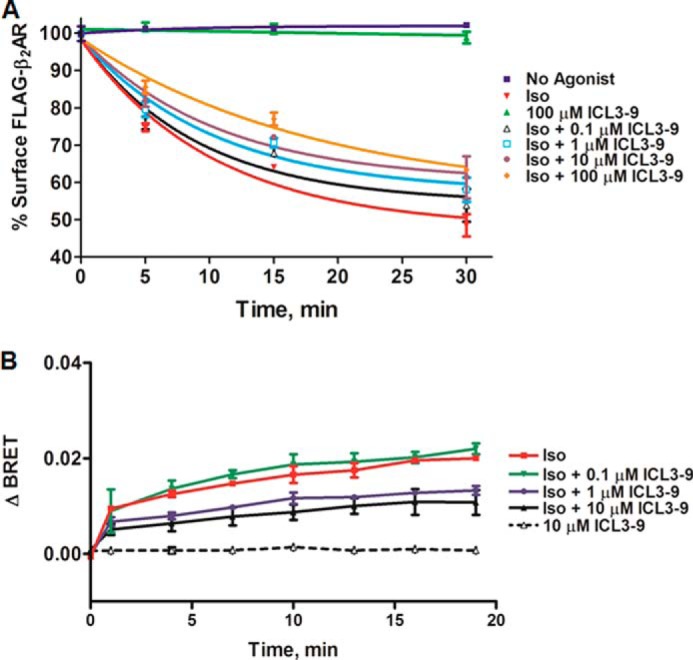
ICL3-9 antagonizes β-agonist promoted β2AR desensitization processes. A, 100 nm isoproterenol-induced β2AR internalization was monitored by cell surface ELISA in HEK 293 cells stably overexpressing a FLAG-β2AR that were pretreated with various concentrations of ICL3-9 for 5 min (0.05% DMSO was included in non-pepducin-treated cells). The data are represented by the mean of three independent experiments ± S.D. B, HEK 293 cells co-transfected with GFP10-β-arrestin2 and β2AR-RLucII (same pair as in Fig. 2B) were preincubated with coelenterazine 400a and various concentrations of ICL3-9 for 2 min and stimulated with 100 nm isoproterenol. BRET was monitored at the indicated times post-addition. Background BRET has been subtracted from the plotted points. The data are represented by the mean of four independent experiments ± S.D.
ICL3-9 Promotes a Unique Conformational Change in the β2AR
The structure of the β2AR-Gs complex suggests a large outward movement of TM6 that is unique to the proposed active state of the receptor (6). Site-specific monobromobimane labeling of Cys265 in TM6 of the β2AR allows detection of TM6 movement because Cys265 moves from a hydrophobic environment to a solvent-exposed position upon receptor activation (5, 45, 46). Monobromobimane is an environment-sensitive fluorophore that exhibits decreased fluorescence intensity and a red shift of peak emission in polar environments (45). Consequently, a decrease in fluorescence and an increase in λmax is indicative of a receptor conformational change at the proximal portion of TM6 (Fig. 10A). The addition of isoproterenol to purified Cys265 monobromobimane-labeled β2AR led to a dose-dependent decrease in fluorescence intensity demonstrating the effect of an orthosteric agonist on TM6 movement. In contrast, the addition of ICL3-9 did not promote any change in monobromobimane fluorescence suggesting that ICL3-9 does not promote significant movement of the proximal portion of TM6 (Fig. 10B). Similar results were observed for ICL3-8 (data not shown).
FIGURE 10.

ICL3-9 promotes unique conformational changes in the β2AR that promote Gs coupling. A, monobromobimane is an environmentally sensitive fluorophore that when chemically conjugated to β2AR-Cys265 can indicate local conformational changes. When the β2AR is in an inactive state, Cys265-monobromobimane is occupying a hydrophobic pocket and fluorescence is high. Upon receptor activation, a large outward movement of TM6 repositions Cys265 to be solvent-exposed resulting in decreased fluorescence and an increase in λmax. B, lipid bicelles containing 50 nm monobromobimane-labeled β2AR were incubated for 10 min at 25 °C with isoproterenol (300 nm or 1 μm) or ICL3-9 (20 or 100 μm) in 20 mm HEPES, pH 7.5, 100 mm NaCl. Fluorescence spectra were gathered by excitation at 370 nm and scanning 430–490 nm at 1.0 nm/s. 0.5% DMSO was included in non-pepducin-stimulated samples. C, lipid bicelles containing 50 nm monobromobimane-labeled β2AR were incubated for 10 min at 25 °C with 20 μm ICL3-9. 200 nm Gs was then incubated for 20 min at 25 °C in co-treatment studies. 0.1% DMSO was included in non-pepducin-stimulated samples. D, lipid bicelles containing 50 nm monobromobimane-labeled β2AR were incubated for 10 min at 25 °C with 20 μm ICL3-9A3. 200 nm Gs was then incubated for 20 min at 25 °C in co-treatment studies. 0.1% DMSO was included in non-pepducin-stimulated samples.
Although Gs binding to the β2AR is enhanced by β-agonists, Gs can also couple to unliganded β2AR and promote TM6 movement similar to agonist-induced changes (46). Indeed, a 4-fold molar excess of Gs led to an ∼10% decrease in fluorescence intensity and an increase in λmax (Fig. 10C). Interestingly, a 10-min preincubation of the β2AR with 20 μm ICL3-9 inhibited Gs-promoted TM6 movement (Fig. 10C), whereas addition of the inactive ICL3-9 variant ICL3-9A3 had no effect on Gs coupling to the β2AR (Fig. 10D). Biochemical analysis and BRET biosensors clearly demonstrate that ICL3-9 promotes β2AR-Gs coupling, but the inability of Gs to induce full TM6 movement on ICL3-9-treated β2AR suggests that the G protein may be coupling to the receptor in a different manner than that induced by isoproterenol.
To further monitor the conformational rearrangement between the β2AR and Gs, we took advantage of a distinct biosensor where the energy donor is positioned at the N terminus of Gαs (RLucII-Gαs) rather than at position 67 (Fig. 11A). Rather than promoting an increase in BRET as is observed with Gαs67-RLucII, isoproterenol stimulation promotes a decrease in BRET between β2AR-GFP10 and RLucII-Gαs. Such a difference in the BRET signal orientation reflects the different position of the energy donor and acceptor in the signaling complex and thus provides a means to assess the conformation of the receptor-Gs complex following activation from two reference points. Similar biosensors with different relative positions of the energy donor and acceptor have previously been used to probe the conformational changes occurring upon activation of Gi by the α2-adrenergic receptor (47). Surprisingly, ICL3-9 induced a steady increase in ΔBRET, in striking contrast to the decreased ΔBRET observed with isoproterenol treatment (Fig. 11B). This result clearly demonstrates that the β2AR conformation promoted by the pepducin is different from that induced by isoproterenol.
FIGURE 11.
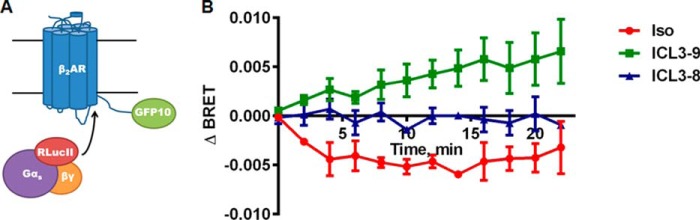
ICL3-9 promotes Gs coupling to the β2AR differently from a β-agonist. A, schematic of Gs engagement analysis by BRET. Upon β-agonist stimulation, RLucII-Gαs is engaged by the β2AR-GFP10 in a manner that causes a decrease in BRET signal. B, HEK 293 cells co-transfected with RLucII-Gαs and β2AR-GFP10 were preincubated with coelenterazine 400a for 2 min and stimulated with 10 μm pepducin or 10 μm isoproterenol. Changes in BRET were monitored over the indicated time points. 0.05% DMSO was included in non-pepducin-stimulated trials. The data are represented by the mean of three independent experiments ± S.D. In contrast to isoproterenol, ICL3-9 promoted an increase in BRET indicating a different mode of engagement of Gs by the pepducin-activated receptor that results in a different conformation of the complex.
DISCUSSION
Conventional stimulation of many GPCRs promotes interaction with heterotrimeric G proteins, GRKs, and β-arrestins to mediate G protein signaling, receptor desensitization and internalization, and β-arrestin-dependent signaling (1, 9). It is now understood that GPCR ligands cannot be simply classified as agonists or antagonists. Functional studies of a diverse set of ligands for the β2AR have demonstrated the complex spectrum of signaling profiles whereby the β2AR can couple to downstream pathways with unbalanced efficacies. This concept of “pluridimensional efficacy” or “ligand-biased signaling” was first observed for the β2AR when compounds that were previously characterized as receptor antagonists were reported to have the ability to stimulate β-arrestin-dependent MAPK signaling (48). Subsequent studies pioneered the re-classification of β2AR ligands. Drake et al. (49) monitored the correlation between indicators of receptor desensitization with G protein activation for a subset of receptor agonists and found that most promoted cAMP production and β-arrestin recruitment. However, the β-agonists N-cyclopentylbutanephrine, isoetharine, and ethyl-norepinephrine demonstrated the ability to more efficiently stimulate β-arrestin-dependent pathways over G protein activation. This β-arrestin-biased profile was attributed to an agonist-promoted increased rate of GRK phosphorylation of the receptor (49). van der Westhuizen et al. (50) continued the “taxonomy” of β2AR ligands by screening a number of full and partial β-agonists, neutral antagonists, and inverse agonists for their ability to modulate cAMP, Ca2+ mobilization, ERK1/2 phosphorylation, and β2AR internalization. Their findings revealed surprising diversity in the signaling profiles of closely related receptor ligands and further support the functional basis of ligand-biased signaling. Ligand biased signaling is believed to depend on an agonist's ability to induce a receptor conformation that either favors or disfavors interaction with downstream effector proteins. The net result of the interaction (or lack thereof) with these signaling molecules leads to the observed diverse signaling profiles. For the β2AR, structural studies further corroborated this notion as distinct ligand-dependent receptor conformations have been observed (5, 7, 8, 45, 51).
Pepducin discovery efforts have yielded a variety of allosteric agonists and antagonists for a diverse set of GPCRs over the past decade (18–25). Most recently, the CXCR4 pepducin ATI-2341 demonstrated receptor-dependent functional selectivity toward Gi over G13, GRK, and β-arrestin (26). To test the concept that pepducins might have a general ability to bias GPCR signaling, we screened 51 pepducins derived from the intracellular regions of the human β2AR for their ability to stimulate cAMP production and β-arrestin binding to the β2AR. This screen identified four distinct classes of pepducins that had agonist-like properties as follows: 1) one that functioned like a partial agonist to activate Gs and promote β-arrestin binding (ICL1-15); 2) a group that was β-arrestin-biased (ICL1-4, ICL1-11, and ICL1-20); 3) one that demonstrated a receptor-dependent Gs-bias (ICL3-9); and 4) a group that was Gs-biased and receptor-independent (e.g. ICL3-8). We further characterized the pepducins that promoted Gs-biased signaling and found that ICL3-8 directly activated Gs, whereas ICL3-9 induced conformational changes in the β2AR that promoted Gs activation.
ICL3-8 is derived from a region of the third intracellular loop that has been proposed to be a critical site of β2AR interaction with Gs. The crystal structure of the β2AR-Gs complex suggests extensive interaction between the proximal portion of the β2AR ICL3 (QKIDKSEGF) and the α5-β4 loop on Gαs (6). Interestingly, all of the pepducins from the proximal portion of ICL3 that activated cAMP production, including ICL3-8, contain this contact region between the receptor and G protein. Thus, this region likely plays a significant role in the guanine nucleotide exchange factor function of an activated receptor. However, it is also worth noting that this sequence alone was not sufficient to activate Gs because ICL3-1 through ICL3-8 all contain the QKIDKSEGF motif but vary in their efficacy from no activation of cAMP production (ICL3-5) to high activation (ICL3-2, -7 and -8) (Fig. 2A). This suggests that the surrounding sequence also contributes to regulating Gs interaction and/or activation. More specifically, the adjacent HV residues following the contact motif (missing in the β2AR-Gs structure) seem to play a role in the activity as pepducins that lack these residues (ICL3-4 and ICL3-5) show a loss of efficacy. In addition, it is possible that the sequence proximal to the QKIDKSEGF motif also contributes to Gs activation. Indeed, previous studies demonstrated that peptides from the proximal portion of the hamster β2AR ICL3 are direct activators of nucleotide exchange on Gs (52). The most effective peptide from these studies was VYSRVFQVAKRQLQK, which is proximal to the QKIDKSEGF motif. It is interesting that this particular sequence is fully contained within the ICL3-2 pepducin, which was the most effective activator of cAMP production (Fig. 2A). In the β2AR-Gs complex structure, this sequence makes extensive contact with the C-terminal tail of the Gαs subunit. The observed increased efficacy of ICL3-2 may be a product of the substantial contact surface between the pepducin and the Gαs subunit and the ability of multiple regions within the pepducin to activate Gs. Although previous studies used unmodified peptides that have a limited ability to cross the cell membrane (52), the N-terminal palmitoylation and C-terminal amidation of a pepducin enable membrane incorporation and effective delivery to the intracellular surface of the cell membrane (18). For ICL3-8, this provides a means of targeting it to the site of action as Gs is localized to the intracellular surface of the plasma membrane.
ICL3-9 targets the β2AR and stimulates interaction with Gs but no apparent interaction with GRKs or β-arrestins. Thus, ICL3-9 functions as a Gs-biased allosteric agonist for the β2AR. Multiple avenues of biophysical analysis have provided insight into the conformational changes that occur upon activation of the β2AR. Crystallographic studies of the activated β2AR-Gs complex demonstrate conformational changes in the β2AR proposed to be indicative of receptor activation. The most notable of these is a 14-Å outward movement of the proximal portion of TM6 and a helical extension of the distal portion of TM5 into the intracellular space (6). NMR analysis of the β2AR confirms mobility in both of these regions (8), whereas monobromobimane labeling of Cys265 of the β2AR confirms movement of the cytoplasmic end of TM6 upon β-agonist treatment (5, 45, 46). In our studies, ICL3-9 did not affect the movement of TM6, and thus its mechanism of receptor-Gs coupling may be different from that induced by conventional β-agonists. As ICL3-9 clearly promotes G protein engagement with the β2AR (BRET biosensors) and receptor-mediated activation of Gs (GTPγS loading in β2AR-containing bicelles) without detectable influence on TM6, conformational changes in TM6 of the β2AR may not be a critical step in adopting the ICL3-9 promoted Gs-biased receptor state. A helical extension of TM5 into the intracellular surface is also associated with agonist-induced β2AR activation (6). This helical extension contains many of the residues in direct contact with Gs in the β2AR-Gs structure and also shares similarity to the peptides that can directly activate Gs. Plausibly, ICL3-9 may have the ability to modulate movement of TM5 and promote a unique active conformation utilizing the helical extension of TM5 to unconventionally activate Gs. It should also be noted that molecular dynamics simulations of the β2AR show a weak relationship between conformational changes in the ligand-binding pocket and TM5/6 movement (8, 53). Thus, the ability of the pepducin to promote conformational changes in this region does not depend on operation through an orthosteric mechanism.
Corroborating the notion that ICL3-9 induces a distinct active state of the β2AR, ICL3-9 appears to promote Gs coupling to the receptor in a different manner than the β-agonist. Although Gs can interact with unliganded mBB-β2AR and promote detectable TM6 movement (46), ICL3-9 possesses the ability to attenuate conventional Gs-promoted TM6 movement, suggesting that ICL3-9 induces a unique coupling of the β2AR and Gs (Fig. 10C). Additionally, BRET analysis monitoring RLucII-Gαs engagement to β2AR-GFP10 demonstrated that the N-terminal region of Gαs may be oriented differently when associated with the ICL3-9 activated receptor as opposed to a β-agonist occupied receptor (Fig. 11). Although not much is known about the conformational changes of the Gα N terminus during G protein activation (54), the observed differences in the BRET signal orientation between RLucII-Gαs and β2AR-GFP10 upon activation with ICL3-9 (increase) versus isoproterenol (decrease) suggest that the position of the N terminus of Gαs differs between the isoproterenol- and ICL3-9-activated receptor states. The loss in BRET upon isoproterenol stimulation reflects a conformational rearrangement of the pre-coupled β2AR-Gs complex (40) that results in an increase in the distance between the GFP at the C terminus of the β2AR and the RLucII at the N terminus of Gαs. In contrast, the ICL3-9-stimulated functional engagement of Gs promotes an increase in BRET and thus reflects a reduction in the distance between the N terminus of Gαs and the C terminus of the β2AR. When a different viewpoint of the complex is monitored using BRET between β2AR-GFP10 and RLucII inserted in the linker 1 region of Gαs between the helical and GTPase domains, comparable BRET changes in direction and efficiency between isoproterenol and ICL3-9 are observed. This suggests that this region of Gαs may be oriented similarly in both the pepducin- and β-agonist-stimulated states.
Although ICL3-9 appears to be the first reported Gs-biased activator of the β2AR, Staus et al. (55) previously reported stimulation of biased signaling from the β2AR using intracellularly expressed nanobodies (intrabodies). In this report, intrabodies against agonist-activated or inactive β2AR were selective for inhibiting G protein activation or GRK and β-arrestin engagement. The expression of intrabodies that block GRK phosphorylation shift the activation profile of a β-agonist from activating G proteins and β-arrestin to one that selectively stimulates G protein signaling. Essentially, these intrabodies transform a balanced agonist to a Gs-biased β2AR modulator, although they do not have the ability to activate signaling on their own. Moreover, it is unknown whether the intrabodies stabilize an agonist-bound conformation that favors Gs activation or sterically hinders GRK interaction with the β2AR, while not affecting Gs activation. Although ICL3-9 can directly promote β2AR-dependent Gs-biased signaling, we also do not know whether this is due to the ICL3-9-induced conformation being unable to mediate GRK/β-arrestin binding or whether ICL3-9 directly inhibits GRK/β-arrestin binding to the β2AR. Identification of the ICL3-9-binding site on the β2AR will be critical in answering these questions.
Because pepducins have been historically thought to be specific for their cognate receptor, another interesting aspect of ICL3-9 was its ability to utilize both the β1AR and β2AR to mediate cAMP production. The β1AR and β2AR are closely related family members and share ∼54% amino acid identity with the transmembrane domains and intracellular loops being the most conserved. For example, ICL1 of the β2AR (Ile55–Leu75) is ∼71% identical with ICL1 of the β1AR (Leu78–Ala101), whereas ICL2 is 77% identical between the β2AR (Val129–Leu155) and β1AR (Leu154–Leu178). The ICL3 of the β2AR (Phe217–Ile288) and the β1AR (Phe241–Ile329) exhibit lower homology with ∼38% identity, although this is partly due to a proline-rich insert between Leu266 and Ala302 in the β1AR. Interestingly, the ICL3 of β1AR lacks multiple residues in the ICL3-9 sequence. The commonality of sequence features on the intracellular surface of both the β1AR and β2AR may be the reason that ICL3-9 can activate Gs through both receptors as highly homologous regions may be indistinguishable to the binding and/or function of ICL3-9. Conversely, uncommon regions between the two receptors (i.e. sequence divergence in ICL3) are unlikely to play a critical role in ICL3-9 operation. For example, the C-terminal tail of the β1AR and β2AR does not exhibit significant homology and thus is unlikely to contribute to the interaction or activity of ICL3-9.
Previous studies on the β2AR also identified a few inverse agonists such as carvedilol and nebivolol that appear to function as β-arrestin-biased agonists by selectively promoting β-arrestin recruitment and signaling over G protein activation (56, 57). It is worth noting that carvedilol is used in the treatment of heart failure, and its ability to function in a β-arrestin-biased manner appears to be cardioprotective (57–59). Although we were not specifically searching for β-arrestin-biased pepducins, we identified a few pepducins from ICL1 that had a clear β-arrestin bias with one being ∼75% as effective as isoproterenol in promoting β-arrestin binding (Fig. 2, C and D). Characterizing the ability of the β2AR to selectively activate β-arrestin-associated pathways (e.g. receptor internalization and β-arrestin-dependent signaling) using our β-arrestin-biased pepducins could be an interesting avenue of future investigation. Moreover, our results suggest that pepducins may prove particularly useful in dissecting the mechanisms involved in biased signaling and the potential links between β-arrestin activation and the treatment of heart failure.
Gs-biased pepducins demonstrated an independence from classical receptor desensitization mechanisms as the induction of functional desensitization of the β2AR was not observed in primary human airway smooth muscle cells. These data serve as an initial “proof-of-concept” that Gs-biased agonists could serve as a potentially advantageous asthma therapeutic. Certainly, ICL3-9 is a potentially attractive lead drug candidate as its receptor dependence leads to a degree of drug targeting and specificity, although its potency must be improved. Direct activators of Gs such as ICL3-2 and ICL3-8 might also prove advantageous in promoting airway smooth muscle relaxation, while targeting these molecules to the proper cell type will be critical. Beyond their application in the treatment of asthma, the pepducins provide additional tools to study β2AR activation and the benefits of Gs-biased signaling.
Acknowledgments
We thank Drs. Christopher So and Ken Carlson for valuable discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants R37 GM047417 and R01 GM044944 (to J. L. B.), P01 HL114471 (to J. L. B. and R. A. P.), P30 ES013508 (to R. A. P.), and T32 GM100836 (to R. C.). This work was also supported by Canadian Institute for Health Research Grant CIHR-11215 (to M. B.).
This article was selected as a Paper of the Week.
- β2AR
- β2-adrenergic receptor
- GPCR
- G protein-coupled receptor
- GRK
- G protein-coupled receptor kinase
- Gs
- Gαsβγ heterotrimer
- ASM
- airway smooth muscle
- CXCR
- CXC chemokine receptor
- ICL
- intracellular loop
- IBMX
- 3-isobutyl-1-methylxanthine
- BRET
- bioluminescence resonance energy transfer
- RLucII
- Renilla reniformis luciferase II
- β1AR
- β1-adrenergic receptor
- EP2R
- prostaglandin E2 receptor
- DDM
- n-dodecyl-β-d-maltoside
- DOPC
- dimyristoyl phosphatidylcholine
- CHAPSO
- 3-([3-cholamidopropyl]dimethylammonio)-2-hydroxy-1-propanesulfonate
- CHS
- cholesterol hemisuccinate
- GTPγS
- guanosine 5′-O-[γ-thio]triphosphate
- mBB
- monobromobimane
- TM
- transmembrane domain
- DPBS
- Dulbecco's phosphate-buffered saline
- HBSS
- Hanks' buffered saline solution.
REFERENCES
- 1. Lefkowitz R. J. (2007) Seven transmembrane receptors: something old, something new. Acta Physiol. 190, 9–19 [DOI] [PubMed] [Google Scholar]
- 2. Cherezov V., Rosenbaum D. M., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Kuhn P., Weis W. I., Kobilka B. K., Stevens R. C. (2007) High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rasmussen S. G., Choi H. J., Rosenbaum D. M., Kobilka T. S., Thian F. S., Edwards P. C., Burghammer M., Ratnala V. R., Sanishvili R., Fischetti R. F., Schertler G. F., Weis W. I., Kobilka B. K. (2007) Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature 450, 383–387 [DOI] [PubMed] [Google Scholar]
- 4. Rosenbaum D. M., Cherezov V., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Yao X. J., Weis W. I., Stevens R. C., Kobilka B. K. (2007) GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science 318, 1266–1273 [DOI] [PubMed] [Google Scholar]
- 5. Rasmussen S. G., Choi H. J., Fung J. J., Pardon E., Casarosa P., Chae P. S., Devree B. T., Rosenbaum D. M., Thian F. S., Kobilka T. S., Schnapp A., Konetzki I., Sunahara R. K., Gellman S. H., Pautsch A., Steyaert J., Weis W. I., Kobilka B. K. (2011) Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature 469, 175–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zocher M., Fung J. J., Kobilka B. K., Müller D. J. (2012) Ligand-specific interactions modulate kinetic, energetic, and mechanical properties of the human β2 adrenergic receptor. Structure 20, 1391–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nygaard R., Zou Y., Dror R. O., Mildorf T. J., Arlow D. H., Manglik A., Pan A. C., Liu C. W., Fung J. J., Bokoch M. P., Thian F. S., Kobilka T. S., Shaw D. E., Mueller L., Prosser R. S., Kobilka B. K. (2013) The dynamic process of β(2)-adrenergic receptor activation. Cell 152, 532–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deshpande D. A., Penn R. B. (2006) Targeting G protein-coupled receptor signaling in asthma. Cell. Signal. 18, 2105–2120 [DOI] [PubMed] [Google Scholar]
- 10. Schellenberg R., Lichtenthal A., Wöhling H., Graf C., Brixius K. (2008) Nebivolol and metoprolol for treating migraine: an advance on β-blocker treatment? Headache 48, 118–125 [DOI] [PubMed] [Google Scholar]
- 11. Baker J. G., Hill S. J., Summers R. J. (2011) Evolution of β-blockers: from anti-anginal drugs to ligand-directed signalling. Trends Pharmacol. Sci. 32, 227–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cazzola M., Calzetta L., Matera M. G. (2011) β2-Adrenoceptor agonists: current and future direction. Br. J. Pharmacol. 163, 4–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Poirier L., Tobe S. W. (2014) Contemporary use of β-blockers: clinical relevance of subclassification. Can. J. Cardiol. 30, S9–S15 [DOI] [PubMed] [Google Scholar]
- 14. Lohse M. J., Benovic J. L., Codina J., Caron M. G., Lefkowitz R. J. (1990) β-arrestin: a protein that regulates beta-adrenergic receptor function. Science 248, 1547–1550 [DOI] [PubMed] [Google Scholar]
- 15. Nelson H. S., Weiss S. T., Bleecker E. R., Yancey S. W., Dorinsky P. M., and SMART Study Group (2006) The Salmeterol Multicenter Asthma Research Trial: a comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest 129, 15–26 [DOI] [PubMed] [Google Scholar]
- 16. McMahon A. W., Levenson M. S., McEvoy B. W., Mosholder A. D., Murphy D. (2011) Age and risks of FDA-approved long-acting β(2)-adrenergic receptor agonists. Pediatrics 128, e1147–e1154 [DOI] [PubMed] [Google Scholar]
- 17. Deshpande D. A., Theriot B. S., Penn R. B., Walker J. K. (2008) β-Arrestins specifically constrain β2-adrenergic receptor signaling and function in airway smooth muscle. FASEB J. 22, 2134–2141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. O'Callaghan K., Kuliopulos A., Covic L. (2012) Turning receptors on and off with intracellular pepducins: new insights into G-protein-coupled receptor drug development. J. Biol. Chem. 287, 12787–12796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Covic L., Gresser A. L., Talavera J., Swift S., Kuliopulos A. (2002) Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc. Natl. Acad. Sci. U.S.A. 99, 643–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Covic L., Misra M., Badar J., Singh C., Kuliopulos A. (2002) Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat. Med. 8, 1161–1165 [DOI] [PubMed] [Google Scholar]
- 21. Kaneider N. C., Agarwal A., Leger A. J., Kuliopulos A. (2005) Reversing systemic inflammatory response syndrome with chemokine receptor pepducins. Nat. Med. 11, 661–665 [DOI] [PubMed] [Google Scholar]
- 22. Lee H. Y., Kim S. D., Shim J. W., Kim H. J., Kwon J. Y., Kim J. M., Baek S. H., Park J. S., Bae Y. S. (2010) Activation of human monocytes by a formyl peptide receptor 2-derived pepducin. FEBS Lett. 584, 4102–4108 [DOI] [PubMed] [Google Scholar]
- 23. Tchernychev B., Ren Y., Sachdev P., Janz J. M., Haggis L., O'Shea A., McBride E., Looby R., Deng Q., McMurry T., Kazmi M. A., Sakmar T. P., Hunt S., 3rd, Carlson K. E. (2010) Discovery of a CXCR4 agonist pepducin that mobilizes bone marrow hematopoietic cells. Proc. Natl. Acad. Sci. U.S.A. 107, 22255–22259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Remsberg J. R., Lou H., Tarasov S. G., Dean M., Tarasova N. I. (2007) Structural analogues of smoothened intracellular loops as potent inhibitors of Hedgehog pathway and cancer cell growth. J. Med. Chem. 50, 4534–4538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Licht T., Tsirulnikov L., Reuveni H., Yarnitzky T., Ben-Sasson S. A. (2003) Induction of pro-angiogenic signaling by a synthetic peptide derived from the second intracellular loop of S1P3 (EDG3). Blood 102, 2099–2107 [DOI] [PubMed] [Google Scholar]
- 26. Quoyer J., Janz J. M., Luo J., Ren Y., Armando S., Lukashova V., Benovic J. L., Carlson K. E., Hunt S. W., 3rd, Bouvier M. (2013) Pepducin targeting the C-X-C chemokine receptor type 4 acts as a biased agonist favoring activation of the inhibitory G protein. Proc. Natl. Acad. Sci. U.S.A. 110, E5088–E5097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Penn R. B., Panettieri R. A., Jr., Benovic J. L. (1998) Mechanisms of acute desensitization of the β2AR-adenylyl cyclase pathway in human airway smooth muscle. Am. J. Respir. Cell Mol. Biol. 19, 338–348 [DOI] [PubMed] [Google Scholar]
- 28. Hamdan F. F., Audet M., Garneau P., Pelletier J., Bouvier M. (2005) High-throughput screening of G protein-coupled receptor antagonists using a bioluminescence resonance energy transfer 1-based β-arrestin2 recruitment assay. J. Biomol. Screen. 10, 463–475 [DOI] [PubMed] [Google Scholar]
- 29. Tran T. M., Friedman J., Qunaibi E., Baameur F., Moore R. H., Clark R. B. (2004) Characterization of agonist stimulation of cAMP-dependent protein kinase and G protein-coupled receptor kinase phosphorylation of the β2-adrenergic receptor using phosphoserine-specific antibodies. Mol. Pharmacol. 65, 196–206 [DOI] [PubMed] [Google Scholar]
- 30. Daunt D. A., Hurt C., Hein L., Kallio J., Feng F., Kobilka B. K. (1997) Subtype-specific intracellular trafficking of α2-adrenergic receptors. Mol. Pharmacol. 51, 711–720 [DOI] [PubMed] [Google Scholar]
- 31. Panettieri R. A., Murray R. K., DePalo L. R., Yadvish P. A., Kotlikoff M. I. (1989) A human airway smooth muscle cell line that retains physiological responsiveness. Am. J. Physiol. 256, C329–C335 [DOI] [PubMed] [Google Scholar]
- 32. Cooper P. R., Mesaros A. C., Zhang J., Christmas P., Stark C. M., Douaidy K., Mittelman M. A., Soberman R. J., Blair I. A., Panettieri R. A. (2010) 20-HETE mediates ozone-induced, neutrophil-independent airway hyper-responsiveness in mice. PLoS One 5, e10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vaughan D. J., Millman E. E., Godines V., Friedman J., Tran T. M., Dai W., Knoll B. J., Clark R. B., Moore R. H. (2006) Role of the G protein-coupled receptor kinase site serine cluster in β2-adrenergic receptor internalization, desensitization, and β-arrestin translocation. J. Biol. Chem. 281, 7684–7692 [DOI] [PubMed] [Google Scholar]
- 34. Nobles K. N., Xiao K., Ahn S., Shukla A. K., Lam C. M., Rajagopal S., Strachan R. T., Huang T. Y., Bressler E. A., Hara M. R., Shenoy S. K., Gygi S. P., Lefkowitz R. J. (2011) Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal. 4, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kang D. S., Tian X., Benovic J. L. (2014) Role of β-arrestins and arrestin domain-containing proteins in G protein-coupled receptor trafficking. Curr. Opin. Cell Biol. 27, 63–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. January B., Seibold A., Allal C., Whaley B. S., Knoll B. J., Moore R. H., Dickey B. F., Barber R., Clark R. B. (1998) Salmeterol-induced desensitization, internalization and phosphorylation of the human β2-adrenoceptor. Br. J. Pharmacol. 123, 701–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kallal L., Gagnon A. W., Penn R. B., Benovic J. L. (1998) Visualization of agonist-induced sequestration and down-regulation of a green fluorescent protein-tagged β2-adrenergic receptor. J. Biol. Chem. 273, 322–328 [DOI] [PubMed] [Google Scholar]
- 38. Hollingsworth J. W., Theriot B. S., Li Z., Lawson B. L., Sunday M., Schwartz D. A., Walker J. K. (2010) Both hematopoietic-derived and non-hematopoietic-derived β-arrestin-2 regulates murine allergic airway disease. Am. J. Respir. Cell Mol. Biol. 43, 269–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Walker J. K., Fong A. M., Lawson B. L., Savov J. D., Patel D. D., Schwartz D. A., Lefkowitz R. J. (2003) β-Arrestin-2 regulates the development of allergic asthma. J. Clin. Invest. 112, 566–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Galés C., Rebois R. V., Hogue M., Trieu P., Breit A., Hébert T. E., Bouvier M. (2005) Real-time monitoring of receptor and G-protein interactions in living cells. Nat. Methods 2, 177–184 [DOI] [PubMed] [Google Scholar]
- 41. Samama P., Cotecchia S., Costa T., Lefkowitz R. J. (1993) A mutation-induced activated state of the β2-adrenergic receptor. Extending the ternary complex model. J. Biol. Chem. 268, 4625–4636 [PubMed] [Google Scholar]
- 42. Luttrell L. M. (2014) Minireview: More than just a hammer: ligand “bias” and pharmaceutical discovery. Mol. Endocrinol. 28, 281–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chidiac P., Hebert T. E., Valiquette M., Dennis M., Bouvier M. (1994) Inverse agonist activity of β-adrenergic antagonists. Mol. Pharmacol. 45, 490–499 [PubMed] [Google Scholar]
- 44. Samama P., Pei G., Costa T., Cotecchia S., Lefkowitz R. J. (1994) Negative antagonists promote an inactive conformation of the β2-adrenergic receptor. Mol. Pharmacol. 45, 390–394 [PubMed] [Google Scholar]
- 45. Yao X., Parnot C., Deupi X., Ratnala V. R., Swaminath G., Farrens D., Kobilka B. (2006) Coupling ligand structure to specific conformational switches in the β2-adrenoceptor. Nat. Chem. Biol. 2, 417–422 [DOI] [PubMed] [Google Scholar]
- 46. Yao X. J., Vélez Ruiz G., Whorton M. R., Rasmussen S. G., DeVree B. T., Deupi X., Sunahara R. K., Kobilka B. (2009) The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proc. Natl. Acad. Sci. U.S.A. 106, 9501–9506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Galés C., Van Durm J. J., Schaak S., Pontier S., Percherancier Y., Audet M., Paris H., Bouvier M. (2006) Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat. Struct. Mol. Biol. 13, 778–786 [DOI] [PubMed] [Google Scholar]
- 48. Azzi M., Charest P. G., Angers S., Rousseau G., Kohout T., Bouvier M., Piñeyro G. (2003) β-Arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc. Natl. Acad. Sci. U.S.A. 100, 11406–11411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Drake M. T., Violin J. D., Whalen E. J., Wisler J. W., Shenoy S. K., Lefkowitz R. J. (2008) β-arrestin-biased agonism at the β2-adrenergic receptor. J. Biol. Chem. 283, 5669–5676 [DOI] [PubMed] [Google Scholar]
- 50. van der Westhuizen E. T., Breton B., Christopoulos A., Bouvier M. (2014) Quantification of ligand bias for clinically relevant β2-adrenergic receptor ligands: implications for drug taxonomy. Mol. Pharmacol. 85, 492–509 [DOI] [PubMed] [Google Scholar]
- 51. Liu J. J., Horst R., Katritch V., Stevens R. C., Wüthrich K. (2012) Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science 335, 1106–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cheung A. H., Huang R. R., Graziano M. P., Strader C. D. (1991) Specific activation of Gs by synthetic peptides corresponding to an intracellular loop of the β-adrenergic receptor. FEBS Lett. 279, 277–280 [DOI] [PubMed] [Google Scholar]
- 53. Dror R. O., Arlow D. H., Maragakis P., Mildorf T. J., Pan A. C., Xu H., Borhani D. W., Shaw D. E. (2011) Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. U.S.A. 108, 18684–18689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Baltoumas F. A., Theodoropoulou M. C., Hamodrakas S. J. (2013) Interactions of the α-subunits of heterotrimeric G-proteins with GPCRs, effectors and RGS proteins: a critical review and analysis of interacting surfaces, conformational shifts, structural diversity and electrostatic potentials. J. Struct. Biol. 182, 209–218 [DOI] [PubMed] [Google Scholar]
- 55. Staus D. P., Wingler L. M., Strachan R. T., Rasmussen S. G., Pardon E., Ahn S., Steyaert J., Kobilka B. K., Lefkowitz R. J. (2014) Regulation of β2-adrenergic receptor function by conformationally selective single-domain intrabodies. Mol. Pharmacol. 85, 472–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Erickson C. E., Gul R., Blessing C. P., Nguyen J., Liu T., Pulakat L., Bastepe M., Jackson E. K., Andresen B. T. (2013) The β-blocker Nebivolol Is a GRK/β-arrestin biased agonist. PloS one 8, e71980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wisler J. W., DeWire S. M., Whalen E. J., Violin J. D., Drake M. T., Ahn S., Shenoy S. K., Lefkowitz R. J. (2007) A unique mechanism of β-blocker action: carvedilol stimulates β-arrestin signaling. Proc. Natl. Acad. Sci. U.S.A. 104, 16657–16662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bril A., Slivjak M., DiMartino M. J., Feuerstein G. Z., Linee P., Poyser R. H., Ruffolo R. R., Jr., Smith E. F., 3rd (1992) Cardioprotective effects of carvedilol, a novel β-adrenoceptor antagonist with vasodilating properties, in anaesthetised minipigs: comparison with propranolol. Cardiovasc. Res. 26, 518–525 [DOI] [PubMed] [Google Scholar]
- 59. Ruffolo R. R., Jr., Bril A., Feuerstein G. Z. (1993) Cardioprotective potential of carvedilol. Cardiology 82, Suppl. 3, 24–28 [DOI] [PubMed] [Google Scholar]