

Background: HCV core induces liver steatosis.
Results: Core inhibits the triglyceride hydrolase activity of ATGL while enhancing its interaction with its cofactor CGI-58 and with lipid droplets.
Conclusion: ATGL is a new functional cofactor of core-induced liver steatosis.
Significance: Defining the disruption of ATGL function mediated by core provides new insight into the mechanisms of ATGL activation.
Keywords: Adipose Triglyceride Lipase (ATGL), Hepatitis C Virus (HCV), Lipid Droplet, Lipolysis, Triglyceride
Abstract
Liver steatosis is a common health problem associated with hepatitis C virus (HCV) and an important risk factor for the development of liver fibrosis and cancer. Steatosis is caused by triglycerides (TG) accumulating in lipid droplets (LDs), cellular organelles composed of neutral lipids surrounded by a monolayer of phospholipids. The HCV nucleocapsid core localizes to the surface of LDs and induces steatosis in cultured cells and mouse livers by decreasing intracellular TG degradation (lipolysis). Here we report that core at the surface of LDs interferes with the activity of adipose triglyceride lipase (ATGL), the key lipolytic enzyme in the first step of TG breakdown. Expressing core in livers or mouse embryonic fibroblasts of ATGL−/− mice no longer decreases TG degradation as observed in LDs from wild-type mice, supporting the model that core reduces lipolysis by engaging ATGL. Core must localize at LDs to inhibit lipolysis, as ex vivo TG hydrolysis is impaired in purified LDs coated with core but not when free core is added to LDs. Coimmunoprecipitation experiments revealed that core does not directly interact with the ATGL complex but, unexpectedly, increased the interaction between ATGL and its activator CGI-58 as well as the recruitment of both proteins to LDs. These data link the anti-lipolytic activity of the HCV core protein with altered ATGL binding to CGI-58 and the enhanced association of both proteins with LDs.
Introduction
Hepatitis C virus (HCV)5 is a global health problem and a major cause of liver-associated mortality. More than 50% of individuals infected with HCV suffer from liver steatosis, an important risk factor for developing liver fibrosis and cancer. Steatosis is caused by the accumulation of triglycerides (TG) in cellular lipid droplets (LDs) (1). LDs consist of a hydrophobic core composed of mainly TG and cholesterol esters surrounded by a phospholipid monolayer. During the fed state, more TG and cholesterol esters are synthesized at the endoplasmic reticulum (ER) and stored in LDs, which increases the number and size of LDs (i.e. lipogenesis). Under energy demands (e.g. fasting, exercise), stored lipids are hydrolyzed (i.e. lipolysis) (2). Thereby, free fatty acids (FFAs) are sequentially removed from the glycerol backbone of the TG molecule by at least three different enzymes. These sequential reactions are initiated by adipose triglyceride lipase (ATGL) and its activator comparative gene identification 58 (CGI-58) and then further carried out by hormone-sensitive lipase (HSL) and monoacylglycerol lipase (3). Under basal conditions, ATGL mostly localizes to ER-related membranes in the cytoplasm (4), although a small fraction of ATGL also complexes with its inhibitor, G0/G1 switch gene 2 (G0S2), on LDs (5). Upon energy demand and the subsequent rise in intracellular cAMP levels, activation of protein kinases and phosphorylation of LD-associated proteins (e.g. PLIN1 and PLIN5) as well as ATGL and HSL leads to the translocation of the two lipases to LDs, where ATGL binds to and is stimulated by CGI-58 to acutely increase lipolysis (6). Conversely, in response to food intake and increasing insulin concentrations, G0S2 directly inhibits ATGL activity to decrease lipolysis (5).
The HCV nucleocapsid core is a major structural component of virions and plays a central role in HCV pathogenesis (7). Core contains a hydrophobic C-terminal signal peptide that anchors it to the ER membrane, where it is cleaved by two host peptidases (8). Processed core remains attached to the outer leaflet of the ER membrane and can access the surface of LDs, a crucial step in successfully assembling new virions (9). Previously we reported that core traffics from the ER to LDs through the activity of diacylglycerol acyltransferase 1 (DGAT1), one of the two enzymes that catalyzes the final step in TG biosynthesis (10). We further showed that core at the surface of LDs impairs LD turnover, thereby inducing steatosis in cell culture and murine livers (11). Core also affects other steatogenic mechanisms, such as inhibiting microsomal triglyceride transfer protein (MTP), a key protein in VLDL assembly and release from hepatocytes (12), down-regulating phosphatase and tensin homolog (PTEN), and insulin receptor substrate 1 (IRS-1), thereby inducing the formation of larger LDs (13).
In addition, core increases lipogenesis by altering expression of genes involved in fatty acid biosynthesis. Specifically, core activates transcription factors, such as sterol regulatory element-binding proteins and peroxisome proliferator-activated receptors α and γ in cultured cells and in core-transgenic mice (14–17). Core directly binds and activates the DNA binding domain of the retinoid X receptor, a transcriptional regulator involved in cellular lipid synthesis (18). Interestingly, lipogenesis induction through sterol regulatory element-binding protein has also recently been linked to the 3′-untranslated region of the HCV single-stranded RNA genome (19).
Although the mechanisms by which core affects lipogenesis are well defined, those explaining how core inhibits lipolysis remain unclear. To address this question, we investigated the anti-lipolytic properties of core in ATGL deficient (ATGL−/−) mice. Core expression in these mice did not decrease lipolysis of isolated LDs pointing to ATGL as a key enzyme mediating core's effect on lipolysis. Notably, the anti-lipolytic function of core was only observed when core and ATGL were located at LDs. Moreover core expression was associated with elevated binding of ATGL to its coactivator CGI-58 and to LDs. Collectively, our data identify the ATGL·CGI-58 complex as a molecular target of the HCV core protein and indicate that dynamics of LD association/dissociation or the stability of the complex at LDs might play a critical new role in the regulation of lipolysis.
EXPERIMENTAL PROCEDURES
Plasmids
Lentiviral and adenoviral constructs of core have been previously described (11). The HA-core construct is described in Camus et al. (20). N-terminal His-tagged ATGL, CGI-58, G0S2, and HSL were generated by inserting the human or mouse cDNA of these genes in the pcDNA4/HisMax C vector (Invitrogen) (21).
Cell Lines and Culture Conditions
NIH/3T3, Huh7, HEK293T, and COS-7 cells were obtained from ATCC. Cells were grown under standard cell culture conditions in DMEM or RPMI (Invitrogen) containing 10% fetal bovine serum or bovine calf serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin (5% CO2, 95% humidity, 37 °C) and transfected with X-tremeGENE 9 DNA Transfection Reagent (Roche Applied Science) or Metafectene (Biontex) according to the manufacturer's protocol. Calcium phosphate-mediated transfection of HEK293T cells was used to produce lentiviral particles. Mouse embryonic fibroblasts (MEFs) were established from ATGL−/− embryos or their control littermates as described previously (22).
Animal Studies
ATGL−/− mice have been described previously (23). 8–10-week-old male ATGL−/− mice and control C57BL6 mice were administered 4.5 × 1010 pfu of adenovirus expressing GFP or core/GFP in 300 μl of normal saline by tail vein injection. Four days later, mice were sacrificed, and livers were excised. All animal experiments were approved by the UCSF Institutional Animal Care and Use Committees.
Antibodies and Reagents
The following antibodies were obtained commercially: anti-core (clone C7–50, Affinity BioReagents), anti-ADRP (GP40, Progen), anti-ATGL (2138S, Cell Signaling), anti-CGI-58 (H00051099-M01, Abnova), anti-tubulin (ab28439, Abcam), anti-His (#27-4710-01, Amersham Biosciences), anti-GFP (FL, Santa Cruz), anti-FLAG M2 (Sigma), anti-FLAG (F7425, Sigma), anti-mouse Ig HRP (eBioscience), and anti-rabbit Ig HRP (eBioscience). Oleic acid-albumin from bovine serum was from Sigma (O3008).
Lentivirus and Adenovirus Production and Transduction
Lentiviral particles were produced as described previously (10). High-titer adenoviral stocks were produced by the Vector Development Laboratory at the Baylor College of Medicine. Cfus were determined by infecting HEK293 cells with serial dilutions of viral stocks and counting GFP-positive foci 2 days post-infection. The efficiency of NIH/3T3 cells transduction with lentiviruses was verified by microscopic analysis of GFP expression.
Lipogenesis in Cultured Cells
To evaluate de novo lipogenesis, Cos-7 cells were loaded with radiolabeled [14C]glucose (d-[U-14C]glucose] from American Radiolabeled Chemicals) (0.1 μCi/ml) in medium containing 1 g/liter glucose (i.e. 7.5 μmol per well of a 6-well plate) for 20 h. Cells were then washed with phosphate-buffered saline (PBS), and lipids were extracted by the Folch method (chloroform:methanol:acetic acid, 2:1:1%). Total lipids were re-dissolved in chloroform and separated by thin-layer chromatography using hexane:diethylether:acetic acid (70:29:1) as solvent. Bands corresponding to TGs were excised, and radioactivity was determined by liquid scintillation counting. Cells were lysed using SDS/NaOH (0.1%/0.3 n), and protein concentrations were determined using bicinchoninic acid (BCA) reagent (Pierce).
In Vivo Lipogenesis
Mice injected with adenovirus expressing core/GFP or GFP were given an intraperitoneal injection of 2H-labeled saline (3.5% volume/body weight of 99.9% 2H2O containing 0.9% NaCl) 4 days after infection. After injection, mice were returned to their cages for 5 h before liver samples were freeze-clamped using Wollenberger tongs, weighed, and subjected to MS analysis. 2H-Labeled plasma water and total palmitate, cholesterol, and glycerol were determined as previously described (24–26). Briefly, 2H-labeled water was determined after H/D exchange with acetone (24). Liver tissue was saponified, and the 2H-labeled glycerol was determined after conversion to its triacetate derivative and analyzed by ammonia chemical ionization; total enrichment was determined from the M+18 adduct ion (25). The 2H-labeled palmitate and cholesterol was determined by reacting samples with TMS-diazomethane (to convert fatty acids to their methyl esters) and then with acetic anhydride (to convert cholesterol to its acetyl derivative). Samples were analyzed under electron impact ionization, and the enrichment was determined by selected ion monitoring m/z 270–272 (palmitate) and 368–370 (cholesterol). Newly synthesized fractions were determined using previously reported equations (26). Briefly, we compared the total labeling of glycerol, palmitate, or cholesterol against that of water assuming constants of 4.5, 22, or 25, respectively (25, 26).
Statistical Analysis
Error is as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001. When one variable differed between experimental groups, unpaired two-tailed Student's t tests were used. When experimental groups differed in two variables, two-way analysis of variance was used with the appropriate post-testing.
Lipolysis of Isolated Liver Lipid Droplets
The measurement of lipid droplet associated lipolysis activity was performed as described (27). Briefly, livers were excised, intensively washed with PBS, and placed on ice. Tissues were homogenized in buffer A (0.25 m sucrose, 1 mm EDTA, 1 mm DTT, pH 7.0, 1 μg/ml pepstatin, 2 μg/ml antipain, and 20 μg/ml leupeptin as protease inhibitors) and centrifuged at 1000 × g and 4 °C for 10 min to remove non-disrupted tissue and nuclei. Post-nuclear fractions were transferred to siliconized tubes overlaid with overlay buffer (50 mm potassium phosphate buffer, pH 7.4, 100 mm KCl, 1 mm EDTA, 1 μg/ml pepstatin, 2 μg/ml antipain, and 20 μg/ml leupeptin as protease inhibitors), and centrifuged for 2 h at 100,000 × g and 4 °C. Floating LDs were collected from the top of the tubes and resuspended in overlay buffer by brief sonication. TG content was determined using TG reagent (Infinity, Thermo Fisher) and glycerol as the standard (Sigma). Protein determination was performed using BCA reagent. 1 mm TG of isolated LD was incubated in the presence of 1% BSA (essentially fatty acid free, Sigma) in 0.1 m potassium phosphate buffer containing protease inhibitors in a final volume of 200 μl for 1 h in a 37 °C water bath under constant shaking. LDs were solubilized by adding 20 μl of 10% Triton X-100 to the samples. After mixing and incubation for 10 min at room temperature, samples were centrifuged for 30 min at 20,000 × g. The FFA content of the underlying solution was determined using NEFA kit (WAKO chemicals). To calculate net FFA release of the tissue LDs, FFA levels were also measured from the LD substrate before incubation.
In Vitro TG Hydrolase Activity Assay
Lysates of Cos-7 cells overexpressing ATGL, CGI-58, G0S2, or β-galactosidase (β-gal) as control were incubated in the presence or in the absence of in vitro translated recombinant core protein (1 μl) or with lysates (20 μg of protein) containing core overexpressed in HEK293T cells with a radiolabeled triolein substrate ([9,10-3H]triolein from PerkinElmer Life Sciences) for 1 h at 37 °C under constant shaking. Radiolabeled triolein substrate was prepared as described (27). Briefly, 0.3 mm 10 μCi/ml [3H]triolein was mixed with phosphatidylcholine/phosphatidylinositol (45 μm; 3:1) in potassium phosphate buffer (100 mm, pH 7) with a sonicator. Radiolabeled FFAs were extracted and quantified as described (27). Briefly, FFAs were extracted by adding 3.25 ml of methanol/chloroform/n-heptane with a ratio of 10/9/7 (v/v/v), 0.1 m potassium carbonate, and 0.1 m boric acid, pH 10.5. Samples were mixed, and after centrifugation for 10 min at 1000 × g, the supernatant was subjected to liquid scintillation counting.
Lipolysis of Radiolabeled Lipid Droplets
To generate intracellular radiolabeled TG stores, Cos-7 cells either transfected with core or a control plasmid were incubated with 0.6 μCi/ml [3H]oleic acid (American Radiolabeled Chemicals) and 560 μm oleic acid complexed to BSA (Sigma) for 22 h. Thereafter, LDs were isolated as described (20). An aliquot of the LD fraction (200,000 cpm) was incubated with Cos-7 lysates (20 μg of protein) overexpressing ATGL, CGI-58, or β-gal as the control in the presence of 4% BSA (essentially FFA free) in a total volume of 200 μl for 1 h at 37 °C. The reaction was terminated as described for the in vitro TG hydrolase activity assay, and the radioactivity present in the FFA extract was determined by liquid scintillation counting.
TG Metabolism in Cultured Cells
To determine TG degradation in MEFs, cells were incubated with 300 μm oleic acid complexed to BSA for 20 h as described (28). Thereafter, medium was changed to medium containing 2% BSA (fatty acid-free) and 10 μm triacsin C (Sigma). At different time points medium was collected, and lipids were extracted using hexane:isopropyl alcohol (3:2). Total lipid extract was evaporated and dissolved in water containing 1% Triton X-100. TG content was determined using Infinity TG reagent (Thermo Fisher) and glycerol (Sigma) as standard. FFA released in the culture medium was determined using a NEFA kit (WAKO). Cells were lysed in SDS, NaOH (0.1%/0.3 n), and protein concentrations were determined using BCA reagent and BSA as standard.
LD Measurements
Cells grown on coverslips were fixed in 4% paraformaldehyde for 30 min at room temperature, washed with phosphate-buffered saline (PBS), and permeabilized in 0.1% Triton X-100 for 5 min. For oil-red-O (ORO)-staining coverslips were incubated for 5 min in 60% isopropyl alcohol and stained with ORO staining solution (stock: 0.5 g ORO (Sigma) in 100 ml of isopropyl alcohol; the stock was diluted 6:4 (stock:water) and filtered before use) and differentiated in 60% isopropyl alcohol for 1 s and embedded in Mowiol (Calbiochem) mounting medium. Cells were analyzed with an Axio observer Z1 microscope (Zeiss) equipped with EC Plan Neofluar 20×/0.5 PHM27, EC Plan Neofluar 40×/0.75 PH, and Plan Apo 63×/1.4 oil differential interference contrast M27 objectives, filter sets 38HE, 43HE, 45, and 50, Optovar 1.25, and 1.6× magnification, and an Axiocam MRM REV 3. For quantification of LDs we used Volocity.
Coimmunoprecipitation Experiments
293T cells were seeded at a density of 106 cells per 10-cm dish and cotransfected with HA-core, ATGL-His, CGI-58-His, G0S2-His, HSL-His, or the appropriate control vectors using X-tremeGene 9 transfection reagent (Roche Applied Science). Alternatively, ATGL-His and CGI-58-HA were cotransfected with or without FLAG-core. Cells were harvested and lysed in lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40) by chemical and mechanical lysis using a 26-gauge needle. Lysates were centrifuged at 20,000 × g and 4 °C for 15 min. Equal amounts of lysate proteins (500 μg) were incubated with 20 μl of antibody-conjugated agarose (monoclonal anti-HA agarose, Sigma; Anti-FLAG M2-agarose, Sigma) overnight at 4 °C. Thereafter, beads were extensively washed with lysis buffer, and heated in 2× Laemmli buffer for 10 min at 95 °C. Input and immunoprecipitation fractions were subjected to Western blotting analysis using anti-FLAG, anti-His, and anti-HA and the appropriate secondary antibodies.
RESULTS
Core Expression Reduces TG Degradation in Murine Liver
To define the molecular mechanism by which core suppresses lipolysis in vivo, we expressed core in mouse livers by injecting the tail veins of mice with adenovirus (Fig. 1A) expressing either bicistronically core with GFP or GFP alone as previously described (11). Four days after injection, livers were harvested and disrupted using a Dounce homogenizer. TG contents were measured using Infinity TG reagent and normalized to total protein. Similar to results previously reported by us and others, we found that expression of core increased intracellular TG levels in the liver by 2-fold (Fig. 1B). To determine the effect of core on LD-associated TG hydrolase activity, we purified LDs from core-expressing or control livers by ultracentrifugation and performed self-digestion experiments in vitro. Core expression reduced the TG hydrolase activity associated with liver LDs by 43% as compared with samples from control mice (Fig. 1C). The expression of core and GFP in liver lysates was verified by Western blotting (Fig. 1D). These results indicate that TG accumulation observed in vivo with core expression is linked to decreased TG hydrolase activity at LDs.
FIGURE 1.
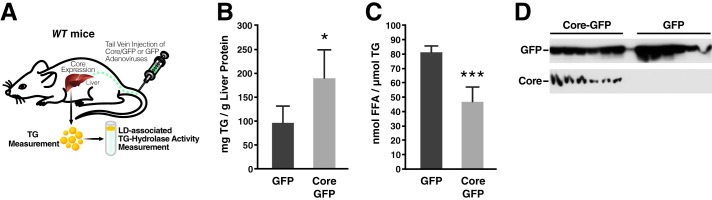
Core expression reduces TG hydrolysis and FFA release in murine liver. A, schematic of the experimental procedure in mice. B–D, WT mice were injected with adenovirus expressing core and GFP or GFP alone. Livers were harvested and lysed 4 days after infection (see “Experimental Procedures” for details). B, TG content was measured and normalized to total liver protein (n = 6 mice/group). C, LDs were isolated using density gradient centrifugation and washed, and equal amounts of TG were incubated for lipolysis 1 h at 37 °C. Samples were then adjusted to 1% Triton X-100 and centrifuged at 4 °C and 20,000 × g for 30 min. FFA in the underlying solution was determined using commercial kits (n = 5). Data are presented as the mean ± S.D. D, expression of core and GFP was confirmed by Western blot with antibodies against core and GFP.
Core Does Not Increase Lipogenesis In Vivo and In Vitro
To investigate whether core affects lipogenesis, we evaluated the incorporation of deuterium into newly generated TGs in control or core-expressing mice. Five hours after an intraperitoneal injection of 2H-labeled saline, we collected liver samples and determined their deuterium content in TG-palmitate, TG-glycerol, and cholesterol by MS. We found that the synthesis rates of glycerol and palmitate were similar in core-expressing and control livers, whereas that of cholesterol was slightly but not significantly decreased in core-expressing mice (38%, p = 0.06) (Fig. 2A). These results indicate that, at least under the short term expression conditions used in these studies, the HCV core protein does not increase the synthesis of neutral lipids in vivo.
FIGURE 2.
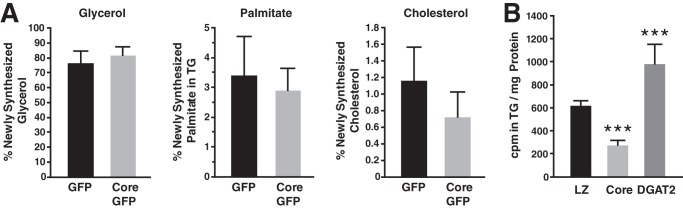
Core does not increase TG lipogenesis. A, mice injected with adenovirus expressing core and GFP or GFP alone were given an intraperitoneal injection of 2H-labeled saline 4 days after infection. After 5 h, livers samples were harvested and subjected to MS. Deuterium content in TG-palmitate, TG-glycerol, and cholesterol was measured (n = 6 mice/group). B, 24 h after transfection with FLAG-core, DGAT2-FLAG, or LacZ, Cos-7 cells were loaded with radiolabeled [14C]glucose. 20 h after loading, medium was removed, and lipids were extracted and subjected to TLC. The bands corresponding to TG were excised, and TG radiolabeling (cpm) was determined by liquid scintillation counting, normalized to total protein. Results are representative of two independent experiments with three replicates in each experiment. Data are presented as the mean ± S.D.
We confirmed this finding by measuring de novo lipogenesis in Cos-7 cells transiently transfected with vectors expressing core, the triglyceride-synthesizing enzyme DGAT2, or β-gal as a control. Transfected cells were loaded with radiolabeled [14C]glucose for 20 h. To evaluate the incorporation of labeled glucose into TG, lipids were extracted and subjected to thin-layer chromatography after which TGs were excised, and their radioisotope incorporation was measured. DGAT2, but not core, increased incorporation of radiolabeled glucose into TGs as compared with β-gal control cells (Fig. 2B). On the contrary, core induced a statistically significant decrease in de novo synthesis of TGs. These results support the model that core expression increases TG content by decreasing lipolysis rather than by increasing de novo lipogenesis.
Core Inhibitory Function on LD-associated TG Hydrolase Activity Is ATGL-dependent
Because ATGL is the main TG hydrolase, we injected adenovirus encoding core and GFP or GFP alone into the tail veins of wild-type and ATGL−/− mice (Fig. 3A). Four days after injection, we harvested livers and measured their TG content. As expected, ATGL deficiency by itself increased TG levels in liver (Fig. 3B). Core expression elevated TG levels in both wild-type and ATGL−/− mice, although to a lesser extent in ATGL−/− mice (2.3- versus 1.5-fold) (Fig. 3B). Next, we isolated LDs by ultracentrifugation and measured the TG hydrolase activity in purified LDs. As anticipated, core expression decreased the LD-associated TG hydrolase activity by 36% in livers of wild-type mice (Fig. 3C). ATGL deficiency reduced LD-associated lipase activity by 50%. Interestingly, core did not affect the already reduced TG hydrolase activity of LDs isolated from ATGL−/− mouse livers (Fig. 3C). We verified core's expression and LD localization by Western blotting in wild-type and ATGL−/− livers (Fig. 3D). These results point to LD-associated ATGL as the main lipase targeted by the inhibitory function of HCV core on lipolysis. However, they also indicate that additional host factors, possibly lipases not present on LDs, may participate in core-induced steatosis in vivo, explaining why core still induced TG accumulation in ATGL−/− mice liver.
FIGURE 3.

Core requires ATGL to inhibit LD-associated hydrolase activity from murine liver. A, schematic of the experimental procedure in mice. B–D, WT and ATGL−/− mice were injected with adenovirus expressing core and GFP or GFP alone. Livers were harvested 4 days after infection. B, TG content of liver lysates was measured and normalized to total liver protein (n = 4–5 mice/group). C, LDs were isolated using density gradient centrifugation. Equal amounts of TG were incubated for 1 h at 37 °C. Thereafter, samples were adjusted to 1% Triton X-100 and centrifuged for 30 min at 20,000 × g and 4 °C. FFA in the underlying solution was determined with commercial kits (n = 4). Data are presented as the mean ± S.D. D, the presence of GFP and core in the cytosolic and LD fractions, respectively, was confirmed by Western blot.
Core Protects TGs from ATGL-mediated Hydrolysis in Cell Culture
We complemented our in vivo studies with lipolysis studies in MEFs. MEFs were isolated from wild-type and ATGL−/− mice and infected with lentiviral vectors expressing core and GFP or GFP alone, as previously described (11). Two days after infection, TGs were isolated and quantified. Core expression increased intracellular TG levels 1.9-fold in WT MEFs. ATGL deficiency led to 3.1-fold elevated intracellular TG levels compared with WT cells (Fig. 4A). However, core expression did not further increase TG levels in ATGL−/− MEFs, supporting that increased TG levels induced by core requires ATGL.
FIGURE 4.

Core requires ATGL to inhibit TG degradation in cell culture. A–C, WT and ATGL−/− MEF were transduced with lentiviruses encoding HA-core and GFP or a control virus encoding GFP alone. A, 48 h after infection, neutral lipids were extracted, and the amount of TG was measured and normalized to total protein. B and C, cells were incubated with oleic acid for 20 h before chasing with medium containing 2% fatty acid-free BSA and 10 μm triacsin C. B, at the indicated time points, neutral lipids were extracted, and the amount of TG was measured. C, FFA levels were measured from the media supernatant. Both TG and FFA amounts were normalized to total protein. Results are representative of two independent experiments with three replicates in each experiment. Data are presented as the mean ± S.D. D–F, Huh7 cells were transduced with lentiviruses encoding HA-core and GFP or a control virus encoding GFP alone. 48 h after infection cells were fixed and stained with oil-red-O. Quantification of: D, number of LDs, mean of > 10 cells ± S.E.; E, LD diameter, mean of >150 lipid droplets ± S.E.; F, LD area, mean of >20 cells ± S.E. ORO, oil-red-O.
Next, we measured intracellular lipolysis in WT and ATGL−/− MEFs expressing core. We performed a pulse-chase experiment and determined the intracellular TG levels as well as the release of FFA into the cell culture supernatant. To do so, cells were loaded with oleic acid to fill up their TG stores (pulse) and chased in the presence of 2% BSA and triacsin C to accelerate lipolysis and to prevent re-esterification of FFAs (29). During the chase period, WT MEFs progressively lost 69% of their intracellular TG content (Fig. 4B). In the presence of core, however, TG stores were reduced only by 30%, demonstrating that core inhibits TG degradation. In ATGL−/− MEFs, TG stores were only reduced by 12% during the chase. Expression of core did not further decelerate TG degradation in ATGL−/− MEFs. Similar results were obtained when we measured the release of FFAs into the cell culture medium (Fig. 4C), further supporting that ATGL is required for core to decrease lipolysis.
To understand how of core affects ATGL-mediated hydrolysis of TG, we first analyzed the LD composition of cells expressing core or a GFP control. In accordance with previous reports (30, 31), no difference in LD number was observed in core-expressing cells, whereas LD area per cell and average LD diameter were significantly increased (Fig. 4, D–F). This difference in size suggests structural and functional differences in LDs induced by core (for review, see Ref. 32).
Proper Localization of Core and ATGL to LDs Is a Prerequisite for Core to Inhibit TG Hydrolysis
To examine the functional association of core with the ATGL·CGI-58 complex, we performed TG hydrolase assays using lysates of Cos-7 cells overexpressing ATGL, CGI-58, G0S2, or β-gal as a control (Fig. 5A). The respective lysates were incubated with an artificial TG substrate in the presence or absence of in vitro translated core (Fig. 5A). ATGL TG hydrolase activity was efficiently induced by CGI-58 and inhibited by G0S2 (Fig. 5B). The addition of in vitro translated core protein did not affect ATGL TG hydrolase activity, implicating that core must associate with LDs to inhibit lipolysis (Fig. 5B). Alternatively, we incubated lysates of Cos-7 cells overexpressing ATGL and CGI-58, ATGL and β-gal, or β-gal alone as a control with core-coated or control LDs isolated from oleate-loaded Cos-7 cells (Fig. 5A). The addition of ATGL-containing lysate slightly increased the release of FFA from isolated LDs, whereas the addition of ATGL and CGI-58 led to a 3-fold increase in FFA released as expected (Fig. 5C). However, no difference in the release of FFAs was observed between core-coated and control LDs (Fig. 5C). This indicates that both ATGL and core proteins need to be localized on the same LD surface for core to inhibit TG hydrolysis.
FIGURE 5.

Core and ATGL association with LDs is necessary to inhibit lipolysis. A, Western blot of Cos-7 cell lysates expressing β-gal, ATGL, CGI-58, or G0S2, in vitro translated core, or isolated LD fractions from Cos-7 cells expressing core or an empty vector. B, cell lysates of Cos-7 cells overexpressing ATGL, CGI-58, G0S2, or β-gal as control were incubated with a radiolabeled triolein substrate in the presence or absence of in vitro translated core protein for 1 h at 37 °C. FFAs were then extracted, and radioactivity was determined by liquid scintillation counting. Results are representative of two independent experiments with three replicates in each experiment. Data are presented as the mean ± S.D. C, radiolabeled LDs were isolated from Cos-7 cells transfected with core or an empty vector and then incubated with Cos-7 cell lysates expressing ATGL, CGI-58, and G0S2 or β-gal as the control for 1 h at 37 °C. FFAs were then extracted, and radioactivity was determined by liquid scintillation counting. Results are representative of two independent experiments with three replicates in each experiment. Data are presented as the mean ± S.D. D–F, NIH/3T3 were transduced with a lentivirus encoding HA-core WT or SPMT mutant and GFP or a control virus encoding GFP alone. D, 24 h after infection, neutral lipids were extracted, and TGs were measured and normalized to total protein. E and F, cells were incubated with oleic acid for 20 h before a 6-h chase using medium with 2% fatty acid-free BSA and 10 μm triacsin C. Neutral lipids were extracted, the amount of TG was measured and normalized to T0 (E). FFA levels were measured in the culture supernatant (F). Both TG and FFA levels were normalized to the amount of cellular protein. Results are representative of three independent experiments with three replicates in each experiment. Data are presented as the mean ± S.D. G, Western blot of cell extracts (left panels) or isolated LD fractions (right panels) from Huh7-Lunet cells expressing core WT or SPMT mutant. Results are representative of two independent experiments.
To further examine the role of core LD localization in TG hydrolysis, we examined a core mutant (SPMT) that is not properly processed and cannot localize to the surface of LDs (8). NIH/3T3 cells were infected with lentiviral vectors expressing GFP, WT core, or the SPMT core mutant. Two days after infection, TGs were isolated. Unlike WT core, expression of the SPMT mutant did not increase TG levels in NIH/3T3 cells (Fig. 5D), supporting that core localization to LDs is important for its TG-protecting properties. We next performed a pulse-chase experiment in transduced NIH/3T3 fibroblasts as described in Fig. 4. After a 6-h chase period in the presence of triacsin C, the SPMT mutant, unlike WT core, did not delay TG hydrolysis and the release of FFA in the cell culture supernatant (Fig. 5, E and F). These results underscore that the localization of core to LDs is important for its anti-lipolytic function.
Because of the central role of LD localization in the functional interaction between core and ATGL, we examined whether core affects the localization of ATGL and its cofactor CGI-58 to LDs. LDs were isolated from Huh7-Lunet cells expressing FLAG-tagged core proteins (WT or SPMT). Interestingly, we observed that WT core enhanced the association of both endogenous ATGL and CGI-58 to LDs (Fig. 5, G and H), which did not occur with the SPMT mutant. These findings further highlight the importance of core localization to LDs in the ATGL engagement.
Core Does Not Bind ATGL but Enhances the Interaction between ATGL and CGI-58
As core increases LD association of ATGL and CGI-58 and diminishes ATGL-mediated activity on LDs we examined whether core directly binds to ATGL or the ATGL·CGI-58 complex. Therefore, we performed coimmunoprecipitation experiments with overexpressed HA-tagged core and His-tagged ATGL, CGI-58, G0S2, or HSL in 293T cells. After immunoprecipitating HA-core protein, no interaction with any of the His-tagged proteins was observed (Fig. 6A). In parallel experiments core did coimmunoprecipitate with FLAG-DGAT1, a known cellular cofactor of core (Fig. 6B) (10).
FIGURE 6.

Enhanced interaction of ATGL with CGI-58 in the presence of core. A and B, His-tagged ATGL, CGI-58, G0S2, HSL, and FLAG-DGAT1 were co-expressed with HA-core or a control vector. 24 h after transfection cells were lysed, and lysates were immunoprecipitated (IP) with HA- or FLAG-agarose overnight at 4 °C. After washes, bound proteins were subjected to SDS-PAGE and analyzed by Western blotting with antibodies against His, HA, or FLAG. C, His-tagged mouse and human ATGL and CGI-58 were co-expressed with FLAG-core or a control vector. 24 h after transfection, cells were lysed, and lysates were immunoprecipitated with FLAG- or His-agarose overnight at 4 °C. After washes, bound proteins were subjected to SDS-PAGE and analyzed by Western blot with antibodies against His and FLAG. D, His-ATGL and HA-CGI-58 were co-expressed with or without FLAG-core in 293T cells. 24 h after transfection, cells were lysed, and lysates were immunoprecipitated with HA-agarose. Bound proteins were subjected to SDS-PAGE and analyzed by Western blot with antibodies against His, HA, and FLAG. Results are representative of two independent experiments. E, a model of core disrupting ATGL function at LDs. Core-coating of LDs leads to a change in the biophysical or proteomic properties of LDs preventing proper access of the ATGL·CGI-58 complex to stored TGs while causing prolonged residence of the complex at LDs. Under normal conditions (bottom panel), the presence of ATGL·CGI-58 complexes at the LD surface induces TG hydrolysis, LD shrinkage, and loss of the complexes from the surface. In the presence of core (top panel), LD-associated ATGL·CGI-58 complexes are not active, the LD content is thus preserved, and ATGL·CGI-58 complexes accumulate at the LD surface. The question remains of whether core primarily manipulates ATGL·CGI-58 activity or mechanistically targets the equilibrium of ATGL·CGI-58 recruitment and dissociation at LDs.
Similar results were observed when mouse ATGL as well as mouse and human CGI-58 (all His-tagged) were co-expressed with the FLAG-tagged core (Fig. 6C). Interestingly, when HA-CGI-58 was co-expressed with His-ATGL and FLAG-core and immunoprecipitated with HA-agarose, more ATGL coimmunoprecipitated with CGI-58 in the presence of core (Fig. 6D). Collectively, these results are consistent with the model that ATGL and CGI-58 strongly interact at LDs in the presence of core, but this interaction does not result in the activation of the ATGL lipase activity as otherwise expected.
DISCUSSION
The steatogenic function of the HCV core protein is a matter of intense research because of its potential clinical relevance. Here, we confirm that core inhibits lipolysis at LDs in vivo and show that this anti-lipolytic activity requires the TG lipase ATGL. Interestingly, adding either exogenous core to ATGL-coated LDs or exogenous ATGL to core-coated LDs did not affect ATGL-mediated lipolysis. This indicates that both core and the ATGL·CGI-58 complex must associate with LDs for core to inhibit LD-associated TG hydrolase activity. Surprisingly, in the absence of any detectable physical interaction between core and ATGL or its activator CGI-58, we observed that expression of core increased the interaction of ATGL with CGI-58 and the localization of the ATGL·CGI-58 complex at LDs. We propose that core coats LDs to alter their biophysical properties or proteomic composition (33), thereby promoting a strong interaction between ATGL and CGI-58 and prolonging their presence at LDs while preventing their access to stored TG (Fig. 6E).
Much of what we know about the regulation of lipolysis stems from studies in adipocytes. In response to hormonal stimulation, protein kinase A phosphorylates two key substrates: the diacylglycerol hydrolase HSL (34) and the LD-associated protein PLIN1 (35, 36). Upon phosphorylation, PLIN1, which tightly coats adipocyte LDs and prevents lipase access, is transformed from an inhibitor into an activator of lipolysis. CGI-58, bound to PLIN1 in the basal state, is released to activate ATGL, which acts upstream of HSL. In non-adipose tissues where PLIN1 is largely absent, other members of the PLIN family take over to coat the LD surface. Among these, PLIN 2, 3, and 5 interact with ATGL (6, 37–40). PLIN5 interacts with ATGL and CGI-58 and forms a barrier to prevent lipolysis (38, 41, 42). However, similarly to PLIN1, PLIN5 is also a target of PKA-mediated phosphorylation, and it was speculated that its phosphorylation might release ATGL and CGI-58 to enable TG hydrolysis (38).
Similarly to PLIN proteins, core tightly coats LDs, but contrary to PLIN1 and PLIN5, we find the viral protein does not interact with ATGL or CGI-58. This finding suggests that core either changes the biophysical environment of LDs or it binds an unknown protein that regulates the interaction between ATGL, CGI-58, and LDs and/or the activity of the lipase complex at LDs. Because core expression perturbs localization of PLIN2 and 3 (also known as ADRP and TIP47) at LDs (33, 43), one possibility is that core expression inhibits ATGL TG hydrolase activity by inducing a disequilibrium of these proteins at the LD surface (Fig. 6E). Alternatively, core could alter the intracellular vesicular trafficking of ATGL to LDs, which occurs through coat proteins (COPI and COPII) that mediate transport from the Golgi to the ER and vice versa (44, 45). Of note, knocking down COPI components increases LD formation, linking vesicular trafficking to ATGL activity (46).
The recruitment of ATGL to LDs is a prerequisite for intracellular TG mobilization (47, 48). Interestingly, a catalytically inactive ATGL mutant localizes to LDs more than active ATGL, implying that ATGL activity does not directly correlate with its association to LDs (48, 49).
Consistent with this observation, we found that in the presence of the inhibitory core protein the association of the ATGL·CGI-58 complex to LDs was increased as compared with LDs without core. The question remains if and how enhanced association of ATGL with LDs is linked to its decreased TG hydrolase activity. It can be assumed that LDs carrying active ATGL complexes at their surface rapidly reduce in size, a process that per se may lead to a progressive release of ATGL molecules from the shrinking LD surface. In contrast, LDs associated with inactive ATGL or coated with core do not decrease in size; thus ATGL molecules do not dissociate and could accumulate on the LD surface. A possibility is that core primarily disrupts the equilibrium between free cytosolic ATGL and ATGL bound to LDs and CGI-58, thus leading to inhibition of proper TG hydrolase activity. A similar regulatory mechanism exists for Rab GTPases, which switch from inactive to active states during vesicular trafficking (for review, see Ref. 50). Alternatively, core may primarily disrupt the catalytic activity of ATGL, e.g. by recruiting a yet unidentified endogenous repressor of ATGL to LDs, or displace a new activator, other than CGI-58, from ATGL, which could then e.g. prevent proper dissociation of ATGL and CGI-58 molecules from the shrinking LD surface. Numerous factors have been identified to interact with ATGL (e.g. UBXD8 (51), PEDF (52), Fsp27 (53)), which could mediate how core affects ATGL activity and/or localization.
Notably, we show that in the absence of ATGL core expression still induced steatosis in murine livers without reducing LD-associated TG hydrolase activity as observed with WT LDs. In contrast, no residual steatogenic effect of core was observed in ATGL−/− MEF cells pointing to the involvement of liver-specific factors not present on LDs that may participate in core-induced steatosis in hepatocytes in vivo. One possibility is that core's reported inhibition of microsomal triglyceride transfer protein and a resulting decrease in VLDL secretion from the liver could contribute to remaining steatosis in vivo but not in vitro (12). Another possibility is that core, in addition to ATGL, functionally targets other lipases, including HSL or patatin-like phospholipase 3 (PNPLA3), a triglyceride hydrolase previously associated with non-alcoholic fatty liver disease (54) potentially before or after they associated with LDs. Because the mechanism of LD hydrolysis is poorly characterized in hepatocytes, future studies are required to address whether core protein interacts with any of the known ATGL-interacting proteins or modulates the recruitment of HSL or patatin-like phospholipase 3 to core-coated liver LDs.
Acknowledgments
We thank members of the Ott, Zechner, and Farese laboratories for support and Yuya Nishida for help with the MEF isolation and immortalization.
This work was supported, in whole or in part, by National Institutes of Health Grants AI069090, AI097552 (to M. O.), and P30 DK026743 (University of California-San Francisco Liver Center). This work was also supported by the Gladstone Institutes.
- HCV
- hepatitis C virus
- TG
- triglyceride
- LD
- lipid droplet
- ER
- endoplasmic reticulum
- FFA
- free fatty acid
- ATGL
- adipose triglyceride lipase
- CGI-58
- comparative gene identification 58
- HSL
- hormone-sensitive lipase
- DGAT
- diacylglycerol acyltransferase
- G0S2
- G0/G1 switch gene 2
- MEF
- mouse embryonic fibroblast.
REFERENCES
- 1. Adinolfi L. E., Gambardella M., Andreana A., Tripodi M. F., Utili R., Ruggiero G. (2001) Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology 33, 1358–1364 [DOI] [PubMed] [Google Scholar]
- 2. Walther T. C., Farese R. V., Jr. (2012) Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 81, 687–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lass A., Zimmermann R., Oberer M., Zechner R. (2011) Lipolysis, a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog. Lipid Res. 50, 14–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Soni K. G., Mardones G. A., Sougrat R., Smirnova E., Jackson C. L., Bonifacino J. S. (2009) Coatomer-dependent protein delivery to lipid droplets. J. Cell Sci. 122, 1834–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang X., Lu X., Lombès M., Rha G. B., Chi Y. I., Guerin T. M., Smart E. J., Liu J. (2010) The G0/G1 switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 11, 194–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Granneman J. G., Moore H. P., Krishnamoorthy R., Rathod M. (2009) Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl). J. Biol. Chem. 284, 34538–34544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheng F. K., Torres D. M., Harrison S. A. (2014) Hepatitis C and lipid metabolism, hepatic steatosis, and NAFLD: still important in the era of direct acting antiviral therapy? J. Viral Hepat. 21, 1–8 [DOI] [PubMed] [Google Scholar]
- 8. McLauchlan J., Lemberg M. K., Hope G., Martoglio B. (2002) Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 21, 3980–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miyanari Y., Atsuzawa K., Usuda N., Watashi K., Hishiki T., Zayas M., Bartenschlager R., Wakita T., Hijikata M., Shimotohno K. (2007) The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9, 1089–1097 [DOI] [PubMed] [Google Scholar]
- 10. Herker E., Harris C., Hernandez C., Carpentier A., Kaehlcke K., Rosenberg A. R., Farese R. V., Jr., Ott M. (2010) Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 16, 1295–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harris C., Herker E., Farese R. V., Jr., Ott M. (2011) Hepatitis C virus core protein decreases lipid droplet turnover: a mechanism for core-induced steatosis. J. Biol. Chem. 286, 42615–42625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perlemuter G., Sabile A., Letteron P., Vona G., Topilco A., Chrétien Y., Koike K., Pessayre D., Chapman J., Barba G., Bréchot C. (2002) Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. FASEB J. 16, 185–194 [DOI] [PubMed] [Google Scholar]
- 13. Clément S., Peyrou M., Sanchez-Pareja A., Bourgoin L., Ramadori P., Suter D., Vinciguerra M., Guilloux K., Pascarella S., Rubbia-Brandt L., Negro F., Foti M. (2011) Down-regulation of PTEN and IRS-1 by HCV 3a core protein triggers the formation of large lipid droplets in hepatocytes. Hepatology 54, 38–49 [DOI] [PubMed] [Google Scholar]
- 14. Yamaguchi A., Tazuma S., Nishioka T., Ohishi W., Hyogo H., Nomura S., Chayama K. (2005) Hepatitis C virus core protein modulates fatty acid metabolism and thereby causes lipid accumulation in the liver. Dig. Dis. Sci. 50, 1361–1371 [DOI] [PubMed] [Google Scholar]
- 15. Kim K. H., Hong S. P., Kim K., Park M. J., Kim K. J., Cheong J. (2007) HCV core protein induces hepatic lipid accumulation by activating SREBP1 and PPARγ. Biochem. Biophys. Res. Commun. 355, 883–888 [DOI] [PubMed] [Google Scholar]
- 16. Moriishi K., Mochizuki R., Moriya K., Miyamoto H., Mori Y., Abe T., Murata S., Tanaka K., Miyamura T., Suzuki T., Koike K., Matsuura Y. (2007) Critical role of PA28γ in hepatitis C virus-associated steatogenesis and hepatocarcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 104, 1661–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tanaka N., Moriya K., Kiyosawa K., Koike K., Gonzalez F. J., Aoyama T. (2008) PPARα activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J. Clin. Invest. 118, 683–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsutsumi T., Suzuki T., Shimoike T., Suzuki R., Moriya K., Shintani Y., Fujie H., Matsuura Y., Koike K., Miyamura T. (2002) Interaction of hepatitis C virus core protein with retinoid X receptor α modulates its transcriptional activity. Hepatology 35, 937–946 [DOI] [PubMed] [Google Scholar]
- 19. Li Q., Pène V., Krishnamurthy S., Cha H., Liang T. J. (2013) Hepatitis C virus infection activates an innate pathway involving IKK-α in lipogenesis and viral assembly. Nat. Med. 19, 722–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Camus G., Herker E., Modi A. A., Haas J. T., Ramage H. R., Farese R. V., Jr., Ott M. (2013) Diacylglycerol acyltransferase-1 localizes hepatitis C virus NS5A protein to lipid droplets and enhances NS5A interaction with the viral capsid core. J. Biol. Chem. 288, 9915–9923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schweiger M., Paar M., Eder C., Brandis J., Moser E., Gorkiewicz G., Grond S., Radner F. P., Cerk I., Cornaciu I., Oberer M., Kersten S., Zechner R., Zimmermann R., Lass A. (2012) G0/G1 switch gene-2 regulates human adipocyte lipolysis by affecting activity and localization of adipose triglyceride lipase. J. Lipid Res. 53, 2307–2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harris C. A., Haas J. T., Streeper R. S., Stone S. J., Kumari M., Yang K., Han X., Brownell N., Gross R. W., Zechner R., Farese R. V., Jr. (2011) DGAT enzymes are required for triacylglycerol synthesis and lipid droplets in adipocytes. J. Lipid Res. 52, 657–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haemmerle G., Lass A., Zimmermann R., Gorkiewicz G., Meyer C., Rozman J., Heldmaier G., Maier R., Theussl C., Eder S., Kratky D., Wagner E. F., Klingenspor M., Hoefler G., Zechner R. (2006) Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 312, 734–737 [DOI] [PubMed] [Google Scholar]
- 24. Shah V., Herath K., Previs S. F., Hubbard B. K., Roddy T. P. (2010) Headspace analyses of acetone: a rapid method for measuring the 2H-labeling of water. Anal. Biochem. 404, 235–237 [DOI] [PubMed] [Google Scholar]
- 25. Bederman I. R., Foy S., Chandramouli V., Alexander J. C., Previs S. F. (2009) Triglyceride synthesis in epididymal adipose tissue: contribution of glucose and non-glucose carbon sources. J. Biol. Chem. 284, 6101–6108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Han S., Akiyama T. E., Previs S. F., Herath K., Roddy T. P., Jensen K. K., Guan H. P., Murphy B. A., McNamara L. A., Shen X., Strapps W., Hubbard B. K., Pinto S., Li C., Li J. (2013) Effects of small interfering RNA-mediated hepatic glucagon receptor inhibition on lipid metabolism in db/db mice. J. Lipid Res. 54, 2615–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schweiger M., Eichmann T. O., Taschler U., Zimmermann R., Zechner R., Lass A. (2014) Measurement of lipolysis. Methods Enzymol. 538, 171–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brasaemle D. L., Rubin B., Harten I. A., Gruia-Gray J., Kimmel A. R., Londos C. (2000) Perilipin A increases triacylglycerol storage by decreasing the rate of triacylglycerol hydrolysis. J. Biol. Chem. 275, 38486–38493 [DOI] [PubMed] [Google Scholar]
- 29. Paar M., Jüngst C., Steiner N. A., Magnes C., Sinner F., Kolb D., Lass A., Zimmermann R., Zumbusch A., Kohlwein S. D., Wolinski H. (2012) Remodeling of lipid droplets during lipolysis and growth in adipocytes. J. Biol. Chem. 287, 11164–11173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lyn R. K., Kennedy D. C., Stolow A., Ridsdale A., Pezacki J. P. (2010) Dynamics of lipid droplets induced by the hepatitis C virus core protein. Biochem. Biophys. Res. Commun. 399, 518–524 [DOI] [PubMed] [Google Scholar]
- 31. Depla M., Uzbekov R., Hourioux C., Blanchard E., Le Gouge A., Gillet L., Roingeard P. (2010) Ultrastructural and quantitative analysis of the lipid droplet clustering induced by hepatitis C virus core protein. Cell. Mol. Life Sci. 67, 3151–3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Suzuki M., Shinohara Y., Ohsaki Y., Fujimoto T. (2011) Lipid droplets: size matters. J. Electron Microsc. (Tokyo) 60, 101–116 [DOI] [PubMed] [Google Scholar]
- 33. Sato S., Fukasawa M., Yamakawa Y., Natsume T., Suzuki T., Shoji I., Aizaki H., Miyamura T., Nishijima M. (2006) Proteomic profiling of lipid droplet proteins in hepatoma cell lines expressing hepatitis C virus core protein. J. Biochem. 139, 921–930 [DOI] [PubMed] [Google Scholar]
- 34. Egan J. J., Greenberg A. S., Chang M. K., Wek S. A., Moos M. C., Jr., Londos C. (1992) Mechanism of hormone-stimulated lipolysis in adipocytes: translocation of hormone-sensitive lipase to the lipid storage droplet. Proc. Natl. Acad. Sci. U.S.A. 89, 8537–8541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Greenberg A. S., Egan J. J., Wek S. A., Garty N. B., Blanchette-Mackie E. J., Londos C. (1991) Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J. Biol. Chem. 266, 11341–11346 [PubMed] [Google Scholar]
- 36. Egan J. J., Greenberg A. S., Chang M. K., Londos C. (1990) Control of endogenous phosphorylation of the major cAMP-dependent protein kinase substrate in adipocytes by insulin and β-adrenergic stimulation. J. Biol. Chem. 265, 18769–18775 [PubMed] [Google Scholar]
- 37. MacPherson R. E., Ramos S. V., Vandenboom R., Roy B. D., Peters S. J. (2013) Skeletal muscle PLIN proteins, ATGL and CGI-58, interactions at rest and following stimulated contraction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 304, R644–R650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang H., Bell M., Sreenivasan U., Sreenevasan U., Hu H., Liu J., Dalen K., Londos C., Yamaguchi T., Rizzo M. A., Coleman R., Gong D., Brasaemle D., Sztalryd C. (2011) Unique regulation of adipose triglyceride lipase (ATGL) by perilipin 5, a lipid droplet-associated protein. J. Biol. Chem. 286, 15707–15715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yamaguchi H., Wang H. G. (2004) CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 279, 45495–45502 [DOI] [PubMed] [Google Scholar]
- 40. Subramanian V., Rothenberg A., Gomez C., Cohen A. W., Garcia A., Bhattacharyya S., Shapiro L., Dolios G., Wang R., Lisanti M. P., Brasaemle D. L. (2004) Perilipin A mediates the reversible binding of CGI-58 to lipid droplets in 3T3-L1 adipocytes. J. Biol. Chem. 279, 42062–42071 [DOI] [PubMed] [Google Scholar]
- 41. Granneman J. G., Moore H. P., Mottillo E. P., Zhu Z., Zhou L. (2011) Interactions of perilipin-5 (Plin5) with adipose triglyceride lipase. J. Biol. Chem. 286, 5126–5135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pollak N. M., Schweiger M., Jaeger D., Kolb D., Kumari M., Schreiber R., Kolleritsch S., Markolin P., Grabner G. F., Heier C., Zierler K. A., Rülicke T., Zimmermann R., Lass A., Zechner R., Haemmerle G. (2013) Cardiac-specific overexpression of perilipin 5 provokes severe cardiac steatosis via the formation of a lipolytic barrier. J. Lipid Res. 54, 1092–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boulant S., Douglas M. W., Moody L., Budkowska A., Targett-Adams P., McLauchlan J. (2008) Hepatitis C virus core protein induces lipid droplet redistribution in a microtubule- and dynein-dependent manner. Traffic 9, 1268–1282 [DOI] [PubMed] [Google Scholar]
- 44. Grönke S., Mildner A., Fellert S., Tennagels N., Petry S., Müller G., Jäckle H., Kühnlein R. P. (2005) Brummer lipase is an evolutionary conserved fat storage regulator in Drosophila. Cell Metab. 1, 323–330 [DOI] [PubMed] [Google Scholar]
- 45. Murugesan S., Goldberg E. B., Dou E., Brown W. J. (2013) Identification of diverse lipid droplet targeting motifs in the PNPLA family of triglyceride lipases. PLoS ONE 8, e64950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Beller M., Sztalryd C., Southall N., Bell M., Jäckle H., Auld D. S., Oliver B. (2008) COPI complex is a regulator of lipid homeostasis. PLoS Biol. 6, e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Listenberger L. L., Ostermeyer-Fay A. G., Goldberg E. B., Brown W. J., Brown D. A. (2007) Adipocyte differentiation-related protein reduces the lipid droplet association of adipose triglyceride lipase and slows triacylglycerol turnover. J. Lipid Res. 48, 2751–2761 [DOI] [PubMed] [Google Scholar]
- 48. Schweiger M., Schoiswohl G., Lass A., Radner F. P., Haemmerle G., Malli R., Graier W., Cornaciu I., Oberer M., Salvayre R., Fischer J., Zechner R., Zimmermann R. (2008) The C-terminal region of human adipose triglyceride lipase affects enzyme activity and lipid droplet binding. J. Biol. Chem. 283, 17211–17220 [DOI] [PubMed] [Google Scholar]
- 49. Smirnova E., Goldberg E. B., Makarova K. S., Lin L., Brown W. J., Jackson C. L. (2006) ATGL has a key role in lipid droplet/adiposome degradation in mammalian cells. EMBO Rep. 7, 106–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hutagalung A. H., Novick P. J. (2011) Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 91, 119–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Olzmann J. A., Richter C. M., Kopito R. R. (2013) Spatial regulation of UBXD8 and p97/VCP controls ATGL-mediated lipid droplet turnover. Proc. Natl. Acad. Sci. U.S.A. 110, 1345–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Notari L., Baladron V., Aroca-Aguilar J. D., Balko N., Heredia R., Meyer C., Notario P. M., Saravanamuthu S., Nueda M. L., Sanchez-Sanchez F., Escribano J., Laborda J., Becerra S. P. (2006) Identification of a lipase-linked cell membrane receptor for pigment epithelium-derived factor. J. Biol. Chem. 281, 38022–38037 [DOI] [PubMed] [Google Scholar]
- 53. Grahn T. H., Kaur R., Yin J., Schweiger M., Sharma V. M., Lee M. J., Ido Y., Smas C. M., Zechner R., Lass A., Puri V. (2014) Fat-specific Protein 27 (FSP27) Interacts with adipose triglyceride lipase (ATGL) to regulate lipolysis and insulin sensitivity in human adipocytes. J. Biol. Chem. 289, 12029–12039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Romeo S., Kozlitina J., Xing C., Pertsemlidis A., Cox D., Pennacchio L. A., Boerwinkle E., Cohen J. C., Hobbs H. H. (2008) Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 40, 1461–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]