

Background: The role of Sestrin2 (Sesn2) in skin squamous cell carcinoma (SCC) and melanoma is unknown.
Results: Sesn2 increases AKT activation and resistance to UVB and chemotherapeutics and is up-regulated in SCC and melanoma.
Conclusion: Sesn2 promotes AKT activation and survival of SCC and melanoma cells.
Significance: Sesn2 may promote tumorigenesis and chemoresistance of SCC and melanoma.
Keywords: Akt PKB, Apoptosis, Cancer, Cancer Biology, Tumor
Abstract
Skin cancer is the most common cancer in the United States and is mainly caused by environmental UV radiation. Reducing skin cancer incidence is becoming an urgent issue. The stress-inducible protein Sestrin2 (Sesn2) plays an important role in maintaining redox and metabolic homeostasis and their related pathologies. However, the role of Sesn2 in cancer remains unclear. Here we show that UVB radiation induces Sesn2 expression in normal human keratinocytes, mouse skin, normal human melanocytes, and melanoma cells. In addition, Sesn2 promotes AKT activation through a PTEN-dependent mechanism. Sesn2 deletion or knockdown sensitizes squamous cell carcinoma (SCC) cells to 5-fluorouracil-induced apoptosis and melanoma cells to UVB- and vemurafenib-induced apoptosis. In mice Sesn2 knockdown suppresses tumor growth from injected human SCC and melanoma cells. Last, as compared with normal skin, Sesn2 is up-regulated in both human skin SCC and melanoma. Our findings demonstrate that Sesn2 promotes AKT activation and survival in response to UVB stress and chemotherapeutics and suggest that Sesn2 is oncogenic in skin SCC and melanoma.
Introduction
Skin cancer, including basal cell carcinoma, squamous cell carcinoma (SCC),2 and melanoma, is the most common cancer in the United States (1, 2). The incidence of skin cancer continues to rise at an alarming rate each year. The major risk factor for skin cancer is ultraviolet radiation in sunlight and artificial tanning beds, including both UVB (280–315 nm) and UVA (315–400 nm) radiation (2). Despite many advances in targeted therapies and surgical removal of early stage skin cancers, advanced stage metastatic skin squamous cell carcinoma and melanoma are difficult to treat due to therapeutic resistance (3, 4).
Sestrins (Sesns) are a family of evolutionarily conserved stress-inducible proteins. Mammalian organisms express three Sestrins, Sesn1, Sesn2, and Sesn3. Sestrins act as antioxidant factors to suppress oxidative stress through their oxidoreductase activity, mTOR inhibition, and Nrf2 activation (5–7). This regulation of the AMP kinase (AMPK)/mTOR pathway by Sestrins plays a broader role in organism health in physiological and pathological contexts. By maintaining metabolic homeostasis, Sestrins protect cells and organisms from age-related physiological abnormalities in Drosophila (8). In Caenorhabditis elegans, cSesn promotes health and lifespan and protects against life stressors (9).
As the target genes of the tumor suppressor p53 (10), Sestrins are considered to have the potential to suppress tumors by detoxifying reactive oxygen species and inhibiting the oncogenic mTOR pathway (6, 11–13). Furthermore, the SESN1 (6q21) and SESN2 (1p35) loci are frequently deleted in several human cancers, including kidney cancer and sarcomas (14–16). However, the role of Sestrins in skin SCC and melanoma remains unknown.
Here we show that UVB radiation induces Sesn2 in normal human keratinocytes and melanocytes, mouse skin, and SCC and melanoma cells. We found that Sesn2 up-regulation is induced by UVB irradiation in association with malignant transformation. Sesn2 promotes AKT activation through regulating PTEN. Loss of Sesn2 sensitizes cells to apoptosis induced by UVB and chemotherapeutic agents. Sesn2 is up-regulated in both human SCC and melanoma. Our findings demonstrate that Sesn2 is a positive regulator of AKT activation and cell survival and suggest an oncogenic role of Sesn2 in SCC and melanoma.
EXPERIMENTAL PROCEDURES
Human Skin Tumor Samples
All human specimens were studied after approval by the University of Chicago Institutional Review Board. Frozen tissues were obtained under consent (Dept. of Medicine, University of Chicago). RNA samples and protein lysates were used to determine Sesn2 levels by real-time PCR and Western blotting. Formalin-fixed, paraffin-embedded tissue blocks were obtained from the archives in the tissue bank of the Section of Dermatology, Department of Medicine, University of Chicago. Non-sun-exposed normal skin, nevus, and malignant and metastatic melanoma tissues were used for immunohistochemical analysis of Sesn2 protein levels.
Cell Culture
WT, Sesn2 KO MEF cells (17), HeLa (human cervical cancer cells), HaCaT (kindly provided by Professor N. Fusenig), A431 (human squamous carcinoma cells), A375 (human amelanotic melanoma cells), and MEL624 melanoma cells were maintained in monolayer cultures in 95% air, 5% CO2 at 37 °C in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin (Invitrogen). Other melanoma cells were generously provided by Dr. Meenhard Herlyn (Wistar Institute, Philadelphia) and cultured as described previously (18). Inducible expression of PTEN in WM793TR-PTEN cells was obtained by treatment of cultures with doxycycline (Sigma) at a final concentration of 100 ng/ml. Cells were maintained in DMEM with GlutaMAX (Invitrogen) supplemented with 10% fetal calf serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 4 μg/ml insulin (Sigma). The HaCaT cell line was cultured for <20 passages. Normal human epidermal keratinocytes (NHEKs) and melanocytes (NHEMs) were obtained from Clonetics (Lonza) and Invitrogen, respectively, and cultured according to the manufacturers' instructions. NHEK and NHEM cells were cultured for <4 passages. No authentication was done.
siRNA or Plasmid Transfection
A375 cells were transfected with negative control (NC) or siRNA (ON-TARGETplus SMARTpool, Dharmacon) targeting p53 or AKT3 using TransIT-siQUEST® Transfection Reagent (Madison, WI) according to the manufacturer's instructions. Plasmid transfection was performed with X-tremeGENE 9 (Roche Applied Science) according to the manufacturer's instructions.
Lentiviral Production and Infection
Lentiviral constructs expressing shNC (shLuc) and shSesn2 were generated as described previously (5, 6). Negative control shRNA (shNC, kindly provided by Dr. Seungmin Hwang, University of Chicago), shPTEN1 (Plasmid #25638), and shPTEN2 (Plasmid #25639) were obtained from Addgene. Lentivirus was produced by cotransfection into 293T cells with lentiviral constructs together with the pCMVdelta8.2 packaging plasmid and pVSV-G envelope plasmid using X-treme 9 (Roche Applied Science) as described previously (19–21). Virus-containing supernatants were collected 24–48 h after transfection and used to infect recipients. Target cells were infected in the presence of Polybrene (8 μg/ml, Sigma) and selected with puromycin at 1 μg/ml for 6 days.
Western Blotting
Protein concentration was determined using the BCA assay (Pierce). Western blotting was performed as described previously (22). Antibodies used included Sesn2 (Proteintech Group, Inc, Chicago, IL), ENO1 (Abcam, Cambridge, MA), GAPDH, p53, p21, PTEN, AKT, poly(ADP-ribose) polymerase (Santa Cruz, Santa Cruz, CA), phosphor-AKT (p-AKT), and cleaved-caspase 3 (Cell Signaling Technology, Danvers, MA).
Cell Fractionation
Cytosol and membrane protein fractions were isolated using a Mem-PER Plus Membrane Protein Extraction kit (Thermo Scientific, Rockford, IL).
Immunohistochemical and Immunofluorescence Analysis
Sesn2 levels were determined using immunohistochemical analysis by the immunohistochemistry core facility at the University of Chicago. The anti-Sesn2 antibody (Proteintech Group, Inc., Chicago, IL) was used, with the protein levels visualized using the alkaline phosphatase anti-alkaline phosphatase (APAAP) method, in which the substrate staining (red) is easily distinguishable from melanin (brown). Immunofluorescence analysis of Sesn2 or PTEN was performed as described previously (23, 24) using a fluorescence microscope (Olympus IX71) or confocal microscope (Olympus F1000 IX81).
Real-time PCR
Total RNA was isolated using the Qiagen RNeasy plus mini kit. Quantitative real time PCR assays were performed using ABI7300 (Applied Biosystems, Foster City, CA) in 96-well plates with the SYBR® Green PCR Master Mix (Applied Biosystems) as in our previous studies (25). The threshold cycle number (CT) for each sample was determined in triplicate. The CT for values for Sesn2 and PTEN were normalized against GAPDH as described previously (19, 20, 25). The sequences of the primers used were: mouse Sesn2 gene, 5′-TAG CCTGCA GCC TCA CCT AT-3′ (forward) and 5′-TAT CTG ATG CCA AAG ACG CA-3′ (reverse); human Sesn2 gene, 5′- GAC CATGGC TAC TCG CTG AT-3′ (forward) and 5′- GCT GCC TGG AAC TTC TCA TC-3′ (reverse).
Analysis of Tumor Growth in Nude Mice
All animal procedures have been approved by the University of Chicago Institutional Animal Care and Use Committee. Athymic Nude mice were obtained from Harlan Sprague-Dawley. 1 × 106 A431-shNC, A431-shSesn2, A375-shNC, and A375-shSesn2 cells were injected subcutaneously into the right flank of 6-week-old female nude mice. Tumors were monitored weekly, and tumor volume (TV) was calculated using the formula, TV (mm3) = d2 × D/2, where d and D are the shortest and the longest diameters, respectively.
Mitochondrial Membrane Potential
Mitochondrial membrane potential was assessed using the potentiometric dye JC-1 (Invitrogen) following the manufacturer's instructions. In brief, after treatment cells were trypsinized and resuspended in 3 ml of medium containing 2 μm JC-1 for 15 min at 37 °C in the dark and then pelleted by centrifugation and resuspended in 500 μl of PBS. Bivariate analysis was performed by flow cytometry with excitation at 488 nm, and mitochondrial function was assessed as JC-1 green (depolarized mitochondria) or red (polarized mitochondria) fluorescence.
In Vitro Cell Proliferation Assay
Cell proliferation of A431 and A375 stable cells was analyzed using the MTS assay (Promega, Madison, WI) according to the manufacturer's instructions as in our recent studies (21). The BrdU assay was monitored using a cell proliferation ELISA BrdU (colorimetric, Roche Applied Science) kit according to the manufacturer's instructions as in our previous studies (26).
Statistical Analyses
Statistical analyses were performed using Prism 5 (GraphPad software, San Diego, CA). Data are expressed as the mean of at least three independent experiments and analyzed by Student's t test. Error bars indicate the standard error of the means (S.E.). A p value of <0.05 was considered statistically significant.
RESULTS
Sesn2 Up-regulation Is Induced by UVB Irradiation in Keratinocytes, Mouse Skin, and SCC Cells and Associated with Keratinocyte Transformation
To determine the role of Sesn2 in skin carcinogenesis, we assessed the effect of UVB irradiation on Sesn2 in human keratinocytes, mouse skin, and SCC cells. UVB radiation increased Sesn2 protein levels in NHEK, mouse skin, non-tumorigenic HaCaT cells, and human SCC A431 cells (Fig. 1, A–D). It also induced Sesn2 expression in immortalized mouse embryonic fibroblasts (MEFs) (Fig. 1E). Immunofluorescence analysis showed that Sesn2 levels were higher in A431 SCC cells than those in NHEK and the non-tumorigenic HaCaT cells. These findings indicate that UVB irradiation increases Sesn2 levels in keratinocytes, mouse skin, and SCC cells and that SCC cells express higher Sesn2 levels than normal and non-tumorigenic keratinocytes.
FIGURE 1.
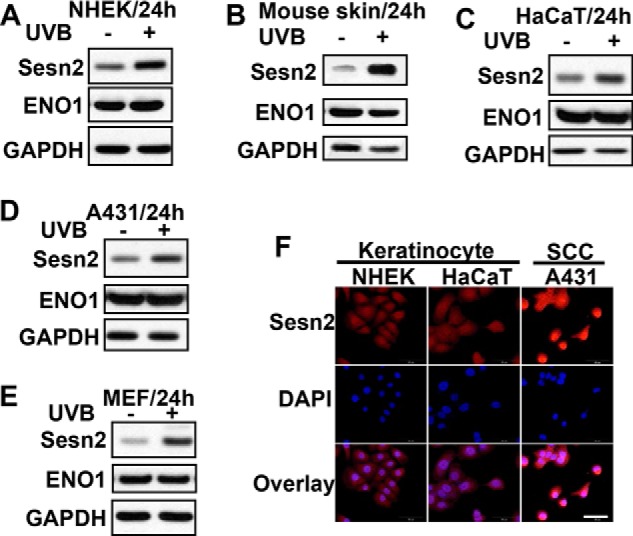
UVB induces Sesn2 up-regulation in normal keratinocytes, mouse skin, HaCaT keratinocytes and SCC cells. A–E, immunoblot analysis of Sesn2, ENO1, and GAPDH in NHEKs (A), mouse skin (B), HaCaT (C), A431 (D), and MEF (E). Cells were irradiated with sham or UVB (20 mJ/cm2). For the mouse skin in B, mice were irradiated with UVB (100 mJ/cm2) three times a week for 23 weeks. F, immunofluorescence analysis of Sesn2 expression in NHEK, HaCaT, and A431 cells. Scale bar, 50 μm.
Sesn2 Up-regulation Is Induced by UVB Irradiation in Normal Melanocytes and Melanoma Cells and Associated with Melanocyte Transformation
In addition, we assessed the effect of UVB on Sesn2 levels in human melanocytes and melanoma cells. UVB irradiation increased Sesn2 protein levels in NHEMs (Fig. 2A) and human melanoma cells at different phases of progression: A375 (Fig. 2B, radial growth phase (RGP)), WM793 (Fig. 2C, vertical growth phase (VGP)), and MEL624 (Fig. 2D, metastatic (Met)) melanoma cells. These results suggest a conserved UVB-induced Sesn2 up-regulation through melanoma genesis and progression. In addition, immunofluorescence analysis showed that Sesn2 expression was increased in A375, WM793, and MEL624 cells as compared with normal melanoma cells (NHEM) (Fig. 2E). UVB increased Sesn2 mRNA levels in NHEM, A375, WM793, and MEL624 melanoma cells (Fig. 2F). In addition to A375 cells, we found that the Sesn2 protein level was increased in other human melanoma cells, including WM266–4, WM115, WM35, WM3670, CHL-1, and HS294T cells, as compared with NHEM cells (Fig. 2G). These findings indicate that in normal melanocytes and melanoma cells, UVB irradiation induces Sesn2 up-regulation and that melanoma cells express higher Sesn2 than normal melanocytes.
FIGURE 2.

UVB induces Sesn2 up-regulation in normal melanocytes and melanoma cells. A–D, immunoblot analysis of Sesn2 and GAPDH in NHEMs (A), A375 (B, radial growth phase (RGP)), WM793 (C, vertical growth phase (VGP)), and MEL624 (D, metastatic (Met)) melanoma cells at 1, 6, or 24 h post-UVB as indicated. Cells were irradiated with sham or UVB (20 mJ/cm2). E, immunofluorescence analysis of Sesn2 expression in NHEM, A375, WM793, and MEL cells. Scale bar, 50 μm. F, real-time PCR analysis of Sesn2 mRNA levels in NHEM, A375, WM793, and MEL624 cells at 24 h post-sham or post-UVB irradiation (20 mJ/cm2). *, p < 0.05; t test, between comparison groups. G, immunoblot analysis of Sesn2 and GAPDH in NHEM, A375, WM266–4, WM115, WM35, WM3670, CHL-1, and HS294T melanoma cells.
Sesn2 Activates AKT through a PTEN-dependent Mechanism
To determine the function of Sesn2 in cancer cells, we analyzed the effect of increasing and decreasing Sesn2 on the oncogenic AKT pathway, which is critical for promoting cancer development and chemoresistance (27). Overexpression of Sesn2 in A431, A375, and HeLa cells increased AKT phosphorylation at serine 473 (Fig. 3A). Sesn2 deletion in MEF cells decreased AKT phosphorylation (Fig. 3B). Similarly, knockdown of Sesn2 in A375 and A431 cells that express PTEN inhibited AKT phosphorylation (Fig. 3C). In contrast, in the PTEN-null U87 cells, Sesn2 knockdown did not affect AKT phosphorylation (Fig. 3C), indicating that the regulation of AKT phosphorylation by Sesn2 is PTEN-dependent. Furthermore, in the PTEN-null WM793 melanoma cells, Sesn2 knockdown had no effect on AKT phosphorylation, whereas upon re-expression of PTEN by doxycycline, Sesn2 knockdown increased PTEN levels and inhibited AKT phosphorylation (Fig. 3D). Sesn2 knockdown moderately increased PTEN protein levels in A375, A431, and doxycycline-treated WM793TR-PTEN cells (Fig. 3, C and D). These data indicate that Sesn2 regulates AKT activation through PTEN.
FIGURE 3.
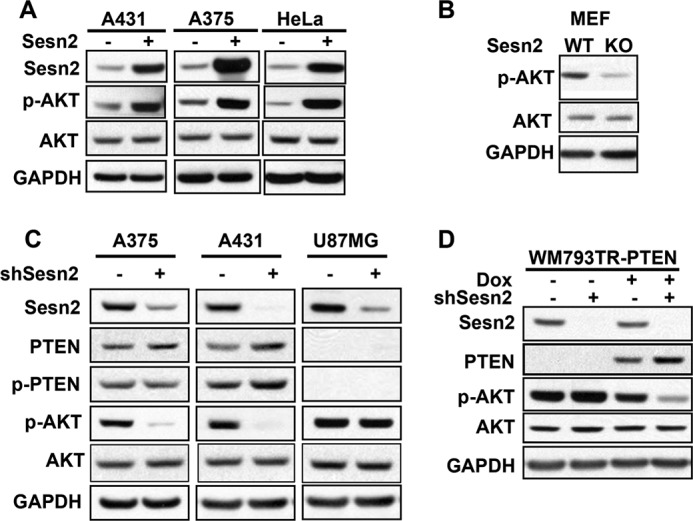
Sesn2 promotes AKT activation through a PTEN-dependent mechanism. A, immunoblot analysis of Sesn2, p-AKT, AKT, and GAPDH in A375 and A431 cells infected with either a lentiviral vector expressing control or Sesn2 (A375 and A431) and HeLa cells transfected with a plasmid expressing control or Sesn2 using X-tremeGENE 9. B, immunoblot analysis of p-AKT, AKT, and GAPDH in WT and Sesn2 KO MEF cells. C, immunoblot analysis of Sesn2, PTEN, p-PTEN, p-AKT, AKT, and GAPDH in A375, A431, and U87MG cells infected with a lentiviral vector expressing shRNA targeting Sesn2 (shSesn2) or negative control (shNC). D, immunoblot analysis of Sesn2, PTEN, p-PTEN, p-AKT, AKT, and GAPDH in WM793TR-PTEN cells infected with a lentiviral vector expressing shNC or shSesn2. Cells were treated with vehicle or doxycycline (Dox; 100 ng/ml) for 48 h before lentiviral transduction.
To determine the role of PTEN in Sesn2 regulation of AKT activation, we investigated the effect of Sesn2 on the PTEN membrane association that is required for PTEN activity. We found that both Sesn2 knockdown in A375 or A431 cells and its deletion in MEF cells increased PTEN membrane localization (Fig. 4, A and F). In Sesn2 KO MEF cells, immunofluorescence of PTEN staining also indicated membrane PTEN accumulation as compared with Sesn2 WT cells (Fig. 4, G–I). These findings suggest that Sesn2 promotes AKT activation through regulating PTEN membrane recruitment.
FIGURE 4.

Sesn2 regulates PTEN membrane association. A–C, immunoblot analysis of PTEN, Na-K ATPase (membrane marker), and GAPDH (cytosolic marker) in cytosolic and membrane protein fractions isolated from A375 (A) and A431 (B) cells stably infected with a lentiviral vector expressing shNC or shSesn2 and WT and Sesn2 KO MEF cells (C). D–F, quantification of PTEN levels in A (D), B (E), and C (F). *, p < 0.05; t test, between comparison groups. NS, not significant. G and H, confocal images from immunofluorescence assay of the localization of PTEN (green) in WT (G) and Sesn2 KO MEF cells (H). Scale bar, 50 μm. The blue is a DAPI nuclear counterstain. I, quantification of PTEN staining intensity along the indicated lines (yellow) in G and H.
Sesn2 Promotes Tumor Cell Survival after Treatment with UVB and Chemotherapeutic Agents
To determine the function of Sesn2 in cell survival, an oncogenic process promoted by AKT activation, we assessed the effect of Sesn2 knock-out and knockdown on apoptosis induced by UVB radiation and chemotherapeutic agents. UVB radiation induced apoptosis in A375, A431, and MEF cells in a dose-dependent manner as shown by the formation of cleaved poly(ADP-ribose) polymerase (PARP) or cleaved caspase 3 (Fig. 5, A and B), whereas Sesn2 knockdown in A375 or A431 cells and Sesn2 knock-out in MEF cells further sensitized cells to UVB-induced apoptosis as measured by the increase in cleaved caspase-3 (Fig. 5, A and B) and by the decrease in mitochondrial potential (Fig. 5C). Sesn2 knock-out reduced UVB-induced AKT activation in MEF cells (Fig. 5D), consistent with the role of Sesn2 in AKT activation shown in Fig. 3. The BRAF inhibitor vemurafenib PLX4032 (PLX), a targeted anti-melanoma agent recently approved by the Food and Drug Administration, resulted in increased Sesn2 levels in BRAF wild-type melanoma cells (B16F10) and BRAF mutant melanoma cells (A375 and MEL624) (Fig. 5, E and F). PLX induced apoptosis in A375 and MEL624 cells as indicated by caspase 3 cleavage (Fig. 5F). Sesn2 knockdown increased the apoptosis induced by BRAF inhibition (Fig. 5F). In A431 cells, 5-fluorouracil (5-FU), a widely used chemotherapeutic agent for SCC, induced Sesn2 expression (Fig. 5G) and induced some cell death as indicated by decreased cell viability (Fig. 5H). Sesn2 knockdown significantly increased the cell death induced by 5-FU (Fig. 5H). These findings indicate that Sesn2 increases cell survival after exposure to UVB irradiation and chemotherapeutics.
FIGURE 5.

Sesn2 deletion or knockdown increases UVB- or chemotherapeutic-induced apoptosis. A, immunoblot analysis of cleaved-caspase3, Sesn2, and GAPDH in sham or UVB-irradiated A375 and A431 cells stably transfected with lentiviral control or shSesn2 plasmid. Cells were irradiated with different doses of UVB and collected at 24 h post-UVB treatment. B, immunoblot analysis of Sesn2, cleaved poly(ADP-ribose) polymerase (PARP), and GAPDH in sham or UVB-irradiated WT and Sesn2 KO MEF cells. Cells were irradiated with different doses of UVB and collected at 24 h post-UVB treatment. C, changes in the mitochondrial membrane potential in sham or UVB-irradiated A375 and A431 cells stably infected with lentivirus expressing control shRNA (shNC) or shSesn2 and WT and Sesn2 KO MEF cells at 18-h post-UVB or -sham irradiation. Flow cytometric histogram shown on the left is from shNC or shSesn2 A375 cells. D, immunoblot analysis of p-AKT, AKT, Sesn2, and GAPDH in sham or UVB (20 mJ/cm2)-treated WT and Sesn2 KO MEF cells at different time points after UVB treatment. E, immunoblot analysis of Sesn2, p-ERK, ERK, and GAPDH in vehicle or PLX4032 (PLX, 10 μm)-treated A375 (BRAF mutant) and B16F10 (BRAF wild type) cells at 24 h post-PLX treatment. F, immunoblot analysis of Sesn2, cleaved-caspase3, and GAPDH in vehicle or PLX-treated A375 and MEL624 (BRAF mutant) cells stably infected with a lentiviral vector expressing shNC or shSesn2. Cells were starved overnight using 0.1% FBS medium and then treated with different doses of PLX and collected at 48 h post-PLX treatment. Sesn2 protein levels in E and F were quantified using ImageJ software (below each band in arbitrary units). G, immunoblot analysis of Sesn2 and GAPDH in vehicle or 5-FU (50 μm)-treated A431 SCC cells at 24 h post-5-FU. H, MTS assay of cell viability of A431 cells stably infected with a lentiviral vector expressing shNC or shSesn2 post-5-FU. Cells were starved overnight using 0.1% FBS medium, treated with different doses of 5-FU, and then analyzed at 48 h post-5-FU treatment. Data are shown as the mean ± S.E. for three independent experiments performed. *, p < 0.05; t test, compared with the shNC group.
Sesn2 Knockdown Inhibits Growth of Xenografted A431 and A375 Human Tumors in Nude Mice
To determine the role of Sesn2 in tumor development in vivo, we analyzed the effect of Sesn2 knockdown on tumor growth using a subcutaneous xenograft mouse model. A431, A375, and MEL624 cells grew tumors over time after subcutaneous inoculation (Fig. 6, A–E). Sesn2 knockdown in these cells inhibited tumor growth (Fig. 6, A–E). However, Sesn2 knockdown had no effect on cell proliferation in vitro (Fig. 6F). There was no difference in either the histology of A431 and A375 tumors (H&E staining) or apoptosis (TUNEL) between shNC and shSesn2 groups (data not shown). It is possible that Sesn2 increases cell survival during subcutaneous inoculation, and thus shSesn2 cells have fewer cells to grow into tumors. These results indicate that Sesn2 is critical for in vivo tumor formation. As compared with control groups, AKT activation was reduced in xenografted A431-shSesn2 human tumors in nude mice (Fig. 6G). These findings are consistent with our in vitro findings (Fig. 3C).
FIGURE 6.

Sesn2 knockdown suppresses growth of xenografted A431 and A375 human tumors in nude mice. A, average volume (mm3) of A431-shNC and A431-shSesn2 tumors at different weeks after subcutaneous injection. B, representative tumors from xenografted A431 group. C, differences in tumor weight in xenografted A431 group. D and E, average volume (mm3) of A375-shNC and A375-shSesn2 (D); MEL624-shNC and MEL624-shSesn2 (E) tumors at different weeks after subcutaneous injection. F, MTS and BrdU assay of in vitro cell proliferation of A431, MEL624, and A375 cells infected with a lentiviral vector expressing shNC or shSesn2. G, immunoblot analysis of Sesn2, p-AKT, AKT, and GAPDH in xenografted A431 human tumors in nude mice. For all of the graphs, data are shown as the mean ± S.E. (n = 3). *, p < 0.05; t test, compared with the control group; all other comparisons were not significant.
Sesn2 Is Up-regulated in Human SCC and Melanoma
To further investigate the specific function of Sesn2 in human skin malignancies, we evaluated Sesn2 protein levels in human SCC and melanoma. As compared with normal human skin, Sesn2 was up-regulated in human SCC at both the mRNA and protein levels (Fig. 7, A and B). Similarly, Sesn2 was up-regulated in mouse SCC as compared with adjacent normal skin derived from UVB-irradiated mice (Fig. 7C).
FIGURE 7.
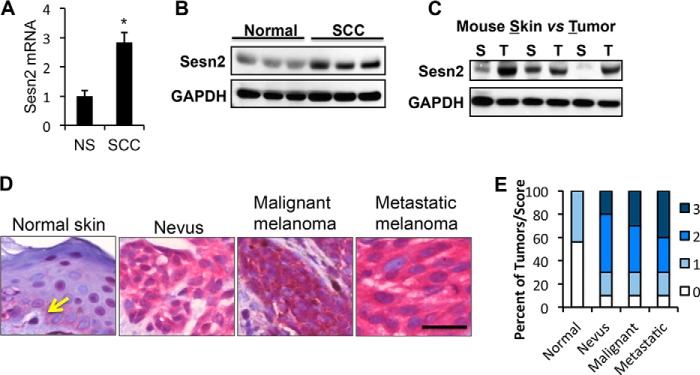
Up-regulation of Sesn2 in both human SCC and melanoma. A, real-time PCR analysis of Sesn2 mRNA levels in normal human skin (NS) and SCC. Data are shown as the mean ± S.E., n = 3. *, p < 0.05; t test, compared with the normal human skin group. B, immunoblot analysis of Sesn2 and GAPDH in normal human skin and SCC. C, immunoblot analysis of Sesn2 and GAPDH in UVB-induced SCC tumors (T) and the corresponding adjacent non-tumor skin (S) in mice. D, representative immunohistochemical analysis of Sesn2 in normal human skin, nevus, malignant melanoma, and metastatic melanoma using the alkaline phosphatase anti-alkaline phosphatase method in which the substrate staining (red) is easily distinguishable from melanin (brown). Scale bar, 25 μm. The arrow indicates a representative melanocyte in normal human epidermis. E, percentage of tumors (in stacked column format) for each score of Sesn2 expression.
To determine the role of Sesn2 in human melanoma, we used immunohistochemical analysis to determine the differences in Sesn2 protein levels in human nevus, malignant melanoma, and metastatic melanoma as compared with melanocytes in sun-protected normal non-lesional skin. A low level of Sesn2 protein was detected in melanocytes in normal skin (Fig. 7D). However, the Sesn2 levels were increased (score 2 or 3) in 70% of nevus (7/10), 70% of malignant melanoma (7/10), and 70% of metastatic melanoma (7/10) samples as compared with none of the normal skin samples (0/16) (Fig. 7E). This up-regulation was statistically significant as analyzed by the Mann-Whitney U test (p = 0.0007 for nevus, malignant melanoma, and metastatic melanoma versus normal skin). These findings demonstrate that Sesn2 is up-regulated in human SCC and the early stage of melanoma and suggest that Sesn2 is an oncogene in human skin malignancies.
DISCUSSION
Sesn2 has been suggested as a tumor suppressor (6, 11–13). Here we have shown that Sesn2 is up-regulated by UVB radiation and is associated with malignant transformation of keratinocytes and melanocytes. Sesn2 is crucial for cell survival of SCC and melanoma cells after UVB radiation and chemotherapeutics, including 5-FU and the BRAF inhibitor vemurafenib. We found that Sesn2 is essential for promoting AKT activation through regulating PTEN. Last, Sesn2 is up-regulated in human skin SCC and melanoma. Our findings suggest an oncogenic role of Sesn2 in skin carcinogenesis and chemoresistance.
Sestrin expression is inducible in response to diverse cellular stresses including genotoxic stress, hypoxia, energy deficiency, and oxidative stress (28). UV radiation activates the expression of Sesn1 and Sesn2 through p53 (10, 14). p53-responsive elements were identified in the first exon and 9.6 kb downstream of the Sesn2 gene (29, 30). These studies suggest that Sesn2 is a tumor suppressor. However, in p53-mutated HaCaT or A431 cells, we found Sesn2 induction by UVB irradiation, suggesting a p53-indpendent mechanism. Indeed, dSesn was found to be up-regulated in Drosophila by loss of PTEN or activation of the mTOR pathway (8), suggesting a potential oncogenic role of Sesn2.
Among all the Sestrin genes, daily UV radiation up-regulates only Sesn2 in human fibroblasts and keratinocytes in reconstructed skin, whereas it down-regulates the other Sestrin genes including Sesn1-T1, Sesn1-T2, and Sesn3 (31). Our findings indicate that when wild-type p53 is expressed, UVB radiation induces Sesn2 expression in normal human epidermal keratinocytes, normal human melanocytes, melanoma cells, and mouse skin and that it also up-regulates Sesn2 in HaCaT and A431 cells that express mutated p53 (32) (Fig. 1). These findings suggest that Sesn2 induction serves as an adaptation mechanism for both normal cells and cancer cells in response to genotoxic stress and anti-cancer treatment in a p53-dependent and -independent manner. The role of other Sestrins in skin cancer remains unknown and requires further investigation.
The role of Sesn2 in human cancers remains unclear. Sestrin up-regulation is crucial for stress adaptation, maintaining redox balance and metabolic homeostasis in physiological and pathological contexts (28, 33). In response to genotoxic stress, p53-mediated Sesn1 and Sesn2 activate AMP kinase and thus inhibit the oncogenic mTOR pathway (6, 34), suggesting a tumor suppressive role. In contrast, AKT-mediated but p53-independent Sesn2 induction promotes cell survival under energetic stress (35), suggesting an oncogenic function of Sesn2. Our findings demonstrate that Sesn2 promotes PTEN-dependent AKT activation and cell survival after UVB irradiation and chemotherapeutic treatment. It is possible that Sesn2 inhibits PTEN activity by suppressing PTEN membrane association (Fig. 4). Future investigations will determine the molecular and biochemical mechanism by which Sesn2 controls PTEN activity. Both vemurafenib and 5-FU were observed to induce Sesn2 expression. Given the crucial role of AKT in carcinogenesis and chemoresistance in SCC and melanoma (3, 4, 27), the regulation of AKT by Sesn2 may not only promote the development of SCC and melanoma but also confer therapeutic resistance, suggesting an oncogenic role of Sesn2 in SCC and melanoma.
Such a role is further supported by the up-regulation of Sesn2 in both human SCC and melanoma and the role of Sesn2 in xenografted tumor growth in nude mice. We found that, as compared with normal skin, Sesn2 is up-regulated in human SCC as well as mouse SCC. The up-regulation of Sesn2 was detected in nevi, malignant melanoma, and metastatic melanoma, suggesting that Sesn2 plays an active role in the early stages of melanomagenesis. UVB-induced Sesn2 may confer a survival advantage for melanocytes and facilitate malignant transformation. Indeed, we found that the pro-survival function of Sesn2 seems to be critical for the development of xenografted human melanoma and SCC tumors, likely through promoting the AKT pathway, as Sesn2 had no effect on cell proliferation in vitro (Fig. 6F). It is likely that increased AMP kinase activation by Sesn2 inhibits mTOR activation to reduce cell proliferation (6, 13), which counteracts the positive regulation of cell proliferation by increased AKT activation caused by Sesn2.
Although the p53-dependent induction of Sesn2 (10) suggests a tumor-suppressive function of Sesn2, our data support an oncogenic function of Sesn2 in SCC and melanoma cells. Indeed, such a pleiotropic function of genes has been detected in another p53 target gene TIGAR (12, 36). Although p53 is a key tumor suppressor, the p53 target gene TIGAR has been demonstrated to possess both an antioxidant function and serve as an oncogene in intestinal cancer (37). Regulation of the antioxidant regulator Nrf2 by Sesn2 may also play a role in the oncogenic function of Sesn2 (7), as the antioxidant Nrf2 is shown to be an oncogene in pancreatic cancer (38) and is implicated as an oncogene in melanoma and SCC (39, 40). Inhibition of Nrf2 by Brusatol enhances the efficacy of chemotherapy (41). Our work here identified Sesn2 as playing an oncogenic role by regulating AKT activation and cell survival.
In summary, our findings have demonstrated a previously unknown role of Sesn2 in UVB-irradiated keratinocytes and melanocytes and in SCC and melanoma cells. Sesn2 is a positive regulator for AKT activation and promotes cell survival after UVB stress and chemotherapeutic treatment. Our results may provide new molecular insights into the signaling network that amplifies and maintains AKT activation through Sesn2 up-regulation and shed light on the critical oncogenic role of Sesn2 in SCC and melanoma.
Acknowledgments
We thank Terri Li for immunohistochemical Sesn2 analysis and Dr. Ann Motten for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants ES016936 (NIEHS; to Y.-Y. H.), CA172660 (NCI; to A. B.), GM067983 (NIGMS; to A. A.), P30 CA014599 (University of Chicago Cancer Research Center, and UL1 TR000430 (Clinical and Translational Science Award). This work was also supported by American Cancer Society Grant RSG-13-078-01 (to Y.-Y. H.) and the University of Chicago Friends of Dermatology Endowment Fund.
- SCC
- squamous cell carcinoma
- Sesn
- Sestrin
- NHEK
- normal human epidermal keratinocyte
- NHEM
- normal human epidermal melanocyte
- NC
- negative control
- shNC
- negative control shRNA
- CT
- cycle number
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- MEF
- mouse embryonic fibroblast
- 5-FU
- 5-fluorouracil
- BRAF
- v-raf murine sarcoma viral oncogene homolog B
- PLX
- PLX4032.
REFERENCES
- 1. Rogers H. W., Weinstock M. A., Harris A. R., Hinckley M. R., Feldman S. R., Fleischer A. B., Coldiron B. M. (2010) Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch. Dermatol. 146, 283–287 [DOI] [PubMed] [Google Scholar]
- 2. D'Orazio J., Jarrett S., Amaro-Ortiz A., Scott T. (2013) UV Radiation and the Skin. Int. J. Mol. Sci. 14, 12222–12248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tsao H., Chin L., Garraway L. A., Fisher D. E. (2012) Melanoma: from mutations to medicine. Genes Dev. 26, 1131–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Claerhout S., Verschooten L., Van Kelst S., De Vos R., Proby C., Agostinis P., Garmyn M. (2010) Concomitant inhibition of AKT and autophagy is required for efficient cisplatin-induced apoptosis of metastatic skin carcinoma. Int. J. Cancer 127, 2790–2803 [DOI] [PubMed] [Google Scholar]
- 5. Budanov A. V., Sablina A. A., Feinstein E., Koonin E. V., Chumakov P. M. (2004) Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 304, 596–600 [DOI] [PubMed] [Google Scholar]
- 6. Budanov A. V., Karin M. (2008) p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134, 451–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bae S. H., Sung S. H., Oh S. Y., Lim J. M., Lee S. K., Park Y. N., Lee H. E., Kang D., Rhee S. G. (2013) Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab. 17, 73–84 [DOI] [PubMed] [Google Scholar]
- 8. Lee J. H., Budanov A. V., Park E. J., Birse R., Kim T. E., Perkins G. A., Ocorr K., Ellisman M. H., Bodmer R., Bier E., Karin M. (2010) Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 327, 1223–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang Y. L., Loh K. S., Liou B. Y., Chu I. H., Kuo C. J., Chen H. D., Chen C. S. (2013) SESN-1 is a positive regulator of lifespan in Caenorhabditis elegans. Exp. Gerontol. 48, 371–379 [DOI] [PubMed] [Google Scholar]
- 10. Budanov A. V., Shoshani T., Faerman A., Zelin E., Kamer I., Kalinski H., Gorodin S., Fishman A., Chajut A., Einat P., Skaliter R., Gudkov A. V., Chumakov P. M., Feinstein E. (2002) Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene 21, 6017–6031 [DOI] [PubMed] [Google Scholar]
- 11. Budanov A. V. (2011) Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Antioxid. Redox Signal. 15, 1679–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sablina A. A., Budanov A. V., Ilyinskaya G. V., Agapova L. S., Kravchenko J. E., Chumakov P. M. (2005) The antioxidant function of the p53 tumor suppressor. Nat. Med. 11, 1306–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cam M., Bid H. K., Xiao L., Zambetti G. P., Houghton P. J., Cam H. (2014) p53/TAp63 and AKT regulate mammalian target of rapamycin complex 1 (mTORC1) signaling through two independent parallel pathways in the presence of DNA damage. J. Biol. Chem. 289, 4083–4094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Velasco-Miguel S., Buckbinder L., Jean P., Gelbert L., Talbott R., Laidlaw J., Seizinger B., Kley N. (1999) PA26, a novel target of the p53 tumor suppressor and member of the GADD family of DNA damage and growth arrest inducible genes. Oncogene 18, 127–137 [DOI] [PubMed] [Google Scholar]
- 15. Ragnarsson G., Eiriksdottir G., Johannsdottir J. T., Jonasson J. G., Egilsson V., Ingvarsson S. (1999) Loss of heterozygosity at chromosome 1p in different solid human tumours: association with survival. Br. J. Cancer 79, 1468–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schwab M., Praml C., Amler L. C. (1996) Genomic instability in 1p and human malignancies. Genes Chromosomes Cancer 16, 211–229 [DOI] [PubMed] [Google Scholar]
- 17. Lee J. H., Budanov A. V., Talukdar S., Park E. J., Park H. L., Park H. W., Bandyopadhyay G., Li N., Aghajan M., Jang I., Wolfe A. M., Perkins G. A., Ellisman M. H., Bier E., Scadeng M., Foretz M., Viollet B., Olefsky J., Karin M. (2012) Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab. 16, 311–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shao Y., Aplin A. E. (2010) Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res. 70, 6670–6681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ming M., Shea C. R., Guo X., Li X., Soltani K., Han W., He Y. Y. (2010) Regulation of global genome nucleotide excision repair by SIRT1 through xeroderma pigmentosum C. Proc. Natl. Acad. Sci. U. S. A. 107, 22623–22628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Han W., Ming M., Zhao R., Pi J., Wu C., He Y. Y. (2012) Nrf1 CNC-bZIP protein promotes cell survival and nucleotide excision repair through maintaining glutathione homeostasis. J. Biol. Chem. 287, 18788–18795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu C. L., Qiang L., Han W., Ming M., Viollet B., He Y. Y. (2013) Role of AMPK in UVB-induced DNA damage repair and growth control. Oncogene 32, 2682–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qiang L., Yang Y., Ma Y. J., Chen F. H., Zhang L. B., Liu W., Qi Q., Lu N., Tao L., Wang X. T., You Q. D., Guo Q. L. (2009) Isolation and characterization of cancer stem like cells in human glioblastoma cell lines. Cancer Lett. 279, 13–21 [DOI] [PubMed] [Google Scholar]
- 23. Qiang L., Wu C., Ming M., Viollet B., He Y. Y. (2013) Autophagy controls p38 activation to promote cell survival under genotoxic stress. J. Biol. Chem. 288, 1603–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qiang L., He Y. Y. (2014) Autophagy deficiency stabilizes TWIST1 to promote epithelial-mesenchymal transition. Autophagy 10, 1864–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Han W., Ming M., He T. C., He Y. Y. (2010) Immunosuppressive cyclosporin A activates AKT in keratinocytes through PTEN suppression: implications in skin carcinogenesis. J. Biol. Chem. 285, 11369–11377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ming M., Feng L., Shea C. R., Soltani K., Zhao B., Han W., Smart R. C., Trempus C. S., He Y. Y. (2011) PTEN positively regulates UVB-induced DNA damage repair. Cancer Res. 71, 5287–5295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hers I., Vincent E. E., Tavaré J. M. (2011) Akt signalling in health and disease. Cell. Signal. 23, 1515–1527 [DOI] [PubMed] [Google Scholar]
- 28. Lee J. H., Budanov A. V., Karin M. (2013) Sestrins orchestrate cellular metabolism to attenuate aging. Cell Metab. 18, 792–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee S. O., Andey T., Jin U. H., Kim K., Singh M., Sachdeva M., Safe S. (2012) The nuclear receptor TR3 regulates mTORC1 signaling in lung cancer cells expressing wild-type p53. Oncogene 31, 3265–3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wei C. L., Wu Q., Vega V. B., Chiu K. P., Ng P., Zhang T., Shahab A., Yong H. C., Fu Y., Weng Z., Liu J., Zhao X. D., Chew J. L., Lee Y. L., Kuznetsov V. A., Sung W. K., Miller L. D., Lim B., Liu E. T., Yu Q., Ng H. H., Ruan Y. (2006) A global map of p53 transcription-factor binding sites in the human genome. Cell 124, 207–219 [DOI] [PubMed] [Google Scholar]
- 31. Marionnet C., Pierrard C., Lejeune F., Sok J., Thomas M., Bernerd F. (2010) Different oxidative stress response in keratinocytes and fibroblasts of reconstructed skin exposed to non extreme daily-ultraviolet radiation. PLoS ONE 5, e12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lehman T. A., Modali R., Boukamp P., Stanek J., Bennett W. P., Welsh J. A., Metcalf R. A., Stampfer M. R., Fusenig N., Rogan E. M. (1993) p53 mutations in human immortalized epithelial cell lines. Carcinogenesis 14, 833–839 [DOI] [PubMed] [Google Scholar]
- 33. Budanov A. V., Lee J. H., Karin M. (2010) Stressin' sestrins take an aging fight. EMBO Mol. Med. 2, 388–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sanli T., Linher-Melville K., Tsakiridis T., Singh G. (2012) Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells. PLoS ONE 7, e32035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ben-Sahra I., Dirat B., Laurent K., Puissant A., Auberger P., Budanov A., Tanti J. F., Bost F. (2013) Sestrin2 integrates Akt and mTOR signaling to protect cells against energetic stress-induced death. Cell Death Differ. 20, 611–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li T., Kon N., Jiang L., Tan M., Ludwig T., Zhao Y., Baer R., Gu W. (2012) Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149, 1269–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheung E. C., Athineos D., Lee P., Ridgway R. A., Lambie W., Nixon C., Strathdee D., Blyth K., Sansom O. J., Vousden K. H. (2013) TIGAR is required for efficient intestinal regeneration and tumorigenesis. Dev. Cell 25, 463–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. DeNicola G. M., Karreth F. A., Humpton T. J., Gopinathan A., Wei C., Frese K., Mangal D., Yu K. H., Yeo C. J., Calhoun E. S., Scrimieri F., Winter J. M., Hruban R. H., Iacobuzio-Donahue C., Kern S. E., Blair I. A., Tuveson D. A. (2011) Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim Y. R., Oh J. E., Kim M. S., Kang M. R., Park S. W., Han J. Y., Eom H. S., Yoo N. J., Lee S. H. (2010) Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J. Pathol. 220, 446–451 [DOI] [PubMed] [Google Scholar]
- 40. Miura S., Shibazaki M., Kasai S., Yasuhira S., Watanabe A., Inoue T., Kageshita Y., Tsunoda K., Takahashi K., Akasaka T., Masuda T., Maesawa C. (2014) A Somatic mutation of the KEAP1 gene in malignant melanoma is involved in aberrant NRF2 activation and an increase in intrinsic drug resistance. J. Invest. Dermatol. 134, 553–556 [DOI] [PubMed] [Google Scholar]
- 41. Ren D., Villeneuve N. F., Jiang T., Wu T., Lau A., Toppin H. A., Zhang D. D. (2011) Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. U. S. A. 108, 1433–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]