

Background: Non-small-cell lung cancer (NSCLC) exhibits EGFR mutation.
Results: Treatment with isoliquiritigenin (ILQ) inhibited growth and induced apoptosis in tyrosine kinase inhibitor-sensitive and -resistant NSCLC cells. ILQ suppressed wild type and mutant (L858R/T790M) EGFR kinase activity and attenuated H1975 lung cancer cell xenograft tumor growth.
Conclusion: ILQ directly targets wild type or mutant EGFR.
Significance: ILQ could be a potential therapeutic agent against NSCLC.
Keywords: Apoptosis, Cell Growth, Epidermal Growth Factor Receptor (EGFR), Lung Cancer, Tyrosine-Protein Kinase (Tyrosine Kinase)
Abstract
Non-small-cell lung cancer (NSCLC) is associated with diverse genetic alterations including mutation of epidermal growth factor receptor (EGFR). Isoliquiritigenin (ILQ), a chalcone derivative, possesses anticancer activities. In the present study, we investigated the effects of ILQ on the growth of tyrosine kinase inhibitor (TKI)-sensitive and -resistant NSCLC cells and elucidated its underlying mechanisms. Treatment with ILQ inhibited growth and induced apoptosis in both TKI-sensitive and -resistant NSCLC cells. ILQ-induced apoptosis was associated with the cleavage of caspase-3 and poly-(ADP-ribose)-polymerase, increased expression of Bim, and reduced expression of Bcl-2. In vitro kinase assay results revealed that ILQ inhibited the catalytic activity of both wild type and double mutant (L858R/T790M) EGFR. Treatment with ILQ inhibited the anchorage-independent growth of NIH3T3 cells stably transfected with either wild type or double-mutant EGFR with or without EGF stimulation. ILQ also reduced the phosphorylation of Akt and ERK1/2 in both TKI-sensitive and -resistant NSCLC cells, and attenuated the kinase activity of Akt1 and ERK2 in vitro. ILQ directly interacted with both wild type and double-mutant EGFR in an ATP-competitive manner. A docking model study showed that ILQ formed two hydrogen bonds (Glu-762 and Met-793) with wild type EGFR and three hydrogen bonds (Lys-745, Met-793, and Asp-855) with mutant EGFR. ILQ attenuated the xenograft tumor growth of H1975 cells, which was associated with decreased expression of Ki-67 and diminished phosphorylation of Akt and ERK1/2. Taken together, ILQ suppresses NSCLC cell growth by directly targeting wild type or mutant EGFR.
Introduction
Lung cancer is the most common cancer throughout the world and is the leading cause of cancer mortality (1). About 80–90% of lung cancers are classified as non-small-cell lung cancer (NSCLC),4 which is usually diagnosed at an advanced stage (2). NSCLC is considered to be a smart cancer that is linked to several genetic alterations including activating mutations of the epidermal growth factor receptor (EGFR) gene, amplification of the Met oncogene and mutation of the Kirsten rat sarcoma viral oncogene homolog (KRAS) gene. About 40–80% of NSCLC patients exhibit elevated EGFR expression (3, 4). Inappropriate overexpression of the EGFR tyrosine kinase leads to amplification of intracellular signaling mediated by mitogen-activated protein (MAP) kinases (5) and phosphatidylinositol 3-kinase (PI3-K)/Akt (6), thereby promoting cancer cell proliferation. Because of the crucial role of the EGFR tyrosine kinase in tumor cell survival, several EGFR tyrosine kinase inhibitors (EGFR-TKIs), including erlotinib and gefitinib, have emerged as first-line therapy for clinical management of NSCLC patients (4). Moreover, treatment with erlotinib exhibited better progression-free survival as well as overall survival among Chinese NSCLC patients with EGFR mutation as compared with those without EGFR mutation (7). However, the clinical success of these EGFR-TKIs is limited by primary or acquired resistance. Although NSCLC harboring mutations in the EGFR respond well to EGFR-TKIs, virtually all patients with a good initial response will relapse due to acquired resistance. The acquired resistance to EGFR-TKIs has been shown to be associated with the secondary gatekeeper mutation, T790M (8, 9). For instance, resistance to gefitinib appears within two years corresponding with mutation of EGFR (L858R/T790M) (10, 11). Thus, for effective treatment of NSCLC, inhibition of both wild type EGFR and mutant EGFR (L858R/T790M) functions might be a promising therapeutic strategy.
A wide variety of anticancer agents isolated from various medicinal plants have shown multi-targeted therapeutic and/or chemopreventive potential. Isoliquiritigenin (ILQ, 4, 2′, 4′-trihydroxychalcone), present in licorice, shallot, and bean sprouts (12), possesses antioxidative (13), anti-inflammatory (14), and anticancer (15) activities. ILQ has been reported to inhibit the proliferation and induce apoptosis in various cancer cell types in culture (16–19). It has also been shown to suppress the growth of xenograft tumors developed from human cancer cells in athymic nude mice (20, 21) and to inhibit tumor angiogenesis (22), diminish migration, invasion (23, 24), and metastasis (25) of cancer cells. Previous studies have shown that ILQ inhibits proliferation of human lung cancer (A549) cells by arresting cells at the G1 and G2/M phase of cell cycle and induces p53- and Fas-FasL-mediated apoptosis in these cells (26, 27). Considering that EGFR tyrosine kinase signaling plays a critical role in NSCLC, we examined the anticancer potential of ILQ in several lung cancer cell lines harboring wild type or mutant EGFR, and elucidated the mechanisms of its possible modulation of EGFR signaling. Our study revealed that ILQ directly interacts with EGFR and its downstream signaling kinases, Akt1 and extracellular signal-regulated kinase (ERK)1/2, and inhibits the catalytic activities of these kinases, thereby suppressing proliferation and inducing apoptosis in EGFR-sensitive and -resistant lung cancer cells. Moreover, ILQ attenuated the in vivo xenograft tumor growth of human lung cancer (NCI-H1975) cells expressing mutant (L858R/T790M) EGFR.
EXPERIMENTAL PROCEDURES
Materials
ILQ (purity >99%) was purchased from Selleckchem (Houston, TX). Erlotinib, gefitinib, and CL-387785 were purchased from Sigma-Aldrich. Antibodies against phosphorylated (p-)EGFR (Tyr-1068), p-Akt (Ser-473), cleaved PARP, caspase-3, Bim, total EGFR, p90RSK, p110, mTOR, and p70RSK were from Cell Signaling Biotechnology (Beverly, MA). Antibodies against p-ERKs (T202/Y204), phosphatidylinositol 3-kinase (PI3-K), Bcl-2, Raf, MEK, MNK, and β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). For immunohistochemistry, the Ki-67 antibody was from Thermo Scientific (Fremont, CA). CNBr-Sepharose 4B and glutathione-Sepharose 4B beads were purchased from GE Healthcare (Piscataway, NJ). The protein assay kit was obtained from Bio-Rad. The DNA construct of wild type EGFR and mutant EGFR (L858R) were from Addgene (Cambridge, MA). The mutant EGFR (T790M/L858R) was produced using the QuickChange lightning site-directed mutagenesis kit (Stratagene, Santa Clara, CA). Active wild type EGFR, mutant T790M/L858R EGFR, phospholipase C-γ. (PLCγ substrate), active ERK1, ERK2, Akt1, and Akt2 were purchased from Millipore. Cell Titer 96 Aqueous One Solution Reagent [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS)] kit for the cell proliferation assay was from Promega.
Cell Culture and Transfection
Human lung adenocarcinoma, HCC827, NCI-H1650, NCI-H1975, and A549 cells, 293T, and NIH3T3 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA). HCC827GR (gefitinib-resistant) cells were kindly provided by Dr. Pasi A. Jänne (Dept. of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA) (28). The HCC827, NCI-H1650, NCI-H1975, and HCC827GR cells were cultured in RPMI 1640 medium containing penicillin (100 units/ml), streptomycin (100 μg/ml), l-glutamine (2 mm), and 10% fetal bovine serum (FBS; Gemini Bio-Products, Calabasas, CA). A549 cells were maintained in F12K medium with 10% FBS. 293T cells were cultured in MEM with 10% FBS. NIH3T3 cells were cultured in DMEM with 10% calf serum. Cells were maintained in a 5% CO2, 37 °C humidified incubator. All cells were cytogenetically tested and authenticated before being frozen. Each vial of frozen cells was thawed and maintained in culture for a maximum of 8 weeks. For stable expression of wild type or mutant EGFR, 293T cells were transfected with the pBabe-mock, pBabe-wild EGFR, or pBabe-mutant EGFR plasmid using the jetPEI poly transfection reagent (Polyplus-transfection SAS, Saint Quentin Yvelines, France), following the manufacturer's suggested protocols. The transfection medium was changed at 4 h after transfection, and then cells were cultured for 36 h. Virus particles were harvested by filtration using a 0.45-mm syringe filter, then combined with 8 mg/ml of polybrene (Millipore) and infected into NIH3T3 cells for 24 h. The cell culture media were replaced with fresh culture medium and cells cultured for 24 h, and then cells were selected with puromycin (1 mg/ml) for 36 h. Selected cells were used in subsequent experiments.
In Vitro Kinase Assay
Active EGFR, EGFR T790M/L858R, ERK1, ERK2, Akt1, or Akt2 (100 ng) protein, and their respective substrates were incubated in the presence or absence of ILQ for 10 min at 30 °C. The mixture was suspended in kinase buffer supplemented with 10 μl of diluted [γ-32P]ATP solution. Incorporated radioactivity was determined using a scintillation counter or autoradiography.
Molecular Modeling
Computer modeling of ILQ with wild type EGFR and T790 mutant EGFR was performed using the Schrödinger Suite 2011 program (29). First an x-ray diffraction structure of wild type EGFR with a resolution of 2.60 Å complexed with erlotinib (PDB ID 1M17) (30) and an x-ray diffraction structure of the EGFR T790M mutant with a resolution of 2.90 Å bound to WZ4002 (PDB ID 3IKA) (31) were obtained from the RCSB Protein Data Bank (32). These structures were prepared under the standard procedure of Protein Preparation Wizard described in Schrödinger Suite 2011. Hydrogen atoms were added consistent with a pH of 7, and all water molecules were removed. ILQ was prepared using LigPrep of Schrödinger for docking by default parameters. Then ILQ-protein docking was performed using the Induced-Fit docking program of Schrödinger that allows for flexibility of ligands to fit at the binding pocket. For Glide docking parameters, the receptor and ligand Van der Waals scaling were both set at 0.5, and the maximum number of poses at 20. For prime refinement, we refined the residues only within 5.0 Å of ligands' poses. Glide re-docking was set to re-dock into structures within 30.0 kcal/mol of the best structure, and the best 20 poses were retained under extra precision (XP). Herein, we could obtain the best-docked representative structure.
Anchorage-independent Cell Transformation Assay
Lung cancer cells (8 × 103 per well) suspended in BME supplemented with 10% FBS and 1% antibiotics were added to 0.3% agar with different doses of each compound in a top layer over a base layer of 0.6% agar with different doses of each compound. The effects of ILQ on EGF-dependent or -independent cell transformation were investigated in NIH3T3 cells stably transfected with wild type or mutant EGFR (33). Cells (8 × 103 per well) were exposed to EGF with or without ILQ in 1 ml of 0.33% BME agar containing 10% FBS or in 3.5 ml of 0.5% BME agar containing 10% FBS. The cultures were maintained at 37 °C in a 5% CO2 incubator for 3 weeks, after which time the cell colonies were counted under a microscope with the aid of the Image-Pro Plus software program (versus version 6.1, Media Cybernetics).
MTS Assay
Cells (1 × 104) were seeded in a 96-well plate and then incubated for 24 h with different concentrations of ILQ (0, 10, 20, or 40 μm) or gefitinib (1 μm) for 24 h. The effect of ILQ on viability was estimated using the Cell-Titer 96 Aqueous MTS-One Solution cell proliferation assay kit (Promega) according to the manufacturer's instructions. The assay solution was added to each well, and absorbance (492 nm) was read with a 96-well plate reader (Labsystem Multiskan MS, Labsystem, Finland).
Annexin V Staining
Cells were seeded in 100-mm dishes and treated with vehicle, ILQ, or gefitinib for 24 h. Cells were then fixed in 70% ethanol and stored at −20 °C for 24 h. After staining with annexin V, apoptosis was determined using a BD FACSCalibur Flow Cytometer (BD Biosciences, San Jose, CA).
Ex Vivo/in Vitro Pull-down Assay
ILQ-Sepharose 4B beads were prepared according to the manufacturer's instructions (Amersham Biosciences). Cellular supernatant fractions (500 μg) and active EGFR or mutant EGFR proteins (200 ng) were incubated with ILQ-Sepharose 4B (or Sepharose 4B alone as a control) beads in reaction buffer. After incubation with gentle rocking overnight at 4 °C, the beads were washed three times with buffer, and proteins bound to the beads were analyzed by Western blotting with an EGFR antibody.
Western Blot Assay
Cell lysates were prepared with RIPA buffer (10 mm Tris, pH 7.5, 150 mm NaCl, 5 mm EDTA, 1% Triton X-100, 1 mm DTT, 0.1 mm PMSF, and protease inhibitor mixture), and the supernatant fractions were boiled for 5 min. The protein concentration was determined using a protein assay kit (Bio-Rad), as described in the manufacturer's manual. Lysates were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride (PVDF) membrane (Amersham Biosciences). After transfer, the membrane was incubated with specific primary antibodies at 4 °C overnight. Protein bands were visualized by chemiluminescence after hybridization with a horseradish peroxidase-conjugated secondary antibody.
Xenograft Assay
Athymic nude mice (6 weeks old) were obtained from Charles River (Seoul, South Korea) and maintained under specific pathogen-free conditions based on the guidelines established by the Seoul National University Animal Care and Use Committee. Mice were divided into 4 groups: 1) vehicle group (n = 10); 2) 1 mg/kg of ILQ (n = 10); 3) 5 mg/kg of ILQ (n = 10); and 4) 5 mg/kg of gefitinib (n = 10)]. NCI-H1975 cells (1 × 106 cells/100 μl) were suspended in RPMI 1640 medium and inoculated with 100 μl matrigel subcutaneously into the right flank of each mouse. Vehicle, ILQ, or gefitinib was injected intraperitoneal three times per week for 12 days. Tumor volume was calculated from measurements of two diameters of the individual tumor base using the following formula: tumor volume (mm3) = (length × width × height × 0.52). Mice were monitored until tumors reached 1 cm3 total volume, at which time mice were euthanized and tumors extracted.
Immunohistochemical Analysis
Tumor tissues were embedded in paraffin and subjected to immunohistochemistry. Tissues were de-paraffinized and hydrated, then permeabilized with 0.5% Triton X-100/1 × PBS for 10 min. Tissues were hybridized with Ki-67 (1:150), p-Akt (1:100), or p-ERK1/2 (1:100) as the primary antibody and biotinylated goat anti-rabbit IgG as the secondary antibody. An ABC kit (Vector Laboratories, Inc., Burlingame, CA) was used to detect target proteins according to the manufacturer's instructions. After developing with 3,3′-diaminobenzidine, the sections were counterstained with hematoxylin and observed by microscope (200×) and Image-Pro Plus software (v. 6.1; Media Cybernetics).
Statistical Analysis
Data are expressed as means ± S.D., and the Student's t test was used to perform statistical analysis for single comparisons. A probability value of p < 0.05 was used as the criterion for statistical significance.
RESULTS
ILQ Inhibits Growth and Induces Apoptosis in Human Lung Cancer Cells
To investigate the anticancer potential of ILQ, we first examined the effect of the compound on the anchorage-independent growth of TKI-sensitive (HCC827 and NCI-H1650) and -resistant (NCI-H1975, HCC827GR and A549) human lung cancer cells. Incubation with ILQ attenuated soft-agar colony formation in human lung cancer (HCC827, NCI-H1650, NCI-H1975, HCC827GR, and A549) cells in a concentration-dependent manner (Fig. 1A). ILQ (40 μm) inhibited colony formation by 90, 70, 95, 40, and 50% in HCC827, NCI-H1650, NCI-H1975, HCC827GR, and A549 cells, respectively. We next examined the effect of ILQ on the induction of apoptosis in these cells. AnnexinV-PI staining revealed that treatment with ILQ caused significant induction of apoptosis in both TKI-sensitive and -resistant cells (Fig. 1B). The induction of apoptosis by ILQ (40 μm) in these cells was in the order of NCI-H1975 > NCI-H1650 > HCC827 > A549 > HCC827GR. A similar pattern of decreased cell viability was observed upon incubation of these lung cancer cells with ILQ (Fig. 1C). Among the gefitinib-sensitive (HCC827 and NCI-H1650) and gefitinib-resistant (NCI-H1975, HCC827GR, and A549) cells, H1975 cells exhibited the highest sensitivity to ILQ-induced growth inhibition and apoptosis. However ILQ up to 80 μm had no effect on growth of normal human lung MRC-5 cells (supplemental Fig. S1). In addition, treatment of HCC827, NCI-H1650, and NCI-H1975 cells with ILQ induced cleavage of caspase-3 and PARP in all 3 cell lines in a concentration-dependent manner (Fig. 1D). ILQ also increased the expression of the proapoptotic protein Bim and reduced the expression of the anti-apoptotic protein Bcl2 in HCC827, NCI-H1650, and NCI-H1975 cells (Fig. 1D).
FIGURE 1.

ILQ inhibits growth and induces apoptosis of lung cancer cell lines. A, TKI-sensitive (HCC827 and H1650) and TKI-resistant (H1975, HCC827GR, and A549) human lung cancer cells were treated with the indicated concentrations of ILQ or gefitinib and anchorage-independent cell growth was assessed as described under “Experimental Procedures.” The number of colonies was counted under a microscope with the aid of the Image-Pro Plus software (versus version 6.1) program. B, cells were incubated with ILQ (40 μm) for 24 h, and the percent of apoptosis was measured by Annexin-V/PI staining. C, cells were incubated with ILQ (40 μm) or gefitinib (1 μm) for 24 h, and cell viability was analyzed using the MTS assay. Results are shown as means ± S.D. (n = 3). The asterisks (*), (**), or (***) indicate a significant difference (p < 0.05), (p < 0.01), or (p < 0.001), respectively, between ILQ-treated and untreated cells. D, cells expressing wild type EGFR (HCC827 and H1650) or L858R/T790M mutant EGFR (HCC827GR) were treated with ILQ for 24 h, and the expression of apoptotic markers was detected by immunoblotting.
The Antiproliferative Effect of ILQ Is Mediated through Inhibition of the Kinase Activity of Both Wild Type and Mutant EGFR
Because ILQ exhibited the most potent anticancer effect in H1975 cells, which have EGFR double mutations, we hypothesized that ILQ might target EGFR. In vitro radioactive kinase assay results showed that ILQ attenuated the kinase activity of both wild type (Fig. 2A) and mutant (L858R/T790M) EGFR (Fig. 2B). The inhibitory effect of ILQ on the catalytic activity of wild type and mutant EGFR was comparable to that elicited by EGFR antagonists, erlotinib or gefitinib (Fig. 2, A and B) or treatment with CL387785, a pharmacological inhibitor of the T790M mutant EGFR (Fig. 2B). Fig. 2C illustrates the stable overexpression of wild type and T790M mutant EGFR in NIH3T3 cells. Cells transfected with wild type EGFR showed significantly increased colony number upon treatment with EGF, whereas cells harboring T790M mutant EGFR elicited marked increases in soft agar colony formation independent of EGF stimulation (Fig. 2D). Treatment with ILQ significantly inhibited the anchorage-independent growth of both wild type and mutant EGFR-transfected-NIH3T3 cells in a concentration-dependent manner (Fig. 2E).
FIGURE 2.

ILQ inhibits wild type and mutant EGFR (L858R/T790M) kinase activity. A, active wild type EGFR was incubated in vitro with [γ-32P]ATP in the presence or absence of the indicated concentrations of ILQ, erlotinib or gefitinib for 30 min and incorporated radioactivity was determined using a scintillation counter. Data are presented as means ± S.D. of three independent experiments each conducted in triplicate. B, active mutant EGFR (L858R/T790M) was incubated in vitro with its substrate PLCγ1 and [γ-32P]ATP in the presence or absence of the indicated concentrations of ILQ, CL-387785, or Gefitinib for 30 min, and the phosphorylation of PLCγ1 was detected in an autoradiogram. Data are representative of three independent experiments showing similar results. C, NIH3T3 cells were stably transfected with plasmids expressing wild type (EGFR W) or double-mutant (EGFR DM) EGFR or Mock. The expression of wild type and mutant (L858R/T790M) EGFR was detected by Western blotting. D, NIH3T3 cells stably transfected with mock, EGFR W or EGFR DM were treated or not treated with EGF and subjected to an anchorage-independent growth assay. The experiment was performed in triplicate, and the number of colonies was counted under a microscope with the aid of the Image-Pro Plus software (versus version 6.1) program. E, NIH3T3 cells transfected with either wild type (EGFR W) or mutant (EGFR DM) EGFR were treated or not treated with the indicated concentrations of ILQ or gefitinib in the presence or absence of EGF in a soft agar colony formation assay. ILQ inhibits EGF-induced colony formation in EGFR W-transfected cells in a concentration-dependent manner (left panel). Treatment with ILQ reduced the colony numbers in EGFR DM-transfected cells without EGF challenge (middle panel) or with EGF treatment (right panel) in a concentration-dependent manner. The colony number is shown as a percentage of each vehicle-treated group (100%). Results are shown as means ± S.D. (n = 3). The asterisks (*) and (**) indicate a significant difference (p < 0.05) and (p < 0.01), respectively, between the ILQ-treated and untreated groups.
ILQ Attenuates the Phosphorylation and/or Catalytic Activity of Akt and ERK1/2
Because Akt and ERK1/2 function as EGFR downstream signaling kinases, we examined the effect of ILQ on the phosphorylation and/or catalytic activities of these kinases. Incubation of TKI-sensitive (HCC827 and H1650) and -resistant (H1975) cells with ILQ inhibited the phosphorylation of Akt and ERK1/2, without affecting that of EGFR (Fig. 3A). In vitro kinase assay results revealed that ILQ attenuated the kinase activity of Akt1 (Fig. 3B) and ERK2 (Fig. 3C). Notably, ILQ-mediated inhibition of Akt1, but not Akt2, activity was less intense than that of Akt 1X, which is a pharmacological inhibitor of Akt (Fig. 3B). Likewise, the inhibitory effect of ILQ on ERK2, but not ERK1, activity was relatively less compared with that elicited by a specific inhibitor (Cay10561) of ERK1/2 (Fig. 3C).
FIGURE 3.
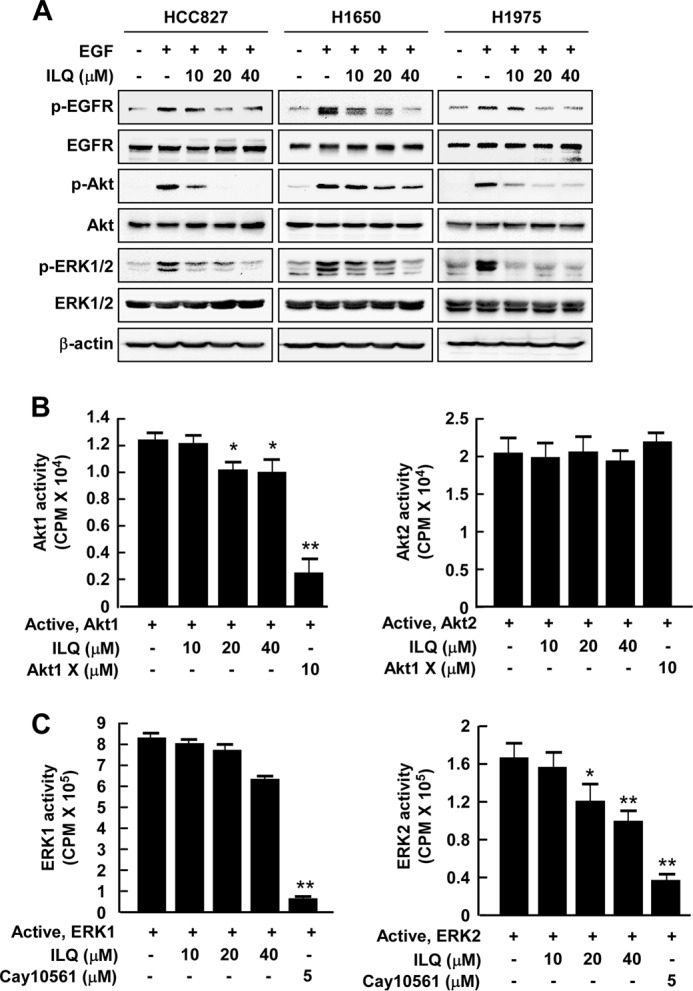
Effects of ILQ on phosphorylation and/or kinase activity of EGFR downstream signaling kinases, Akt and ERK1/2, in lung cancer cells. A, human lung cancer (HCC827, H1650, and H1975) cells, which were starved, were incubated with the indicated concentrations of ILQ and EGF (10 ng/ml) for 30 min and the expression of pEGFR, EGFR, p-Akt, Akt, p-ERK1/2, ERK1/2, and β-actin was detected by immunoblotting. Data are representative of three independent experiments showing similar results. B, active Akt1 (left panel) or Akt2 (right panel) was incubated in vitro with [γ-32P]ATP in the presence or absence of the indicated concentrations of ILQ or Akt inhibitor, Akt1X, for 30 min and radioactive incorporation was determined using a scintillation counter. Data are presented as means ± S.D. of three independent experiments each conducted in triplicate. (*, p < 0.05 and **, p < 0.01 compared with untreated control). C, active ERK1 (left panel) or ERK2 (right panel) was incubated in vitro with [γ-32P]ATP in the presence or absence of the indicated concentrations of ILQ or ERK inhibitor, Cay10561, for 30 min and radioactive incorporation was determined using a scintillation counter. Data are presented as means ± S.D. of three independent experiments each conducted in triplicate. The asterisks (*) and (**) indicate a significant difference (p < 0.05) and (p < 0.01), respectively, between the ILQ-treated and untreated groups.
ILQ Interacts with Both Wild Type and Mutant EGFR
We next determined whether ILQ could interact with EGFR and/or its downstream signaling kinases. Incubation of an NCI-H1975 cell lysate with ILQ-Sepharose 4B beads followed by pull-down and Western blotting revealed that ILQ interacted with EGFR, Akt, and ERK1/2, but did not bind with p110, mTOR, Raf, MEK, p70S6K, MNK, or p90RSK (Fig. 4A). Moreover, the interaction of ILQ with wild type (Fig. 4B) or mutant EGFR (Fig. 4C) was abrogated upon co-incubation with ATP, suggesting that ILQ binds with wild type or mutant EGFR in an ATP-competitive manner. To better understand the mechanisms of the ILQ interaction with the wild type or T790M mutant EGFR, we performed a computational docking model study using Induced-Fit docking module of Schrödinger Suite 2011 (29). In the docked models, ILQ showed good binding affinity with both the wild type and T790M mutant EGFR. From these results, we found that ILQ docked nicely within the ATP-binding sites of wild type as well as T790M mutant EGFR. According to the docking model, ILQ formed hydrogen bonds with amino acid residues located at the ATP-binding pocket of wild type and mutant EGFR. ILQ formed hydrogen bonds with Glu-762 and Met-793 of wild type EGFR (Fig. 4, D and E) and with Lys-745, Met-793, and Asp-855 of T790M mutant EGFR (Fig. 4, F and G). These results suggested that ILQ might be a potential inhibitor of both wild type and T790M mutant EGFR. (Images were generated with UCSF Chimera program (34)).
FIGURE 4.

ILQ directly interacts with wild type or mutant EGFR. A, lysate from H1975 cells was incubated with Sepharose 4B-conjugated ILQ or Sepharose 4B-only beads and a pull-down assay was performed as described under “Experimental Procedures.” Immunoprecipitates were subjected to Western blotting to examine the interaction between ILQ and the indicated proteins. Data are representative of three separate experiments eliciting similar results. B, active wild type EGFR (1 μg) or C, active mutant (L858R/T790M) EGFR (1 μg) was incubated in vitro with unlabeled ATP (10 or 100 μm) and 50 μl of ILQ-Sepharose 4B or 50 μl of Sepharose 4B (as a negative control) in reaction buffer at a final volume of 500 μl. The mixtures were incubated at 4 °C overnight with shaking. After washing, the pulled-down proteins were detected by Western blotting. Data are representative of three independent experiments. D–G, computational docking model predicts the interaction between ILQ and wild type or mutant EGFR. D and E, ILQ directly binds to wild type EGFR. From the Induced Fit Docking results, the ILQ binding mode with EGFR is similar to that of erlotinib with EGFR. ILQ binds to EGFR at the ATP binding site. Hydrogen bonds are formed between ILQ and EGFR at Glu-762 and Met-793. F and G, ILQ directly binds to T790M mutant EGFR. Hydrogen bonds are formed between ILQ and the T790M mutant EGFR at Lys-745, Met-793, and Asp-855.
ILQ Inhibits the Growth of Tumor Xenografts from NCI-H1975 Cells in Nude Mice
To evaluate the effect of ILQ on lung tumor growth in vivo, we generated tumor xenografts from NCI-H1975 cells in nude mice by subcutaneous administration and treated the tumor-bearing mice with vehicle or ILQ. Treatment with ILQ retarded the growth of NCI-H1975 tumor xenografts as evidenced by significantly decreased tumor volume and mass (Fig. 5, A and B). ILQ, given intraperitoneal at doses of 1 or 5 mg/kg body weight, exhibited tumor volumes of 676.1 ± 80.4 mm3 and 579.9 ± 70.9 mm3, respectively, whereas the tumor volume in vehicle-treated group was 1,210 ± 190.7 mm3. The effect of ILQ on tumor growth was compared with that of gefitinib, which failed to inhibit the growth of NCI-H1975 cells as xenograft tumors. Immunohistochemical analysis of xenograft tumor tissues from vehicle- or ILQ (5 mg/kg body weight)-treated animals showed that treatment with ILQ inhibited the expression of the Ki-67 cell proliferation marker and the phosphorylation of Akt and ERK1/2 (Fig. 5C). However, gefitinib failed to affect the expression of Ki-67, p-Akt, or p-ERK1/2 (Fig. 5C).
FIGURE 5.

ILQ inhibits xenograft tumor growth of NCI-H1975 cells. NCI-H1975 cells were subcutaneously injected into athymic nude mice for the development of xenograft tumors. Mice were untreated (Cont) or treated with ILQ (1 or 5 mg/kg body weight) or gefitinib (5 mg/kg body weight) intraperitoneally as detailed under “Experimental Procedures.” Effect of ILQ on H1975 cells xenograft (A) tumor volume and (B) tumor weight. The asterisks (*), (**), and (***) indicate a significant difference (p < 0.05), (p < 0.01), and (p < 0.001), respectively, between the ILQ-treated and untreated groups. C, immunohistochemical analysis of Ki-67, p-Akt, and p-ERK1/2 expression in xenograft tumors formed by NCI-H1975 cells in mice treated with vehicle, ILQ or gefitinib. Stained cells were counted from four separate areas on the slide and an average of three samples was calculated per group. Data are expressed as mean percent of control. The asterisk indicates a significant difference (*, p < 0.05 or **, p < 0.01) compared with control.
DISCUSSION
Over the last several years, extensive efforts have been made to find a cure for lung cancer, especially NSCLC, which is a leading cause of cancer death today (2–4). Although in current clinical settings, EGFR-TKIs are considered as first-line therapy (4), these drugs are less effective in NSCLC patients overexpressing wild type EGFR (7). The use of EGFR-TKIs also leads to the development of resistance, thereby limiting their long-term clinical success (8, 9). Moreover, the genetic alterations in NSCLC are not only limited to EGFR but also mutation and/or amplification of several other genes, including Met, KRAS, and ErbB3, is associated with disease progression (3, 4, 6). Met- or ErbB3-amplified or KRAS-mutated NSCLC cells have been reported to be resistant to EGFR-TKIs (28, 35–37). Thus, the search for a multi-targeted therapy is a timely need for reducing the incidence and mortality from NSCLC. In the present study, we examined the anticancer effects of ILQ, a dietary chemopreventive agent (15), in a series of NSCLC cells exhibiting a different status of EGFR, Met, ErbB3, or KRAS, and elucidated the underlying molecular mechanisms. Our study revealed that treatment with ILQ reduced viability, induced apoptosis and inhibited anchorage-independent growth of TKI-sensitive (HCC827, H1650) and TKI-resistant (H1975, HCC827GR and A549) lung cancer cells. H1975 cells, which harbor EGFR double mutations (L858R and T790M), were the most sensitive to ILQ treatment. Compared with its effects on H1975 cells, ILQ showed significant anticancer effects in A549 (KRAS-mutated), HCC827GR (Met- and ErbB3-amplified), and TKI-sensitive (HCC827 and H1650) cells, suggesting that ILQ might act as a multi-targeted therapy against NSCLC.
ILQ reportedly inhibits the growth of A549 lung cancer cells by inducing G1 or G2/M phase cell cycle arrest (26, 27). Besides cell cycle arrest, we found that ILQ induced cleavage of PARP and caspase-3, expression of the pro-apoptotic protein Bim and inhibited expression of anti-apoptotic Bcl2 in HCC827, H1650, and H1975 cells. These results suggest that the compound is effective in inducing apoptosis in NSCLC cells expressing either wild type or mutant EGFR. Whereas ILQ (10, 20, 40 μm) induced cytotoxicity in NSCLC cells, our study revealed that the compound at a concentration of as high as 80 μm failed to induce cell death in human normal lung MRC-5 cells. Zhang et al. (18) also reported that ILQ induced apoptosis selectively in human prostate cancer (C4–2) cells without affecting that of normal prostate epithelial cells.
Because ILQ exhibited the most prominent anti-proliferative and apoptotic effects in H1975 cells having EGFR double mutations (L858R and T790M), we hypothesized that ILQ might interfere with the EGFR signaling pathway. In vitro kinase assay data revealed that ILQ attenuated the catalytic activity of both wild type and double mutant (L858R and T790M) EGFR. To further confirm this finding, we stably transfected NIH3T3 cells with wild type and double mutant (L858R and T790M) EGFR and examined the responsiveness of these cells to EGF. Whereas wild type EGFR-transfected NIH3T3 cells showed increased colony formation upon treatment with EGF, NIH-3T3 cells harboring double mutant EGFR formed anchorage-independent colonies even in absence of exogenous EGF treatment. In response to EGF, the EGFR double mutant NIH3T3 cells showed significantly increased colony numbers compared with cells without EGF challenge. The finding that ILQ inhibits EGF-induced colony formation in NIH3T3 cells expressing wild type EGFR as well as in cells harboring EGFR double mutations with or without EGF treatment suggests that the compound targets both wild type and mutant EGFR as the underlying mechanisms of its anticancer effects in NSCLC.
The EGFR tyrosine kinase transmits activating signals to various downstream signaling molecules, including Akt and ERK1/2. We therefore examined the effect of ILQ on the activation of these kinases in NSCLC cells expressing either wild type or mutant EGFR. Although treatment with ILQ diminished the constitutive phosphorylation of Akt and ERK1/2 in TKI-sensitive (HCC827 and H1650) and -resistant (H1975) cells, the compound failed to affect the phosphorylation of EGFR. Our findings are in good agreement with several previous studies reporting the inhibitory effects of ILQ on Akt and ERK1/2 activation. ILQ-mediated inhibition of Akt phosphorylation was associated with the induction of apoptosis (21) and suppression of migration and invasion (24) of human breast cancer cells. ILQ induced apoptosis in interleukin-6-stimulated human multiple myeloma cells (20) and inhibited proliferation of human prostate cancer (C4–2) cells (18) partly through the inhibition of ERK1/2 activation. To examine the effect of ILQ on the catalytic activities of these kinases, we performed an in vitro kinase assay. We found that ILQ significantly inhibited the catalytic activity of ERK2 and Akt1, but failed to affect that of ERK1 and Akt2. The reasons and consequences of the differential effects of ILQ on ERK1 and ERK2 activities, and similarly between Akt1 and Ak2 activities, are still unclear. The exact roles of ERK and Akt isoforms in NSCLC cellular proliferation merit further investigation.
Because ILQ attenuated the catalytic activity of EGFR without affecting EGFR phosphorylation, we determined whether the compound can interact with EGFR or its downstream signaling molecules, and modulate the phosphorylation and/or catalytic activities of components of the EGFR signaling pathway. Because H1975 cells showed the highest sensitivity to ILQ, we incubated lysates of these cells with Sepharose-4B-conjugated ILQ to examine the possible interaction of ILQ with EGFR or its downstream signaling molecules, such as p110, Raf, MEK, ERK1/2, Akt, mTOR, p70S6K, and MNK. The pull-down assay results revealed that ILQ directly binds with EGFR. In addition, ILQ showed a weak interaction with Akt and ERK1/2. However, the actual mode of interaction of ILQ with Akt or that of ERK1/2, and the consequences of such interactions merit further investigation. The abrogation of ILQ binding with wild type or mutant EGFR in vitro in the presence of ATP suggests that the ILQ interaction with EGFR (wild type or mutant) is ATP-dependent. Because ILQ showed strong binding with EGFR in the pull-down assay, we used a docking model study to further clarify the pattern of interaction between ILQ and EGFR. The docking model indicated that ILQ formed hydrogen bonds with wild type EGFR and T790M mutant EGFR, suggesting that ILQ targets both wild type and mutant EGFR. Because ILQ formed two hydrogen bonds (Glu-762 and Met-793) with wild type EGFR and 3 hydrogen bonds (Lys-745, Met-793, and Asp-855) of mutant EGFR, ILQ retained its functional activity against the mutant EGFR compared with gefitinib.
The findings that ILQ, but not gefitinib, significantly inhibits the volume and weight of TKI-resistant H1975 xenograft tumor growth in nude mice and down-regulates the expression of cell proliferation markers Ki-67, p-Akt, and p-ERK1/2 in xenograft tumor tissues, suggesting that ILQ exerts anticancer effects in NSCLC cells by modulating the EGFR signaling pathway. Li et al. have also demonstrated that the inhibition of human breast cancer (MDA-MB-231) xenograft tumor growth by ILQ is associated with the down-regulation of Akt phosphorylation (21). In conclusion, our study provides novel findings showing that ILQ induces apoptosis and inhibits proliferation of NSCLC cells by directly binding with both wild type and double-mutant (L858R and T790M) EGFR and suppressing the EGFR signaling mediated through downstream Akt and ERK1/2. Based on our study, ILQ may be considered as a potential therapeutic choice for NSCLC treatment.
Supplementary Material
Acknowledgments
We thank Todd Schuster for supporting experiments and Nicki Brickman at The Hormel Institute, University of Minnesota for assistance in submitting our manuscript.
This work was supported by grants from National Institutes of Health CA1669011, CA172457, R37 CA081064, and ES016548, by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 2010–0029233), Republic of Korea, and by the Korea Food Research Institute, Republic of Korea.

This article contains supplemental Fig. S1.
- NSCLC
- non-small-cell lung cancer
- ILQ
- isoliquiritigenin
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- TKI
- tyrosine kinase inhibitor.
REFERENCES
- 1. Jemal A., Bray F., Center M. M., Ferlay J., Ward E., Forman D. (2011) Global cancer statistics. CA Cancer J. Clin. 61, 69–90 [DOI] [PubMed] [Google Scholar]
- 2. Sacco P. C., Maione P., Rossi A., Barecshino M. A., Sgambato A., Casaluce F., Napolitano A., Palazzolo G., Rossi E., Ferrara C., Gridelli C. (2014) The PI3k inhibitors: new hopes in the battle against advanced NSCLC. Front Biosci. 19, 259–271 [DOI] [PubMed] [Google Scholar]
- 3. John T., Liu G., Tsao M. S. (2009) Overview of molecular testing in non-small-cell lung cancer: mutational analysis, gene copy number, protein expression and other biomarkers of EGFR for the prediction of response to tyrosine kinase inhibitors. Oncogene 28, S14–23 [DOI] [PubMed] [Google Scholar]
- 4. Pallis A. G., Syrigos K. N. (2013) Epidermal growth factor receptor tyrosine kinase inhibitors in the treatment of NSCLC. Lung Cancer 80, 120–130 [DOI] [PubMed] [Google Scholar]
- 5. Nguyen T. T., Tran E., Nguyen T. H., Do P. T., Huynh T. H., Huynh H. (2004) The role of activated MEK-ERK pathway in quercetin-induced growth inhibition and apoptosis in A549 lung cancer cells. Carcinogenesis 25, 647–659 [DOI] [PubMed] [Google Scholar]
- 6. West L., Vidwans S. J., Campbell N. P., Shrager J., Simon G. R., Bueno R., Dennis P. A., Otterson G. A., Salgia R. (2012) A novel classification of lung cancer into molecular subtypes. PLoS One 7, e31906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li R. C., Zheng L. J., Fang M. H., Yu S. Y. (2014) The presence of EGFR mutations predicts the response in Chinese non-small cell lung cancer patients treated with erlotinib. Int. J. Biol. Markers 29, e112–e119 [DOI] [PubMed] [Google Scholar]
- 8. Carrera S., Buque A., Azkona E., Aresti U., Calvo B., Sancho A., Arruti M., Nuno M., Rubio I., de Lobera A. R., Lopez C., Vivanco G. L. (2014) Epidermal growth factor receptor tyrosine-kinase inhibitor treatment resistance in non-small cell lung cancer: biological basis and therapeutic strategies. Clin. Transl. Oncol. 16, 339–350 [DOI] [PubMed] [Google Scholar]
- 9. Ma C., Wei S., Song Y. (2011) T790M and acquired resistance of EGFR TKI: a literature review of clinical reports. J. Thorac. Dis. 3, 10–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kobayashi S., Boggon T. J., Dayaram T., Jänne P. A., Kocher O., Meyerson M., Johnson B. E., Eck M. J., Tenen D. G., Halmos B. (2005) EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352, 786–792 [DOI] [PubMed] [Google Scholar]
- 11. Pao W., Miller V. A., Politi K. A., Riely G. J., Somwar R., Zakowski M. F., Kris M. G., Varmus H. (2005) Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2, e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Haraguchi H., Ishikawa H., Mizutani K., Tamura Y., Kinoshita T. (1998) Antioxidative and superoxide scavenging activities of retrochalcones in Glycyrrhiza inflata. Bioorg. Med. Chem. 6, 339–347 [DOI] [PubMed] [Google Scholar]
- 13. Chin Y. W., Jung H. A., Liu Y., Su B. N., Castoro J. A., Keller W. J., Pereira M. A., Kinghorn A. D. (2007) Anti-oxidant constituents of the roots and stolons of licorice (Glycyrrhiza glabra). J. Agric. Food Chem. 55, 4691–4697 [DOI] [PubMed] [Google Scholar]
- 14. Kim J. Y., Park S. J., Yun K. J., Cho Y. W., Park H. J., Lee K. T. (2008) Isoliquiritigenin isolated from the roots of Glycyrrhiza uralensis inhibits LPS-induced iNOS and COX-2 expression via the attenuation of NF-κB in RAW 264.7 macrophages. Eur. J. Pharmacol. 584, 175–184 [DOI] [PubMed] [Google Scholar]
- 15. Cuendet M., Guo J., Luo Y., Chen S., Oteham C. P., Moon R. C., van Breemen R. B., Marler L. E., Pezzuto J. M. (2010) Cancer chemopreventive activity and metabolism of isoliquiritigenin, a compound found in licorice. Cancer Prev. Res. 3, 221–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hsu Y. L., Kuo P. L., Lin C. C. (2005) Isoliquiritigenin induces apoptosis and cell cycle arrest through p53-dependent pathway in Hep G2 cells. Life Sci. 77, 279–292 [DOI] [PubMed] [Google Scholar]
- 17. Yuan X., Zhang B., Chen N., Chen X. Y., Liu L. L., Zheng Q. S., Wang Z. P. (2012) Isoliquiritigenin treatment induces apoptosis by increasing intracellular ROS levels in HeLa cells. J. Asian Nat. Prod. Res. 14, 789–798 [DOI] [PubMed] [Google Scholar]
- 18. Zhang X., Yeung E. D., Wang J., Panzhinskiy E. E., Tong C., Li W., Li J. (2010) Isoliquiritigenin, a natural anti-oxidant, selectively inhibits the proliferation of prostate cancer cells. Clin. Exp. Pharmacol. Physiol. 37, 841–847 [DOI] [PubMed] [Google Scholar]
- 19. Zhou G. S., Song L. J., Yang B. (2013) Isoliquiritigenin inhibits proliferation and induces apoptosis of U87 human glioma cells in vitro. Mol. Med. Rep. 7, 531–536 [DOI] [PubMed] [Google Scholar]
- 20. Chen X., Wu Y., Jiang Y., Zhou Y., Wang Y., Yao Y., Yi C., Gou L., Yang J. (2012) Isoliquiritigenin inhibits the growth of multiple myeloma via blocking IL-6 signaling. J. Mol. Med. 90, 1311–1319 [DOI] [PubMed] [Google Scholar]
- 21. Li Y., Zhao H., Wang Y., Zheng H., Yu W., Chai H., Zhang J., Falck J. R., Guo A. M., Yue J., Peng R., Yang J. (2013) Isoliquiritigenin induces growth inhibition and apoptosis through downregulating arachidonic acid metabolic network and the deactivation of PI3K/Akt in human breast cancer. Toxicol. Appl. Pharmacol. 272, 37–48 [DOI] [PubMed] [Google Scholar]
- 22. Wang Z., Wang N., Han S., Wang D., Mo S., Yu L., Huang H., Tsui K., Shen J., Chen J. (2013) Dietary compound isoliquiritigenin inhibits breast cancer neoangiogenesis via VEGF/VEGFR-2 signaling pathway. PLoS One 8, e68566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kwon G. T., Cho H. J., Chung W. Y., Park K. K., Moon A., Park J. H. (2009) Isoliquiritigenin inhibits migration and invasion of prostate cancer cells: possible mediation by decreased JNK/AP-1 signaling. J. Nutr. Biochem. 20, 663–676 [DOI] [PubMed] [Google Scholar]
- 24. Wang K. L., Hsia S. M., Chan C. J., Chang F. Y., Huang C. Y., Bau D. T., Wang P. S. (2013) Inhibitory effects of isoliquiritigenin on the migration and invasion of human breast cancer cells. Expert Opin. Ther. Targets 17, 337–349 [DOI] [PubMed] [Google Scholar]
- 25. Yamazaki S., Morita T., Endo H., Hamamoto T., Baba M., Joichi Y., Kaneko S., Okada Y., Okuyama T., Nishino H., Tokue A. (2002) Isoliquiritigenin suppresses pulmonary metastasis of mouse renal cell carcinoma. Cancer Lett. 183, 23–30 [DOI] [PubMed] [Google Scholar]
- 26. Hsu Y. L., Kuo P. L., Chiang L. C., Lin C. C. (2004) Isoliquiritigenin inhibits the proliferation and induces the apoptosis of human non-small cell lung cancer a549 cells. Clin. Exp. Pharmacol. Physiol. 31, 414–418 [DOI] [PubMed] [Google Scholar]
- 27. Ii T., Satomi Y., Katoh D., Shimada J., Baba M., Okuyama T., Nishino H., Kitamura N. (2004) Induction of cell cycle arrest and p21(CIP1/WAF1) expression in human lung cancer cells by isoliquiritigenin. Cancer Lett. 207, 27–35 [DOI] [PubMed] [Google Scholar]
- 28. Engelman J. A., Zejnullahu K., Mitsudomi T., Song Y., Hyland C., Park J. O., Lindeman N., Gale C. M., Zhao X., Christensen J., Kosaka T., Holmes A. J., Rogers A. M., Cappuzzo F., Mok T., Lee C., Johnson B. E., Cantley L. C., Jänne P. A. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316, 1039–1043 [DOI] [PubMed] [Google Scholar]
- 29. Schrödinger. (2011) Schrödinger Suite LLC, New York [Google Scholar]
- 30. Stamos J., Sliwkowski M. X., Eigenbrot C. (2002) Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 277, 46265–46272 [DOI] [PubMed] [Google Scholar]
- 31. Zhou W., Ercan D., Chen L., Yun C. H., Li D., Capelletti M., Cortot A. B., Chirieac L., Iacob R. E., Padera R., Engen J. R., Wong K. K., Eck M. J., Gray N. S., Jänne P. A. (2009) Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462, 1070–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Godin-Heymann N., Bryant I., Rivera M. N., Ulkus L., Bell D. W., Riese D. J., 2nd, Settleman J., Haber D. A. (2007) Oncogenic activity of epidermal growth factor receptor kinase mutant alleles is enhanced by the T790M drug resistance mutation. Cancer Res. 67, 7319–7326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 35. Ladanyi M., Pao W. (2008) Lung adenocarcinoma: guiding EGFR-targeted therapy and beyond. Mod. Pathol. 21, S16–S22 [DOI] [PubMed] [Google Scholar]
- 36. Trusolino L., Bertotti A., Comoglio P. M. (2010) MET signalling: principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 11, 834–848 [DOI] [PubMed] [Google Scholar]
- 37. Turke A. B., Zejnullahu K., Wu Y. L., Song Y., Dias-Santagata D., Lifshits E., Toschi L., Rogers A., Mok T., Sequist L., Lindeman N. I., Murphy C., Akhavanfard S., Yeap B. Y., Xiao Y., Capelletti M., Iafrate A. J., Lee C., Christensen J. G., Engelman J. A., Jänne P. A. (2010) Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 17, 77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.