

Background: von Willebrand factor recruits platelets into a thrombus via interactions with GPIb.
Results: The cleavage of fibrinopeptides A/B is critical for the incorporation of VWF in a fibrin network. VWF incorporates independently from FXIII via its C domains.
Conclusion: Fibrin-bound VWF is involved in platelet adhesion at a high shear rate.
Significance: The role of VWF in platelet-dependent thrombus formation is explained.
Keywords: Fibrin, Fibrinogen, Platelet, Platelet Glycoprotein Ib, Surface Plasmon Resonance (SPR), von Willebrand Factor, Arvin, Ellipsometry
Abstract
Attachment of platelets from the circulation onto a growing thrombus is a process involving multiple platelet receptors, endothelial matrix components, and coagulation factors. It has been indicated previously that during a transglutaminase reaction activated factor XIII (FXIIIa) covalently cross-links von Willebrand factor (VWF) to polymerizing fibrin. Bound VWF further recruits and activates platelets via interactions with the platelet receptor complex glycoprotein Ib (GPIb). In the present study we found proof for binding of VWF to a fibrin monomer layer during the process of fibrinogen-to-fibrin conversion in the presence of thrombin, arvin, or a snake venom from Crotalus atrox. Using a domain deletion mutant we demonstrated the involvement of the C domains of VWF in this binding. Substantial binding of VWF to fibrin monomers persisted in the presence of the FXIIIa inhibitor K9-DON, illustrating that cross-linking via factor XIII is not essential for this phenomenon and suggesting the identification of a second mechanism through which VWF multimers incorporate into a fibrin network. Under high shear conditions, platelets were shown to adhere to fibrin only if VWF had been incorporated. In conclusion, our experiments show that the C domains of VWF and the E domain of fibrin monomers are involved in the incorporation of VWF during the polymerization of fibrin and that this incorporation fosters binding and activation of platelets. Fibrin thus is not an inert end product but partakes in further thrombus growth. Our findings help to elucidate the mechanism of thrombus growth and platelet adhesion under conditions of arterial shear rate.
Introduction
Fibrin formation, adhesion of platelets, and activation of coagulation factors are important processes associated with the formation of a thrombus under conditions of high hydrodynamic shear stress. Adhesion of platelets from blood circulation onto a growing thrombus is a complicated and not well understood process requiring multiple receptors on platelets and coagulation proteins from plasma. von Willebrand factor (VWF)3 is a key adhesive protein in capturing platelets from the circulation under influence of high shear rate (1–3). This polymeric plasma protein, composed of subunits of ∼250 kDa, circulates at low wall shear rates in a globular non-adhesive conformation. Under the influence of increasing hydrodynamic shear forces, VWF changes into an elongated form that provides an adhesive surface for platelets via binding to glycoprotein Ib (GPIb).
The formation of a stable thrombus relies on the presence of VWF and a fibrin network. The latter is formed by the thrombin-catalyzed conversion of plasma protein fibrinogen into fibrin monomers. The fibrin monomers polymerize and are cross-linked to a shear-resistant fibrin network. Interestingly, VWF appears to be an important player in the generation of thrombin and thus in fibrin formation. Both normal and fibrin-enhanced thrombin generation are markedly reduced in VWF-deficient platelet-rich plasma (4), and fibers formed in solutions of polymerizing fibrin are larger in the presence than in the absence of platelets (5). Moreover, patients with a VWF deficiency produce smaller thrombi compared with healthy individuals (6).
Previous data have shown that von Willebrand protein can serve as a link between polymerizing fibrin and platelet surface glycoprotein Ib (7). Although, the latter interaction has been studied in much detail, the mechanism by which VWF interacts with polymerized fibrin and how it alters its platelet adhesive properties is less well investigated. It is reported that VWF via its C1C2 domain binds to fibrin (6, 8, 9). Fibrin may directly interact with VWF given the observation that thrombin-activated factor XIII (FXIIIa) cross-links VWF to fibrin via covalent bonds between glutamine in VWF and lysine in fibrin α-chains (10).
In this study we investigated the binding of VWF with fibrinogen, fibrin monomers, and polymerized fibrin using ellipsometry and surface plasmon resonance (SPR). In addition, we performed flow experiments in which VWF-fibrin surfaces were perfused with washed platelets. Our results indicate that VWF incorporates via its C1C2 domain into an evolving fibrin network only after the thrombin-catalyzed removal of fibrinopeptides A (FpA) and/or B (FpB). In addition, we have demonstrated that fibrin-bound VWF plays an important role in the adhesion of platelets to fibrin at high shear rate.
EXPERIMENTAL PROCEDURES
Materials
HEPES buffer (20 mm Hepes, 100 mm NaCl, 0.02% NaN3, pH 7.4) was prepared with de-ionized water (Milli-Q3 system, Millipore, Etten-Leur, The Netherlands). Arvin (Ancrod) was from National Institute for Biological Standards and Control (Hertfordshire, UK). Snake venom from Crotalus atrox was obtained from Latoxan (Valence, France). Human thrombin was prepared in-house as described before (11). von Willebrand factor (Hemate P 500IE) was obtained from CSL Behring GmbH (Marburg, Germany), human fibrinogen (plasminogen and VWF-depleted) was from Kordia (Leiden, The Netherlands), human fibrinogen E-domain was from Hematologic Technologies (Essex Junction), and factor XIIIa inhibitor (K9-DON) was from Zedira GmbH (Darmstadt, Germany). Apyrase was obtained from Sigma, DiOC6(3) (3,3″-dihexyloxacarbocyanine iodide) was from Anaspec (Fremont), and Alexa Fluor 647-fibrinogen conjugate was from Molecular Probes (Eugene, OR). Abciximab was from Janssen Biologics B.V. (Reopro; Leiden, The Netherlands). Production of the ΔC1C2VWF was described before (9). 38 overlapping peptides spanning the C1C2 domain of VWF were obtained from JPT Peptide Technologies GmbH (Berlin, Germany). All peptides were 15 amino acids in length with an overlap of 10 amino acids within the consecutive peptides.
Ellipsometric Determination of Protein-Protein Interactions
Measurement of protein adsorption to planar bilayers by ellipsometry has been described extensively (12–16). Briefly, the technique determines the change in reflection coefficients of reflecting surfaces, e.g. silicon surfaces, as a consequence of the adsorption of thin (0.1–10 nm) protein film. Such changes result in an alteration of the polarization state of reflected light that can be accurately measured using an ellipsometer. The (complex) refractive index of the silicon slides was obtained from measurements of bare slides in buffer. Thickness d and refractive index n of the adsorbed proteins were calculated for an optical 3-layer system as before (14, 15). The change in surface mass Γ of the adsorbed proteins, expressed in μg/cm2, was calculated from the layer thickness d and refractive index n using the relation,
The Lorentz factor L is defined as L(n) = (n2 − 1)/(n2 + 2), d is expressed in nm, n1 is the refractive index of the buffer as determined by refractometry, A and M are the molar refractivity and molecular weight of the proteins, and v is the specific volume of proteins at room temperature. Values of A/M and v were in these calculations 0.241 and 0.729, respectively (15, 16).
For this study we used an 8-channel ellipsometer (Synapse BV, Maastricht, The Netherlands). Polished silicon wafers (WaferNet GmbH, Eching, Germany) were used as reflecting surfaces (cut to 0.5 × 4-cm slides). The slides were cleaned with sparkleen and chromic acid at 80 °C. After 30 min the slides were extensively flushed with MilliQ water and dried. To prevent binding of fibrinogen to the walls of the measurement cuvettes, incubations with fibrinogen were performed in an external holder without stirring. Fibrinogen was adsorbed onto the surfaces by incubating the slides with a HEPES buffer solution containing 1.2 μm fibrinogen for 1 h at room temperature. After adsorption of fibrinogen, the surface was flushed with fresh HEPES buffer and blocked with HEPES buffer containing 15 μm BSA.
Transmittance Experiment
To study the incorporation of VWF into a fibrin network, optical transmittance was applied based on the detection of incident light passing through a sample. A decrease in transmittance reflects the formation of a fibrin network. Changes in transmittance were measured at 37 °C in a 96-well plate at a wavelength of 450 nm using a Spectra Max (Molecular Devices, Sunnyvale, CA). Experiments were performed in HEPES buffer; curves represent the average from three independent experiments. Raw curves were fitted using the formula,
where τ is the velocity of the fibrin network formation, y0 is the final transmittance (T) of the fibrin clot, and parameter y0 + a reflects the initial level of transmittance before any fibrin network is formed.
Biacore
Surface plasmon resonance experiments were performed to investigate the interaction between fibrinogen and VWF using a Biacore T100 biosensor system (GE Healthcare). Fibrinogen was immobilized on a C1-sensor chip using amine-coupling chemistry according to the instructions of the manufacturer (Enzyme Research Laboratories, Swansea, UK). BSA was immobilized on a control channel to measure nonspecific interactions and correct for bulk refractive index changes. Final immobilization levels ranged between 900 and 1200 refractive units. The binding of VWF (final concentration 0.2–0.8 μm) in the absence or presence of thrombin (final concentration 20 nm) was studied in HBS-EP buffer (10 mm HEPES, 150 mm NaCl, 3 mm EDTA, and 0.005% Tween 20, pH 7.4) at a flow rate of 5 μl/min for 60 min at 25 °C followed by a 15-min dissociation with HBS-EP buffer. Regeneration of the sensor chip surface was performed by 1–3 pulses of 50 mm NaOH for 6 s at a flow rate of 20 μl/min. Data were analyzed using Scrubber2 (Version 2.0 Center for Biomolecular Interactions Analysis, University of Utah).
Perfusion Experiments
To study the effect of incorporation of VWF into a fibrin network on platelet adhesion, we performed flow perfusion experiments. Washed platelets were prepared from whole blood anticoagulated 1:6 with acid-citrate-dextrose solution (80 mm trisodium citrate, 52 mm citric acid, 183 mm d-(+)-glucose) as described before (17). The platelet pellet was resuspended in HEPES buffer, pH 7.45, to reach a final concentration of 2.6 × 108 platelets/ml. Platelets were labeled with DiOC6 (1.7 μm). Washed glass coverslips were coated with a solution containing 0.3 μm fibrinogen and 3 nm Alexa Fluor 647 fibrinogen conjugate for 90 min at room temperature. Glass coverslips were washed with saline buffer, blocked with HEPES solution, pH 7.45, containing 1% BSA for 1 h, and washed again. Subsequently, a second incubation step of 30 min was performed with fibrinogen (Kordia, 6 nm) in the absence or presence of thrombin (20 nm) and/or VWF (0.4 μm). Coated coverslips were mounted in a parallel plate flow chamber as described before (18, 19). Platelets in suspension were perfused for 7.5 min through the flow chamber at a shear rate of 1500 s−1 using a pulse-free pump. Immediately thereafter the chamber was flushed with rinse buffer (HEPES buffer, pH 7.45, 2 mm CaCl2, 1 units/ml heparin). Phase contrast and fluorescent images were recorded with an EVOS microscope (Advanced Microscopy Group, Bothell, WA) equipped with a 60× oil ultra-transparent objective (Olympus, Hamburg, Germany). Phase contrast and fluorescence (green fluorescent protein (GFP) and cyanine 5 (Cy5)) images were captured from 10 arbitrarily chosen microscopic fields per flow experiment.
RESULTS
VWF Interacts with a Fibrin(ogen) Monolayer in the Presence of Thrombin
In a first set of experiments we studied the interaction between VWF and immobilized fibrinogen on a silicon surface under stirring conditions using ellipsometry (Fig. 1A). The surface mass and thickness of the fibrinogen monolayer deposited on the slide was 0.32 ± 0.05 μg/cm2 and 18 ± 11 nm, respectively, suggesting that fibrinogen is oriented mainly side-on on the surface. It appeared that VWF hardly binds to this layer of immobilized fibrinogen. The increase in surface mass and thickness was 0.013 ± 0.007 μg/cm2 and 18.8 nm, respectively. The addition of fibrinogen (6 nm) resulted in a similar increase in mass and thickness (Fig. 1C,D). However, when VWF (0.04 μm) was added simultaneously with thrombin (20 nm), the surface mass (0.061 ± 0.017 μg/cm2) and the thickness (31 ± 11 nm) increased significantly (p < 0.05) when compared with VWF in the absence of thrombin (Fig. 1, C and D). Scanning electron microscopy visualization of the thrombin-treated slides confirmed the conversion of fibrinogen into fibrin fibers (data not shown). These findings suggest that the cleavage of FpA and/or FpB from fibrinogen is required to allow interaction between VWF and fibrinogen.
FIGURE 1.

Interaction of VWF with fibrin(ogen). The interaction of VWF (0.04 (null-ellipsometry) or 0.2 (SPR) μm) with immobilized fibrinogen in the presence or absence of human thrombin (20 nm) was measured with null-ellipsometry (A) and SPR (B). RU, Change in surface mass (C) and thickness (D) of fibrinogen- and fibrin-coated ellipsometry slides after the addition of fibrinogen (Fg) (6 nm) or VWF (0.04 μm) in the presence and absence of 20 nm thrombin (FIIa) was recorded by ellipsometry. *, p < 0.05 (Mann-Whitney U test).
The very same observations were obtained applying SPR. Here, fibrinogen was covalently immobilized on a dextran surface using a NHS/EDC (sulfo-N-hydroxysuccinimide/l-ethyl-3-(3-dimethylaminopropyl)carbodiimide) method before the perfusion of a solution of VWF (0.2 μm). VWF failed to bind to covalently bound fibrinogen. However, simultaneous perfusion of VWF (0.2 μm) and thrombin (20 nm) did result in binding of VWF to the fibrinogen-coated surface (Fig. 1B). Control experiments using either fibrinogen or VWF in the absence of thrombin resulted in a marginal increase in the surface mass, indicating that a very small amount of each protein binds to immobilized fibrinogen.
To investigate the ability of VWF to bind to an in situ polymerizing fibrin network, thrombin and fibrinogen were added simultaneously to a fibrinogen-coated slide. The surface mass increased with 0.054 ± 0.016 μg/cm2 and the average thickness with 22 ± 11 nm (Fig. 1, A, C, and D). When VWF was added together with fibrinogen and thrombin the surface mass significantly increased (0.11 ± 0.012 μg/cm2, p = 0.012). The thickness of the layer (29.8 ± 6.1 nm), however, did not differ significantly (Fig. 1D). Finally, we verified the binding capacity of VWF to an already formed fibrin layer. Therefore, fibrinogen was adsorbed on the surface and converted into fibrin (monomers) by incubation with thrombin (50 nm) for 10 min. Samples were flushed and incubated with a fibrinogen solution (1.2 μm) in HEPES buffer for another 10 min. After 10 min the slides were flushed again with fresh HEPES buffer to remove any excess of fibrinogen from the solution. This resulted in the formation of a polymerized fibrin monolayer on the surface. The surface mass and thickness of this polymerized fibrin layer on the surface was 0.734 ± 0.11 μg/cm2 and 47 ± 17 nm, respectively. Adding VWF in the presence of thrombin on such a polymerized fibrin layer did not change the surface mass or thickness (Fig. 1, C and D). Taken together, these results indicate that VWF can only interact with fibrin(ogen) during the process of fibrin polymerization. Additionally, these findings confirm the notion that FpA and/or FpB prohibit the interaction between VWF and fibrinogen.
To further investigate the interaction between VWF and fibrinogen in the presence of thrombin (20 nm), dose-response experiments were performed. Using ellipsometry (Fig. 2A) and SPR (Fig. 2B), application of increasing amounts of VWF resulted in an increased surface mass.
FIGURE 2.
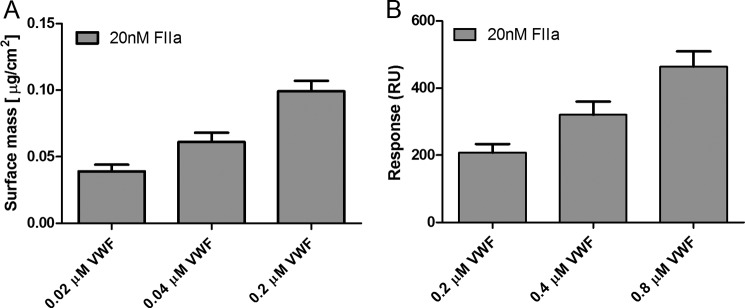
Dose-dependence of the VWF-fibrin(ogen) interaction. Panel A shows the change in surface mass upon the addition of increasing concentrations of VWF (0.02, 0.04, and 0.2 μm) and a fixed amount of thrombin (20 nm) to fibrinogen-coated ellipsometry slides. Panel B shows response units (RU) for increasing amounts of VWF (0.2, 0.4, and 0.8 μm) and a fixed amount of thrombin (20 nm) on covalently immobilized fibrinogen measured with SPR. Results represent the mean of 6 (A) or 3 (B) measurements ± S.D.
Cleavage of FpA or FpB Drives the Interaction of VWF with a Fibrin Monolayer
To further explore the precise role of thrombin in facilitating the interaction between VWF and fibrinogen, we used arvin and protease III, enzymes that convert fibrinogen into fibrin. Arvin only cleaves off FpA, whereas protease III from the venom of C. atrox cleaves the 42-amino-terminal residues of the fibrinogen Bβ chain, including FpB. Using ellipsometry and SPR, we observed a similar stimulating effect of these enzymes on the binding of VWF to fibrinogen, suggesting that removal of either FpA or FpB is sufficient for the incorporation of VWF into the fibrin network (Table 1). The SPR experiments performed with VWF and C. atrox snake venom did not show binding of VWF to fibrin monomers, most probably due to the different coupling procedure used to bind the fibrinogen to the surface. These results further rule out the possibility that fibrin-bound thrombin acts as the mediator through which VWF binds indirectly to fibrin.
TABLE 1.
Influence of cleavage of FpA and/or FpB on the interaction of VWF with fibrin(ogen)
RU, refractive units.
| Conditions | Ellipsometrya |
SPRa |
||
|---|---|---|---|---|
| Surface mass | S.D. | Response | S.D. | |
| μg/cm2 | RU | |||
| VWF +thrombin | 0.065 | 0.005 | 119.20 | 9.49 |
| VWF + arvin | 0.058 | 0.011 | 60.20 | 13.97 |
| VWF+ C. atrox venom | 0.105 | 0.008 | 9.96 | 6.19 |
a The adsorption of VWF (0.04 (null-ellipsometry) or 0.2 (SPR) μm) on a fibrin monolayer in the presence of thrombin (20 nm), arvin (1 unit/ml), and snake venom from C. atrox (2 units/ml) was determined with null-ellipsometry and SPR.
C1C2 Domain of VWF Interacts Independently of FXIIIa with the E-domain of Fibrinogen
Considering that removal of FpA/FpB stimulates the binding of VWF, we hypothesized an interaction of VWF with the E-domain of fibrinogen. To confirm our hypothesis, further ellipsometry experiments were performed. VWF and E-domain of fibrinogen were preincubated to verify an effect on VWF binding to thrombin-treated immobilized fibrinogen. Preincubation with the E-domain significantly inhibited the binding of VWF (Fig. 3).
FIGURE 3.
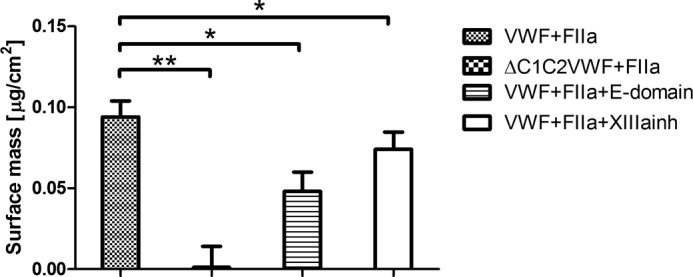
FXIIIa-independent interaction of C domain containing VWF with the E-domain of fibrinogen. The binding of C1C2 deleted VWF (0.2 μm) to a thrombin-treated fibrinogen monolayer was compared with the binding of VWF. The effect of preincubating VWF with the E-domain of fibrinogen (0.2 μm) in the presence of thrombin (FIIa) (20 nm) and the addition of FXIIIa inhibitor K9-DON (20 μm) was investigated. Mean surface mass ± S.E. for the different conditions of three independent experiments is shown. *, p < 0.05 (Mann-Whitney U test).
As it has been suggested previously that the C1C2 domain of VWF is implicated in the interaction with fibrin (9), we wished to confirm this notion using ellipsometry. We compared the ability of full-length VWF and a C1C2 deletion mutant of VWF (ΔC1C2VWF) to adsorb on a fibrin monolayer. Interestingly, ΔC1C2VWF failed to interact with the fibrin layer (Fig. 3), illustrating that the C1C2 domain is involved in the interaction between VWF and fibrin.
Given that FXIIIa is able to cross-link VWF to fibrin (20, 21), we checked for the presence of a FXIII contamination in fibrinogen. Using ELISA measurements, the FXIII concentration proved to be insignificant. To assess this further, the fibrinogen monolayer was preincubated with a saturating amount of FXIIIa inhibitor K9-DON (20 μm) for 10 min in the presence of 3 mm CaCl2 and 20 nm thrombin. FXIII, if present as a contaminant of the fibrinogen preparation, will be activated by thrombin, but its transglutaminase activity will be blocked by K9-DON (22, 23). In the presence of K9-DON, substantial (although 10% lower) binding of VWF to the fibrin-coated layer was observed (Fig. 3), indicating that the observed interaction between VWF and fibrin occurs largely independent of FXIIIa.
Binding of VWF Does Not Hinder the Development of a Fibrin Network
To determine whether the incorporation of VWF into a fibrin monolayer affects fibrin polymerization, fibrin formation was studied by transmittance recording; the decrease in transmittance corresponds to the density of the formed fibrin network. In these experiments, thrombin (1 nm) and fibrinogen (6 μm) were incubated in the absence/presence of VWF (8 μm), and the transmittance was followed over time. In the presence of VWF, a higher transmittance was obtained compared with the control condition, suggesting the formation of a less dense network (Fig. 4A). A similar increase in transmittance by VWF was observed in the presence of arvin or the snake venom of C. atrox, confirming that also in solution the cleavage of either FpA or FpB is sufficient for the interaction of VWF with fibrin. This decrease in density upon interaction with VWF is further illustrated in Fig. 4B, demonstrating a lower τ value in the presence of VWF regardless of the cleavage of FpA/B by either thrombin, arvin, or the snake venom from C. atrox. Together, these findings indicate that the formation of a fibrin clot is not hindered by fibrin-VWF interaction.
FIGURE 4.
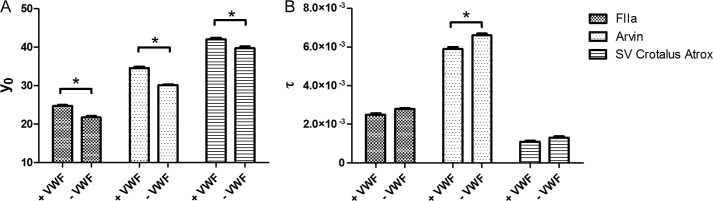
Effect of VWF on fibrin clot formation. Effect of VWF on fibrin clot formation. Transmittance experiments were performed in the presence of the indicated combinations of VWF (8 μm), fibrinogen (6 μm), thrombin (FIIa) (1 nm), arvin (1 unit/ml), and snake venom (SV) from C. atrox (2 units/ml). Transmittance curves were fitted using the equation T= y0 + a(exp(−τ*t)). The parameters y0 (associated with the transmittance of the fibrin clot) (A) and τ (corresponding to the rate of fibrin network formation) (B) are shown for the different conditions. *, p < 0.05 (Mann-Whitney U test).
Identification of Binding Sites of the C1C2 Domain of VWF Involved in the Interaction with Fibrin Monomers
To identify the binding sites of the C domains of VWF that interact with a fibrin monolayer we studied the inhibitory effect of 38 overlapping peptides spanning the C1C2 domain of VWF, as originally described by Sadler et al. (24). The binding of VWF (0.04 μm) to a fibrinogen monolayer was followed by ellipsometry in the presence of thrombin (20 nm) and the different peptides (0.04 mm) at high shear rate. Four of the tested peptides (H-PCEDSCRSGFTYVLH-OH, H-RSGFTYVLHEGECCG-OH, H-TTCNPCPLGYKEENN-OH, and H-PLGYKEENNTGECCG-OH) significantly (p < 0.05) decreased the amount of fibrin-bound VWF (Fig. 5A). However, peptides with partly overlapping sequences, including H-AQCSQKPCEDSCRSG-OH, H-VLHEGECCGRCLPSA-OH, H-KLECRKTTCNPCPLG-OH, and H-PLGYKEENNTGECCG-OH did not show inhibitory properties. These results indicate that sequences 2481FTY2483 and 2631YKE2633 are important for the interaction of VWF with fibrin monomers. These sequences are located in the VWC3 and VWC5 domain of VWF (Fig. 5B) according to annotation of Zhou et al. (25) and most probably exposed in VWF concatemers due to the high hydrodynamic flow (26). Interestingly, both Tyr-2483 and Tyr-2631 are well conserved in the VWF sequence from humans to zebrafish (Fig. 5C). This evolutionary conservation suggests that both amino acids have important roles in the function of VWF.
FIGURE 5.

Identification of the binding sites of the C domains of VWF that interact with a fibrin monolayer. Panel A shows the % inhibition of adsorption of VWF (0.04 μm) on immobilized fibrinogen in the presence of human thrombin (20 nm) and the indicated peptides of the C domains (0.04 mm) measured with null-ellipsometry. The percent inhibition compared with the control condition was calculated, and the mean ± S.E. of three independent experiments is shown for the different peptides. *, p < 0.05 (Mann-Whitney U test). Panel B shows the localization of Tyr-2483 and Tyr-2631 on VWC3 and VWC5 domains, respectively. Panel C demonstrates the presence of Tyr-2483 and Tyr-2631 in the C domains of the VWF sequence of different species.
The Interaction between VWF and Fibrin Enables the Incorporation of Platelets into an Evolving Fibrin Network
We next determined whether the incorporation of VWF into a fibrin monolayer enables the recruitment of platelets at high wall shear rate (1500 s−1). Therefore, experiments were performed in which washed platelets were perfused over a fibrin monolayer formed in the presence or absence of VWF. To only observe rolling of platelets, indicative for the presence of VWF, we performed the experiments in the presence of abciximab, a fibrinogen receptor GPIIb/IIIa blocking agent (27). We observed that VWF increased the capability of platelets (p = 0.0049) to adhere to the fibrin surface (Fig. 6A). To further certify that the platelet adhesion is dependent on platelet-GPIb interactions, we added a blocking anti-GPIb antibody (0.06 μm) to the washed platelets. We observed almost complete blocking of platelet adhesion to the surface. In the presence of Abciximab we observed reduced binding of platelets to VWF bound to fibrin monolayer, suggesting that both GPIb and GPIIb/IIIa are important (Fig. 6B) (25, 26).
FIGURE 6.
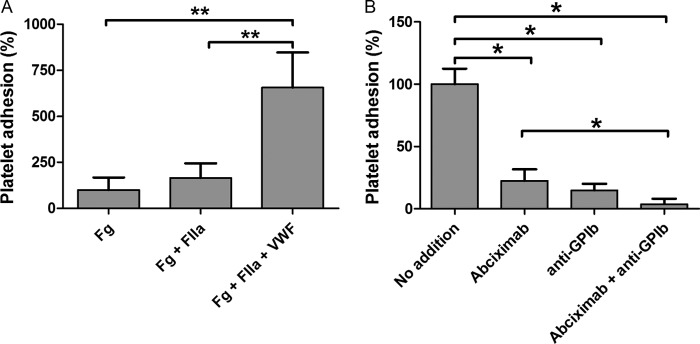
VWF-dependent platelet adhesion to thrombin-treated fibrinogen. In panel A washed platelets in the presence of abciximab (0.4 μm) were perfused over coverslips coated with fibrinogen (Fg), fibrinogen and thrombin (Fg+FIIa), or fibrinogen, thrombin, and VWF (Fg+FIIa+VWF) at 1500 s−1 for 7.5 min. In panel B washed DiOC6-labeled platelets were perfused over coverslips coated with fibrinogen, thrombin, and VWF at 1500 s−1 for 7.5 min. Representative phase contrast and fluorescence (GFP 395/509 nm; Cy5 650/670 nm) images were taken from 10 arbitrarily chosen microscopic fields (image size 150 × 110 μm). The number of platelets adhering to the fibrin(ogen) was normalized to the control condition (100%), and for each condition the mean ± S.E. (n = 10) is shown. *, p < 0.05; **, p < 0.01 (Mann-Whitney U test).
DISCUSSION
Incorporation of platelets in a growing thrombus under high shear stress, as in arteries, is dependent on VWF. In the literature interaction between VWF and fibrin(ogen) has been described, although the mechanism has not been elucidated (9). In the present study we applied ellipsometry, SPR, transmittance, and in vitro perfusion experiments to reveal the mechanisms involved in the binding of VWF to fibrin. We demonstrated that VWF interacts likely through its C1C2 domain with the E-domain of fibrinogen after the enzymatic release of fibrinopeptides FpA and FpB. VWF bound to fibrin supports platelet adhesion under high shear stress, resulting in a growing thrombus.
Cleavage of the fibrinopeptides is a central event in the reactions leading to fibrin polymerization. The rapid cleavage of FpA by thrombin can result in polymerization of fibrin without cleavage of FpB (28). Using arvin we have demonstrated that the cleavage of FpA is sufficient for the interaction of VWF with fibrinogen adsorbed on a solid surface. Cleavage of FpB has been shown to occur slowly and exposes a secondary polymerization site that contributes to lateral organization of fibrin protofibrils (29, 30). Furthermore our data indicate that the cleavage of FpB by thrombin or protease III from snake venom C. atrox enables the interaction between VWF and fibrinogen. The strong adsorption of VWF in the presence of snake venom C. atrox is consistent with previous findings where cleavage of FpB and exposure of β15–42 residues by protease III from snake venom of C. atrox induces the release of VWF from endothelial cells (31–33). Importantly, this release is known to be independent of the cleavage of FpA and of FXIIIa-mediated fibrin cross-linking (32, 33).
Previous studies indicated that FXIIIa is able to cross-link VWF to fibrin (10, 34). During this process FXIIIa creates covalent bonds between glutamine in VWF and lysine in fibrin α-chains (10). In our assays the contribution of FXIIIa in the interaction of VWF with fibrin proved to be minimal, as incorporation of VWF into a fibrin monolayer persisted in the presence of a saturating amount of FXIIIa (K9-DON) inhibitor (22). Our results thereby point to a second mechanism through which VWF multimers incorporate into a fibrin network.
The action of an enzyme that releases FpA and/or FpB from fibrinogen suggests that these peptides hinder (prohibit) the binding of VWF. The data also suggest that the E domain of fibrinogen is a potential VWF binding domain, confirmed by the observation that preincubation of VWF with the E domain of fibrinogen completely impeded the binding of VWF to fibrinogen. Because the cleavage of FpA/B exposes fibrinogen polymerization sites and the binding of VWF to fibrinogen appears to be enabled by the same enzymatic reaction, we investigated a possible inhibitory effect of VWF on the polymerization of fibrin monomers. However, using either ellipsometry or light transmission measurements, we found no evidence that binding of VWF impairs fibrin clot formation.
Additionally we localized the site of the C1C2 domain via which VWF interacts with fibrin monomers. Peptides containing Tyr-2483 and Tyr-2631 were found to inhibit the binding of VWF to a fibrin monolayer. The presence of both amino acids in the VWF sequence of many species underscores their important role in the function of VWF.
We confirmed the hypothesis that the incorporation of VWF into a fibrin network results in the recruitment of platelets by performing platelet perfusion experiments under conditions of high shear rate. A significantly increased adherence of platelets to the surface coated with fibrinogen was established in the conditions with VWF and thrombin compared with conditions containing fibrinogen and/or thrombin. Importantly, the addition of a GPIb blocking antibody almost completely abolished the adhesion of platelets to the fibrinogen surface. Taken together, we have provided evidence that both GPIb and GPIIb/IIIa are necessary for sustaining platelet adhesion under high shear. At high shear rate platelets are captured by fibrin-bound VWF. The initial binding occurs between VWF and GPIb, which is further supported by interaction of platelets receptor GPIIb/IIIa with fibrinogen.
It has been shown that the interaction of platelets with fibrin and VWF renders them procoagulant (4). This mechanism, therefore, not only promotes passive growth of the thrombus mass but also provides foci of further thrombin production (35) and thus promotes active growth of the thrombus.
In conclusion, we have demonstrated that VWF can be incorporated into a fibrin network during the process of thrombin-dependent cleavage of fibrinogen. This process is dependent on the C1C2 domain of VWF. Of note, VWF might have a dual role in the hemostatic process. Fibrin-bound VWF stimulates thrombin formation, which in turn rapidly exposes the VWF binding site on fibrinogen through the removal of fibrinopeptides A/B. This interaction likely contributes to thrombus growth and platelet adhesion under high shear stress. Additionally it may help to explain the observed phenotype in types I and III of von Willebrand disease and the Bernard-Soulier syndrome. Low amounts of (functional) VWF (and thus of FVIII) or a deficiency in GPIb-IX-V, here, lead to a disruption in thrombus formation and wound healing (4, 36–38).
Acknowledgments
We thank Roy Schrijver for technical help during SPR experiments and Silvie Sebastian for purifying the VWF deletion mutant. Dr. Frauke Swieringa is gratefully acknowledged for helping to set up the perfusion system.
This work was supported by The Netherlands Heart Foundation Project 2006T5301.
- VWF
- von Willebrand factor
- GPIb
- glycoprotein Ib
- FXIIIa
- factor XIII
- SPR
- surface plasmon resonance
- FpA
- fibrinopeptide A
- FpB
- fibrinopeptide B
- DiOC6
- 3,3″-dihexyloxacarbocyanine iodide.
REFERENCES
- 1. Siedlecki C. A., Lestini B. J., Kottke-Marchant K. K., Eppell S. J., Wilson D. L., Marchant R. E. (1996) Shear-dependent changes in the three-dimensional structure of human von Willebrand factor. Blood 88, 2939–2950 [PubMed] [Google Scholar]
- 2. Schneider S. W., Nuschele S., Wixforth A., Gorzelanny C., Alexander-Katz A., Netz R. R., Schneider M. F. (2007) Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc. Natl. Acad. Sci. U.S.A. 104, 7899–7903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ruggeri Z. M., Orje J. N., Habermann R., Federici A. B., Reininger A. J. (2006) Activation-independent platelet adhesion and aggregation under elevated shear stress. Blood 108, 1903–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Béguin S., Kumar R., Keularts I., Seligsohn U., Coller B. S., Hemker H. C. (1999) Fibrin-dependent platelet procoagulant activity requires GPIb receptors and von Willebrand factor. Blood 93, 564–570 [PubMed] [Google Scholar]
- 5. Niewiarowski S., Regoeczi E., Stewart G. J., Senyl A. F., Mustard J. F. (1972) Platelet Interaction with Polymerizing Fibrin. J. Clin. Invest. 51, 685–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Turitto V. T., Weiss H. J., Baumgartner H. R. (1984) Platelet interaction with rabbit subendothelium in von Willebrand's disease: altered thrombus formation distinct from defective platelet adhesion. J. Clin. Invest. 74, 1730–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Loscalzo J., Inbal A., Handin R. I. (1986) von Willebrand protein facilitates platelet incorporation in polymerizing fibrin. J. Clin. Invest. 78, 1112–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Endenburg S. C., Hantgan R. R., Lindeboom-Blokzijl L., Lankhof H., Jerome W. G., Lewis J. C., Sixma J. J., de Groot P. G. (1995) On the role of von Willebrand factor in promoting platelet adhesion to fibrin in flowing blood. Blood 86, 4158–4165 [PubMed] [Google Scholar]
- 9. Keuren J. F., Baruch D., Legendre P., Denis C. V., Lenting P. J., Girma J.-P., Lindhout T. (2004) Von Willebrand factor C1C2 domain is involved in platelet adhesion to polymerized fibrin at high shear rate. Blood 103, 1741–1746 [DOI] [PubMed] [Google Scholar]
- 10. Hada M., Kaminski M., Bockenstedt P., McDonagh J. (1986) Covalent cross-linking of von Willebrand factor to fibrin. Blood 68, 95–101 [PubMed] [Google Scholar]
- 11. Church F. C., Whinna H. C. (1986) Rapid sulfopropyl-disk chromatographic purification of bovine and human thrombin. Anal. Biochem. 157, 77–83 [DOI] [PubMed] [Google Scholar]
- 12. Machán R., Miszta A., Hermens W., Hof M. (2010) Real-time monitoring of melittin-induced pore and tubule formation from supported lipid bilayers and its physiological relevance. Chem. Phys. Lipids 163, 200–206 [DOI] [PubMed] [Google Scholar]
- 13. Miszta A., Machán R., Benda A., Ouellette A. J., Hermens W. T., Hof M. (2008) Combination of ellipsometry, laser scanning microscopy and Z-scan fluorescence correlation spectroscopy elucidating interaction of cryptdin-4 with supported phospholipid bilayers. J. Pept. Sci. 14, 503–509 [DOI] [PubMed] [Google Scholar]
- 14. Benes M., Billy D., Benda A., Speijer H., Hof M., Hermens W. T. (2004) Surface-dependent transitions during self-assembly of phospholipid membranes on mica, silica, and glass. Langmuir 20, 10129–10137 [DOI] [PubMed] [Google Scholar]
- 15. Cuypers P. A., Corsel J. W., Janssen M. P., Kop J. M., Hermens W. T., Hemker H. C. (1983) The adsorption of prothrombin to phosphatidylserine multilayers quantitated by ellipsometry. J. Biol. Chem. 258, 2426–2431 [PubMed] [Google Scholar]
- 16. Cuypers P. A., Hermens W. T., Hemker H. C. (1978) Ellipsometry as a tool to study protein films at liquid-solid interfaces. Anal. Biochem. 84, 56–67 [DOI] [PubMed] [Google Scholar]
- 17. Auger J. M., Kuijpers M. J., Senis Y. A., Watson S. P., Heemskerk J. W. (2005) Adhesion of human and mouse platelets to collagen under shear: a unifying model. FASEB J. 19, 825–827 [DOI] [PubMed] [Google Scholar]
- 18. Van Kruchten R., Cosemans J. M., Heemskerk J. W. (2012) Measurement of whole blood thrombus formation using parallel-plate flow chambers: a practical guide. Platelets 23, 229–242 [DOI] [PubMed] [Google Scholar]
- 19. Kuijpers M. J., Schulte V., Bergmeier W., Lindhout T., Brakebusch C., Offermanns S., Fässler R., Heemskerk J. W., Nieswandt B. (2003) Complementary roles of glycoprotein VI and α2β1 integrin in collagen-induced thrombus formation in flowing whole blood ex vivo. FASEB J. 17, 685–687 [DOI] [PubMed] [Google Scholar]
- 20. Smith E. L., Cardinali B., Ping L., Ariëns R. A., Philippou H. (2013) Elimination of coagulation factor XIII from fibrinogen preparations. J. Thromb. Haemost. 11, 993–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. La Corte A. L. C., Philippou H., Ariëns R. A. S. (2011) Role of fibrin structure in thrombosis and vascular disease. In Advances in Protein Chemistry and Structural Biology (Rossen D., ed.) pp. 75–127, Academic Press, New York: [DOI] [PubMed] [Google Scholar]
- 22. Sabo T. M., Brasher P. B., Maurer M. C. (2007) Perturbations in Factor XIII resulting from activation and inhibition examined by solution based methods and detected by MALDI-TOF MS. Biochemistry 46, 10089–10101 [DOI] [PubMed] [Google Scholar]
- 23. Matlung H. L., VanBavel E., van den Akker J., de Vries C. J., Bakker E. N. (2010) Role of transglutaminases in cuff-induced atherosclerotic lesion formation in femoral arteries of ApoE3 Leiden mice. Atherosclerosis 213, 77–84 [DOI] [PubMed] [Google Scholar]
- 24. Sadler J. E. (1998) Biochemistry and genetics of von Willebrand factor. Annu. Rev. Biochem. 67, 395–424 [DOI] [PubMed] [Google Scholar]
- 25. Zhou Y.-F., Eng E. T., Zhu J., Lu C., Walz T., Springer T. A. (2012) Sequence and structure relationships within von Willebrand factor. Blood 120, 449–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou Y.-F., Springer T. A. (2014) Highly reinforced structure of a C-terminal dimerization domain in von Willebrand factor. Blood 123, 1785–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Coller B. S., Anderson K., Weisman H. F. (1995) New antiplatelet agents: platelet GPIIb/IIIa antagonists. Thromb. Haemost. 74, 302–308 [PubMed] [Google Scholar]
- 28. Bailey K., Bettelheim F. R., Lorand L., Middlebrook W. R. (1951) Action of thrombin in the clotting of fibrinogen. Nature 167, 233–234 [DOI] [PubMed] [Google Scholar]
- 29. Weisel J. W. (1986) Fibrin assembly. Lateral aggregation and the role of the two pairs of fibrinopeptides. Biophys. J. 50, 1079–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weisel J. W., Litvinov R. I. (2013) Mechanisms of fibrin polymerization and clinical implications. Blood 121, 1712–1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ribes J. A., Francis C. W., Wagner D. D. (1987) Fibrin induces release of von Willebrand factor from endothelial cells. J. Clin. Invest. 79, 117–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ribes J. A., Ni F., Wagner D. D., Francis C. W. (1989) Mediation of fibrin-induced release of von Willebrand factor from cultured endothelial cells by the fibrin β chain. J. Clin. Invest. 84, 435–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bunce L. A., Sporn L. A., Francis C. W. (1992) Endothelial cell spreading on fibrin requires fibrinopeptide B cleavage and amino acid residues 15–42 of the β chain. J. Clin. Invest. 89, 842–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ribes J. A., Francis C. W. (1990) Multimer size dependence of von Willebrand factor binding to cross-linked or noncross-linked fibrin. Blood 75, 1460–1465 [PubMed] [Google Scholar]
- 35. Cosemans J. M., Schols S. E., Stefanini L., de Witt S., Feijge M. A., Hamulyák K., Deckmyn H., Bergmeier W., Heemskerk J. W. (2011) Key role of glycoprotein Ib/V/IX and von Willebrand factor in platelet activation-dependent fibrin formation at low shear flow. Blood 117, 651–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Béguin S., Keularts I., Al Dieri R., Bellucci S., Caen J., Hemker H. C. (2004) Fibrin polymerization is crucial for thrombin generation in platelet-rich plasma in a VWF-GPIb-dependent process, defective in Bernard-Soulier syndrome. J. Thromb. Haemost. 2, 170–176 [DOI] [PubMed] [Google Scholar]
- 37. Huizinga E. G., Tsuji S., Romijn R. A., Schiphorst M. E., de Groot P. G., Sixma J. J., Gros P. (2002) Structures of glycoprotein Ibα and its complex with von Willebrand factor A1 domain. Science 297, 1176–1179 [DOI] [PubMed] [Google Scholar]
- 38. Ruggeri Z. M., De Marco L., Gatti L., Bader R., Montgomery R. R. (1983) Platelets have more than one binding site for von Willebrand factor. J. Clin. Invest. 72, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]