

Background: Cdc7 kinase and protein phosphatase 2A are important in DNA replication initiation.
Results: DNA unwinding element (DUE)-binding protein (DUE-B) is an essential replication factor that is regulated by Cdc7 and PP2A.
Conclusion: The phosphorylation of DUE-B determines its ability to bind the minichromosome maintenance helicase and regulate Cdc45 chromatin loading.
Significance: PP2A and Cdc7 regulate DUE-B mediated loading of Cdc45 to the prereplication complex.
Keywords: Cell Cycle, Cell Division, DNA Replication, DNA Synthesis, Protein Phosphatase 2 (PP2A), Cdc7, DUE-B, MCM, cdc45
Abstract
The DNA unwinding element (DUE)-binding protein (DUE-B) binds to replication origins coordinately with the minichromosome maintenance (MCM) helicase and the helicase activator Cdc45 in vivo, and loads Cdc45 onto chromatin in Xenopus egg extracts. Human DUE-B also retains the aminoacyl-tRNA proofreading function of its shorter orthologs in lower organisms. Here we report that phosphorylation of the DUE-B unstructured C-terminal domain unique to higher organisms regulates DUE-B intermolecular binding. Gel filtration analyses show that unphosphorylated DUE-B forms multiple high molecular weight (HMW) complexes. Several aminoacyl-tRNA synthetases and Mcm2–7 proteins were identified by mass spectrometry of the HMW complexes. Aminoacyl-tRNA synthetase binding is RNase A sensitive, whereas interaction with Mcm2–7 is nuclease resistant. Unphosphorylated DUE-B HMW complex formation is decreased by PP2A inhibition or direct DUE-B phosphorylation, and increased by inhibition of Cdc7. These results indicate that the state of DUE-B phosphorylation is maintained by the equilibrium between Cdc7-dependent phosphorylation and PP2A-dependent dephosphorylation, each previously shown to regulate replication initiation. Alanine mutation of the DUE-B C-terminal phosphorylation target sites increases MCM binding but blocks Cdc45 loading in vivo and inhibits cell division. In egg extracts alanine mutation of the DUE-B C-terminal phosphorylation sites blocks Cdc45 loading and inhibits DNA replication. The effects of DUE-B C-terminal phosphorylation reveal a novel S phase kinase regulatory mechanism for Cdc45 loading and MCM helicase activation.
Introduction
Unwinding of eukaryotic DNA for replication initiation requires the sequential binding of multiple proteins onto replication origins, ultimately resulting in activation of the minichromosome maintenance (MCM)3 replicative helicase. This process is separated between the M/G1 and S phases of the cell cycle such that the origin recognition complex recognizes replication origins in late M/G1 phase to serve as a platform for the recruitment of additional replication proteins (1). Chromatin-bound origin recognition complex enlists Cdc6 onto replication origins and these factors load the Cdt1-bound Mcm2–7 replicative helicase around double stranded origin DNA to form the pre-replication complex (pre-RC) (1–4). MCM complexes in the pre-RC lack strong helicase activity, which is activated during the G1/S phase transition by the Cdc7/Dbf4-dependent kinase (DDK) and cyclin-dependent kinase (CDK) (5, 6) that trigger the association of Cdc45 and the GINS complex with the pre-RC. Binding of Cdc45 and GINS to Mcm2–7 stimulates the helicase activity ∼100-fold in the CMG (Cdc45-MCM-GINS) complex, converting the pre-RC to the preinitiation complex (reviewed in Ref. 7). DDK directly phosphorylates each MCM subunit, except for Mcm5. Phosphorylation by DDK changes conformation of the MCM complex to alleviate the replication inhibitory activity of the N-terminal serine/threonine-rich domain of Mcm4 (8, 9). The requirement of DDK for cell survival can be bypassed by mutation of the MCM subunits, suggesting that the Mcm2–7 complex is an essential target of DDK (9, 10). However, these bypass mutants show growth defects in the absence of DDK activity, implying that there may be other factors phosphorylated by DDK for efficient DNA replication.
In addition to S phase kinase activity, protein phosphatase 2A (PP2A) has been shown to be essential for chromatin binding and helicase activation by Cdc45 (11, 12). PP2A blocks the inhibitory effect of ATM/ATR on Cdc45 chromatin loading (13), however, the target of PP2A that is essential for Cdc45 loading was not indicated. The global effect of PP2A has been shown to antagonize Cdk1 activity in controlling cell cycle progression (14). The balance of PP2A and Cdk1 activities coordinates with the oscillation of Cdk1 levels and controls cell cycle progression (15).
DUE-B is a 209-amino acid protein identified in a yeast one-hybrid screen using the DNA unwinding element from the c-myc replication origin as bait (16). The N-terminal 150-residue domain of DUE-B shows strong evolutionary conservation with bacterial and eukaryotic aminoacyl-tRNA proofreading enzymes (17, 18). In higher eukaryotes DUE-B additionally contains a highly conserved 59-amino acid C-terminal domain necessary for binding to Cdc45 in vitro and upon co-expression in insect cells (19). siRNA knockdown of DUE-B in HeLa cells slows the G1/S transition (17). Moreover, DUE-B and Cdc45 coordinately bind to the DUE regions of active replication origins in vivo dependent on the presence of a DUE (19–21). In Xenopus egg extracts, DUE-B and Cdc45 binding to replicating sperm chromatin both depend on pre-RC formation and Cdk2 activity. The observation that DUE-B immunodepletion blocks Cdc45 loading (19) suggests that DUE-B facilitates Cdc45 binding to the MCM complex.
In Saccharomyces cerevisiae Sld3 binds Cdc45, and Sld3 phosphorylation promotes its interaction with Dpb11, which is essential for recruitment of Cdc45 to the MCM complex (22–24). Three proteins, GEMC1, DUE-B, and Treslin/TICRR, have been reported to be necessary for Cdc45 loading and CMG formation (19, 25–27), but the detailed contributions of GEMC1, DUE-B, and Treslin/TICRR to Cdc45 loading are not yet well defined.
Based on the interplay between DUE-B, MCMs, and Cdc45 (17), the goal of the present experiments was to determine how DUE-B phosphorylation affects its interaction with MCM and Cdc45 proteins. Here we show that DUE-B is de-phosphorylated in M phase and phosphorylated in G1/S phase. Phosphorylated DUE-B forms homodimers, whereas de-phosphorylated DUE-B interacts with the Mcm2–7 complex and aminoacyl-tRNA synthetases. In Xenopus egg extracts this interaction is blocked by Cdc7 phosphorylation and enhanced by PP2A dephosphorylation. We find that CKII can prime DUE-B for Cdc7 phosphorylation. Confirming the importance of DUE-B phosphorylation in replication initiation, a C-terminal Ser/Thr to Ala mutant blocks Cdc45 and RPA loading on sperm chromatin and inhibits DNA replication. The same mutant increases MCM binding but inhibits Cdc45/RPA loading onto chromatin and cell division in HeLa cells.
EXPERIMENTAL PROCEDURES
DUE-B Expression and Purification
Human DUE-B cDNA was PCR amplified and expressed as a His6-tagged protein in HeLa cells (DUE-BHeLa) or in Sf9 insect cells (DUE-BSf9) as described (19). Baculovirus-infected cells were harvested 48 h after infection and lysed on ice in lysis buffer (50 mm NaH2PO4, pH 8.0, 300 mm NaCl, 20 mm imidazole, 0.5% Nonidet P-40, 1× protease inhibitor mixture (Sigma)). The lysate was clarified by centrifugation, and DUE-BSf9 was isolated by Ni-nitrilotriacetic acid (NTA)-agarose (Qiagen) chromatography. The resin was washed with washing buffer (50 mm NaH2PO4, pH 8.0, 500 mm NaCl, 40–60 mm imidazole, 0.1% Nonidet P-40, 1× protease inhibitor mixture). DUE-BSf9 was eluted with elution buffer (50 mm NaH2PO4, pH 8.0, 100 mm NaCl, 200 mm imidazole, 1× protease inhibitor mixture) and concentrated by centrifugal ultrafiltration.
Phosphorylation Site Mutations
DUE-B C-terminal phosphorylation sites (serine 179, 181, 182, 194, 196, 197, 204, 205, and threonine 187) were mutated to unphosphorylatable alanine (DUE-B(S/T)-A) or phosphomimic aspartic acid (DUE-B(S/T)-D).
Chromatography
Gel filtration columns were packed with Sephacryl S-400 HR resin (GE Healthcare) equilibrated in modified egg extract buffer (50 mm KCl, 50 mm HEPES-KOH, pH 7.6, 5 mm MgCl2) at 0.1 ml/min. Protein samples were loaded and fractionated at 0.1 ml/min with modified egg extract buffer. Eluted fractions were collected and analyzed by Western blotting.
Xenopus Egg Extracts
Low-speed Xenopus interphase egg extracts were prepared according to Lebofsky et al. (28). Low-speed Xenopus metaphase egg extracts were prepared according to Chong et al. (29). One hundred and eighty nanomolar recombinant DUE-BSf9 was added to interphase Xenopus egg extract and incubated at room temperature for 30 min. The egg extract incubated with recombinant DUE-BSf9 was clarified by centrifugation at 16,000 × g for 10 min at 4 °C, diluted with equal volume of modified egg extract buffer, and fractionated by gel filtration.
Mass Spectrometry
To identify DUE-B interacting proteins in high molecular weight complexes, G1 phase egg extract was incubated with or without DUE-BSf9 and fractionated by gel filtration. Fractions containing DUE-BSf9 in the high molecular weight complexes were combined and DUE-BSf9 was isolated together with other interaction proteins using Ni-NTA-agarose.
Exactly the same procedure was carried out on a sample without DUE-BSf9. The Ni-NTA-agarose bead-bound proteins from both samples were analyzed by MALDI-TOF mass spectrometry (Ohio State University Proteomics Facility). Protein fragments were identified using the MASCOT peptide mass fingerprint algorithm.
A total of 282 proteins was identified from egg extract incubated with DUE-BSf9. Many proteins were also found from samples without DUE-BSf9. By subtracting the same hits present in both samples, a list of proteins unique to the sample incubated with DUE-BSf9 was obtained.
Immunoblotting
Anti-DUE-B antibody was raised in rabbits against a His6-tagged recombinant human DUE-B expressed in bacteria. Mcm3 (ab4460) and Mcm5 (ab17967) antibodies were purchased from Abcam.
Kinase and Phosphatase Inhibitors
Forty μm Cdc7 inhibitor PHA-767491 (30, 31) (Santa Cruz) or 100 μm to 2 mm casein kinase II inhibitor 4,5,6,7-tetrabromo-2-azabenzimidazole (TBB) (EMD Millipore) was used in Xenopus egg extracts. HeLa cells were treated with 5 μm PHA-767491 for 20 h. Okadaic acid (OA) was purchased from Sigma.
DNA Replication Assay with [α-32P]dCTP
Recombinant human WT or DUE-B(S/T)-D were expressed in HeLa cells and purified by Ni-NTA chromatography. The proteins were dephosphorylated in vitro with 0.07 mg/ml of PP2A (Cayman chemical) in EB buffer (50 mm KCl, 50 mm HEPES, pH 7.6, 5 mm MgCl2) at 37 ºC for 1 h and reactions were stopped by adding 100 nm OA. Alternatively, recombinant WT and DUE-B(S/T)-D were treated with PP2A in the presence of 100 nm OA as a control. The dephosphorylated or unmodified WT and DUE-B(S/T)-D was added to interphase Xenopus egg extract supplemented with 0.1 μCi/μl of [α-32P]dCTP and 0.75 μm OA.
Demembranated sperm was added to the egg extract (1500 sperm/μl). DNA replication reactions were stopped at 60 min and the proteins were digested with protease K. Labeled DNA was resolved on a 0.8% TBE-agarose gel and exposed to x-ray film overnight.
Co-immunoprecipitation
Antibody-conjugated protein G beads were incubated with cleared protein lysate from HeLa cells or Xenopus egg extract at 4 °C for 4 h on a rotator in the presence of 1× protease inhibitor mixture (Sigma) and 1× phosphatase inhibitor mixture (Thermo Scientific). The beads were washed 5 times with IP buffer (20 mm HEPES, pH 7.4, 0.5% Triton X-100, 150 mm NaCl, 1.5 mm MgCl2, 2 mm EGTA, 10 mm NaF, 2 mm DTT, 1 mm NaVO4, 1 mm PMSF, 20 μm aprotinin). The bead-bound proteins were detected by SDS-PAGE and Western blotting.
RESULTS
DUE-B Phosphorylation Level Changes with the Cell Cycle
The level of total cellular DUE-B protein and its distribution between cytoplasmic and nuclear fractions does not vary detectably over the cell cycle (17). To capture the dynamics of DUE-B phosphorylation during an unperturbed cell cycle, cells were blocked in mitosis with nocodazole, released into fresh medium. The level of DUE-B phosphorylation was examined by Western blotting immunoprecipitated DUE-B with an anti-phosphoserine/phosphothreonine antibody (Fig. 1, A–C). DUE-B phosphorylation levels were lowest during mitosis, and increased after cells were released from the nocodazole block (Fig. 1, B and C), suggesting that the level of DUE-B phosphorylation changes during cell cycle progression. It is possible that fluctuation of DUE-B phosphorylation level affects its function in DNA replication initiation.
FIGURE 1.

DUE-B is dephosphorylated and incorporated into high molecular weight complexes in M phase. A, HeLa A1 cells expressing ∼4-fold levels of His6-tagged WT DUE-BHeLa were released from mitotic synchrony and sampled at the indicated times. DNA content was analyzed by flow cytometry. B, cells collected at each time point were lysed and His6-tagged DUE-B was affinity purified from whole cell extracts and probed using anti-phospho-Ser/Thr antibody. Whole cell extract of HeLa cell without expressed His6-tagged DUE-B was used in the control pull down. C, quantitation of Western blot. DUE-B phosphorylation signal was normalized to the total DUE-B signal. D, HeLa cell whole cell extracts from asynchronous cells, M-phase cells, and cells 2 h after nocodazole release were fractionated by gel filtration. Approximate sizes of proteins were indicated by BSA and thyroglobulin. E, M-phase and interphase Xenopus egg extracts were fractionated by gel filtration.
DUE-B Is Incorporated into High Molecular Weight (HMW) Complexes during M Phase
To test the hypothesis that phosphorylation of DUE-B could change its ability to interact with additional proteins, the formation of complexes containing DUE-B was assayed by size exclusion chromatography of cell extracts. The level of DUE-B in each fraction was monitored by Western blotting. When whole cell protein extract from asynchronous HeLa cells was analyzed, almost all DUE-B eluted as a single peak slightly after the BSA standard (molecular mass ∼66 kDa) (Fig. 1D, asynchronous). The predicted size of molecules in the DUE-B peak is about 50 kDa, consistent with the molecular mass of the DUE-B homodimer (∼47 kDa) (17).
When cells were synchronized in M phase, a substantially increased portion of DUE-B was detected in the HMW fractions, suggesting that dephosphorylated DUE-B is able to bind other biomolecules during mitosis. As cells moved out of M into G1 the majority of DUE-B in the HMW complexes moved back to the homodimer form. To confirm that DUE-B HMW complexes that form during M phase are not specific to human cell extract we analyzed Xenopus egg extracts by gel filtration. In interphase egg extract, almost all DUE-B existed as homodimers (Fig. 1E). However, in M phase Xenopus egg extracts a nominal fraction of DUE-B was found in HMW complexes. These results suggest that the formation of DUE-B HMW complexes is conserved between human and frog.
Importantly, the formation of high molecular weight complexes happens in the M phase of the cell cycle, when DUE-B has the lowest phosphorylation level. In a previous study, we reported that the C terminus unphosphorylated recombinant human DUE-B expressed in Sf9 insect cells is able to form high molecular weight complexes when incubated with Xenopus egg extract or HeLa cell extract (17). Therefore, we tested whether phosphorylation plays a role in forming DUE-B high molecular weight complexes.
Phosphorylation Decreases DUE-B Incorporation into HMW Complexes
To assess the relationship between DUE-B phosphorylation and the formation of HMW complexes, recombinant human DUE-B was expressed in Sf9 insect cells (DUE-BSf9) and isolated. When purified DUE-BSf9 was subjected to size exclusion chromatography it eluted exclusively as homodimers (Fig. 2A). In contrast, when DUE-BSf9 was incubated with interphase egg extract, 35–40% of the total DUE-BSf9 was detected in HMW complexes (Fig. 2B). Therefore, DUE-BSf9 was modified when added to egg extract to yield DUE-B populations that eluted with endogenous DUE-BXl homodimers, and in the form of HMW complexes.
FIGURE 2.

Phosphorylation controls DUE-B incorporation into high molecular weight complexes. A, recombinant human DUE-B expressed and purified from Sf9 insect cells (DUE-BSf9) was fractionated by gel filtration. B, DUE-BSf9 was incubated with the interphase egg extract and fractionated by gel filtration. C, interphase egg extract was supplemented with phosphatase inhibitor mixture and incubated with DUE-BSf9. Proteins were separated by gel filtration. D, DUE-BSf9 was phosphorylated by casein kinase II prior to incubation with interphase egg extract. Proteins were separated using gel filtration after incubation. E, recombinant His6-tagged human DUE-B C-terminal (amino acids 160–209, see Fig. 5) (S/T)-A mutant (all C-terminal Ser and Thr residues mutated to Ala) was synthesized in vitro and incubated with interphase Xenopus egg extracts. F, recombinant His6-tagged human DUE-B C-terminal (amino acids 160–209, see Fig. 5) Ser/Thr-Asp mutant (all C-terminal Ser and Thr residues mutated to Asp) was synthesized in vitro and incubated with interphase Xenopus egg extracts. G, recombinant His6-tagged human DUE-BWT was synthesized in vitro and incubated with interphase Xenopus egg extracts. H, DUE-B from interphase egg extract was treated with λ-phosphatase with or without phosphatase inhibitor. Replicate samples were analyzed by SDS-PAGE and Western blot.
When the interphase egg extract was supplemented with phosphatase inhibitor mixture before the addition of DUE-BSf9, the percentage of DUE-BSf9 present in the HMW complexes decreased to 24% of the total DUE-BSf9 (Fig. 2C). To test directly whether the formation of DUE-B HMW complexes is related to its phosphorylation, DUE-BSf9 was phosphorylated by CKII in vitro, which only phosphorylates the DUE-B C-terminal domain (18). The kinase-treated DUE-BSf9 was purified and incubated with interphase Xenopus egg extract. As shown in Fig. 2D, preincubation of DUE-BSf9 with CKII significantly decreased the amount of DUE-BSf9 in HMW complexes. To confirm that the release of DUE-B from HMW complexes was a result of C-terminal phosphorylation, all Ser and Thr residues in the DUE-B C terminus were mutated to non-phosphorylatable alanine (DUE-B(S/T)-A) or the phosphomimic aspartate (DUE-B(S/T)-D). The corresponding wild type (WT) and mutant proteins were expressed by in vitro transcription/translation and added to egg extracts. The unphosphorylatable DUE-B(S/T)-A formed a large amount of HMW complexes (55–60%), whereas the DUE-B(S/T)-D phosphomimic mutant formed significantly fewer HMW complexes (14–15%) (Fig. 2, E–G). These results indicate that unphosphorylated C-terminal DUE-B preferentially forms HMW complexes, and that phosphorylation strongly disfavors, but does not completely block, the formation of DUE-B HMW complexes.
These results also predict that endogenous DUE-BXl in interphase Xenopus egg extract is phosphorylated because most interphase DUE-BXl is in dimer form (Fig. 1E). To test this hypothesis, the endogenous protein was purified by immunoprecipitation from interphase Xenopus egg extracts and treated with λ-protein phosphatase. Virtually all DUE-B purified from the interphase Xenopus egg extract was sensitive to phosphatase treatment, which resulted in an upward mobility shift when separated by SDS-PAGE (Fig. 2H).
DUE-B Phosphorylation Is Controlled by Cdc7 and PP2A
To identify the kinases and phosphatases that control DUE-B phosphorylation several inhibitors were added to the egg extracts. First, we used OA, which specifically inhibits PP2A in egg extracts (11, 32, 33), to examine the effect of PP2A activity on DUE-B. When DUE-BSf9 was incubated in OA-treated egg extracts, the amount of DUE-BSf9 detected in HMW complexes was dramatically decreased to about 9% of total DUE-BSf9 (Fig. 3A), a more substantial decrease than was observed after treatment with phosphatase inhibitor mixture treatment (Fig. 2C). The decrease of DUE-BSf9 in HMW complexes in response to OA treatment suggests that PP2A counteracts unidentified kinases in the egg extract by dephosphorylating DUE-B.
FIGURE 3.

Inhibition of Cdc7 and PP2A in Xenopus egg extract changes the amount of DUE-B incorporated in high molecular weight complex. A, DUE-BSf9 was incubated with interphase egg extract supplemented with the PP2A inhibitor OA. B, DUE-BSf9 was incubated with interphase egg extract supplemented with OA and the Cdc7 inhibitor PHA-767491. C, DUE-BSf9 was incubated with interphase egg extract supplemented with OA and p27. D, DUE-BSf9 was incubated with interphase egg extract supplemented with OA and casein kinase II inhibitor TBB. E, DUE-BSf9 was incubated with interphase egg extract supplemented with PHA-767491.
To determine which kinases were counterbalancing PP2A, S phase kinase inhibitors were tested. As shown in Fig. 3B, in the presence of OA the highly selective DDK inhibitor PHA-767491 (30, 31) reversed the effect of OA treatment, with over 32% DUE-BSf9 detected in HMW complexes. In contrast, inhibition of CDK2 by p27 did not affect the DUE-BSf9 distribution (Fig. 3C), nor did inhibition of CKII by TBB (34) (Fig. 3D). In keeping with this finding, DDK inhibition resulted in an increase of both DUE-BSf9 and endogenous DUE-BXl in HMW (presumably unphosphorylated) fractions in the absence of OA (Fig. 3E). These results suggest that DDK rather than CDK2 or CKII regulates DUE-B phosphorylation and intermolecular binding.
To test whether regulation of DUE-B HMW complex formation by phosphorylation is only seen with proteins expressed in Sf9 insect cells in Xenopus egg extracts, we purified recombinant His6-tagged DUE-B from HeLa cells (DUE-BHeLa). DUE-BHeLa was treated with λ-protein phosphatase and incubated with whole cell extracts from asynchronous HeLa cells. As shown in Fig. 4, A and B, only the phosphatase-treated DUE-BHeLa was incorporated into HMW complexes, whereas untreated DUE-BHeLa exclusively formed dimers. Also, when HeLa cells were treated with the Cdc7 inhibitor PHA-767491, the endogenous DUE-B accumulated in HMW complexes that eluted similarly to DUE-BSf9 complexes (Fig. 4, C and D). Thus, dephosphorylation of DUE-BHeLa promotes HMW complex formation.
FIGURE 4.

The regulation of DUE-B phosphorylation and high molecular weight complex formation is not specific to Xenopus egg extract. A, recombinant DUE-BHeLa was expressed and purified from HeLa cells. Untreated recombinant DUE-BHeLa (His6 tagged) was added to HeLa whole cell extract. Proteins were fractionated by gel filtration. B, recombinant DUE-BHeLa was dephosphorylated by λ-phosphatase and incubated with HeLa cell whole cell extract. C, whole cell lysate from asynchronous HeLa cells was fractionated by gel filtration. D, HeLa cells were treated with 5 μm PHA-767491 for 20 h. Whole cell extracts were fractionated by gel filtration. Interphase egg extract was chromatographed before (E) or after (F) addition of sperm chromatin. G, DUE-B can be phosphorylated by Cdc7 to form homodimers. Phosphorylated DUE-B is dephosphorylated by PP2A and is incorporated into high molecular weight complexes. The kinase and phosphatase activities at equilibrium control DUE-B phosphorylation levels in vivo.
Moreover, we added sperm chromatin to Xenopus interphase egg extract and detected a small but reproducible fraction of endogenous DUE-BXl eluting similarly to DUE-BSf9 HMW complexes during S phase (Fig. 4, E and F). These results show that endogenous DUE-Bxl shifts between homodimeric and HMW-bound forms during unperturbed replication.
Collectively, these data show that inhibition of PP2A results in accumulation of DUE-B in phosphorylated homodimer form due to Cdc7 kinase phosphorylation, whereas inhibition of Cdc7 kinase results in PP2A-dependent dephosphorylation of DUE-B and the formation of high molecular weight complexes. These activities are predicted to form an equilibrium in which the DUE-B phosphorylation status and intermolecular binding are regulated by the dynamic balance of kinase and phosphatase activities (Fig. 4G). The formation of high molecular weight complexes is not unique to DUE-BSf9, as dephosphorylated DUE-BHeLa and S phase DUE-BXl produce complexes that elute similarly.
Cdc7 Phosphorylation on DUE-B Can Be Primed by Other Kinases
CKII does not phosphorylate sites in DUE-B in vitro other than those in the C terminus (18) and CKII is able to decrease the amount of DUE-BSf9 incorporated into HMW complexes (Fig. 2D); however, inhibition of CKII by TBB in the presence of OA did not change the DUE-BSf9 distribution compared with the OA-treated sample (Fig. 3D), possibly because CKII does not directly phosphorylate DUE-B in egg extracts. Alternatively, CKII phosphorylation may not directly control DUE-BSf9 distribution but may potentiate DUE-B phosphorylation by Cdc7. As shown in Fig. 5A, the C-terminal region of DUE-B is rich in serine and threonine. The C-terminal region is predicted to be disordered, and does not appear as a stable structure upon DUE-B crystallization (18). Among those serine residues, there are several conserved target sequences for CKII and Cdc7. The Cdc7 sites fall into two different classes: intrinsic sites and phosphorylation-generated sites (35–38) (Fig. 5A). The intrinsic sites are characterized by serine or threonine followed by an acidic residue at the +1 position ((S/T)(D/E)). The phosphorylation-generated sites consist of serine or threonine followed by a phosphoserine or phosphothreonine at the +1 position (S/T)S(P)/T(P). Phosphorylation of the Ser/Thr at the +1 position by another kinase could prime the (S/T)S(P)/T(P) for Cdc7 phosphorylation. In the C terminus of DUE-B there are two intrinsic Cdc7 sites and three phosphorylation-generated sites that could be primed by CKII phosphorylation of the serines at the +1 position.
FIGURE 5.
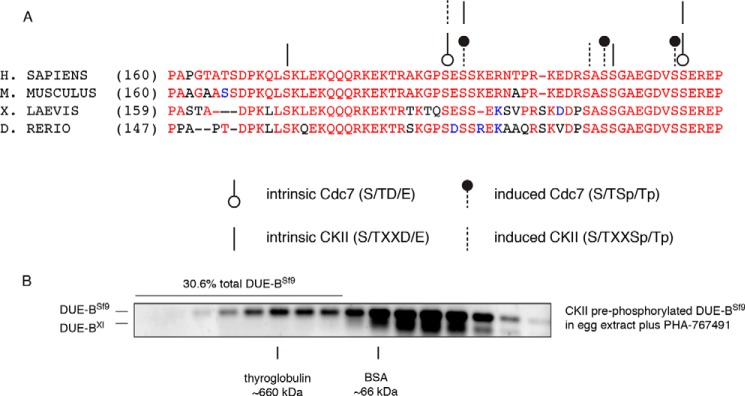
Casein kinase II can prime DUE-B for phosphorylation by Cdc7 kinase. A, multiple organism alignment of the DUE-B C terminus tail amino acid sequences. Circles and lines point out the potential phosphorylation sites by Cdc7 and CKII, respectively. The open circles and dashed lines indicate the predicted intrinsic kinase phosphorylation sites. The closed circles and solid lines show the potential phosphorylation induced kinase sites. B, DUE-BSf9 was pre-phosphorylated by casein kinase II in vitro. The phosphorylated DUE-BSf9 was incubated with interphase egg extract supplemented with Cdc7 inhibitor.
To test the hypothesis that CKII could prime Cdc7 phosphorylation, DUE-BSf9 was phosphorylated with CKII in vitro. The CKII-phosphorylated DUE-BSf9 was then incubated with PHA-767491-treated egg extracts. In contrast to the decrease in the HMW complexes when DUE-BSf9 was pre-phosphorylated by CKII and added to Cdc7-competent extracts (Fig. 2D), pre-phosphorylation of DUE-BSf9 by CKII was not sufficient to remove DUE-BSf9 from HMW complexes when Cdc7 was inhibited (Fig. 5B). This result suggests that CKII, and possibly other kinases, can prime DUE-B for phosphorylation by Cdc7
DUE-B Interacts with Subunits of the MCM and Multisynthetase Complexes
To identify the proteins that interact with DUE-B, interphase egg extract was incubated with DUE-BSf9 and fractionated by gel filtration.
Fractions containing HMW complexes were pooled (Fig. 6A) and DUE-BSf9 was isolated together with its interacting proteins by Ni-NTA bead pull down. The interacting proteins were identified by mass spectrometry. All six subunits of the Mcm2–7 complex and a group of aminoacyl-tRNA synthetases (ARS proteins (39)) were identified as DUE-BSf9 interacting proteins (Table 1). To test whether DUE-B and MCMs co-eluted, the chromatographic fractions were blotted for Mcm7, but the elution profile of DUE-BSf9 incubated with interphase egg extract did not exactly overlap with that of the Mcm7 marker for the MCM heterohexamer (Fig. 6A). However, when DUE-BSf9 was added to interphase egg extract that had been treated with RNase A (Fig. 6B) or benzonase nuclease (not shown), the distribution of DUE-BSf9 in the HMW complexes closely overlapped with that of the Mcm2–7 complex.
FIGURE 6.

DUE-B interacts with the MCM complex. A, DUE-BSf9 was incubated with interphase egg extract and fractionated by gel filtration. Mcm7 and DUE-B were detected using specific antibodies. B, interphase egg extract was treated with RNase A and then incubated with DUE-BSf9. C, DUE-BSf9 was incubated with benzonase nuclease-treated interphase egg extract and then immunoprecipitated (IP) by affinity purified DUE-B antibody or normal rabbit IgG. The immunoprecipitates were resolved by SDS-PAGE and probed for specific proteins. D, DUE-BSf9 was incubated with benzonase nuclease-treated interphase egg extract. Mcm3 or Mcm5 were immunoprecipitated by specific antibodies. The immunoprecipitates were resolved by SDS-PAGE and Western blotted for DUE-B.
TABLE 1.
Mass spectrometry identification of proteins pulled down by DUE-BSf9 in HMW complexes
Proteins pulled down from HMW fractions of control extracts (no DUE-BSf9 added) are excluded. Scores are ranked as low (<75), good (75–100), and excellent (>100) confidence.
| Protein | Score |
|---|---|
| Valyl-tRNA synthetase (VRS) | 359 |
| Lysyl-tRNA synthetase (KRS) | 232 |
| Glutamyl-prolyl tRNA synthetase (EPRS) | 146 |
| Prolyl-tRNA synthetase (PRS) | 146 |
| Aspartyl-tRNA synthetase (DRS) | 89 |
| Phenylalanyl-tRNA synthetase (FRS) α chain | 66 |
| Leucyl-tRNA synthetase | 65 |
| Phenylalanyl-tRNA synthetase (FRS) b chain | 43 |
| Asparaginyl-tRNA synthetase (NRS) | 43 |
| Glutaminyl-tRNA synthetase | |
| Mcm2 | 521 |
| Mcm3 | 213 |
| Mcm4 | 230 |
| Mcm5 | 230 |
| Mcm6 | 286 |
| Mcm7 | 87 |
We used co-immunoprecipitation to confirm the interaction between DUE-B and the MCM complex. We found that DUE-BSf9 was able to co-immunoprecipitate with Mcm3 and Mcm5 (Fig. 6C) from benzonase nuclease-treated extracts. Reciprocally, Mcm3 and Mcm5 could individually co-immunoprecipitate DUE-BSf9 from these egg extracts (Fig. 6D).
Although Mcm2, -3, -4, -5, -6, and -7 subunits were identified by mass spectrometry as interacting with DUE-B, we could not detect co-immunoprecipitation of DUE-BSf9 with other MCM subunits possibly due to weaker or indirect interactions. Nevertheless, these data support the contention that DUE-B is able to bind to the Mcm2–7 complex in Xenopus egg extracts.
These results reflect the dual nature of DUE-B; i.e. not only does DUE-B participate in DNA replication initiation, but the N-terminal 159-amino acid domain of DUE-B is highly conserved as an aminoacyl-tRNA editing enzyme with close structural homology to aminoacyl-tRNA synthetases (18). Several aminoacyl-tRNA synthetases form a multi-synthetase complex (∼1.0–1.2 × 106 Da), some components of which are inherently loosely bound or dissociated from the complex following specific signals (39). Multi-synthetase complex constituents interact with multiple proteins including ATM, ATR, p53, and display non-canonical activities in translation initiation, cell migration, intra- and intercellular signaling and other cellular processes (40, 41). The RNase A digestion data suggests that the interaction of DUE-B with the ARS proteins is mediated by RNA, whereas interaction with the MCM complex is RNase A insensitive.
DUE-B Phosphorylation Affects MCM Binding and Cdc45 Loading in Vivo
To address the significance of DUE-B phosphorylation for DNA replication, we chose to characterize the effects of the DUE-B C-terminal nonphosphorylatable Ser/Thr-Ala and phosphomimic Ser/Thr-Asp mutants on replication in vitro and in vivo.
Notwithstanding the observation that dephosphorylated DUE-BHeLa, DUE-BXl, and DUE-BSf9 form different HMW complexes in interphase than mitotic DUE-B, the foregoing chromatography results imply that C-terminal phosphorylation of DUE-BSf9 controls its interaction with replication proteins in egg extracts. To bridge these results, we analyzed the effect on DNA replication of adding DUE-B C-terminal mutant protein to egg extracts, or expressing DUE-B C-terminal mutants in HeLa cells.
DUE-BWT, DUE-BΔCT (a C-terminal residue 160–209 deletion mutant), DUE-B(S/T)-A, and DUE-B(S/T)-D were purified from HeLa cells and added to egg extracts (Fig. 7A). After the addition of [α-32P]dCTP and sperm chromatin, it was observed that only the DUE-B(S/T)-A mutant dominantly inhibited DNA replication (Fig. 7B). The inhibition of Cdc45 and RPA loading onto chromatin (Fig. 7C) by the DUE-B(S/T)-A mutant indicates that the nonphosphorylatable DUE-B(S/T)-A mutant blocks replication prior to origin unwinding. We note as well that each of the DUE-B constructs was able to bind the chromatin template, irrespective of the presence or absence of the C-terminal tail.
FIGURE 7.

The C-terminal DUE-B(S/T)-A mutant dominantly inhibits Cdc45 loading. A, His6-tagged DUE-B C-terminal constructs were expressed in HeLa cells and isolated using Ni-NTA beads. The isolated proteins were added to egg extracts and detected by Western blotting. B, sperm chromatin was added to extracts shown in panel A. DNA synthesis was monitored by [α-32P]dCTP incorporation (autoradiogram). C, sperm chromatin was purified from replication reactions and analyzed by Western blotting.
We next added the DUE-BWT, DUE-BΔCT, DUE-B(S/T)-A, and DUE-B(S/T)-D proteins to DUE-B immunodepleted extracts (Fig. 8A). As reported previously, DUE-B immunodepletion blocks DNA replication (Fig. 8B) and Cdc45 loading (Fig. 8C), which could be restored by the addition of DUE-BWT (19). However, in depleted extract neither DUE-BΔCT nor DUE-B(S/T)-A could restore replication.
FIGURE 8.

C-terminal DUE-B mutants inhibit Cdc45 loading in DUE-B-depleted egg extracts. A, His6-tagged DUE-B C-terminal constructs were expressed in HeLa cells and isolated using Ni-NTA beads. The isolated proteins were added to anti-DUE-B immunodepleted (▵) egg extracts and detected by Western blotting. Mock depleted, mock ▵. B, sperm chromatin was added to extracts shown in panel A. DNA synthesis was monitored by [α-32P]dCTP incorporation (autoradiogram). C, sperm chromatin was purified from replication reactions and analyzed by Western blotting.
When expressed in HeLa cells, the non-phosphorylatable DUE-B(S/T)-A mutant showed stronger affinity for Mcm3 than DUE-BWT (Fig. 9A). In contrast, the phosphomimic DUE-B(S/T)-D mutant showed decreased interaction with Mcm3. This finding is consistent with the previous chromatographic results indicating that DUE-B dephosphorylation increases MCM binding, and also implies that the phosphorylation status of the DUE-B C terminus can regulate the interaction between DUE-B and the MCM complex in vivo.
FIGURE 9.

DUE-B C terminus phosphorylation status is critical for its function in vivo. A, HeLa cells were transiently transfected with empty vector or vectors that express His6-tagged DUE-B C terminus mutants. The DUE-B mutants were pulled down by Ni-NTA beads and the precipitated proteins were analyzed by Western blotting. Note that the His6-tagged DUE-B(S/T)-D migrated faster than the His6-tagged DUE-BWT and is indistinguishable from the endogenous DUE-B in HeLa cell by Western blot. HeLa cells transfected with empty vector were used in control pull downs. B, endogenous DUE-B was knocked down using DUE-B siRNA in HeLa cells. siRNA-resistant WT or C terminus mutated DUE-B were expressed. C, HeLa cells described in B were fractionated. Chromatin-bound Cdc45 was analyzed by Western blotting. D, WT DUE-B or the C terminus mutants were expressed in HeLa cells. E, cells that express the unphosphorylatable DUE-B(S/T)-A mutant lost the ability to proliferate in a colony forming assay.
To test the hypothesis that the change of interaction between DUE-B and MCM complex might alter the ability of DUE-B to support DNA replication, HeLa cells were treated with siRNA to knock down ∼75% of endogenous DUE-B (Fig. 9B), and siRNA-resistant forms of the DUE-B C-terminal mutants were expressed. DUE-B is a highly abundant protein (∼3 × 106 dimers/cell), and partial knockdown resulted in a nominal decrease in Cdc45 loading (Fig. 9, B and C). Cells expressing DUE-BWT restored Cdc45 loading, whereas DUE-B(S/T)-D did not significantly affect Cdc45 chromatin binding. In contrast, expression of DUE-B(S/T)-A or DUE-BΔCT dramatically decreased Cdc45 loading, exerting a dominant-negative effect, possibly by heterodimerization with the reduced amount of residual endogenous DUE-B (Fig. 9, B and C). When the C-terminal DUE-B mutants were expressed in HeLa cells and selected for transgene expression without knocking down endogenous DUE-B, we observed that only the C-terminal unphosphorylatable DUE-B(S/T)-A dominantly inhibited cell growth (Fig. 9, D and E). We note that there are comparable amounts of MCM proteins (∼1–3 × 106) (42) and DUE-B dimers (∼3 × 106) (43) per HeLa cell.
These are the results expected if a dynamic equilibrium between phosphorylated DUE-B and dephosphorylated DUE-B is required for replication initiation; overexpression of the nonphosphorylatable DUE-B(S/T)-A would sequester the MCM complex but not transfer Cdc45, whereas DUE-B(S/T)-D would not bind the MCM complex.
Together, these results suggest that DUE-BΔCT does not efficiently bind MCMs and that the endogenous DUE-BHeLa/DUE-BΔCT heterodimer blocks Cdc45 loading when endogenous DUE-B is depleted. In contrast, DUE-B(S/T)-A strongly interacts with MCMs and forms high molecular complexes, whereas homodimeric DUE-B(S/T)-A or heterodimeric DUE-BHeLa/DUE-B(S/T)-A dominantly block Cdc45 loading. Thus decreased DUE-B phosphorylation tightens its interaction with the MCM complex and decreases its activity in supporting DNA replication initiation by Cdc45 loading.
DISCUSSION
DUE-B is required for Cdc45 binding to chromatin in Xenopus egg extract, and the loading of endogenous DUE-B and Cdc45 depends on pre-RC assembly and S phase kinase activity (19). In vivo, tethering of origin recognition complex to an inactive form of the human c-myc replicator leads to coordinated binding of the MCM complex, DUE-B, and Cdc45, and origin activation (21). Taken together, these findings suggest that the interaction between DUE-B, Cdc45, and MCM proteins is physiologically relevant. Here we show that DUE-B forms high molecular weight complexes with the MCM proteins and with a set of aminoacyl-tRNA synthetases in egg extracts in the absence of DNA. The formation of DUE-B high molecular weight complexes is dependent on phosphorylation. Inhibition of either Cdc7 or PP2A shifts the steady state DUE-BSf9 phosphorylation level and modulates the formation of these HMW complexes. The HMW complexes formed by mitotic DUE-BHeLa or DUE-BXl elute at a larger apparent size than DUE-BSf9 or DUE-BXl HMW complexes formed in interphase egg extracts, and may contain quantitatively or qualitatively different DUE-B-bound components if M phase and interphase extracts contain diverse binding factors, or because DUE-B is differentially modified, or both. Thus, the HMW complexes may reflect the interaction between DUE-B and MCMs on or off the chromatin.
C-terminal phosphorylation of DUE-BSf9 interferes with the formation of the interphase HMW complexes and returns DUE-BSf9 to the homodimer form. At least two types of HMW DUE-BSf9 complexes are formed in interphase; complexes containing the Mcm2–7 hexamer, and complexes containing aminoacyl-tRNA synthetases and RNA. In view of the differences between the DUE-B HMW complexes formed in HeLa or Xenopus extracts in vitro versus during passage through M phase in vivo, it is important to note that the extent of HMW complex formation in egg extracts by DUE-B(S/T)-A and DUE-B(S/T)-D parallels the ability of these mutants to bind MCM, load Cdc45, and promote cell division in vivo. Moreover, during DNA replication in egg extracts, a fraction of endogenous DUE-BXl shifts from the phosphorylated dimer form to the interphase HMW form. The simplest interpretation of these results is that phosphorylation of the DUE-B C terminus directly affects its ability to deliver Cdc45 to the pre-RC. However, this does not exclude the possibility that DUE-B phosphorylation indirectly affects Cdc45 loading through aminoacyl-tRNA synthetase signaling.
Cdc7 promotes DNA replication initiation by changing the conformation of the MCM complex to facilitate recruitment of Cdc45 (6, 7). Mutations of MCM subunits can bypass the Cdc7 requirement, however, Cdc7 bypass strains show defects in DNA replication or cell growth suggesting that additional Cdc7 targets might be required for efficient replication initiation (9, 10). The effect of Cdc7 on DUE-B binding to Mcm2–7 implies that Cdc7 phosphorylation of DUE-B is important for efficient replication initiation.
We do not yet understand the detailed mechanism of DUE-B, GEMC1, and Treslin/TICRR loading of Cdc45. Each of these proteins has been reported to bind to Cdc45 and TopBP1, but in contrast to the origin binding of DUE-B and Treslin, GEMC1 loading requires prior assembly of origin recognition complex, but not MCMs. Treslin and TopBP1 bind independently to chromatin and associate only in nuclei after Treslin phosphorylation, whereas DUE-B and GEMC1 bind Cdc45 and TopBP1 in unfractionated egg extracts. Although the phosphorylation of DUE-B is Cdc7 dependent, GEMC1 and Treslin are targets for CDK phosphorylation, and the unphosphorylatable forms of each of these three proteins inhibit Cdc45 loading and chromatin replication (19, 27, 44). The common binding of DUE-B, GEMC1, and Treslin/TICRR with Cdc45 and TopBP1 suggest that these proteins may physically network, whereas the effects of phosphorylation imply that the S phase kinases regulate these interactions during replication initiation.
Protein phosphatase 2A counteracts Cdc7 activity to regulate DUE-BSf9 HMW complex formation. PP2A is known to be an essential enzyme regulating DNA replication and cell cycle progression (14, 45). The balance between PP2A and Cdk1 maintains the dynamics of cell cycle transitions (14). Similarly, the equilibrium of Cdc7 and PP2A in controlling DUE-BSf9 binding to other molecules suggests that PP2A can influence DNA replication initiation by controlling the activity of DUE-B. Chou et al. (11) showed that PP2A controls Cdc45 chromatin binding during DNA replication in Xenopus egg extracts, but the target of PP2A that regulates Cdc45 loading was not identified at that time. Our data argue that PP2A affects DNA replication by controlling the dynamic balance between phosphorylated and dephosphorylated forms of DUE-B.
The OA inhibition of dephosphorylation of DUE-B, which we have observed is reminiscent of the model by Peterson et al. (13) of two pathways for PP2A regulation of Cdc45 loading. In this model, under OA, PP2A was unable to dephosphorylate a kinase substrate in the pathway of Cdc45 loading. We suggest that DUE-B may be a key PP2A and kinase substrate involved in Cdc45 loading.
In the present experiments, conditions that block the conversion between phosphorylated (homodimer) and dephosphorylated (HMW) forms of DUE-B block replication. The decreased chromatin loading of Cdc45 in DUE-B knockdown cells expressing unphosphorylatable DUE-B, and the dominant-negative effect of unphosphorylatable DUE-B on cell division support the view that unphosphorylatable DUE-B (mutant DUE-B(S/T)-A homodimer or mutant-wild type heterodimer) is locked in the HMW form, whereas the DUE-B phosphomimic mutant-wild type heterodimer retains the ability to load Cdc45.
Immunodepletion of DUE-B from Xenopus egg extracts blocks Cdc45 loading and replication of sperm chromatin (19). Because unphosphorylated DUE-B binds with stronger affinity than phosphorylated DUE-B to the MCM complex and Cdc45 (19), we propose that unphosphorylated DUE-B chaperones Cdc45 to the inactive pre-RC. In concert with Cdc7 phosphorylation of the MCM complex during the G1/S phase transition, Cdc7 kinase activity phosphorylates DUE-B to release it from Cdc45 and the pre-RC, leaving Cdc45 on the MCM complex. The transient binding of DUE-B to chromatin and its rapid release upon RPA loading and origin unwinding (17–19) is consistent with this idea. Interestingly, earlier work in budding yeast identified the DDK docking domain (DDD) of the Mcm4175–333 fragment (46) that recruits DDK and facilitates Cdc7 phosphorylation of the Mcm4 N terminus. We note the close overlap between the crystal structure of the N terminus of DUE-B (18) and the predicted structure of the Mcm4175–333 DDD fragment (46).
The simultaneous increase of Cdc7 kinase and PP2A phosphatase activities during S phase (14, 47) is consistent with the rapid turnover of DUE-B phosphorylation and dephosphorylation during replication. The outcome of such turnover would be that DUE-B becomes incorporated into and released from HMW complexes during S phase. Indeed, a fraction of endogenous DUE-BXl shifts from dimeric to HMW forms upon sperm chromatin replication. We have observed as well that a small amount of DUE-B (<10% of the total ∼3 million DUE-B dimers) is sufficient to support DNA replication initiation in Xenopus egg extracts and human cells.4 These observations suggest that DUE-B rapidly cycles between dephosphorylated and rephosphorylated forms during S phase. The regulation of DUE-B protein binding by Cdc7 and PP2A provides another level of control over Cdc45 loading and replication initiation.
Acknowledgments
We thank Yong-jie Xu for generously sharing advice and equipment and Todd Lewis and Joanna Barthelemy for their advice.
This work was supported, in whole or in part, by National Institutes of Health Grant GM53819 from the NIGMS (to M. L.).
Y. Gao and M. Leffak, unpublished data.
- MCM
- minichromosome maintenance
- DUE
- DNA unwinding element
- pre-RC
- pre-replication complex
- DDK
- Dbf4-dependent kinase
- CDK
- cyclin-dependent kinase
- PP2A
- protein phosphatase 2A
- Ni-NTA
- nickel-nitrilotriacetic acid
- TBB
- 4,5,6,7-tetrabromo-2-azabenzimidazole
- OA
- okadaic acid
- HMW
- high molecular weight.
REFERENCES
- 1. Bell S. P., Stillman B. (1992) ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature 357, 128–134 [DOI] [PubMed] [Google Scholar]
- 2. Hoang M. L., Leon R. P., Pessoa-Brandao L., Hunt S., Raghuraman M. K., Fangman W. L., Brewer B. J., Sclafani R. A. (2007) Structural changes in Mcm5 protein bypass Cdc7-Dbf4 function and reduce replication origin efficiency in Saccharomyces cerevisiae. Mol. Cell. Biol. 27, 7594–7602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Evrin C., Fernández-Cid A., Zech J., Herrera M. C., Riera A., Clarke P., Brill S., Lurz R., Speck C. (2013) In the absence of ATPase activity, pre-RC formation is blocked prior to MCM2–7 hexamer dimerization. Nucleic Acids Res. 41, 3162–3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fernández-Cid A., Riera A., Tognetti S., Herrera M. C., Samel S., Evrin C., Winkler C., Gardenal E., Uhle S., Speck C. (2013) An ORC/Cdc6/MCM2–7 complex is formed in a multistep reaction to serve as a platform for MCM double-hexamer assembly. Mol. Cell. 50, 577–588 [DOI] [PubMed] [Google Scholar]
- 5. Walter J. C. (2000) Evidence for sequential action of cdc7 and cdk2 protein kinases during initiation of DNA replication in Xenopus egg extracts. J. Biol. Chem. 275, 39773–39778 [DOI] [PubMed] [Google Scholar]
- 6. Heller R. C., Kang S., Lam W. M., Chen S., Chan C. S., Bell S. P. (2011) Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell 146, 80–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Labib K. (2010) How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev. 24, 1208–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Masai H., Taniyama C., Ogino K., Matsui E., Kakusho N., Matsumoto S., Kim J.-M., Ishii A., Tanaka T., Kobayashi T., Tamai K., Ohtani K., Arai K. (2006) Phosphorylation of MCM4 by Cdc7 kinase facilitates its interaction with Cdc45 on the chromatin. J. Biol. Chem. 281, 39249–39261 [DOI] [PubMed] [Google Scholar]
- 9. Sheu Y.-J., Stillman B. (2010) The Dbf4-Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature 463, 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hardy C. F., Dryga O., Seematter S., Pahl P. M., Sclafani R. A. (1997) mcm5/cdc46-bob1 bypasses the requirement for the S phase activator Cdc7p. Proc. Natl. Acad. Sci. U.S.A. 94, 3151–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chou D. M., Petersen P., Walter J. C., Walter G. (2002) Protein phosphatase 2A regulates binding of Cdc45 to the prereplication complex. J. Biol. Chem. 277, 40520–40527 [DOI] [PubMed] [Google Scholar]
- 12. Lin X. H., Walter J., Scheidtmann K., Ohst K., Newport J., Walter G. (1998) Protein phosphatase 2A is required for the initiation of chromosomal DNA replication. Proc. Natl. Acad. Sci. U.S.A. 95, 14693–14698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Petersen P., Chou D. M., You Z., Hunter T., Walter J. C., Walter G. (2006) Protein phosphatase 2A antagonizes ATM and ATR in a Cdk2- and Cdc7-independent DNA damage checkpoint. Mol. Cell. Biol. 26, 1997–2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krasinska L., Domingo-Sananes M. R., Kapuy O., Parisis N., Harker B., Moorhead G., Rossignol M., Novák B., Fisher D. (2011) Protein phosphatase 2A controls the order and dynamics of cell-cycle transitions. Mol. Cell 44, 437–450 [DOI] [PubMed] [Google Scholar]
- 15. Coudreuse D., Nurse P. (2010) Driving the cell cycle with a minimal CDK control network. Nature 468, 1074–1079 [DOI] [PubMed] [Google Scholar]
- 16. Liu G., Bissler J. J., Sinden R. R., Leffak M. (2007) Unstable spinocerebellar ataxia type 10 (ATTCT)*(AGAAT) repeats are associated with aberrant replication at the ATX10 locus and replication origin-dependent expansion at an ectopic site in human cells. Mol. Cell. Biol. 27, 7828–7838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Casper J. M., Kemp M. G., Ghosh M., Randall G. M., Vaillant A., Leffak M. (2005) The c-myc DNA-unwinding element-binding protein modulates the assembly of DNA replication complexes in vitro. J. Biol. Chem. 280, 13071–13083 [DOI] [PubMed] [Google Scholar]
- 18. Kemp M., Bae B., Yu J. P., Ghosh M., Leffak M., Nair S. K. (2007) Structure and function of the c-myc DNA-unwinding element-binding protein DUE-B. J. Biol. Chem. 282, 10441–10448 [DOI] [PubMed] [Google Scholar]
- 19. Chowdhury A., Liu G., Kemp M., Chen X., Katrangi N., Myers S., Ghosh M., Yao J., Gao Y., Bubulya P., Leffak M. (2010) The DNA unwinding element binding protein DUE-B interacts with Cdc45 in preinitiation complex formation. Mol. Cell. Biol. 30, 1495–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ghosh M., Kemp M., Liu G., Ritzi M., Schepers A., Leffak M. (2006) Differential binding of replication proteins across the human c-myc replicator. Mol. Cell. Biol. 26, 5270–5283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen X., Liu G., Leffak M. (2013) Activation of a human chromosomal replication origin by protein tethering. Nucleic Acids Res. 41, 6460–6474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kamimura Y., Tak Y. S., Sugino A., Araki H. (2001) Sld3, which interacts with Cdc45 (Sld4), functions for chromosomal DNA replication in Saccharomyces cerevisiae. EMBO J. 20, 2097–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muramatsu S., Hirai K., Tak Y.-S., Kamimura Y., Araki H. (2010) CDK-dependent complex formation between replication proteins Dpb11, Sld2, Polϵ, and GINS in budding yeast. Genes Dev. 24, 602–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zegerman P., Diffley J. F. (2007) Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature 445, 281–285 [DOI] [PubMed] [Google Scholar]
- 25. Kumagai A., Shevchenko A., Shevchenko A., Dunphy W. G. (2010) Treslin collaborates with TopBP1 in triggering the initiation of DNA replication. Cell 140, 349–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sansam C. L., Cruz N. M., Danielian P. S., Amsterdam A., Lau M. L., Hopkins N., Lees J. A. (2010) A vertebrate gene, ticrr, is an essential checkpoint and replication regulator. Genes Dev. 24, 183–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Balestrini A., Cosentino C., Errico A., Garner E., Costanzo V. (2010) GEMC1 is a TopBP1-interacting protein required for chromosomal DNA replication. Nat. Cell Biol. 12, 484–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lebofsky R., Takahashi T., Walter J. C. (2009) DNA replication in nucleus-free Xenopus egg extracts. Methods Mol. Biol, 521, 229–252 [DOI] [PubMed] [Google Scholar]
- 29. Chong J. P., Thömmes P., Rowles A., Mahbubani H. M., Blow J. J. (1997) Characterization of the Xenopus replication licensing system. Methods Enzymol. 283, 549–564 [DOI] [PubMed] [Google Scholar]
- 30. Montagnoli A., Valsasina B., Croci V., Menichincheri M., Rainoldi S., Marchesi V., Tibolla M., Tenca P., Brotherton D., Albanese C., Patton V., Alzani R., Ciavolella A., Sola F., Molinari A., Volpi D., Avanzi N., Fiorentini F., Cattoni M., Healy S., Ballinari D., Pesenti E., Isacchi A., Moll J., Bensimon A., Vanotti E., Santocanale C. (2008) A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat. Chem. Biol. 4, 357–365 [DOI] [PubMed] [Google Scholar]
- 31. Gambus A., Khoudoli G. A., Jones R. C., Blow J. J. (2011) MCM2–7 form double hexamers at licensed origins in Xenopus egg extract. J. Biol. Chem. 286, 11855–11864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bialojan C., Takai A. (1988) Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases: specificity and kinetics. Biochem. J. 256, 283–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cohen P., Klumpp S., Schelling D. L. (1989) An improved procedure for identifying and quantitating protein phosphatases in mammalian tissues. FEBS Lett. 250, 596–600 [DOI] [PubMed] [Google Scholar]
- 34. Ruzzene M., Penzo D., Pinna L. A. (2002) Protein kinase CK2 inhibitor 4,5,6,7-tetrabromobenzotriazole (TBB) induces apoptosis and caspase-dependent degradation of haematopoietic lineage cell-specific protein 1 (HS1) in Jurkat cells. Biochem. J. 364, 41–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cho W. H., Lee Y. J., Kong S. I., Hurwitz J., Lee J. K. (2006) CDC7 kinase phosphorylates serine residues adjacent to acidic amino acids in the minichromosome maintenance 2 protein. Proc. Natl. Acad. Sci. U.S.A. 103, 11521–11526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Montagnoli A., Valsasina B., Brotherton D., Troiani S., Rainoldi S., Tenca P., Molinari A., Santocanale C. (2006) Identification of Mcm2 phosphorylation sites by S-phase-regulating kinases. J. Biol. Chem. 281, 10281–10290 [DOI] [PubMed] [Google Scholar]
- 37. Charych D. H., Coyne M., Yabannavar A., Narberes J., Chow S., Wallroth M., Shafer C., Walter A. O. (2008) Inhibition of Cdc7/Dbf4 kinase activity affects specific phosphorylation sites on MCM2 in cancer cells. J. Cell. Biochem. 104, 1075–1086 [DOI] [PubMed] [Google Scholar]
- 38. Randell J. C., Fan A., Chan C., Francis L. I., Heller R. C., Galani K., Bell S. P. (2010) Mec1 is one of multiple kinases that prime the Mcm2–7 helicase for phosphorylation by Cdc7. Mol. Cell 40, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim J. H., Han J. M., Kim S. (2014) Protein-protein interactions and multi-component complexes of aminoacyl-tRNA synthetases. Top. Curr. Chem. 344, 119–144 [DOI] [PubMed] [Google Scholar]
- 40. Lee S. W., Cho B. H., Park S. G., Kim S. (2004) Aminoacyl-tRNA synthetase complexes: beyond translation. J. Cell Sci. 117, 3725–3734 [DOI] [PubMed] [Google Scholar]
- 41. Hausmann C. D., Ibba M. (2008) Aminoacyl-tRNA synthetase complexes: molecular multitasking revealed. FEMS Microbiol. Rev. 32, 705–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ishimi Y., Okayasu I., Kato C., Kwon H.-J., Kimura H., Yamada K., Song S.-Y. (2003) Enhanced expression of Mcm proteins in cancer cells derived from uterine cervix. Eur. J. Biochem. 270, 1089–1101 [DOI] [PubMed] [Google Scholar]
- 43. Kemp M. (2006) Regulation of DNA Replication Initiation by Histone Acetylation and the DNA Unwinding Element Binding Protein DUE–B. Ph.D. thesis, Wright State University, Dayton, OH [Google Scholar]
- 44. Kumagai A., Shevchenko A., Shevchenko A., Dunphy W. G. (2011) Direct regulation of Treslin by cyclin-dependent kinase is essential for the onset of DNA replication. J. Cell Biol. 193, 995–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aladjem M. I., Rodewald L. W., Kolman J. L., Wahl G. M. (1998) Genetic dissection of a mammalian replicator in the human β-globin locus. Science 281, 1005–1009 [DOI] [PubMed] [Google Scholar]
- 46. Sheu Y.-J., Stillman B. (2006) Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol. Cell 24, 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Weinreich M., Stillman B. (1999) Cdc7p-Dbf4p kinase binds to chromatin during S phase and is regulated by both the APC and the RAD53 checkpoint pathway. EMBO J. 18, 5334–5346 [DOI] [PMC free article] [PubMed] [Google Scholar]