

Background: Anti- and pro-apoptotic B cell lymphoma 2 (Bcl-2) proteins possess Bcl-2 homology 3 (BH3) domains generally associated with cell death induction.
Results: Features of anti-apoptotic BH3 domains were identified that limit their killing activity but are important for protein stability.
Conclusion: Pro- and anti-apoptotic BH3 domains are distinct, which affects their function.
Significance: Differences in BH3 domains have significant consequences in apoptotic signaling.
Keywords: Apoptosis, B cell Lymphoma 2 (Bcl-2) Family, Cell Death, Peptides, Protein Structure
Abstract
Bcl-2 homology 3 (BH3) domains are short sequence motifs that mediate nearly all protein-protein interactions between B cell lymphoma 2 (Bcl-2) family proteins in the intrinsic apoptotic cell death pathway. These sequences are found on both pro-survival and pro-apoptotic members, although their primary function is believed to be associated with induction of cell death. Here, we identify critical features of the BH3 domains of pro-survival proteins that distinguish them functionally from their pro-apoptotic counterparts. Biochemical and x-ray crystallographic studies demonstrate that these differences reduce the capacity of most pro-survival proteins to form high affinity “BH3-in-groove” complexes that are critical for cell death induction. Switching these residues for the corresponding residues in Bcl-2 homologous antagonist/killer (Bak) increases the binding affinity of isolated BH3 domains for pro-survival proteins; however, their exchange in the context of the parental protein causes rapid proteasomal degradation due to protein destabilization. This is supported by further x-ray crystallographic studies that capture elements of this destabilization in one pro-survival protein, Bcl-w. In pro-apoptotic Bak, we demonstrate that the corresponding distinguishing residues are important for its cell-killing capacity and antagonism by pro-survival proteins.
Introduction
Apoptosis is a form of programmed cell death that is essential for the development and survival of all multicellular organisms. In mammals, cellular stresses activate the intrinsic (mitochondrial) apoptotic pathway that is regulated by the Bcl-22 family of proteins. This family consists of ∼15 different members with opposing functions, either promoting or inhibiting cell death. These proteins are all related to each other by up to four distinct regions of sequence homology called “Bcl-2 homology domains” (BH1–4).
The pro-survival proteins, which include Bcl-2, Bcl-xL, Bcl-w, Mcl-1, and Bfl-1, possess BH1–4 domains, and these inhibit cell death by binding directly to the two classes of pro-apoptotic proteins: the Bax/Bak proteins and the BH3-only proteins. Bax and Bak, like the pro-survival proteins, possess BH1–4 domains. These proteins are the essential mediators of apoptosis (1, 2) and, following a death stimulus, adopt a conformation that allows them to oligomerize on the outer mitochondrial membrane and assemble into structures that form pores that enable components of the intermitochondrial membrane space to be released into the cytoplasm. Released factors, such as cytochrome c, can then interact with downstream components of the pathway to activate the cellular demolitionists, the caspases. The BH3-only proteins (of which there are eight in humans: Bim, Puma, Bid, Noxa, Bad, and others) possess just a single BH domain (BH3) (3) and initiate the apoptotic cascade either by directly engaging Bax and Bak, resulting in their activation (4–9) and/or by engaging pro-survival proteins to unleash Bax/Bak molecules that are already in an active conformation (8, 10).
Interactions between pro-survival and pro-apoptotic proteins are mediated by the BH3 domain on the pro-apoptotic protein (Bax/Bak or BH3-only) binding into a large hydrophobic groove on the pro-survival protein (11–17). The BH3 domains of Bax and Bak also mediate homodimerization (and possibly heterodimerization) between Bax and Bak molecules (18–22). These dimers are then believed to assemble into higher order oligomers required for mitochondrial pore formation (23, 24). The BH3 domain of all pro-apoptotic proteins possess several conserved features, including four hydrophobic residues, designated positions h1–h4 (of which h2 is always a leucine), a completely conserved aspartic acid residue, and smaller amino acids at certain positions (h1 + 1 and h3 + 1) (Fig. 1A). Subtle differences in some of the conserved residues (i.e. different hydrophobic residues at h1, h3, or h4) as well as in the intervening residues contribute to the significant differences in selectivity in interactions between BH3 domains and pro-survival proteins (15, 25–29). Indeed, some BH3 sequences are promiscuous and engage all pro-survival proteins with high (low nanomolar) affinity, whereas others are more selective, only engaging subsets of pro-survival proteins avidly (30–32).
FIGURE 1.

Pro-survival BH3 domains bind pro-survival proteins poorly. A, alignment of BH3 domains from pro-apoptotic and pro-survival Bcl-2 family members. The four conserved hydrophobic residues h1–h4 are indicated (blue). Residues that distinguish pro-survival versus pro-apoptotic BH3 domains, h1 + 1 and h3, are shaded gray. Shown is killing activity of BimS chimeras with the Bcl-xL (B), Bcl-2 (C), or Bcl-w BH3 (D) domains and their h1 + 1/h3 (mt3) mutants in MEFs. E, Western blot showing relative expression of FLAG-tagged BimS and BimS chimeras expressed in Bax−/−/Bak−/− MEFs. F, killing activity of the Bcl-2 BH3 domain, BimS (positive control), or BimS4E (negative control) in MEFs co-expressing Noxa (to neutralize Mcl-1) or BimS4E as controls. Colonies were scored, and numbers were expressed as a percentage of the number observed in cells transduced with BimS4E alone. G, co-immunoprecipitation of endogenous Mcl-1 from Bax−/−/Bak−/− MEFs expressing FLAG-tagged BimS pro-survival BH3 chimeras. Western blots of immunoprecipitates (IP) and whole cell lysates (WCL) were probed with anti-Mcl-1 and anti-FLAG antibodies. H, BimS chimeras with mutant Bak BH3 domains fail to kill wild-type MEFs, unlike wild-type BimS or a chimera with the wild-type Bak BH3 sequence. Error bars, S.D. of n = 2–3 separate assays. In B–D, F, and H, colonies were scored 7 days after transduction, and numbers are expressed as a percentage of the number observed in cells transduced with an inert BimS mutant (BimS4E).
The BH3 domain on Bax and Bak is located on the α2 helix (33, 34). This packs against the core of the protein, burying all of the key residues required for engaging the binding groove of another Bax/Bak molecule or a pro-survival protein. As such, activation or inhibition of Bax and Bak involves the BH3 domain somehow being displaced (19, 20, 35). Somewhat paradoxically, pro-survival proteins adopt an overall three-dimensional structure almost identical to that of inactive Bax and Bak (33, 34, 36–39). They also possess a BH3 domain, located on the α2 helix. Interestingly, caspase cleavage has been shown to convert some pro-survival proteins (such as Bcl-xL or Bcl-2) to a pro-apoptotic form (40–42). In addition, the BCL-X and MCL-1 genes can be alternatively spliced to give rise to shorter forms (BCL-XS and MCL-1S) that either induce cell death or sensitize cells to apoptosis (43–47). Mutagenesis studies have shown that this pro-apoptotic activity is associated with the BH3 domain (44, 48–50). However, a number of studies using a variety of approaches, including direct affinity measurements, have also shown that the BH3 domains from certain pro-survival proteins (including those that are present in the pro-apoptotic splice variants and caspase cleavage products) have only weak capacity to bind the pro-survival molecule from which they were derived or to bind other pro-survival proteins or have little or no capacity to induce cell death or mitochondrial outer membrane permeabilization in some contexts (8, 44, 48, 51–53).
Although the BH3 domains from pro-apoptotic proteins have been extensively studied biochemically and structurally, the corresponding sequences from pro-survival proteins have received significantly less attention. Here, we examine the structure-function relationships associated with pro-survival protein BH3 domains, provide biochemical and functional data on how they differ from their pro-apoptotic counterparts, and report the first structural information on how they could exert a pro-apoptotic effect. Our results show that key distinguishing features of pro-survival protein BH3 domains reduce their capacity to interact with themselves or other pro-apoptotic molecules. In addition, we show that these key residues in the BH3 domain of pro-survival proteins have a critical role in stabilizing these proteins in cells.
EXPERIMENTAL PROCEDURES
Recombinant Proteins and Peptides
All recombinant Bcl-2 pro-survival proteins with N- and/or C-terminal truncations for binding studies (Bcl-2ΔC22, Bcl-xLΔC24, Bcl-w C29S/A128EΔC29, Mcl-1ΔN170ΔC23, and Bfl-1ΔC19) and the loop-deleted form of human Bcl-xL (Δ27–82ΔC24) used for crystallization were expressed and purified exactly as described previously (16, 25, 31). The Bcl-w BH3 mutant (H43G/M46L/A49I) was expressed and purified exactly as for the non-mutated form. Synthetic peptides were synthesized by Mimotopes and purified by reverse-phase HPLC to >90% purity.
Surface Plasmon Resonance Solution Competition Assay
Solution competition assays were performed using a Biacore 3000 instrument exactly as described previously (54).
Cell Lines
To generate cell lines expressing pro-survival proteins or mutants, mouse embryonic fibroblasts (MEFs) deficient in Bax and Bak (Bax−/−/Bak−/−) were transduced with retroviruses in which expression of each protein was linked to GFP expression via an internal ribosome entry site sequence in the pMIG-IRES vector. Following infection, GFP+ve cells were sorted, and protein expression was monitored by Western blotting. Cells were maintained in DME KELSO medium supplemented with 10% (v/v) fetal bovine serum, 250 μm l-asparagine, and 50 μm 2-mercaptoethanol.
Cytochrome c Release Assay
The cytochrome c assay was performed as described recently (54). Briefly, cells were permeabilized by resuspending in 20 mm HEPES, pH 7.2, 100 mm KCl, 5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 250 mm sucrose, 0.05% (w/v) digitonin (Calbiochem) supplemented with protease inhibitors (Roche Applied Science), and then incubated with peptides for 1 h at 30 °C before pelleting. The supernatant was retained and analyzed for cytochrome c release by Western blotting using an anti-cytochrome c antibody (clone 7H8.2C12; BD Biosciences).
Protein Half-life Assay
Cells (1.5 × 106) expressing FLAG-tagged pro-survival proteins or mutants were plated and grown overnight. The following day, the medium was replaced with fresh medium containing cycloheximide (50 μg/ml), MG132 (10 μm), or both. Cells were then harvested at the indicated times and lysed in Triton X-100-containing lysis buffer (20 mm Tris, pH 7.4, 135 mm NaCl, 1.5 mm MgCl2, 1 mm EDTA, 10% (v/v) glycerol, 1% (v/v) Triton X-100, supplemented with protease inhibitors (Roche Applied Science)). The lysate was then analyzed by Western blotting using an anti-FLAG antibody (Sigma). Blots were reprobed with anti-β-actin antibody (Sigma) to control for sample loading.
Co-immunoprecipitation Assays
Cells expressing FLAG-tagged BimSBH3 chimeras were lysed in the Triton-X-100-containing lysis buffer described above, and then the supernatant was removed and incubated with anti-FLAG antibody coupled to agarose (Sigma) for 2 h at 4 °C with constant mixing. The resin was then washed three times with the lysis buffer before resuspending in SDS-PAGE loading buffer and Western blotting for associated endogenous Mcl-1 that was detected using an anti-Mcl-1 antibody (Rockland).
Cell Killing Assays
For long term clonogenic survival assays, MEFs were transduced with the indicated constructs in which expression of each protein was linked to GFP or mCherry expression via an internal ribosome entry site sequence. GFP+ve or GFP+ve plus mCherry+ve cells (in cases where the effect of gene co-expression was examined) were then sorted and plated at 150 cells/well in 6-well plates and incubated for 7 days. Colonies were then stained with Coomassie Blue and counted.
Protein Crystallization
The structure of the Bcl-xL·Bcl-xL BH3 peptide complex employed a “loop-deleted” form of human Bcl-xL (Δ27–82 and without membrane anchor, residues 210–233), which forms an α1 domain-swapped dimer yet retains BH3 domain binding activity (16, 54–56). Crystals were obtained by mixing Bcl-xL with the BH3 peptide at a molar ratio of 1:1.3 and then concentrating the sample to 10 mg/ml. Crystallization trials were performed at the Bio21 Collaborative Crystallization Centre. Crystals were grown by the sitting drop method at room temperature in 40% (v/v) polyethylene glycol 400, 0.2 m calcium acetate, 0.1 m HEPES, pH 7.5. For the Bcl-w mutant BH3 structure, the protein was concentrated to 10 mg/ml, and crystals were grown by the sitting drop method at room temperature in 1 m trisodium citrate, 0.1 m imidazole, pH 8.0. Prior to cryo-cooling in liquid N2, crystals were equilibrated into cryoprotectant consisting of reservoir solution containing 15% (v/v) ethylene glycol. Crystals were mounted directly from the drop and plunge-cooled in liquid N2.
Crystal Diffraction Data Collection and Structure Determination
Diffraction data were collected at 100 K at the Australian Synchrotron MX2 beamline (Victoria, Australia) (wavelength for both structures was 0.954 Å). The diffraction data were integrated and scaled with either HKL2000 (57) for the Bcl-xL structure or XDS (58) for the Bcl-w structure. The structure was obtained by molecular replacement with PHASER (59–61) using the previous crystal structures of Bcl-xL from the BeclinBH3·Bcl-xL complex (56) (PDB entry 2P1L) with the Beclin peptide removed for the Bcl-xL structure, or a reconstructed Bcl-w monomer from PDB entry 2Y6W (62) for the Bcl-w mutant BH3 structure, as search models. Multiple rounds of building in COOT (63) and refinement in PHENIX (64) led to the final model.
RESULTS
Pro-survival Proteins Do Not Bind Their Own BH3 Domain or Those from Other Pro-survival Proteins Avidly
The BH3 sequences of pro-survival proteins have been implicated in cell killing activity, although some studies have suggested they can only engage pro-survival proteins weakly (8, 48, 50, 52). To better characterize these interactions, we determined the relative affinity (IC50) of synthetic peptides corresponding to pro-survival BH3 domains for the protein from which they were derived. In the case of Bfl-1, we tested two different BH3-like sequences (see Fig. 1A and Table 1 for details of sequences of all peptides used in this study). In one previous report, binding of a Bfl-1 BH3 domain synthetic peptide for Mcl-1 was characterized (52). However, although the sequence used in that study (referred to here as Bfl-1 BH3–2) possesses many of the features of a BH3 sequence motif, it did not correspond to the actual Bfl-1 BH3 domain (referred to here as Bfl-1 BH3–1), which is identifiable by comparison of the Bfl-1 x-ray crystal structures (PDB entries 2VM6, 3MQP, and 3I1H) with other pro-survival protein structures. Nevertheless, we tested both sequences in our binding assay.
TABLE 1.
Sequences of peptides used in this study
Mutated residues are indicated in red.
The tightest of these interactions were the Bcl-xL and Mcl-1 interactions with their cognate BH3 domains (IC50 values of 636 nm and ∼5 μm, respectively), consistent with previous reports that these proteins can bind to their own BH3 sequences (Table 2). These interactions are significantly weaker than those for pro-apoptotic BH3 domain sequences (e.g. from Bim, Bak, and Bax (Table 2) and others (30–32) that are typically in the sub-100 nm range. Very weak (IC50 > 10 μm) or no binding was observed for Bcl-2 and Bcl-w to their own BH3 sequences (Table 2). Similarly, Bfl-1 did not bind either of its two BH3-like sequences (Table 2). Interactions with non-cognate pro-survival BH3 domains were in a similar relative weak affinity range, with only Bcl-xL binding to the Bcl-2 BH3 domain (IC50 ∼1 μm), Bcl-w to the Bcl-xL BH3 domain (IC50 ∼3.5 μm), and Bfl-1 to both the Bcl-2 and Bcl-xL BH3 domains (IC50 of 257 nm and ∼2.3 μm, respectively) (Table 2). No non-cognate interactions were observed with either Bfl-1 BH3 sequence. Hence, these data suggest that pro-survival proteins in general have only weak capacity to interact with themselves or other pro-survival proteins via their BH3 domains. By contrast, BH3 domains from pro-apoptotic proteins typically bind significantly tighter and, in many cases, to a wider range of pro-survival proteins.
TABLE 2.
Relative binding affinities of native and mutant pro-survival and pro-apoptotic protein BH3 domains for pro-survival proteins measured by solution competition assays
Boldface entries highlight interactions of pro-survival proteins with their own BH3 domain. Shown are are IC50 values in nmol ± S.D. from n = 2–4 separate assays measured by solution competition. ND, not determined; in the case of Bcl-2 mt1, this was due to the peptide being only poorly soluble.
| Bcl-xL | Bcl-2 | Bcl-w | Mcl-1 | Bfl-1 | |
|---|---|---|---|---|---|
| nmol | nmol | nmol | nmol | nmol | |
| Pro-survival BH3 domain | |||||
| Bcl-xL | |||||
| Wild type | 636 ± 63 | >10,000 | 3480 ± 102 | >10,000 | 2370 ± 138 |
| mt1 (Kh1 + 1A) | 48 ± 26 | 816 ± 168 | 146 ± 39 | 363 ± 1 | 29 ± 10 |
| mt2 (Ah3I) | 7110 ± 448 | >10,000 | 8000 ± 185 | 647 ± 100 | 3250 ± 213 |
| mt3 (Kh1 + 1A/Ah3I) | 48 ± 1 | 406 ± 97 | 45 ± 4 | 24 ± 3 | 7.6 ± 3 |
| Bcl-2 | |||||
| Wild type | 1130 ± 137 | >10,000 | >10,000 | >10,000 | 257 ± 3 |
| mt1 (Hh1 + 1A) | ND | ND | ND | ND | ND |
| mt2 (Ah3I) | 1700 ± 67 | 6500 ± 500 | >10,000 | 142 ± 2 | 9.3 ± 4 |
| mt3 (Hh1 + 1A/Ah3I) | 186 ± 16 | 1680 ± 76 | 2140 ± 31 | 70 ± 9 | 16 ± 2 |
| Bcl-w | |||||
| Wild type | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| mt1 (Hh1 + 1G) | 152 ± 13 | 94 ± 3 | 128 ± 10 | >10,000 | 182 ± 3 |
| mt2 (Ah3I) | 5900 ± 16 | 4350 ± 30 | 1970 ± 182 | 1150 ± 32 | 679 ± 130 |
| mt3 (Hh1 + 1G/Ah3I) | 125 ± 10 | 50 ± 8 | 46 ± 6 | 509 ± 18 | 30 ± 14 |
| Mcl-1 | |||||
| Wild type | >10,000 | >10,000 | >10,000 | 5300 ± 1210 | >10,000 |
| mt1 (Lh1 + 1A) | >10,000 | >10,000 | >10,000 | >10,000 | ND |
| mt2 (Vh3I) | >10,000 | >10,000 | >10,000 | 3250 ± 116 | ND |
| mt4 (Ah1L/Lh1 + 1A) | >10,000 | >10,000 | >10,000 | 3780 ± 222 | 303 ± 9 |
| mt5 (Ah1L/Lh1 + 1A/Vh3I) | >10,000 | >10,000 | >10,000 | 664 ± 49 | 150 ± 19 |
| Bfl-1 | |||||
| Wild type | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| BH3-1 | |||||
| mt3 (Sh1 + 1A/Vh3I) | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| mt4 (Sh1 + 1A/Vh3I/Dmut) | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| Bfl-1 BH3–2 | |||||
| Wild type | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 |
| Pro-apoptotic | |||||
| Bim | |||||
| Wild type | 16 ± 7 | 7.7 ± 1 | 18 ± 2 | 15 ± 3 | 7.4 ± 1 |
| Bak | |||||
| Wild type | 53 ± 4 | 2960 ± 604 | 929 ± 80 | 20 ± 1 | 5.2 ± 1 |
| mt1 (Gh1 + 1K) | >10,000 | ND | ND | 800 ± 73 | ND |
| mt2 (Ih1 + 1A) | >10,000 | ND | ND | >10,000 | ND |
| Bax | |||||
| Wild type | 124 ± 8 | 92 ± 1 | 33 ± 2 | 76 ± 5 | 22 ± 1 |
The h1 + 1 and h3 Residues of Pro-survival BH3 Domains Account for Their Weak Affinity
To determine whether there were critical features of pro-survival BH3 domains that distinguish them from their pro-apoptotic counterparts, we compared sequence alignments of pro-survival and pro-apoptotic (BH3-only and Bax/Bak) BH3 domains. Based on previous mutagenesis studies (12, 14, 19, 25), we predicted that two major differences at the h1 + 1 and h3 positions (Fig. 1A) in most of the pro-survival versus pro-apoptotic sequences could impact their binding affinity for pro-survival proteins. In pro-survival proteins there is a large residue (histidine in Bcl-2 and Bcl-w, lysine in Bcl-xL, leucine in Mcl-1) at the h1 + 1 position, which is normally occupied by a small residue (typically glycine or alanine) in pro-apoptotic BH3 sequences. At the h3 position, the situation is reversed; in pro-survival proteins, there is always a smaller residue (Bcl-xL, Bcl-2, and Bcl-w have an alanine; Bfl-1 and Mcl-1 have a valine), whereas pro-apoptotic BH3 sequences generally all have a larger hydrophobic residue here (although Bid, a potent pro-apoptotic protein, also has a valine at this position). There are also some sequence-specific features that could be detrimental to binding for certain pro-survival BH3 sequences. For example, unlike nearly all pro-apoptotic BH3 domains, Mcl-1 has an alanine and Bfl-1 has a threonine at h1; this residue is typically a large hydrophobic residue in nearly every other BH3 sequence. In the structurally identified Bfl-1 BH3 domain (Bfl-1 BH3–1), the aspartic acid residue conserved in every other BH3 sequence is a phenylalanine. This residue has previously been shown in a number of studies to be very important for Bcl-2 protein interactions (12, 14, 19, 25).
To test whether the h1 + 1 and h3 residues account for the weak affinity of pro-survival BH3 domains for themselves or other pro-survival proteins, we measured the binding of mutant peptides in which h1 + 1 was switched to an alanine, h3 was switched to isoleucine (a residue commonly found at this position in pro-apoptotic BH3 domains), or both residues were switched. (For clarity, in the figures we have referred to the h1 + 1 mutation as mt1, the h3 mutation as mt2, and the h1 + 1/h3 combination mutant as mt3; see Table 1 for further details of sequences.) In the case of the Bcl-2, Bcl-xL, and Bcl-w BH3 domains, the binding affinities for most pro-survival proteins increased with either mutation, although the h1 + 1 (mt1) mutation usually had a greater impact, whereas combination mutants (mt3) further increased binding, and in some cases, these affinities approached those determined for BH3 domains from potent pro-apoptotic proteins (Table 2) (30–32).
The Bfl-1 and Mcl-1 BH3 domains were different. In the case of Mcl-1, the h1 + 1 (mt1) or h3 (mt2) substitutions on their own had little impact or were detrimental. However, when we switched the unusual alanine at h1 and leucine at h1 + 1 (mt4, which on its own had little effect) together with exchanging the h3 valine for isoleucine (mt5), a more significant improvement in binding was observed for Mcl-1, although not for other pro-survival proteins (Table 2). These results are consistent with the crystal structure of Mcl-1 bound to its own (stapled) BH3 peptide (52). The Bfl-1 BH3 was similar to the Mcl-1 BH3 in that no improvements in binding for any pro-survival protein were observed following mutation of the h1 + 1 plus h3 residues (mt3). Additional mutation of the unusual phenylalanine (mt4) to the highly conserved aspartate generally found at this position also had no impact, even in the context of the h1 + 1 and h3 swaps (Table 2).
Hence, the physiochemical properties of the h1 + 1 and h3 residues are critical for pro-survival protein-BH3 domain interactions and largely account for the low affinity of the Bcl-2, Bcl-xL, and Bcl-w BH3 sequences. However, the unusual nature of these residues in the Mcl-1 and Bfl-1 BH3 domains does not completely account for their weak affinity; hence, other residues that impair binding must also be involved.
Mutation of h1 + 1 and h3 Residues in Pro-survival BH3 Domains Increases Their Cell Killing Activity
To gain further insights into the specificity of pro-survival protein BH3 domains as well as their cell-killing potential, we next examined their behavior in MEFs of different genotypes (wild type, Bcl-x−/−, Mcl-1−/−, and Bax−/−/Bak−/−). Here, we replaced the BH3 domain of the BH3-only protein BimS with different pro-survival BH3 domain sequences or the mutant in which both h1 + 1 and h3 residues (mt3) were replaced with residues found in pro-apoptotic BH3 sequences. Previous studies have shown that similar BimS chimeras adopt the binding profile of the replacement BH3 sequence (31). We focused on the Bcl-xL, Bcl-2, and Bcl-w sequences because specificity studies have previously been reported for the Mcl-1 BH3 domain (44, 52). In long term (7-day) assays, the wild-type BimS sequence was able to potently suppress colony formation of all MEF cell lines examined except for the Bax−/−/Bak−/− cells, consistent with its ability to engage all pro-survival proteins with high affinity and the fact that it also requires Bax and/or Bak for its cell killing activity. By contrast, none of the BimS chimeras with the wild-type pro-survival BH3 sequences had any activity on any cell line except for the Bcl-2 BH3 chimera that killed Mcl-1−/− MEFs (Fig. 1, B–D). Because neutralization of both Bcl-xL and Mcl-1 is required for MEF cell killing (10), this result agrees with the binding data showing that the Bcl-2 BH3 domain only targets Bcl-xL (Table 2). In further agreement with the requirement for neutralization of multiple pro-survival family members for MEF cell killing, the BimS Bcl-xL, Bcl-2, and Bcl-w BH3 chimeras with the mutated BH3 domain were significantly more active across the range of cell lines, consistent with their increased affinity for multiple pro-survival protein targets (Table 2). Notably, all BimS BH3 chimeras expressed at levels equivalent to wild-type BimS except for the Bcl-xL BH3 chimera, which expressed very poorly (Fig. 1E); hence, we cannot draw any conclusion about the pro-death activity of this BH3 domain from these experiments. This weak expression, however, explains why it failed to impact Mcl-1−/− MEF survival despite its possessing a binding affinity for Bcl-xL similar to that of the Bcl-2 BH3 domain, which displayed potent activity on the same cells.
To further examine the requirement for Bak and/or Bax in the activity of the wild-type Bcl-2 BH3 sequence, we generated wild-type, Bax−/−, Bak−/−, or Bax−/−/Bak−/− MEF cell lines that overexpress Noxa to neutralize Mcl-1 or an inert Bim mutant (BimS4E) as a control and then co-expressed the BimS Bcl-2 BH3 chimera. Consistent with the data above showing that this chimera could only kill Mcl-1−/− MEFs, wild-type cells only died when Noxa was present (Fig. 1F). Moreover, this activity is essentially entirely associated with Bak rather than Bax because the Bax−/− cells died to a much greater extent than those deficient in Bak. Because the Bcl-2 BH3 domain did not show any appreciable binding to Bcl-2 itself, these data agree with published data showing that neutralization of Bcl-2 is required for Bax-mediated cell death, whereas Bak only requires inhibition of Mcl-1 and Bcl-xL (10, 65).
The relatively weaker activity of the Bcl-w mutant chimera on wild-type and Bcl-x−/− cells (but not Mcl-1−/− cells) is consistent with the somewhat weaker binding affinity of this mutant BH3 domain to Mcl-1 compared with the corresponding Bcl-xL and Bcl-2 BH3 mutants (Table 2). This binding selectivity was confirmed in co-immunoprecipitation experiments where the Mcl-1 binding capability of the BimS chimeras reflected the relative affinities of the BH3 peptides (i.e. wild-type Bim BH3 > Bcl-xL BH3 mutant > Bcl-2 BH3 mutant) (Fig. 1G), but the Bcl-w BH3 mutant chimera was not found to be associated with any detectable Mcl-1, as also seen with the wild-type Bcl-2, Bcl-xL, and Bcl-w BH3 domain chimeras.
These functional data strongly support the binding data for the pro-survival protein BH3 domains showing that they generally bind weakly or highly selectively to pro-survival proteins, and this is probably due to the unusual features of their h1 + 1 and h3 residues.
To further support this notion, we next examined pro-apoptotic BH3 sequences. Our data would predict that the opposite mutations at h1 + 1 and h3 in this context should reduce pro-survival protein binding and impair cell killing activity. Indeed, switching the small h1 + 1 residue (Gly75) of the pro-apoptotic Bak BH3 domain to a larger residue (lysine, mt1), as found in Bcl-xL, or switching h3 (Ile81) to alanine (mt2) had a detrimental effect on binding of Bak BH3 synthetic peptides to both Mcl-1 and Bcl-xL, the two key regulators of Bak (Table 2) (10). Similarly, a BimS chimera with its BH3 domain switched for the wild-type Bak BH3 sequence was a potent killer of MEFs; however, BimSBak chimeras with the same h1 + 1 (G75K) and h3 (I81A) mutations were inert (Fig. 1H). Combined, these data confirm our hypothesis that the h1 + 1 and h3 residues of BH3 domains are critical for interactions with pro-survival proteins and, hence, cell killing activity.
Cytochrome c Release Induction by the Bcl-xL BH3 Domain Is Enhanced by h1 + 1/h3 Mutations
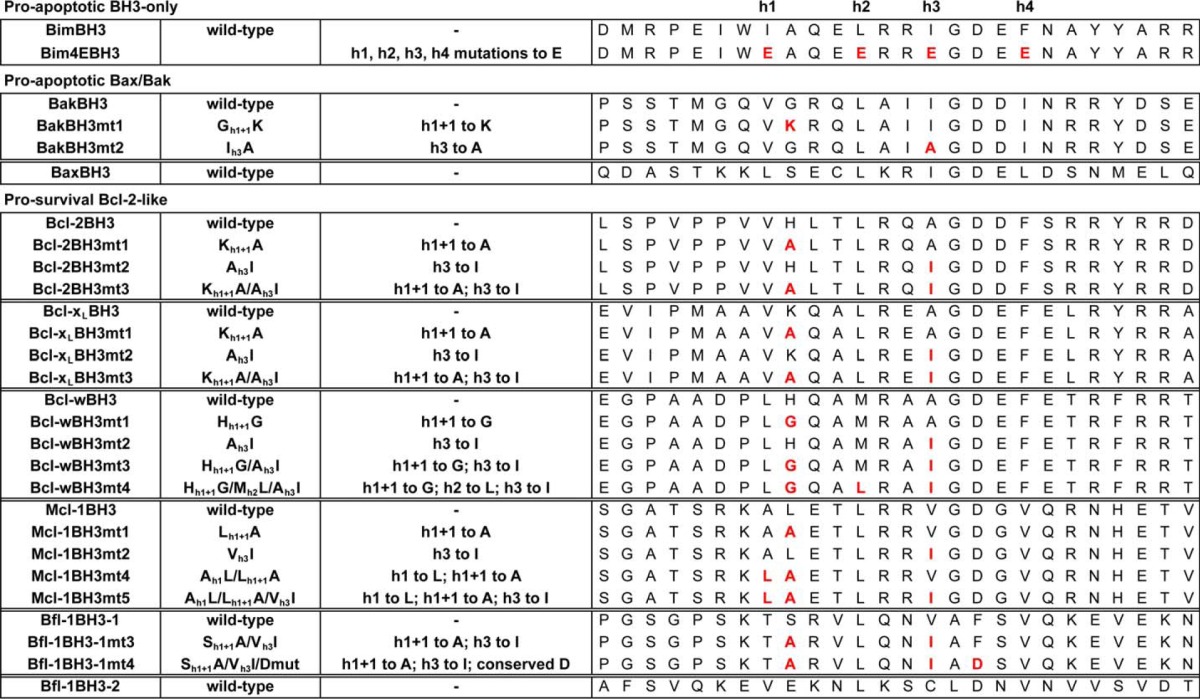
The failure of the BimS Bcl-xL BH3 chimera to express well prevented us gaining further insights into the binding specificity of that BH3 sequence in a cellular environment. Because most of the published work on the cell killing activity of pro-survival proteins has been on Bcl-x with both the Bcl-xS splice variant and the caspase-cleaved form of Bcl-xL, we examined the capacity of synthetic peptides corresponding to this sequence to elicit cytochrome c release from permeabilized MEFs of different genotypes. This approach allows the ligand to be precisely titrated; hence, the relative potencies of different sequences can be more accurately established than when using transduction/overexpression methods. Here, the Bcl-xL BH3 domain peptide demonstrated no capacity to elicit cytochrome c release when tested at a high concentration (10 μm) in any cell line (Fig. 2). In contrast, Bim BH3 was effective at the same concentration in all cell lines except for Bax−/−/Bak−/− MEFs, consistent with its ability to directly activate Bax and Bak and to bind all pro-survival proteins as well as the essential role of Bax/Bak in mitochondrial outer membrane permeabilization. Because the transmembrane domain of Bcl-xS is required for its pro-apoptotic activity (49, 50), we tested the Bcl-xL BH3 peptide fused to a short mitochondrial targeting sequence (66) and now observed cytochrome c release at 10 μm in Mcl-1−/− MEFs but not wild-type or Bcl-x−/− cells. Due to the requirement for neutralization of Bcl-xL and Mcl-1 to induce MEF cell apoptosis, these data support the binding affinity data showing that the Bcl-xL BH3 domain targets Bcl-xL but not Mcl-1 and that membrane localization increases pro-apoptotic activity, probably by increasing the local concentration at the mitochondria, as previously observed with other BH3 sequences (67). A similarly mitochondrially targeted Bim BH3 peptide was significantly more potent (>100-fold) on all cell lines (Fig. 2), consistent with the higher affinity of Bim BH3 for all pro-survival proteins. Finally, the Bcl-xL BH3 h1 + 1/h3 mutant (mt3) sequence fused to the mitochondrial targeting domain elicited cytochrome c release from Mcl-1−/−, Bcl-x−/−, and wild-type MEFs at 1 μm, but not Bax−/−/Bak−/− MEFs, consistent with the increased affinity of the mutant sequence for all pro-survival proteins. Combined, these data further support the binding data showing that the unusual h1 + 1 and h3 residues of the Bcl-xL BH3 domain impair its binding affinity and reduce its selectivity for pro-survival protein binding partners.
FIGURE 2.
Cytochrome c release assay with Bcl-xL BH3 peptides. Permeabilized MEFs were treated with the indicated concentrations of synthetic peptides corresponding to the wild-type Bcl-xL BH3 domain (wt), the wild-type Bcl-xL BH3 domain fused to a mitochondrial targeting sequence (wt-MTS), or the Bcl-xL h1 + 1/h3 mutant BH3 domain fused to a mitochondrial targeting sequence (mt3-MTS) (left) or the wild-type Bim BH3 domain (wt) or the wild-type Bim BH3 domain fused to a mitochondrial targeting sequence (wt-MTS) (right). The presence of cytochrome c released into the soluble fraction following peptide treatment was determined by Western blotting (WB) with an anti-cytochrome c antibody.
Mechanism of Cell Killing by the Caspase-cleaved, N-terminally Deleted Form of Bcl-2
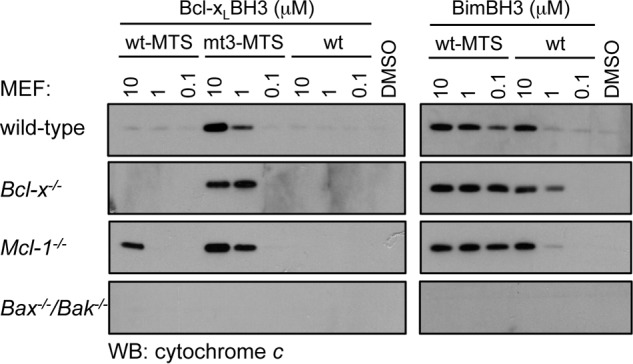
Previous studies have shown that caspase cleavage of Bcl-2 converts it to a pro-apoptotic form (ΔN34) that is dependent on its BH3 domain for cell killing (42). Our binding (Table 2) and functional data (Fig. 1C) would predict that this is probably due to its ability to inhibit Bcl-xL. We therefore tested Bcl-2ΔN34 in similar assays to those used for the BimSBcl-2 BH3 chimeras (Fig. 1C), and consistent with those data, we only observed an effect on Mcl-1−/− MEF survival but no other MEFs tested (Fig. 3). These data confirm that both the BH3 peptide binding and BimSBH3 chimera data reflect the activity of this pro-survival BH3 domain in its native context.
FIGURE 3.
Killing activity of Bcl-2ΔN34 in MEFs. Bcl-2ΔN34 only kills MEFs in the absence of Mcl-1. Colonies were scored 7 days after transduction, and numbers were expressed as a percentage of the number observed in cells transduced with the vector alone. Error bars, S.D. of n = 2–3 separate assays.
Structural Basis for the Weak Affinity of Pro-survival BH3 Domains for Pro-survival Proteins
To gain structural insights into why pro-survival BH3 domains bind pro-survival proteins weakly compared with pro-apoptotic BH3 sequences, we obtained x-ray crystallographic data for the complex of Bcl-xL with its own BH3 domain. This interaction is the tightest of the pro-survival BH3 complexes (IC50 of 636 nm; Table 2) and hence represented the most stable complex for structural studies. It is also a biologically relevant interaction because the bcl-x splice variant and caspase-cleaved forms of Bcl-xL are pro-apoptotic, and this activity is associated with the Bcl-xL BH3 domain (48, 49). Moreover, because the h1 + 1 residue of pro-apoptotic BH3 ligands projects into a tightly constrained region of the canonical binding groove, it was of particular interest to determine how large side chains (such as the lysine in the Bcl-xL BH3 domain) could be accommodated or whether the pro-apoptotic activity of this sequence was associated with it binding at an alternative, distal site.
We were able to obtain crystals of this complex and solved the structure at 2.7 Å resolution (Table 3, PDB entry 4CIN). To aid crystallization we used a “loop deletion” construct of Bcl-xL, which has the large unstructured loop between the α1 and α2 helices removed and which has been used in several previous structural studies on Bcl-xL BH3 ligand binding (16, 54, 56). This construct forms a domain swap dimer involving the α1 helix but has no impact on the canonical ligand binding groove.
TABLE 3.
Data collection and refinement statistics
Values in parentheses represent statistics for the highest resolution shell. Ramachandran statistics are as follows: Bcl-xL·Bcl-xL BH3 structure (PDB code 4CIN), 94.77% in preferred regions, 4.9% in allowed regions, 0.33% in outlier regions; Bcl-w BH3 mutant structure (PDB code 4CIM), 97.35% in preferred regions, 1.99% in allowed regions, 0.66% in outlier regions.
| Bcl-xL·Bcl-xL BH3 (PDB code 4CIN) | Bcl-w BH3 mutant (PDB code 4CIM) | |
|---|---|---|
| Data collection | ||
| Space group | P3121 | P21 |
| Cell dimensions | ||
| a, b, c (Å) | 73.26, 73.26, 136.86 | 41.92, 60.73, 100.4 |
| α, β, γ (degrees) | 90.0, 90.0, 120.0 | 90, 105.85, 90.0 |
| Resolution (Å) | 50.00-2.70 (2.80-2.70) | 50.00-1.50 (1.59-1.50) |
| Rsym or Rmerge | 0.093 (0.939) | 0.072 (0.849) |
| I/σI | 10.90 (1.03) | 9.93 (1.57) |
| Completeness (%) | 98.0 (97.7) | 99.4 (96.5) |
| Redundancy | 2.90 (3.00) | 3.78 (3.64) |
| Refinement | ||
| Resolution (Å) | 46.525-2.69 (2.964-2.69) | 44.229-1.50 (1.53-1.50) |
| No. of reflections | 12,063 (1177) | 51,625 (4996) |
| Rwork/Rfree | 0.193/0.271 (26.7/37.0) | 0.186/0.210 (31.2/34.9) |
| No. of atoms | 2619 | 2938 |
| Protein | 2214 | 2345 |
| Ligand/ion | 329 | 314 |
| Water | 76 | 279 |
| B-Factors | 47.4 | 26.5 |
| Protein | 47.4 | 24.6 |
| Ligand/ion | 54.8 | 48.3 |
| Water | 42.1 | 39.6 |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.008 | 0.007 |
| Bond angles (degrees) | 1.15 | 1.01 |
The crystallographic asymmetric unit contains a whole dimer, offering two independent views of how the ligand engages its target. In this present structure, the general features of both views of how the Bcl-xL BH3 domain engages Bcl-xL are similar, and it occurs via the canonical binding groove, like the binding of pro-apoptotic BH3 domains, such as Bim BH3 (Fig. 4A). In one of the molecules (molecule B), however, there are some regions (e.g. α3 helix residues 105–113 of Bcl-xL and the N terminus of the Bcl-xL BH3 ligand) where the electron density was very poor, so we have restricted our analysis to molecule A, where the entire structure is more clearly resolved, including easily discernable density for the h1 + 1 residue of the ligand (Fig. 4B). In this molecule, this unusual lysine residue extends from the buried face of the BH3 helix toward the helix α4 and out of the groove (Fig. 4, A and C). The N-terminal end of the BH3 helix up to the h1 + 1 lysine residue of the ligand is also forced out of the groove (Fig. 4C), probably as a consequence of having to accommodate the large lysine residue in a tightly restricted region of the binding site. This movement of the ligand out of the groove, in turn, leads to the h1 hydrophobic residue (a valine) preceding the lysine residue no longer being buried, as occurs in most other BH3 complexes (Fig. 4D). Hence, although the large lysine residue at h1 + 1 can be accommodated, this appears to significantly disrupt interactions between the BH3 domain and the canonical binding groove, probably accounting for the reduced binding affinity of the interaction compared with pro-apoptotic BH3 domains.
FIGURE 4.
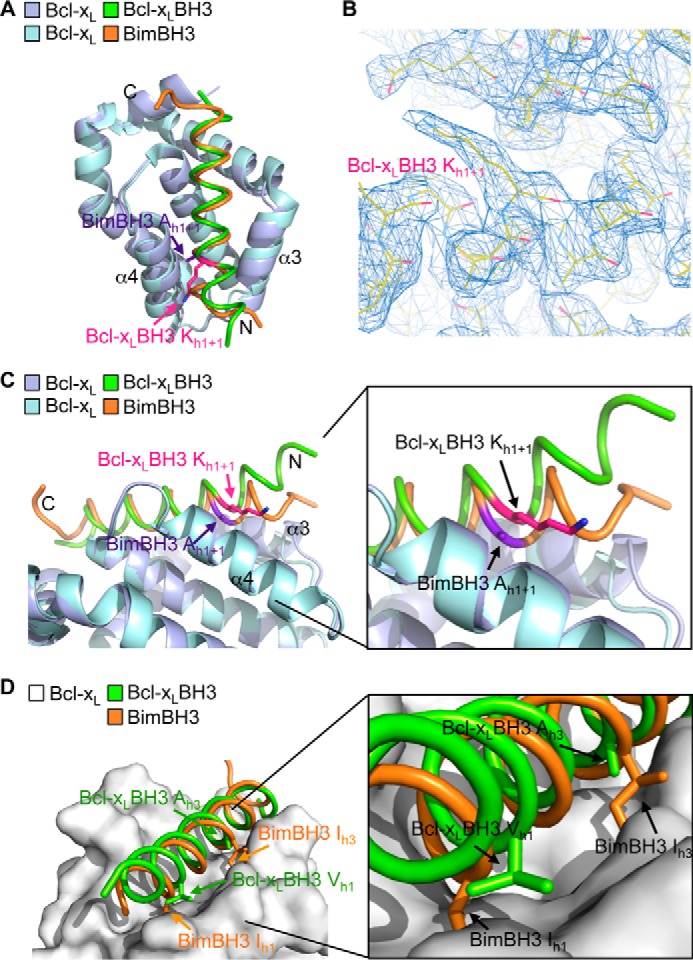
Crystal structure of Bcl-xL bound to its own BH3 domain. A, overlay of the crystal structures of Bcl-xL bound to its own BH3 domain (Bcl-xL molecule A (mauve) and Bcl-xL BH3 (green)) and bound to Bim BH3 (Bcl-xL (light blue) and Bim BH3 (orange)). B, region of the electron density map showing the lysine at h1 + 1 in the Bcl-xL BH3 peptide ligand. C, the N-terminal end of the Bcl-xL BH3 peptide ligand (green) is displaced out of the binding groove relative to the Bim BH3 domain (orange). D, the h1 hydrophobic residue in the Bcl-xL BH3 domain (green) is not buried in the binding groove, unlike the corresponding residue in Bim (orange). Similarly, hydrophobic contacts between Bcl-xL and the h3 residue of the Bcl-xL BH3 ligand are greatly reduced due to the small alanine residue here compared with the larger hydrophobic residue found in all pro-apoptotic BH3 domains (such as the isoleucine in Bim).
Mutations in the BH3 Domain Influence Cellular Pro-survival Protein Stability
Our data on the unusual residues found in the BH3 domains of pro-survival proteins could account for their inability to readily form homodimers (21) and, therefore, why they are unable to act like Bax and Bak to permeabilize the outer mitochondrial membrane. Our data also suggest that pro-survival proteins could be converted to Bax/Bak-like molecules if their BH3 domains were mutated at the h1 + 1 and h3 positions to enhance potential homodimerization. We therefore tested the ability of h1 + 1, h3, or h1 + 1/h3 mutants of Bcl-xL, Bcl-2, and Bcl-w to kill MEFs deficient in both Bax and Bak. No activity was observed for any mutant expressed on its own or in combination with BimS (to neutralize endogenous pro-survival protein activity), unlike Bak when tested similarly (Fig. 5A). Hence, mutations that make the BH3 and groove compatible for association were not sufficient to convert pro-survival proteins to pro-apoptotic proteins.
FIGURE 5.

BH3 domain mutations destabilize pro-survival proteins. A, pro-survival proteins with “pro-apoptotic” BH3 domains do not kill Bax−/−/Bak−/− MEFs, even when co-expressed with BimS. Colonies were scored, and numbers were expressed as a percentage of the number observed in cells transduced with the pMIG vector only. Error bars, S.D. of n = 3 assays. B, mutation of the h1 + 1, h3 or both residues in pro-survival proteins significantly impacts steady-state levels of FLAG-tagged pro-survival proteins in Bax−/−/Bak−/−MEFs. C, the reduced levels of pro-survival BH3 mutants are due to their shorter half-life as a result of them being more rapidly degraded by the proteasome following cycloheximide (CHX) treatment. In B and C, Western blots (WB) of equivalent cell lysates were probed with anti-FLAG antibody and then reprobed with anti-β-actin antibody to control for sample loading. The asterisk in C indicates a nonspecific band that becomes apparent due to the longer exposure of this blot.
Among the possible explanations for these results are the following: (i) the mutant pro-survival proteins are unable to unlatch (21, 22); (ii) the affinities of the mutant pro-survival proteins for their own (mutated) BH3 sequences are too low; and (iii) expression levels of the mutant proteins are too low to display a killing effect. Interestingly, when we examined the expression of each of the pro-survival protein mutants, their steady-state levels were significantly reduced compared with the wild-type protein, with the h1 + 1 substitution generally having a greater effect than the h3 mutant and the combination of h1 + 1 plus h3 mutations showing a further reduction in expression, especially for Bcl-xL and Bcl-w (Fig. 5B). Because none of the constructs could kill cells, we could exclude the possibility that there was selective pressure for low expression in the cells to prevent them from dying. We therefore examined whether the lower protein levels observed for the mutants was due to a general expression defect or to the proteins being destabilized/degraded by monitoring their half-lives following treatment of the cells with cycloheximide to inhibit protein synthesis (Fig. 5C). In all cases, mutation of either h1 + 1 or h3 significantly reduced cellular half-life for each pro-survival protein, with the h1 + 1 mutation again having the greater impact, although less than the combination of both substitutions. This rapid degradation of the protein was mediated by the proteasome because cellular levels were significantly higher in the presence of the proteasome inhibitor MG132 (Fig. 5C). Hence, not only do pro-survival protein BH3 domains influence their capacity to engage themselves, but the stability of their structures is also dependent on the physicochemical properties of the residues that distinguish them from pro-apoptotic BH3 sequences.
Multiple Interactions between α1 and α2 Stabilize the Bcl-xL Structure
Our data showing low cellular levels of the h1 + 1 mutants of pro-survival proteins suggested that contacts between this residue and residues on the α1 helix might be important for protein stability. In fact, in all pro-survival proteins, there are similar hydrogen bonds or hydrophobic contacts involving this residue (Fig. 6). We therefore further examined the importance of this and other interactions between the α1 and α2 helices on pro-survival protein stability. Here, we focused on Bcl-xL because removal of the N-terminal domain following caspase cleavage (40) disengages all contacts between the α1 and α2 helices; hence, the importance of these interactions has a physiologically relevant context.
FIGURE 6.
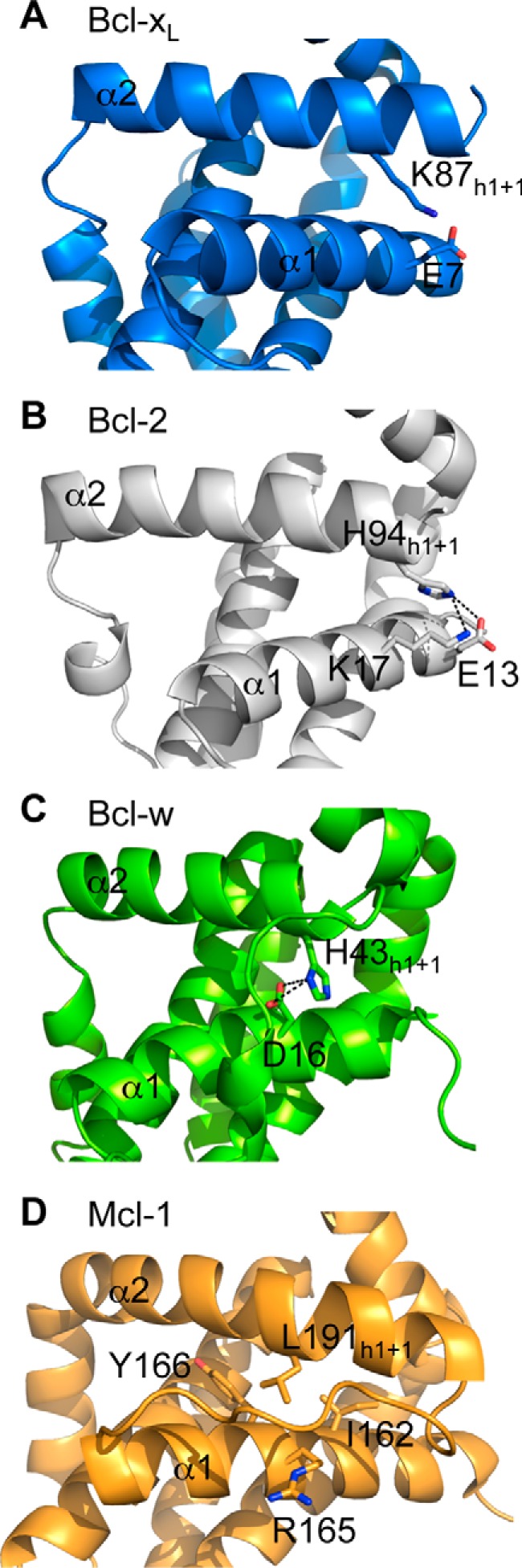
Structures showing interactions between the h1 + 1 residue of the BH3 domain on pro-survival proteins and the α1 helix. In Bcl-xL (PDB code 1PQ0) (A), Bcl-2 (PDB code 2XA0) (B), and Bcl-w (PDB code 1O0L) (C), the h1 + 1 residue makes hydrogen bond/salt bridges with residues on the α1 helix, whereas in Mcl-1 (PDB code 1WSX) (D), the h1 + 1 leucine makes hydrophobic contacts.
The crystal structure of Bcl-xL reveals a network of several interactions between α1 and α2 helices (Fig. 7A). The h1 + 1 residue, Lys87, hydrogen-bonds to Glu7 on α1. Accordingly, an E7A mutation also leads to lower steady state levels of Bcl-xL (Fig. 7B) and reduced Bcl-xL half-life (Fig. 7C), although the effect is significantly less pronounced compared with the K87A mutation (mt1) (Fig. 5, B and C). Inspection of the crystal structure of Bcl-xL suggested that Lys87 could also potentially interact with Asp11 if the interaction with Glu7 was lost (e.g. through the E7A mutation). Indeed, a D11A mutation had an impact similar to that of the E7A mutation on Bcl-xL steady state levels (Fig. 7B) and half-life (Fig. 7C), although this could also reflect the loss of an additional interaction with the α2 (BH3) helix, the Asp11-Arg91 salt bridge (Fig. 7A). Perhaps not surprisingly, the R91A mutation had an effect similar to that of the D11A mutation, but an E7A/D11A combination mutation was more disruptive than the K87A mutation alone (Fig. 7B), consistent with the importance of multiple interactions in this region maintaining protein stability. In addition, Tyr15 makes similarly important contacts; it hydrogen-bonds to Asp95, although the effect of a Y15A mutation was less dramatic compared with D95A (Fig. 7B), where steady state levels were even lower than observed with the K87A mutant. Interestingly, the Asp95 mutation did not have such a severe effect on protein half-life; hence, the low steady state levels perhaps reflect some alternative mechanism involved in controlling Bcl-xL levels.
FIGURE 7.

Interactions between α1 and α2 helices stabilize Bcl-xL. A, close-up view of the Bcl-xL crystal structure (PDB code 1PQ0) highlighting the network of interactions between residues on the α1 and α2 helices. B, mutation of residues on α1 or α2 helices impacts steady-state levels of Bcl-xL in MEFs. A long (dark) exposure is provided so that expression of the E7A/D11A mutant can be seen. C, the reduced levels of the mutants are due to their shorter half-life as they are more rapidly degraded by the proteasome following cycloheximide (CHX) treatment. In B and C, Western blots (WB) of equivalent cell lysates were probed with anti-FLAG antibody and then reprobed with anti-β-actin antibody to control for sample loading. The asterisk in C indicates a nonspecific band that becomes apparent due to the longer exposure of this blot. D, Western blot of lysates of cells expressing full-length FLAG-tagged Bcl-xL and FLAG-tagged Bcl-xLΔN61. The asterisk indicates a nonspecific band.
Combined, the above mutagenesis data suggest that the caspase-cleaved (ΔN61) form of Bcl-xL must be very unstable and poorly expressed due to the loss of multiple interactions between the α1 and α2 helices. Indeed, we observed dramatically reduced levels of this protein compared with wild-type Bcl-xL when expressed in Bax/Bak-deficient MEFs (Fig. 7D). This suggests that caspase cleavage could also serve to reduce Bcl-xL levels in cells and thereby reduce its pro-survival effect rather than converting it into a pro-apoptotic molecule.
Crystal Structure of a Destabilized Bcl-w BH3 Mutant
As demonstrated above, the h1 + 1 residue is important for pro-survival protein stability through interactions with the α1 helix (Figs. 5–7). The h3 residue is also important for pro-survival protein stability (Fig. 5) probably because the methyl group(s) of the h3 alanine (in the case of Bcl-xL, Bcl-2, and Bcl-2) or valine (in the case of Mcl-1) project toward the core of the protein. We therefore attempted to obtain further structural data on how the h1 + 1 and h3 residues contribute to pro-survival protein stability and why switching them to a pro-apoptotic-type sequence leads to protein destabilization and degradation. Although other pro-survival proteins were screened, we were only able to obtain crystals of Bcl-w with its BH3 domain mutated: His to Gly at h1 + 1 and Ala to Ile at h3. Bcl-w is also unusual in that it has a methionine at h2 in its BH3 domain; this was therefore switched to the canonical leucine typically found at this position (Bcl-w mt4; Table 1).
Following purification of the Bcl-w mt 4 protein by gel filtration chromatography, we observed an elution profile different from that for the wild-type protein (62). In the Bcl-w mt4 profile, there was no monomeric protein apparent; instead, all of the protein eluted at retention times consistent with a tetramer and dimer. Despite the relatively poor resolution of these peaks, we attempted to crystallize both forms and were able to obtain crystals for the dimer and solved the structure at 1.5 Å resolution (Table 3; PDB entry 4CIM).
Unexpectedly, the structure shows a dimer of two mutant Bcl-w molecules, each bound to an extended peptide corresponding to the α2 helix containing the mutated Bcl-w BH3 domain (residues 38–58) plus part of the α1-α2 connecting loop (Fig. 8, A and B). This demonstrates that in a subpopulation of Bcl-w mt4 molecules, the N terminus of the protein has undergone extensive proteolysis, which was apparent in SDS-PAGE analysis of the sample (Fig. 8C). This is unlike wild-type Bcl-w, which migrates predominantly as a single band following identical expression and purification protocols (Fig. 8C). The dimer interface is unusual in that it is formed by the excised mt4 BH3 peptides facing each other in an anti-parallel arrangement with reciprocal salt bridges forming between Arg47 on one peptide and Asp51 on the other (Fig. 8A). We suspect that this dimer is a crystallographic artifact because it could not be recapitulated by mixing purified monomeric wild-type Bcl-w with a synthetic peptide corresponding to the ligand seen in the structure (data not shown). Nevertheless, the structure is very informative because it shows that, consistent with the mammalian cell expression data, the Bcl-w protein is destabilized when its BH3 domain is switched with one that looks more like a pro-apoptotic BH3 sequence. This destabilization allows the BH3 domain to be excised away from the core of the protein. We suspect that binding of an excised α2 helix to an intact/uncleaved mutant Bcl-w then stabilizes that protein against proteolysis. Otherwise, the overall structure of the uncleaved Bcl-w mt4 in the crystal structure is similar to the native Bcl-w (37, 68), although with some differences. Most notably, the α3 helix is very disordered in the mutant protein (although with clearly visible electron density) (Fig. 8D), and the α2 helix is also extended by one turn in the mutant (Fig. 8D). Because this structure is the first complex of Bcl-w with a bound ligand, there are no comparator structures to establish whether any of the other differences observed are due to the mutated BH3 domain, although the general features of the interaction with the BH3 peptide are very similar to those seen, for example, in Bcl-xL or Mcl-1 complexes with BH3 peptides (11–14, 36).
FIGURE 8.
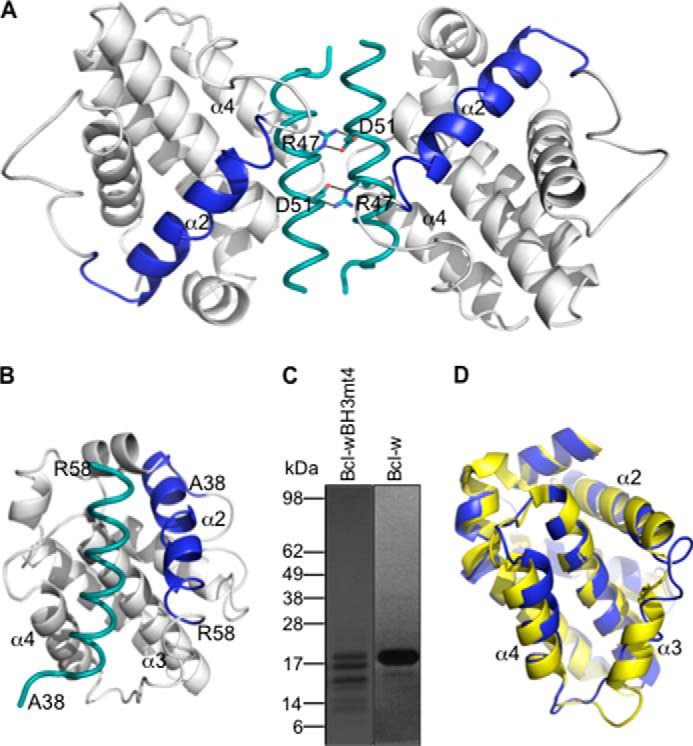
Crystal structure of Bcl-w with a “pro-apoptotic” BH3 domain. A, crystal structure of the Bcl-w dimer formed by reciprocal salt bridges between Arg-47 and Asp-51 on the excised Bcl-w BH3 domains (teal). The position of the excised Bcl-w BH3 domain (residues 38–58) within the intact Bcl-w protein is shown in blue. B, view of the excised BH3 domain (teal) within the canonical binding groove formed by helices α3-α4. C, Coomassie-stained SDS-polyacrylamide gel showing that Bcl-w with a mutant BH3 domain is extensively degraded following expression and purification from E. coli, unlike the wild-type protein. D, the overall structure of the Bcl-w BH3 mutant (mauve) is similar to native Bcl-w (yellow; PDB entry 1O0L without the C-terminal “tail” region removed) (37), although the α3 helix is highly disordered and the α2 helix is extended by one extra turn.
DISCUSSION
The BH3 domains of the pro-apoptotic BH3 have been extensively characterized. All bind to at least several pro-survival proteins with high affinity to either trigger the cell death cascade (in the case of the BH3-only proteins) or to inhibit cell death progression (in the case of Bax/Bak). By contrast, little was known about the BH3 domain of pro-survival protein prior to this report. Our studies of the isolated BH3 sequences show that they significantly differ biochemically and functionally from their pro-apoptotic counterparts, providing key insights into their capacity to elicit a cell death response. Our data on their role in their native context also reveal an important structural role, being critical for pro-survival protein stability.
Physiological Significance of the Low Affinity and Restricted Specificity of Pro-survival BH3 Domains
Our binding analysis of all pro-survival BH3 sequences with each pro-survival target (i.e. 25 different combinations in total) revealed that they are all significantly less capable of engaging either the pro-survival protein from which they were derived or other pro-survival molecules, compared with their pro-apoptotic counterparts. Indeed, the few pro-survival BH3 interactions that were measurable were typically in the hundreds nanomolar to low micromolar range, whereas high affinity pro-apoptotic BH3 sequences generally bind with IC50 values that are <100 nm (30–32) (Table 2). These data are very consistent with the limited published binding data on pro-survival protein BH3 domains (although only affinities for Mcl-1 BH3·Mcl-1 and Bcl-xLBH3·Bcl-xL have been reported previously) as well as other studies showing that interactions with pro-apoptotic splice variants such as Bcl-xS can be difficult to detect using approaches such as co-immunoprecipitation (51, 52). It also partly explains why chimeric pro-apoptotic proteins with pro-survival protein BH3 domains fail to elicit cell death in certain cell types (8).
The low affinity of these interactions and their specificity profiles that we revealed have several implications for the biological functioning of pro-survival proteins and their variants, such as splice variants or enzymatically cleaved forms. First, the low affinity necessarily means that apoptosis is likely to only ensue if intracellular concentrations of the BH3-exposed form are high, particularly when the competing antagonizing interactions (i.e. Bak/Bax-pro-survival protein) are typically of much higher affinity. Indeed, even after we targeted the Bcl-xL BH3 domain to the mitochondrial membrane, it required >100-fold more peptide to induce comparable levels of cytochrome c release into the cytosol (a hallmark of apoptosis) than the BH3 domain derived from the pro-apoptotic protein Bim (Fig. 2). Nevertheless, it could be envisaged that high levels of pro-survival BH3 domain exposure might occur where expression of a splice variant is significantly up-regulated, when a large pool of pro-survival proteins undergo caspase cleavage during an apoptotic response, or following enforced overexpression from vectors with strong promoters, such as would be used in in vitro studies. As such, our binding data highlight that apparently weak BH3 domain interactions (i.e. in the 100 nm to 1 μm range) that have often previously been disregarded as being biologically relevant can have an impact on determining cell fate.
The pro-apoptotic potential of pro-survival proteins is further reduced by the high specificity seen in the interactions of their BH3 domains with pro-survival proteins (i.e. Mcl-1 BH3 only binds Mcl-1, Bcl-2 BH3 only binds Bcl-xL, and Bcl-xL BH3 only binds Bcl-xL and Bcl-w). Hence, in cells where multiple pro-survival proteins are expressed, the pro-apoptotic forms of these proteins might be completely ineffective at inducing cell death on their own. We saw this in our studies with the BimS chimeras containing the Bcl-2 and Bcl-xL BH3 domains (Figs. 1 and 2) as well as with the physiologically important truncated (caspase-cleaved) form on Bcl-2 (Fig. 3), which only impacted MEF cell survival or mitochondrial integrity when Mcl-1 was genetically deleted or neutralized. Our affinity and specificity data on the Bcl-xL BH3 sequence also provide an explanation for why several studies on Bcl-xS showed it could only sensitize cells to apoptotic stimuli rather than kill on its own (48, 51) Similarly, a synthetic cell-penetrating “stapled” form of the Mcl-1 BH3 domain (Mcl-1 SAHB) was only able to induce mitochondrial outer membrane permeabilization or kill cells following co-treatment with other pro-apoptotic proteins, such as tBid (the caspase-8 cleavage product of Bid) or TRAIL (52), consistent with its restricted pro-survival protein binding profile and relatively weak affinity (Table 2).
A more general insight that our affinity data provide is that the weak capacity of pro-survival proteins to engage other pro-survival proteins is potentially an important safety mechanism to prevent unwanted apoptosis following events such as protein unfolding or proteolysis that would expose the BH3 sequence in the pro-survival molecules.
Distinguishing Features of Pro-survival Versus Pro-apoptotic BH3 Domains Account for Differences in Binding Affinities
Our structure-function studies show that the low affinity of the pro-survival protein BH3 domains is largely due to their possessing several distinguishing features compared with pro-apoptotic BH3 sequences, namely a large residue at the h1 + 1 position and a small residue at h3, especially in the case of the Bcl-2, Bcl-xL, Bcl-w, and, to a lesser degree, Mcl-1 BH3 sequences. The Bfl-1 BH3 domain probably possesses other unusual features that reduce its capacity to bind Bcl-2 proteins, although these were not explored in the current study. Interestingly, in an early review, Kelekar and Thompson postulated that these residues might be of functional importance in distinguishing pro-survival and pro-apoptotic Bcl-2 family members (69), although no experiments were performed to explore this possibility. In our studies, the importance of these residues was demonstrated when they were mutated to the corresponding residues in pro-apoptotic BH3 domains (i.e. h1 + 1 substituted for a small residue and h3 substituted for a larger hydrophobic residue). Moreover, these substitutions enabled most pro-survival BH3 domains to now bind to the parental protein from which they were derived with high affinity as well as to most other pro-survival proteins, and in many cases, the affinities were close to that seen for pro-apoptotic BH3 sequences (Table 2). Moreover, these mutations also converted some isolated pro-survival BH3 sequences from being mostly inert in cell killing assays to potent inducers of apoptosis across a range of MEF cell lines, consistent with a gain in capacity to engage pro-survival proteins in the cell (Figs. 1 and 2).
Consistent with this finding, the corresponding residues in Bak were also shown to be critical for its cell killing function, in part because they have reciprocal biophysical properties; h1 + 1 is a small residue (glycine), whereas h3 is larger hydrophobic residue (isoleucine instead of an alanine present in most pro-survival BH3 sequences) (Fig. 1A). In this context, switching h1 + 1 for larger residues and h3 for small residues was detrimental to Bak BH3 killing activity (Fig. 1H). Both h1 + 1 and h3 residues in the BH3 domain of Bak are also important for its antagonism by pro-survival proteins because the same mutations lead to a dramatic loss in affinity for its key regulators, Mcl-1 and Bcl-xL (Table 2).
Structural Basis for the Weak Affinity of Pro-survival Protein BH3 Domains
Published structures of BH3 domains bound to pro-survival proteins predict that the large h1 + 1 residue in pro-survival protein BH3 domains could be incompatible with a canonical BH3-groove interaction due to the necessity to accommodate this residue in a highly sterically restricted location. This suggests that alternative interaction sites (e.g. such as seen in interactions between activator pro-apoptotic BH3 domains and Bax (70)) might account for the cell killing activity reported to be associated with the Bcl-xL and Bcl-2 BH3 domains.
Our crystal structure of the Bcl-xL·Bcl-xL BH3 peptide complex, however, reveals that the canonical binding groove is the most likely interaction site and therefore that pro-survival protein BH3 domains induce cell death signaling by mechanisms similar to their pro-apoptotic counterparts. However, due to the necessity to accommodate the large residue at the h1 + 1 position, the interaction is relatively poorly fitting, with this lysine side chain extending out of the groove and preventing the N-terminal end of the BH3 ligand from fully engaging the binding site. This unusual binding mode prevents formation of a number of contacts with the protein that are otherwise seen in pro-apoptotic BH3-pro-survival protein interactions (e.g. burial of the h1 hydrophobic residue), which probably contributes to the relatively weak affinity of the interaction. The more extensive burial of a larger hydrophobic h3 residue in pro-apoptotic BH3 domains compared with the smaller alanine residue found in the majority of pro-survival BH3 sequences also probably accounts for the higher affinity of pro-apoptotic sequences for pro-survival proteins. Interestingly, in the one other structural example of a pro-survival protein bound to its own BH3 sequence, that for Mcl-1 bound to a stapled Mcl-1 BH3 peptide, a different manner of accommodating the large h1 + 1 residue was observed (52). In that structure, the BH3 ligand is non-helical at its N terminus, allowing the bulky leucine residue at position h1 + 1 to engage the pocket on Mcl-1 that is employed for binding by the h1 residue of pro-apoptotic BH3 sequences (11).
Most Pro-survival Proteins Are Unlikely to Form BH3-in-groove Homodimers Like Bax/Bak
A conundrum in apoptotic signaling regulated by the Bcl-2 pathway is why the multidomain pro-survival and pro-apoptotic proteins have opposing functions yet possess similar three-dimensional structures (34). One possible explanation based on the results in this study is that the distinguishing features of the pro-survival BH3 domains prevent most of them from efficiently assembling into dimers (both homo- and heterodimers). Because dimerization via a “BH3-in-groove” interaction is a critical first step toward higher order oligomerization of Bax and Bak (19, 20), the inability of most pro-survival proteins to readily form similar dimers would thereby preclude them from forming pores in the mitochondria in the same manner as Bax and Bak. Interestingly, the replacement of the h1 + 1 and h3 residues within the BH3 domains of pro-survival proteins is not sufficient to convert them to pro-apoptotic Bax/Bak-like molecules, despite their increased capacity to engage themselves via their BH3 domain, suggesting that additional sequence differences in other regions that are important for pore formation (e.g. the α5 helix) may also contribute to the inherent lack of mitochondrial pore forming capacity. Indeed, it has been reported that replacement of the Bcl-xL α5 with the corresponding sequence in Bax converts it to a pro-apoptotic form (71). By contrast, another study showed that caspase-mediated N-terminal cleavage of Bcl-xL was sufficient to cause it to induce cell death in Bax/Bak-doubly deficient MEFs, indicating that it had been converted to a Bax/Bak-like molecule (41), although we have been unable to produce similar results.3 Interestingly, of the pro-survival proteins, only Bcl-xL can bind its own native BH3 domain with submicromolar affinity. Although this affinity is significantly lower (10–100-fold weaker) than for typical pro-apoptotic BH3 domain-pro-survival protein interactions (30–32), it is in the same affinity range as recently reported for the interaction of Bax with its own BH3 domain (affinity information of the Bak-Bak BH3 domain interaction is not yet available) (21), suggesting that Bcl-xL could potentially form similar BH3-in-groove homodimers, which is also supported by our structural data (Fig. 4). In addition, other structural studies have shown that stimuli such as heating or high pH can induce Bcl-xL to undergo the types of conformational changes (i.e. dissociation of core and latch domains) recently shown to occur when Bax and Bak are activated (21, 22, 72). Hence, if cellular stresses downstream of caspase activation could produce the same conformational changes, then this might explain how the N-terminally truncated Bcl-xL could be converted to a Bax/Bak-like molecule, notwithstanding the aforementioned report (71) indicating the requirement for the α5 replacement.
Very early studies on the caspase-cleaved form of Bcl-2 also suggested that its pro-apoptotic activity was associated with its conversion to a Bax-like death effector protein (42), although at that time, Bax/Bak-doubly deficient cells were not available to dissect the mechanism for this cell killing. Indeed, our results show that this N-terminally deleted form of Bcl-2 has no activity in Bax−/−/Bak−/− cells (Fig. 3) and more likely functions like a BH3-only protein that specifically antagonizes Bcl-xL and becomes activated when the BH3 domain is exposed following caspase-3 cleavage, analogous to tBid activation by caspase-8. This is consistent with our binding data showing that Bcl-2 is unlikely to readily form BH3-in-groove homodimers and function like Bax and Bak, due to its weak affinity for its own BH3 domain.
BH3 Domains Are Critical for Pro-survival Protein Stability
A further explanation for why the BH3 h1 + 1 and h3 mutations were not sufficient to convert pro-survival proteins to Bax/Bak-like proteins is that those mutations appear to be destabilizing to the protein, leading to its proteasomal degradation and low cellular levels (Fig. 5). This instability is reflected in aspects of our crystal structure of a Bcl-w mutant with a “pro-apoptotic” BH3 sequence (Fig. 8). Unlike native Bcl-w, the mutant protein was sensitive to bacterial proteases during expression and purification from Escherichia coli. Hence, it appears that the presence of a pro-apoptotic BH3 sequence led to some degree of protein unfolding, where cleavage sites within the α1-α2 interconnecting loop as well as at the end of the BH3-containing α2 helix became exposed. These proteolytic cleavages resulted in the excision of a Bcl-w (mutant) BH3 domain, which then bound into the groove of another intact mutant Bcl-w molecule, as well as removal of the N-terminal fragment containing the BH4 domain. A similar phenomenon is observed in Bak, where, upon its activation via different apoptotic stimuli, calpain cleavage sites are exposed at positions analogous to the cleavage sites we observed in the Bcl-w mutant, leading to fragmentation into similar BH3 and BH4 domain cleavage products (8). Hence, these data demonstrate that the overall stability of pro-survival protein structure is not compatible with possession of a “pro-apoptotic” type BH3 domain that would enable it to form a stable BH3-in-groove dimer.
One reason for this destabilization following the h1 + 1 substitution, at least in the case of Bcl-xL, is that this residue is one of a number that participate in interactions between the α1 helix and the core of the structure (Figs. 5–7). It appears from our data that the “release” of the α1 exposes epitopes (perhaps on either α1 or α2) that facilitate proteasomal targeting of the protein. Indeed, this could be a mechanism to prevent unwanted cell death induction by Bcl-xL via its BH3 domain following events such as protein unfolding or proteolysis. Previously, Mason et al. (73) reported that a point mutation of a tyrosine residue (Y15C mutation) on α1 that engages α2 on Bcl-xL resulted in decreased Bcl-xL half-life, which concurs with our data on the Y15A substitution (Fig. 7). Critically, this mutation had significant biological consequences because it resulted in the premature death of platelets in vivo (73). Because platelets are predominantly dependent on Bcl-xL for their survival, its rapid elimination caused their accelerated demise. Hence, the apparent pro-apoptotic effect of N-terminally truncated Bcl-xL could also be partly a result of Bcl-xL destabilization and degradation, providing less effective “buffering” against pro-apoptotic proteins within the cell, rather than direct activation of the apoptotic cascade. Consistent with this idea, we show that this form of Bcl-xL is expressed at significantly lower levels than the full-length form (Fig. 7D). Similar low level expression of N-terminally deleted Bcl-2 has also been reported previously (42).
A Distinct Class of BH3 Domains
Currently, BH3 domains can be viewed as falling into two categories. First, there are those that act purely as ligands. These include the BH3 domains of BH3-only proteins but also those on proteins such as Mule (74) and Beclin-1 (75), which have roles in mediating protein-protein interactions that are not directly involved in the apoptotic cascade. Those in the second category have dual roles as ligand and receptor. These include the BH3 domains of Bax and Bak. These BH3 domains are critical for mediating interactions with pro-survival proteins as well as in Bax/Bak homodimerization, the critical first step in apoptotic mitochondrial pore formation. The BH3 domain in Bax/Bak also has a critical structural role whereby it is an integral part of the structure of the inactive protein, the receptor for transient engagement with activator BH3-only proteins. The data in this paper demonstrate that BH3 domains from pro-survival proteins form a third distinct category. Although they have a weak capacity to act as ligands, their primary function is structural. They maintain the integrity of the receptor site on the pro-survival protein through critical contacts with the α1 helix that prevent exposure of epitopes that signal their proteosomal degradation.
Note Added in Proof
Grace J. Gold's contributions to this article fulfill the JBC authorship criteria, but her authorship was inadvertently omitted from the version of the article that was published on November 3, 2014 as a Paper in Press.
This work was supported by grants and fellowships from the National Health and Medical Research Council (NHMRC) of Australia (Program Grant 1016701 (to P. M. C.), Project Grants 1041936 and 1008329 (to W. D. F. and P. M. C.), Career Development Fellowship 1024620 (to E. F. L.), and SPRF 1022618 (to P. M. C.)), Australian Research Council Fellowship FT100100791 (to G. D.), and Cancer Council of Victoria Grant-in-aid 1057949. Infrastructure support was provided by NHMRC IRIISS Grant 361646 and the Victorian State Government OIS grant.
The atomic coordinates and structure factors (codes 4CIN and 4CIM) have been deposited in the Protein Data Bank (http://wwpdb.org/).
E. F. Lee and W. D. Fairlie, unpublished data.
- Bcl-2
- B cell lymphoma-2
- BH
- Bcl-2 homology domain
- Bak
- Bcl-2 homologous antagonist/killer
- Bax
- Bcl-2-associated X protein
- Bcl-xL
- B cell lymphoma extra large
- Mcl-1
- myeloid cell leukaemia protein
- MEF
- mouse embryonic fibroblast
- PDB
- Protein Data Bank.
REFERENCES
- 1. Lindsten T., Ross A. J., King A., Zong W. X., Rathmell J. C., Shiels H. A., Ulrich E., Waymire K. G., Mahar P., Frauwirth K., Chen Y., Wei M., Eng V. M., Adelman D. M., Simon M. C., Ma A., Golden J. A., Evan G., Korsmeyer S. J., MacGregor G. R., Thompson C. B. (2000) The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 6, 1389–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wei M. C., Zong W. X., Cheng E. H., Lindsten T., Panoutsakopoulou V., Ross A. J., Roth K. A., MacGregor G. R., Thompson C. B., Korsmeyer S. J. (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Youle R. J., Strasser A. (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59 [DOI] [PubMed] [Google Scholar]
- 4. Lovell J. F., Billen L. P., Bindner S., Shamas-Din A., Fradin C., Leber B., Andrews D. W. (2008) Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135, 1074–1084 [DOI] [PubMed] [Google Scholar]
- 5. Billen L. P., Kokoski C. L., Lovell J. F., Leber B., Andrews D. W. (2008) Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol. 6, e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuwana T., Bouchier-Hayes L., Chipuk J. E., Bonzon C., Sullivan B. A., Green D. R., Newmeyer D. D. (2005) BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 17, 525–535 [DOI] [PubMed] [Google Scholar]
- 7. Letai A., Bassik M. C., Walensky L. D., Sorcinelli M. D., Weiler S., Korsmeyer S. J. (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2, 183–192 [DOI] [PubMed] [Google Scholar]
- 8. Llambi F., Moldoveanu T., Tait S. W., Bouchier-Hayes L., Temirov J., McCormick L. L., Dillon C. P., Green D. R. (2011) A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell 44, 517–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim H., Rafiuddin-Shah M., Tu H. C., Jeffers J. R., Zambetti G. P., Hsieh J. J., Cheng E. H. (2006) Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat. Cell Biol. 8, 1348–1358 [DOI] [PubMed] [Google Scholar]
- 10. Willis S. N., Chen L., Dewson G., Wei A., Naik E., Fletcher J. I., Adams J. M., Huang D. C. (2005) Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 19, 1294–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Czabotar P. E., Lee E. F., van Delft M. F., Day C. L., Smith B. J., Huang D. C., Fairlie W. D., Hinds M. G., Colman P. M. (2007) Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc. Natl. Acad. Sci. U.S.A. 104, 6217–6222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Day C. L., Smits C., Fan F. C., Lee E. F., Fairlie W. D., Hinds M. G. (2008) Structure of the BH3 domains from the p53-inducible BH3-only proteins Noxa and Puma in complex with Mcl-1. J. Mol. Biol. 380, 958–971 [DOI] [PubMed] [Google Scholar]
- 13. Liu X., Dai S., Zhu Y., Marrack P., Kappler J. W. (2003) The structure of a Bcl-xL/Bim fragment complex: implications for Bim function. Immunity 19, 341–352 [DOI] [PubMed] [Google Scholar]
- 14. Sattler M., Liang H., Nettesheim D., Meadows R. P., Harlan J. E., Eberstadt M., Yoon H. S., Shuker S. B., Chang B. S., Minn A. J., Thompson C. B., Fesik S. W. (1997) Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275, 983–986 [DOI] [PubMed] [Google Scholar]
- 15. Smits C., Czabotar P. E., Hinds M. G., Day C. L. (2008) Structural plasticity underpins promiscuous binding of the prosurvival protein A1. Structure 16, 818–829 [DOI] [PubMed] [Google Scholar]
- 16. Lee E. F., Sadowsky J. D., Smith B. J., Czabotar P. E., Peterson-Kaufman K. J., Colman P. M., Gellman S. H., Fairlie W. D. (2009) High-resolution structural characterization of a helical α/β-peptide foldamer bound to the anti-apoptotic protein Bcl-xL. Angew. Chem. Int. Ed. Engl. 48, 4318–4322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Petros A. M., Nettesheim D. G., Wang Y., Olejniczak E. T., Meadows R. P., Mack J., Swift K., Matayoshi E. D., Zhang H., Thompson C. B., Fesik S. W. (2000) Rationale for Bcl-xL/Bad peptide complex formation from structure, mutagenesis, and biophysical studies. Protein Sci. 9, 2528–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dewson G., Kluck R. M. (2009) Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J. Cell Sci. 122, 2801–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dewson G., Kratina T., Sim H. W., Puthalakath H., Adams J. M., Colman P. M., Kluck R. M. (2008) To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol. Cell 30, 369–380 [DOI] [PubMed] [Google Scholar]
- 20. Dewson G., Ma S., Frederick P., Hockings C., Tan I., Kratina T., Kluck R. M. (2012) Bax dimerizes via a symmetric BH3:groove interface during apoptosis. Cell Death Differ. 19, 661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Czabotar P. E., Westphal D., Dewson G., Ma S., Hockings C., Fairlie W. D., Lee E. F., Yao S., Robin A. Y., Smith B. J., Huang D. C., Kluck R. M., Adams J. M., Colman P. M. (2013) Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152, 519–531 [DOI] [PubMed] [Google Scholar]
- 22. Brouwer J. M., Westphal D., Dewson G., Robin A. Y., Uren R. T., Bartolo R., Thompson G. V., Colman P. M., Kluck R. M., Czabotar P. E. (2014) Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol. Cell 55, 938–946 [DOI] [PubMed] [Google Scholar]
- 23. Dewson G., Kratina T., Czabotar P., Day C. L., Adams J. M., Kluck R. M. (2009) Bak activation for apoptosis involves oligomerization of dimers via their α6 helices. Mol. Cell 36, 696–703 [DOI] [PubMed] [Google Scholar]
- 24. Ma S., Hockings C., Anwari K., Kratina T., Fennell S., Lazarou M., Ryan M. T., Kluck R. M., Dewson G. (2013) Assembly of the Bak apoptotic pore: a critical role for the bak protein α6 helix in the multimerization of homodimers during apoptosis. J. Biol. Chem. 288, 26027–26038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee E. F., Czabotar P. E., van Delft M. F., Michalak E. M., Boyle M. J., Willis S. N., Puthalakath H., Bouillet P., Colman P. M., Huang D. C., Fairlie W. D. (2008) A novel BH3 ligand that selectively targets Mcl-1 reveals that apoptosis can proceed without Mcl-1 degradation. J. Cell Biol. 180, 341–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Foight G. W., Ryan J. A., Gullá S. V., Letai A., Keating A. E. (2014) Designed BH3 peptides with high affinity and specificity for targeting Mcl-1 in cells. ACS Chem. Biol. 9, 1962–1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dutta S., Chen T. S., Keating A. E. (2013) Peptide ligands for pro-survival protein Bfl-1 from computationally guided library screening. ACS Chem. Biol. 8, 778–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen T. S., Palacios H., Keating A. E. (2013) Structure-based redesign of the binding specificity of anti-apoptotic Bcl-x(L). J. Mol. Biol. 425, 171–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dutta S., Gullá S., Chen T. S., Fire E., Grant R. A., Keating A. E. (2010) Determinants of BH3 binding specificity for Mcl-1 versus Bcl-xL. J. Mol. Biol. 398, 747–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Certo M., Del Gaizo Moore V., Nishino M., Wei G., Korsmeyer S., Armstrong S. A., Letai A. (2006) Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9, 351–365 [DOI] [PubMed] [Google Scholar]
- 31. Chen L., Willis S. N., Wei A., Smith B. J., Fletcher J. I., Hinds M. G., Colman P. M., Day C. L., Adams J. M., Huang D. C. (2005) Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 17, 393–403 [DOI] [PubMed] [Google Scholar]
- 32. Ku B., Liang C., Jung J. U., Oh B. H. (2011) Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Res. 21, 627–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moldoveanu T., Liu Q., Tocilj A., Watson M., Shore G., Gehring K. (2006) The x-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol. Cell 24, 677–688 [DOI] [PubMed] [Google Scholar]
- 34. Suzuki M., Youle R. J., Tjandra N. (2000) Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103, 645–654 [DOI] [PubMed] [Google Scholar]
- 35. Oh K. J., Singh P., Lee K., Foss K., Lee S., Park M., Lee S., Aluvila S., Park M., Singh P., Kim R. S., Symersky J., Walters D. E. (2010) Conformational changes in BAK, a pore-forming proapoptotic Bcl-2 family member, upon membrane insertion and direct evidence for the existence of BH3-BH3 contact interface in BAK homo-oligomers. J. Biol. Chem. 285, 28924–28937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Day C. L., Chen L., Richardson S. J., Harrison P. J., Huang D. C., Hinds M. G. (2005) Solution structure of prosurvival Mcl-1 and characterization of its binding by proapoptotic BH3-only ligands. J. Biol. Chem. 280, 4738–4744 [DOI] [PubMed] [Google Scholar]
- 37. Hinds M. G., Lackmann M., Skea G. L., Harrison P. J., Huang D. C., Day C. L. (2003) The structure of Bcl-w reveals a role for the C-terminal residues in modulating biological activity. EMBO J. 22, 1497–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Muchmore S. W., Sattler M., Liang H., Meadows R. P., Harlan J. E., Yoon H. S., Nettesheim D., Chang B. S., Thompson C. B., Wong S. L., Ng S. L., Fesik S. W. (1996) X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 381, 335–341 [DOI] [PubMed] [Google Scholar]
- 39. Petros A. M., Medek A., Nettesheim D. G., Kim D. H., Yoon H. S., Swift K., Matayoshi E. D., Oltersdorf T., Fesik S. W. (2001) Solution structure of the antiapoptotic protein bcl-2. Proc. Natl. Acad. Sci. U.S.A. 98, 3012–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jonas E. A., Hickman J. A., Chachar M., Polster B. M., Brandt T. A., Fannjiang Y., Ivanovska I., Basañez G., Kinnally K. W., Zimmerberg J., Hardwick J. M., Kaczmarek L. K. (2004) Proapoptotic N-truncated BCL-xL protein activates endogenous mitochondrial channels in living synaptic terminals. Proc. Natl. Acad. Sci. U.S.A. 101, 13590–13595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ofengeim D., Chen Y. B., Miyawaki T., Li H., Sacchetti S., Flannery R. J., Alavian K. N., Pontarelli F., Roelofs B. A., Hickman J. A., Hardwick J. M., Zukin R. S., Jonas E. A. (2012) N-terminally cleaved Bcl-xL mediates ischemia-induced neuronal death. Nat. Neurosci. 15, 574–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cheng E. H., Kirsch D. G., Clem R. J., Ravi R., Kastan M. B., Bedi A., Ueno K., Hardwick J. M. (1997) Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 278, 1966–1968 [DOI] [PubMed] [Google Scholar]
- 43. Boise L. H., González-García M., Postema C. E., Ding L., Lindsten T., Turka L. A., Mao X., Nuñez G., Thompson C. B. (1993) bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74, 597–608 [DOI] [PubMed] [Google Scholar]
- 44. Bae J., Leo C. P., Hsu S. Y., Hsueh A. J. (2000) MCL-1S, a splicing variant of the antiapoptotic BCL-2 family member MCL-1, encodes a proapoptotic protein possessing only the BH3 domain. J. Biol. Chem. 275, 25255–25261 [DOI] [PubMed] [Google Scholar]
- 45. Bingle C. D., Craig R. W., Swales B. M., Singleton V., Zhou P., Whyte M. K. (2000) Exon skipping in Mcl-1 results in a bcl-2 homology domain 3 only gene product that promotes cell death. J. Biol. Chem. 275, 22136–22146 [DOI] [PubMed] [Google Scholar]
- 46. Lindenboim L., Kringel S., Braun T., Borner C., Stein R. (2005) Bak but not Bax is essential for Bcl-xS-induced apoptosis. Cell Death Differ. 12, 713–723 [DOI] [PubMed] [Google Scholar]
- 47. Clarke M. F., Apel I. J., Benedict M. A., Eipers P. G., Sumantran V., González-García M., Doedens M., Fukunaga N., Davidson B., Dick J. E., Minn A. J., Boise L. H., Thompson C. B., Wicha M., Núñez G. (1995) A recombinant bcl-x s adenovirus selectively induces apoptosis in cancer cells but not in normal bone marrow cells. Proc. Natl. Acad. Sci. U.S.A. 92, 11024–11028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chang B. S., Kelekar A., Harris M. H., Harlan J. E., Fesik S. W., Thompson C. B. (1999) The BH3 domain of Bcl-x(S) is required for inhibition of the antiapoptotic function of Bcl-x(L). Mol. Cell. Biol. 19, 6673–6681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mitra R. S., Benedict M. A., Qian D., Foreman K. E., Ekhterae D., Nickoloff B. J., Nuñez G. (2001) Killing of sarcoma cells by proapoptotic Bcl-X(S): role of the BH3 domain and regulation by Bcl-X(L). Neoplasia 3, 437–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lindenboim L., Borner C., Stein R. (2001) Bcl-x(S) can form homodimers and heterodimers and its apoptotic activity requires localization of Bcl-x(S) to the mitochondria and its BH3 and loop domains. Cell Death Differ. 8, 933–942 [DOI] [PubMed] [Google Scholar]
- 51. Minn A. J., Boise L. H., Thompson C. B. (1996) Bcl-x(S) anatagonizes the protective effects of Bcl-x(L). J. Biol. Chem. 271, 6306–6312 [DOI] [PubMed] [Google Scholar]
- 52. Stewart M. L., Fire E., Keating A. E., Walensky L. D. (2010) The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat. Chem. Biol. 6, 595–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cosulich S. C., Worrall V., Hedge P. J., Green S., Clarke P. R. (1997) Regulation of apoptosis by BH3 domains in a cell-free system. Curr. Biol. 7, 913–920 [DOI] [PubMed] [Google Scholar]
- 54. Smith B. J., Lee E. F., Checco J. W., Evangelista M., Gellman S. H., Fairlie W. D. (2013) Structure-guided rational design of α/β-peptide foldamers with high affinity for BCL-2 family prosurvival proteins. Chembiochem 14, 1564–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kvansakul M., Yang H., Fairlie W. D., Czabotar P. E., Fischer S. F., Perugini M. A., Huang D. C., Colman P. M. (2008) Vaccinia virus anti-apoptotic F1L is a novel Bcl-2-like domain-swapped dimer that binds a highly selective subset of BH3-containing death ligands. Cell Death Differ. 15, 1564–1571 [DOI] [PubMed] [Google Scholar]
- 56. Oberstein A., Jeffrey P. D., Shi Y. (2007) Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J. Biol. Chem. 282, 13123–13132 [DOI] [PubMed] [Google Scholar]
- 57. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 58. Kabsch W. (2010) Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McCoy A. J., Grosse-Kunstleve R. W., Storoni L. C., Read R. J. (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr. D Biol. Crystallogr. 61, 458–464 [DOI] [PubMed] [Google Scholar]
- 60. Read R. J. (2001) Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr. D Biol. Crystallogr. 57, 1373–1382 [DOI] [PubMed] [Google Scholar]
- 61. Storoni L. C., McCoy A. J., Read R. J. (2004) Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 60, 432–438 [DOI] [PubMed] [Google Scholar]
- 62. Lee E. F., Dewson G., Smith B. J., Evangelista M., Pettikiriarachchi A., Dogovski C., Perugini M. A., Colman P. M., Fairlie W. D. (2011) Crystal structure of a BCL-W domain-swapped dimer: implications for the function of BCL-2 family proteins. Structure 19, 1467–1476 [DOI] [PubMed] [Google Scholar]
- 63. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 64. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 65. Willis S. N., Fletcher J. I., Kaufmann T., van Delft M. F., Chen L., Czabotar P. E., Ierino H., Lee E. F., Fairlie W. D., Bouillet P., Strasser A., Kluck R. M., Adams J. M., Huang D. C. (2007) Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315, 856–859 [DOI] [PubMed] [Google Scholar]
- 66. Weber A., Ausländer D., Häcker G. (2013) Mouse Noxa uses only the C-terminal BH3-domain to inactivate Mcl-1. Apoptosis 18, 1093–1105 [DOI] [PubMed] [Google Scholar]
- 67. Oh K. J., Barbuto S., Pitter K., Morash J., Walensky L. D., Korsmeyer S. J. (2006) A membrane-targeted BID BCL-2 homology 3 peptide is sufficient for high potency activation of BAX in vitro. J. Biol. Chem. 281, 36999–37008 [DOI] [PubMed] [Google Scholar]
- 68. Denisov A. Y., Madiraju M. S., Chen G., Khadir A., Beauparlant P., Attardo G., Shore G. C., Gehring K. (2003) Solution structure of human BCL-w: modulation of ligand binding by the C-terminal helix. J. Biol. Chem. 278, 21124–21128 [DOI] [PubMed] [Google Scholar]
- 69. Kelekar A., Thompson C. B. (1998) Bcl-2-family proteins: the role of the BH3 domain in apoptosis. Trends Cell Biol. 8, 324–330 [DOI] [PubMed] [Google Scholar]
- 70. Gavathiotis E., Suzuki M., Davis M. L., Pitter K., Bird G. H., Katz S. G., Tu H. C., Kim H., Cheng E. H., Tjandra N., Walensky L. D. (2008) BAX activation is initiated at a novel interaction site. Nature 455, 1076–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. George N. M., Evans J. J., Luo X. (2007) A three-helix homo-oligomerization domain containing BH3 and BH1 is responsible for the apoptotic activity of Bax. Genes Dev. 21, 1937–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. O'Neill J. W., Manion M. K., Maguire B., Hockenbery D. M. (2006) BCL-XL dimerization by three-dimensional domain swapping. J. Mol. Biol. 356, 367–381 [DOI] [PubMed] [Google Scholar]
- 73. Mason K. D., Carpinelli M. R., Fletcher J. I., Collinge J. E., Hilton A. A., Ellis S., Kelly P. N., Ekert P. G., Metcalf D., Roberts A. W., Huang D. C., Kile B. T. (2007) Programmed anuclear cell death delimits platelet life span. Cell 128, 1173–1186 [DOI] [PubMed] [Google Scholar]
- 74. Zhong Q., Gao W., Du F., Wang X. (2005) Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 121, 1085–1095 [DOI] [PubMed] [Google Scholar]
- 75. Pattingre S., Tassa A., Qu X., Garuti R., Liang X. H., Mizushima N., Packer M., Schneider M. D., Levine B. (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939 [DOI] [PubMed] [Google Scholar]