

Background: The Fic domain mediates AMPylation and is highly conserved in various species.
Results: BiP is identified as a substrate of Fic, and its AMPylation state is modulated by ER stress.
Conclusion: AMPylation of BiP presents a regulatory mechanism for cells to achieve ER homeostasis.
Significance: BiP is the first known substrate for AMPylation by a eukaryotic Fic protein.
Keywords: Adenosine, AMP, Chaperone, Drosophila, Unfolded Protein Response (UPR), AMPylation, BiP, ER Stress, grp78
Abstract
Drosophila Fic (dFic) mediates AMPylation, a covalent attachment of adenosine monophosphate (AMP) from ATP to hydroxyl side chains of protein substrates. Here, we identified the endoplasmic reticulum (ER) chaperone BiP as a substrate for dFic and mapped the modification site to Thr-366 within the ATPase domain. The level of AMPylated BiP in Drosophila S2 cells is high during homeostasis, whereas the level of AMPylated BiP decreases upon the accumulation of misfolded proteins in the ER. Both dFic and BiP are transcriptionally activated upon ER stress, supporting the role of dFic in the unfolded protein response pathway. The inactive conformation of BiP is the preferred substrate for dFic, thus endorsing a model whereby AMPylation regulates the function of BiP as a chaperone, allowing acute activation of BiP by deAMPylation during an ER stress response. These findings not only present the first substrate of eukaryotic AMPylator but also provide a target for regulating the unfolded protein response, an emerging avenue for cancer therapy.
Introduction
AMPylation is a posttranslational modification involving a covalent attachment of an AMP moiety from ATP to hydroxyl side chains of target substrates (1). This modification was first discovered in Escherichia coli in the 1960s from a study characterizing glutamine synthetase (GS)2 adenylyl transferase (2, 3). As a bifunctional enzyme, GS adenylyl transferase catalyzes both the addition and removal of AMP on glutamine synthetase contingent on changes in nitrogen metabolism in E. coli. Whether this modification was a general regulatory mechanism used by other organisms remained elusive until the recent study of VopS, a bacterial effector protein from Vibrio parahaemolyticus, showed that it modifies Rho family GTPases in the host cell with AMP during infection (4). Adding AMP to a threonine in the switch I region of Rho GTPases blocks their binding to downstream effectors resulting in actin cytoskeleton disruption, consequent cell rounding, and inhibition of multiple signaling pathways (5). AMPylation is mediated by the Fic (filamentation induced by cyclic AMP) domain of VopS, which is also conserved in many higher eukaryotes. Similarly, IbpA from Histophilus somni, which contains two Fic domains, also targets Rho GTPases, although it modifies a tyrosine instead of threonine (6). Another bacterial AMPylator SidM/DrrA, found in Legionella pneumophila, modifies Rab1 GTPase to manipulate host membrane trafficking. Interestingly, this protein catalyzes the modification using a nucleotidyltransferase domain, which instead of the Fic domain is the active site domain found in the aforementioned E. coli GS-ATPase (7).
AMPylation, similar to most posttranslational modifications, can be a reversible process involving counteracting enzymes. For example, the Legionella effector protein SidD is a deAMPylator that acts on specific targets (8). It removes AMP added by SidM/DrrA on Rab GTPases, which occurs in a spatially and temporally regulated manner during infection. It is interesting to note that the active site of SidD resembles a phosphatase-like fold from members of the metal-dependent protein phosphatase (PPM) family.
Further studies revealed that Fic domains are capable of mediating more than just AMPylation. AnkX, a Legionella effector that contains a Fic domain, was shown to be a phosphocholine transferase that targets a serine residue of Rab1 GTPase (9). From its substrate CDP-choline, AnkX transfers phosphocholine instead of the NMP moiety to the target side chain. AvrAC from the plant pathogen Xanthomonas campestris adds UMP to the host kinases BIK1 and RIPK to suppress the host immune response (10). The bacteriophage toxin Doc, which belongs to a distant subfamily of Fic proteins, is a kinase that inhibits bacterial translation by phosphorylating the translation elongation factor EF-Tu (11, 12). Structural analysis has shown that the versatility of the Fic domain for AMPylation, UMPylation, phosphorylation, and phosphocholination occurs by changing the orientation of the nucleotide-based substrates in the active site (13). This yields a remarkable divergence of catalytic mechanisms while maintaining the conserved catalytic core.
The Fic domain is highly conserved across species, including higher eukaryotes (albeit, only in a few fungi; Refs. 4 and 14), which raises the possibility that AMPylation serves a critical role in cellular function. However, all AMPylators characterized thus far have been bacterial proteins, most of which are involved in pathogenesis. In an attempt to understand the physiological function of AMPylation in eukaryotes, we knocked out the gene encoding the Drosophila FicD protein (dfic). We found that dFic enzymatic activity is required in glial cells for visual neurotransmission in flies (15). In addition, this study demonstrated that the catalytic domain of dFic resides in the lumen of the ER, where it is glycosylated.
Another protein that resides in the ER is the molecular chaperone BiP/GRP78, a highly conserved heat shock 70 family protein and a crucial molecular chaperone in the ER (16–18). BiP binds and assists the folding and assembly of newly synthesized proteins. It is also essential for translocation of secretory precursors across the ER membrane (19, 20). Aberrant or misfolded proteins bind to BiP for assembly or to be directed to the ER-associated degradation pathway (ERAD) when the folding attempts continuously fail (21–25). Thereby, BiP contributes to quality control for protein homeostasis in the ER (26–28). BiP has been shown to be phosphorylated on threonine residues in vitro and in vivo (29–31), although how this modification affects the molecular or biological function on BiP is unclear. In addition, BiP undergoes ADP-ribosylation, which is thought to affect substrate binding and release (32–35). Misregulation of BiP is implicated in numerous diseases including neurodegenerative disorders and many types of cancers (36–40).
Here, we report that BiP is a novel substrate for dFic-mediated AMPylation. BiP was predominantly labeled with AMP by dFic in S2 cell lysate. AMPylation of BiP decreases during ER stress but increases upon the reduction of unfolded proteins. Both dFic and BiP are transcriptionally activated upon ER stress induction, implicating a role for dFic in the unfolded protein response (UPR). We identified a conserved threonine residue, Thr-366, as the AMPylation site, which is in close proximity to the ATP binding site of the BiP ATPase domain. Our study presents the first substrate of AMPylation by a eukaryotic protein and proposes a new mode of posttranslational regulation of BiP, which is likely to serve a crucial role in maintaining ER protein homeostasis.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
Drosophila melanogaster Schneider 2 (S2) cells were grown according to standard protocols (41). Cells were maintained at 27 °C in Schneider's medium supplemented with 10% heat-inactivated fetal bovine serum and antibiotics. Cells were transfected using X-tremeGENE HP DNA transfection reagent (Roche Applied Science) according to the manufacturer's protocol and grown for 3 days before harvesting.
Plasmid Constructs
All Drosophila dFic Δ70 constructs for bacterial expression were made with pGex 4T-3-derived vector that has a Tev (tobacco etch virus) cleavage site inserted after the GST coding sequence. dFicΔ70 was inserted into the vector using BamHI and NotI sites. For bacterial expression, Drosophila BiP was cloned into pET28a via BamHI and NotI sites. For transfection in S2 cells, BiP was cloned into pAc5.1 V5-His vector using EcoRI and NotI sites. All BiP constructs for expression in S2 cells contained a C-terminal FLAG followed by a KDEL sequence. Human Fic (hFic) Δ47 was inserted into pE-SUMO using overlap extension PCR cloning at the BsaI site, maintaining the SUMO protease site. Human hBiPΔ19 was obtained from DNASU (Arizona State University) and was transferred from pDONR201 into pDEST17 using Gateway cloning.
Protein Purification
GST-dFic Δ70 constructs were transformed into E. coli Rosetta (DE3) cells, and single colonies were grown to an A600 of 0.6–0.8 and expressed with 0.4 mm isopropyl β-d-thiogalactopyranoside for 20 h at 22 °C. Cells were then lysed by a cell disrupter (EmulsiFlex-C3, Avestin Inc.), and the protein was purified using a standard protocol for GST affinity chromatography (Pierce). His-BiP constructs and His-SUMO-hFicΔ47 were expressed as described above and purified using a standard nickel-affinity purification protocol (Thermo Fisher Scientific, Bremen, Germany). After nickel purification, His-hBiPΔ19 and His-SUMO-hFicΔ47 were further purified on HiLoad 16/60 Superdex 75 (GE Healthcare) attached to an AKTA FPLC.
Concanavalin A Pulldown
S2 cells were harvested and lysed with ConA radioimmune precipitation assay buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 5 mm MgCl2, 5 mm MnCl2, 5 mm CaCl2, 1 mm PMSF, protease inhibitor mixture (Roche Applied Science)), and the soluble fraction was collected after 1000 × g centrifugation for 10 min at 4 °C. The fraction was then loaded onto the equilibrated concanavalin A beads (Sigma) and incubated overnight at 4 °C. The beads were washed 3 times with equilibration buffer (200 mm Tris, pH 7.5, 500 mm NaCl, 5 mm MgCl2, 5 mm MnCl2, 5 mm CaCl2). The fraction bound to the beads was directly used for in vitro AMPylation assays or eluted with SDS sample buffer for SDS-PAGE and Western blot analysis.
Immunoprecipitation
S2 cells transfected with FLAG-tagged BiP constructs were lysed as described above. The lysate was incubated with anti-FLAG M2-agarose (Sigma) overnight at 4 °C, and the beads were washed three times with TBS (50 mm Tris, pH 7.5, 150 mm NaCl). Proteins bound to beads were eluted with SDS sample buffer for SDS-PAGE and Western blot analysis.
In Vitro AMPylation Assay
In vitro AMPylation assays were performed using GST-dFicΔ70 as an enzyme and either recombinant His-BiP protein or S2 cell lysate as substrate. Each reaction contained 0.2 μm GST-dFicΔ70, 250 μm cold ATP, 0.1–0.2 μCi of [α-32P]ATP, and 2 μm His-BiP in 30 μl of AMPylation buffer (20 mm Tris, pH 7.5, 100 mm NaCl, 10 mm MgCl2, 5 mm MnCl2, 5 mm CaCl2, except where exclusion of CaCl2 is indicated). When S2 cell lysate was used as substrate, 20 μg of cell lysate was added to the reaction with 1 mg/ml RNase A. The reactions were incubated for 45 min at 30 °C and stopped by the addition of SDS sample buffer. Samples were analyzed by autoradiography after SDS-PAGE. For the assays where AMPylation was detected by the anti-AMP-Thr antibody (42), the experiment was performed as described above but excluding radiolabeled ATP.
ATPase Assay
ATPase assay using His-BiP constructs were performed on nickel-nitrilotriacetic acid beads (Pierce). 500 nm BiP was bound to the beads for 1 h at 4 °C, and 100 μm ATP was added to the bound proteins in ATPase assay buffer (20 mm Hepes, pH 7.0, 2 mm MgCl2, 25 mm KCl). After 3 h of ATP hydrolysis reaction at 30 °C, supernatant was collected, and the remaining ATP was measured using the ATP bioluminescent assay kit (Sigma) according to the manufacturer's protocol. Luminescence was measured by FLUOstar OPTIMA (BMG Labtech).
Quantitative Real-time PCR
Total RNA was extracted from treated and untreated S2 cells using TRIzol reagent (Ambion). 2 μg of RNA were reverse-transcribed into cDNA using High-Capacity cDNA Reverse Transcription (Applied Biosystems) utilizing random hexamers. RT-PCR was performed using Fast SYBR Green master mix (Applied Biosystems) on a 7500 Fast Real-Time PCR System (Applied Biosystems). Amplification was carried out in 20-μl reaction mixtures containing 100 ng of cDNA (in 5 μl), 7.5 pmol of each primer (in 5 μl), and 10 μl of 2× SYBR Green master mix. Rp49 (ribosomal protein) was used as an internal control, and all reactions were performed in triplicate. Quantification of relative gene expression was analyzed using the ΔΔCt method. The primers used for amplification were dFic (forward (F), 5′-CAGAACAACCGAGTCCACCT-3′; reverse (R), 5′-CGCAGTTTCCAGACCAGATT-3′), BiP (F, 5′-GCTATTGCCTACGGTCTGGA-3′; R, 5′-CATCACACGCTGATCGAAGT-3′), ER oxidoreductin-1-like (Ero1L; F, 5′-ATGAGGCGGAAGAGGACTTT-3′; R, 5′-TGTTAGCCGTCTCGTTGTTG-3′), and Rp49 (F, 5′-ATCGGTTACGGATCAAACAA-3′; R, 5′-GACAATCTCCTTGCGCTTC-3′). For hFic/HYPE and CHOP mRNA analysis, total RNA was extracted from HEK293T cells instead of S2 cells, and GAPDH was used as an internal control. The primers used were hFic/HYPE (F, 5′-ATTGACCATCTCACCCTACCA-3′; R, 5′-ATGTGCCTGATTTCCGAGAGG-3′), CHOP (F, 5′-GCACCTCCCAGAGCCCTCACTCTCC-3′; R, 5′-GTCTACTCCAAGCCTTCCCCCTGCG-3′), and GAPDH (F, 5′-CCATGAGAAGTATGACAACAGCC-3′; R, 5′-GGGTGCTAAGCAGTTGGTG-3′).
LC-MS/MS
Proteins contained in SDS-PAGE gel bands were reduced with DTT and alkylated with iodoacetamide (Sigma) followed by digestion overnight with trypsin (Promega, Madison WI). After concentration and de-salting on an HLB μ-elution plate (Waters Corp., Milford, MA), LC-MS/MS analysis was performed using an Ultimate3000 RSLCnano liquid chromatography system (Dionex, Sunnyvale, CA) coupled to a Q Exactive mass spectrometer (Thermo Fisher Scientific). Samples were loaded onto a 50-μm inner diameter, 15-cm length ES801 PepMap RSLC C18 EASY-Spray column (Thermo Fisher Scientific). A 60-min linear analytical gradient of 2–28% acetonitrile in water with 0.1% formic acid was used to elute peptides into the MS. The Q Exactive was operated using a data-dependent acquisition method. MS spectra were acquired at 70K resolution. Up to 10 MS/MS fragment spectra were acquired per cycle at 17.5K resolution using higher energy collision-induced dissociation.
Raw data were converted to peak lists with ProteoWizard msconvert (Version 3.0.3535) using vendor centroiding of MS2 spectra and the MS2Denoise filter. Peptide and Protein ID was performed using the Central Proteomics Facilities Pipeline (CPFP) Version 2.0.3, in which database searches using X!Tandem (Version 2008.12.01.1) and OMSSA (Version 2.1.8) were combined with iProphet and ProteinProphet (from the Trans-Proteomic Pipeline Version 4.5.2). Searches were performed against release 2012_07 of the UniProtKB D. melanogaster whole proteome sequence database, with common contaminants from cRAP and reversed decoy sequences appended. Tryptic enzyme specificity was used with up to three missed cleavages permitted per peptide. Precursor and fragment tolerances were 20 ppm and 0.1 Da, respectively. Carbamidomethylation of Cys was selected as a fixed modification, and oxidation of Met plus AMPylation of Thr were specified as a variable modifications (Unimod definition phosphoadenosine (T)). Protein identifications were grouped by ProteinProphet to resolve sequence ambiguity and filtered to a 1% false discovery rate at the protein level using decoy identifications. Furthermore, two unique peptide sequences were required per protein.
RESULTS
BiP Is Identified as a Substrate for dFic
Our previous finding that dfic deletion in Drosophila results in blindness suggests that the molecular target of dFic could be a component of a visual signaling pathway. However, as dFic is localized in the ER (15), we postulated that there might be more common and ubiquitous substrates for dFic and used S2 cells to identify substrates using a biochemical approach. In an attempt to enrich the substrates that are also located in the ER, we performed a pulldown assay with S2 cell lysate using the lectin concanavalin A, a procedure commonly used to purify glycoproteins or glycolipids. We observed a protein of ∼72 kDa in glycoprotein-enriched lysate that could be AMPylated by recombinant dFic carrying a constitutive activating mutation, E247G, in an in vitro labeling assay with [α-32P]ATP (Fig. 1A). The E247G mutation releases the intramolecular autoinhibition within the catalytic Fic motif, resulting in a protein with significantly enhanced enzymatic activity including robust autoAMPylation (43).
FIGURE 1.
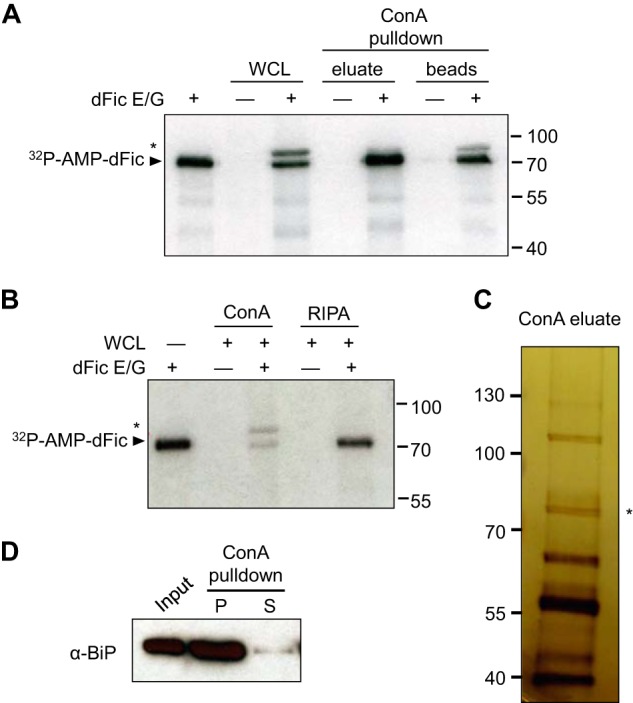
BiP is identified as a substrate for dFic. A, recombinant GST-dFicΔ70 E247G (dFic E/G) was incubated with S2 whole cell lysate and the ConA-bound fraction of lysate in the presence of α-32P-labeled ATP. The arrowhead indicates the autoAMPylated dFic, and the asterisk marks the putative substrate of the ∼72-kDa from the lysate labeled with dFic. WCL, whole cell lysate. B, GST-dFicΔ70 E247G was incubated with S2 whole cell lysate prepared with either standard radioimmune precipitation assay buffer (RIPA) or RIPA buffer added with 5 mm of Mg2+, Mn2+, and Ca2+ (ConA), which is required for concanavalin A binding. C, concanavalin A-bound proteins were eluted from the beads, concentrated, and analyzed by silver stain after SDS-PAGE. The asterisk indicates a single band with the size corresponding to the putative substrate of dFic observed from previous AMPylation assays. D, S2 cell lysate was incubated with ConA-agarose, and the bound (P) and unbound (S) proteins were analyzed by anti-BiP.
We also observed the labeling of a ∼72-kDa band in raw whole cell lysates that had not been previously observed. It is possible that the addition of high cation concentration (Mg2+, Mn2+, and Ca2+) to the lysate, which is required for concanavalin A binding, increases the catalytic reaction by better mimicking the ER environment. Indeed, the AMPylation of the ∼72-kDa protein from the whole cell lysate was observed only when the buffer with high cation contents (ConA buffer) was used to lyse the cells but not with standard radioimmune precipitation assay buffer (Fig. 1B). To identify this protein, glycoprotein-enriched S2 lysate was visualized by silver stain SDS-PAGE, and a single band was observed around 72 kDa (Fig. 1C). MS analysis revealed this protein as heat shock protein 70 cognate 3, also known as BiP. We confirmed that BiP was indeed highly enriched during concanavalin A purification (Fig. 1D). Because BiP is not a glycoprotein itself, the detection of BiP from concanavalin A beads is likely due to its indirect binding to other glycosylated proteins.
Recombinant BiP Is AMPylated by dFic in Vitro
Due to their shared ER localization, BiP appeared to be a possible substrate candidate for dFic. To test whether BiP can be AMPylated by dFic, recombinant BiP with the His6 tag at the N terminus was purified from E. coli and used as a substrate in the in vitro AMPylation assay. We observed significant radiolabeling of both BiP and dFic E247G (Fig. 2A). The catalytic reaction was enhanced in the presence of calcium ions, which was consistent with the previous finding that BiP undergoes in vitro phosphorylation in a calcium-dependent manner (29). BiP was also AMPylated when wild-type dFic was used, albeit with significantly weaker activity (Fig. 2B). This suggests that the labeling mediated by dFic is not likely to be caused by an artifact due to the E247G mutation. Mutating the conserved histidine residue in the Fic motif to an alanine (H375A) abolished the catalytic activity of dFic. Furthermore, human Fic (FicD or HYPE) E234G was also able to efficiently modify human BiP (Fig. 2C).
FIGURE 2.
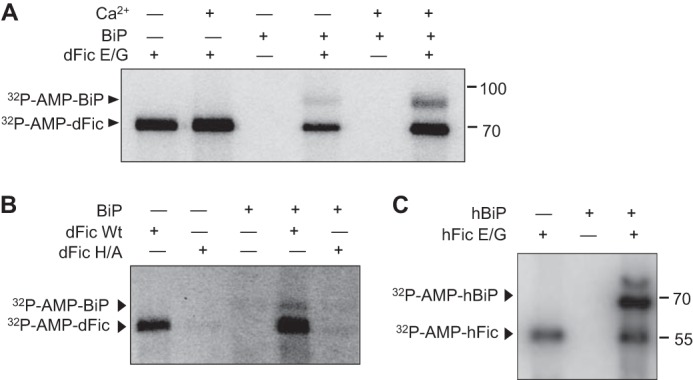
Recombinant BiP is AMPylated by dFic in vitro. A, recombinant His-BiP was used as a substrate for dFic in the AMPylation assay in the presence or absence of 5 mm Ca2+. Arrowheads indicate the AMPylated proteins. B, BiP is AMPylated by wild-type dFic. GST-dFicΔ70 (dFic Wt) and GST-dFicΔ70 H375A (dFic H/A) were incubated with His-BiP in the presence of α-32P-labeled ATP. Arrowheads indicate the AMPylated proteins. C, human version of His-SUMO-FicDΔ47 E234G (hFic) and His-BiPΔ19 (hBiP) were used for the AMPylation assay. dFic E/G, GST-dFicΔ70 E247G.
AMPylation of BiP Is Modulated by ER Stress
As BiP is an important sensor and regulator for the UPR, we analyzed whether the AMPylation status of BiP is differentially regulated when cells are undergoing ER stress. We treated S2 cells with tunicamycin, a pharmacological inducer of ER stress, for different times and blotted the whole cell lysate with anti-AMP-Thr antibody (42). A protein with the size of BiP was AMPylated in S2 cells, but AMPylation started to decrease within 30 min of tunicamycin treatment and was completely absent after 24 h (Fig. 3A). The same trend was observed when DTT was used to trigger ER stress (Fig. 3B), supporting the hypothesis that the level of AMPylated protein recognized by anti-AMP-Thr antibody decreases as more misfolded proteins accumulate in the ER. We then treated the cells with cycloheximide, which reduces the load of unfolded proteins in the ER by decreasing the amount of newly synthesized proteins. The signal of the ∼72-kDa band detected by anti-AMP-Thr antibody was increased upon cycloheximide treatment, in contrast to the effect observed with the ER stress inducers (Fig. 3C).
FIGURE 3.

AMPylation of BiP is modulated by ER stress. A, pharmacological ER stress inducer tunicamycin (Tm) was treated to S2 cells for the indicated times at 2 μg/ml. Whole cell lysates from different time points were analyzed by anti-BiP and anti-Thr-AMP. Anti-tubulin was used as a loading control. The asterisk marks the AMPylated protein from cell lysate with ∼72 kDa. B, 5 mm DTT was used to trigger ER stress in S2 cells for 1 h, and the whole cell lysates were analyzed with the indicated antibodies. Anti-phospho-eIF2α was used as a marker for ER stress. C, 5 mm DTT and 100 μg/ml cycloheximide (CHX), a potent inhibitor of protein synthesis, was added to S2 cells for 4 h. Whole cell lysates were analyzed with indicated antibodies. D, cells were transfected with BiP containing a C-terminal FLAG tag followed by DTT and cycloheximide treatment for 4 h. BiP was then immunoprecipitated with anti-FLAG-agarose and analyzed with anti-BiP and anti-Thr-AMP. Immunoprecipitation was confirmed by anti-FLAG. E, cells were treated with 5 mm DTT and/or 100 μg/ml cycloheximide in the presence or absence of 20 μm MG132 for 4 h.
To confirm that the protein being regulated by AMPylation was BiP, we treated cells transfected with FLAG-tagged BiP with DTT or cycloheximide, immunoprecipitated BiP, and analyzed using the anti-AMP-Thr antibody. As predicted, BiP was AMPylated in untreated cells, whereas lower and higher levels of AMPylation were observed in DTT-treated and cycloheximide-treated cells, respectively (Fig. 3D). The results followed the same pattern of modification shown in Fig. 3C for the S2 whole cell lysate, confirming BiP as the relevant band observed. The level of AMPylated BiP was low when cells were treated with both DTT and cycloheximide (Fig. 3E). Blocking protein degradation with MG132 did not affect the level of AMPylated BiP, which indicates that the decrease of BiP AMPylation upon ER stress is not due to protein degradation. BiP needs to be acutely activated upon accumulation of misfolded proteins caused by ER stress, whereas it needs to stay inactive in the absence of unfolded protein loads. Our results show that the AMPylation status of BiP inversely correlates with its active state during ER homeostasis, suggesting the potential inhibitory mechanism of AMPylation on BiP.
AMPylation of BiP Is a Reversible Event
Our data support the hypothesis that AMPylation occurs upon the reduction of unfolded protein load. To explore the reversibility of AMPylation upon different cellular states, we induced ER stress in cells, removed the stress inducer, and monitored the AMPylation state of BiP over time as the cells recovered from stress. After 1 h of DTT treatment the cells were washed and replated with fresh media to eliminate DTT. As cells slowly recovered from the potent induction of ER stress, we observed that the AMPylation of BiP became visible again after 8 h (Fig. 4A). When cycloheximide was added to the media so that only the existing BiP could be monitored, we still observed a recovery of AMPylated BiP at 8 h (Fig. 4B). This shows that AMPylation is a reversible modification that readily adapts to the fluctuation of unfolded proteins in the ER.
FIGURE 4.
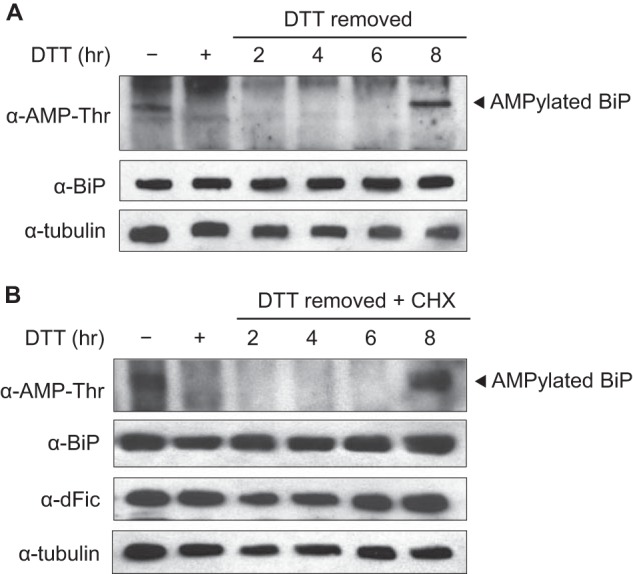
AMPylation of BiP is a reversible event. S2 cells were treated with 5 mm DTT for 1 h for ER stress induction. The cells were then washed and replated with fresh media to remove DTT and left untreated (A) or treated with 100 μg/ml cycloheximide (CHX; B). AMPylation of BiP, indicated by an arrowhead, was then monitored over time from the whole cell lysate using anti-AMP-Thr antibody.
dFic Is Transcriptionally Up-regulated by ER Stress
Upon ER stress, general translation is attenuated to reduce the synthesis of new proteins, whereas a number of chaperones and ER-associated degradation pathway-associated genes are up-regulated to cope with the accumulation of misfolded proteins. BiP is also transcriptionally activated along with many other chaperone genes during ER stress. By modifying this important regulator of UPR, dFic is likely to be involved in the same pathway. To test this we measured the mRNA level of dFic and BiP in S2 cells upon ER stress induction using DTT, tunicamycin, or thapsigargin. Both dFic and BiP mRNAs were significantly increased by ER stress (Fig. 5A). Ero1L, which encodes a protein that maintains the oxidative environment of the ER, was also induced consistent with previous studies (44). Human Fic from HEK293T cells was also induced by ER stress along with CHOP (Fig. 5B), a UPR gene that induces apoptotic signaling under prolonged cellular stress (45). These results further support the proposal that AMPylation mediated by dFic plays a conserved role in the UPR pathway.
FIGURE 5.

dFic is transcriptionally up-regulated by ER stress. A, S2 cells were treated with 2 μg/ml tunicamycin (Tm) or 5 mm DTT for 4 h, and mRNA levels of dFic, BiP, and Ero1L were measured by quantitative real-time PCR. B, HEK293T cells were treated with 2 μg/ml tunicamycin or 1 μm thapsigargin (Tg) for 6 h, and the mRNA levels of hFic and CHOP were measured by quantitative real-time PCR.
AMPylation Site Maps to Thr-366 in the ATPase Domain of BiP
To elucidate where BiP is modified with AMP, three different truncation mutants of BiP were generated. Constructs containing an N-terminal signal sequence-deleted (Δ26) BiP, which mimics the processed form of BiP in the ER (46), the BiP ATPase domain (ATPase), or the BiP substrate binding domain were expressed and purified from E. coli. We found that the ATPase domain could be AMPylated more efficiently than BiPΔ26, whereas no AMPylation was observed with the substrate binding domain (Fig. 6). In addition to the radiolabeling assay, the anti-AMP-Thr antibody was used to confirm the AMPylation of the BiP ATPase domain (Fig. 7A). To identify the AMPylated residue in BiP, we conducted LC-MS/MS analysis on recombinantly AMPylated BiP. Threonine 166 and 366 appeared to be the most probable candidates based on the preliminary MS analysis (Table 1).
FIGURE 6.
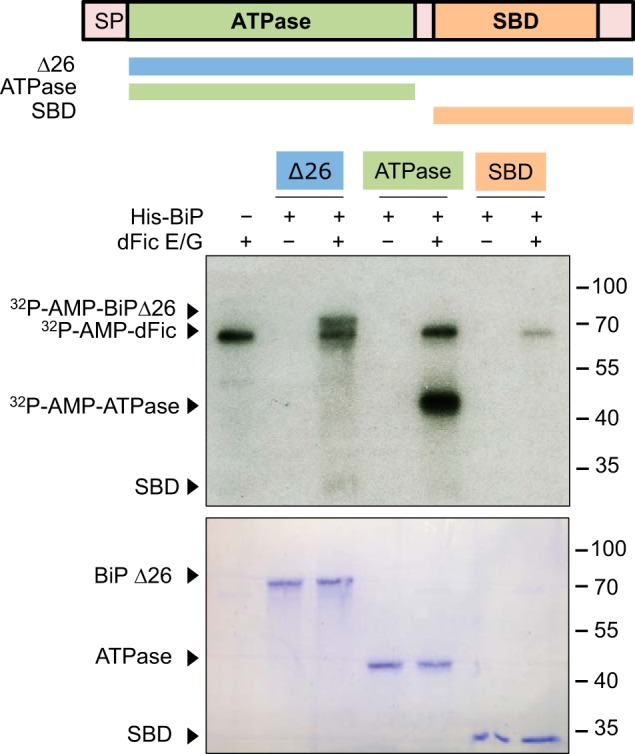
AMPylation occurs on the ATPase domain of BiP. Domain structure of BiP and different protein constructs generated are shown. These constructs were purified from E. coli with N-terminal His6 tag and used as substrates for GST-dFicΔ70 E247G (dFic E/G) in the AMPylation assay. SP, signal peptide. SBD, substrate binding domain.
FIGURE 7.

AMPylation site maps to Thr-366 in the ATPase domain of BiP. A, His-BiP ATPase was incubated with GST-dFicΔ70 E247G (dFic E/G) and cold ATP and analyzed by anti-AMP-Thr. Arrowheads mark the AMPylated proteins. B, various constructs of BiP with a C-terminal FLAG tag followed by ER retention signal KDEL were transfected to S2 cells and immunoprecipitated (IP) using anti-FLAG-agarose. Purified BiP was analyzed by anti-Thr-AMP. Immunoprecipitation was confirmed by anti-FLAG. WB, Western blot. C, BiP wild-type and ADP-ribosylation mutant R470K were expressed and immunoprecipitated from S2 cells and analyzed by anti-AMP-Thr antibody. D, ATPase activity of the recombinant BiP ATPase domain (27–417) wild-type and T366A was measured. E, multiple sequence alignment of BiP homologs from different species. Thr-366 identified as a putative AMPylation site is highly conserved in all homologs. F, structural view of the ATPase domain of human BiP reveals that Thr-366 is located nearby the ATP binding site (PDB code 3LDL).
TABLE 1.
Putative modification sites for AMPylation detected from ES-MS/MS
Full-length His-BiP was coexpressed with GST-dFicΔ70 E247G in E. coli, and BiP was purified using nickel affinity chromatography and analyzed with LC-MS/MS. Peptides containing Thr-166 and Thr-366 showed a mass increase of 329 daltons, which corresponds to the size of AMP. Peptide spectrum match shows the peptides where posttranslational modification was identified. The last column shows a score for the peptide match and the probability for the peptide indentification that supports the localization of the posttranslational modification.

To establish the in vivo site of AMPylation on BiP, we transfected S2 cells with C-terminally FLAG-tagged BiP containing threonine mutations and used the anti-AMP-Thr antibody to monitor the AMPylation of immunoprecipitated BiP. Mutation of Thr-366 to alanine completely abolished the AMPylation of BiP, suggesting it is the sole in vivo site of modification (Fig. 7B). BiP was previously reported to undergo ADP-ribosylation, so to rule out the possibility that the anti-AMP-Thr antibody was detecting ADP-ribosylation we made an ADP-ribosylation defective R470K mutant (35). The anti-AMP-Thr still recognized the transfected R470K mutant of BiP, confirming the specificity of the antibody for AMPylation (Fig. 7C). In support of Thr-366 being a legitimate AMPylation site rather than a cause of structural perturbation, BiP T366A was shown to be a functional ATPase (Fig. 7D). Thr-366 was also found to be highly conserved in all heat shock protein 70 homologs of other species (Fig. 7E). The published structure of BiP reveals that Thr-366 is in close proximity to the ATP binding pocket, raising the possibility that AMPylation, a bulky post-translational modification, might hinder ATP hydrolysis (Fig. 7F). This mechanism would be similar to that observed for glutamine synthetase, the first protein observed to be regulated by AMPylation (2).
AMPylation of BiP by dFic Correlates with the Inactive State of BiP
We next assessed the relationship between AMPylation and the molecular activity of BiP. Due to the complexity of protein folding assays and the fact that both enzymes use ATP as a substrate, definitively proving that BiP is inhibited by dFic AMPylation is beyond the scope of this study. Previous investigators demonstrated that the ATPase domain alone exhibits poor ATPase activity, whereas the addition of a short hydrophobic interdomain linker to the ATPase domain results in robust ATPase activity (47). The ATPase domain of BiP undergoes cycles of ATP binding, hydrolysis, and nucleotide exchange that are tightly coupled to substrate binding and release (28). The linker binding mimics this allosteric coupling of the two domains of BiP. Without the linker, BiP loses the intradomain interaction crucial for its functional cycle and remains inactive. Because the ATPase domain we previously used for the AMPylation assay did not include the linker region (BiP-(27–407)), we created another construct that carries the linker by adding 10 additional amino acids (BiP-(27–417)). These proteins were tagged with N-terminal His6 and purified from E. coli. In line with the previous finding, BiP-(27–417) showed significantly increased ATPase activity compared with BiP-(27–407) (Fig. 8A). We then used these recombinant proteins as substrates for the AMPylation assay. The inactive ATPase domain, BiP-(27–407), was efficiently modified by dFic, whereas the active BiP-(27–417) was a very poor substrate for dFic (Fig. 8B). The observation that Fic AMPylation occurs efficiently on the inactive form of the ATPase domain and inefficiently with the active state of BiP correlates to our finding in cells that the level of AMPylation was high in normal cells and low upon the increased load of unfolded proteins.
FIGURE 8.
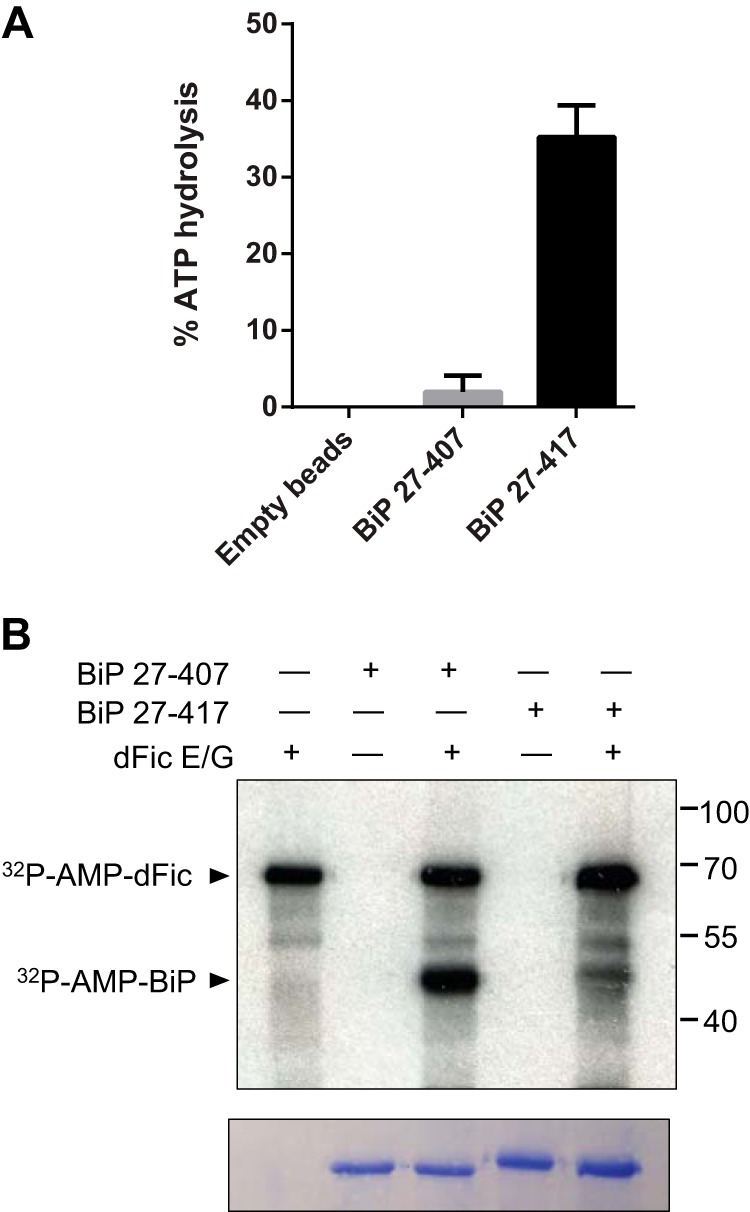
AMPylation of BiP by dFic correlates with the inactive state of BiP. A, ATPase activity of the recombinant BiP-(27–407) and BiP-(27–417). His-BiP-(27–407) and His-BiP-(27–417) (carrying an interdomain linker) proteins were bound to nickel-nitrilotriacetic acid beads and assessed for the ATPase activity. Empty beads were used as a negative control. B, His-BiP-(27–407) and His-BiP-(27–417) were used as substrates for dFic-mediated AMPylation. Arrowheads mark the AMPylated proteins by dFic. dFic E/G, GST-dFicΔ70 E247G.
AMPylation of BiP Is Reversibly Regulated during ER Homeostasis
To gain insight into the amount of modified BiP in cells, we used lysates from unstressed cells and cells stressed with DTT as substrates for the AMPylation assay. As shown above, the level of in vivo AMPylated BiP declines in the presence of ER stress as detected by the anti-AMP-Thr antibody (Fig. 9A). The same amounts of lysates used in this immunoblotting were then used as substrates for the AMPylation assay (Fig. 9B). Consistent with the high degree of in vivo AMPylation, only minimal additional in vitro labeling was observed for BiP from unstressed cells, indicating a low availability of free BiP. By contrast, considerable in vitro labeling was observed for BiP from stressed cells (Fig. 9B), consistent with our observations that BiP is deAMPylated in DTT-treated cells (Fig. 9A). Altogether, we show that when cells are undergoing ER stress and misfolded proteins accumulate, BiP is rapidly deAMPylated; however, in the absence of newly synthesized proteins or upon low unfolded protein load, BiP is AMPylated. Hence, AMPylation is likely to regulate the activity of BiP depending on the state of newly synthesized/misfolded protein loads in the ER (Fig. 9C).
FIGURE 9.
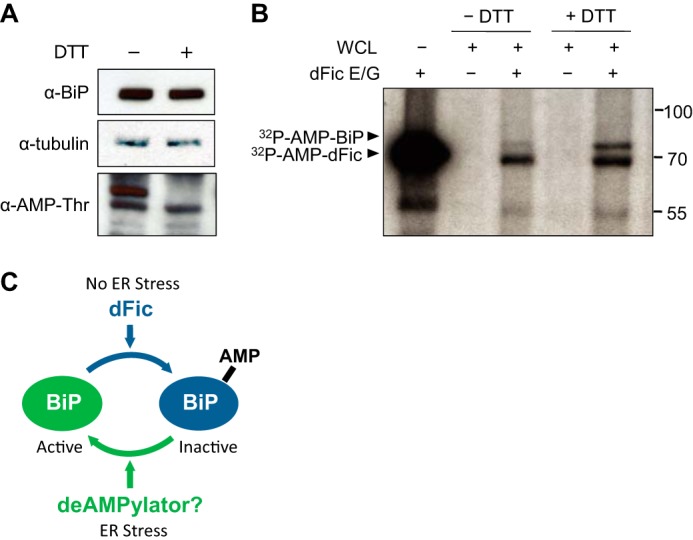
AMPylation of BiP is reversibly regulated during ER homeostasis. A, in vivo AMPylation of BiP was measured in unstressed cells and cells stressed with 5 mm DTT for 1 h using anti-AMP-Thr antibody. B, equal amounts of lysates used for immunoblotting in A were used as substrates for the AMPylation assay with GST-dFicΔ70 E247G (dFic E/G). Arrowheads mark the AMPylated proteins. C, model figure of AMPylation of BiP modulated by ER stress. When cells undergo ER stress and accumulate misfolded proteins, BiP gets deAMPylated and becomes active as the unmodified form. In the absence of ER stress and upon the reduction of unfolded protein load, BiP undergoes AMPylation by dFic, which renders it inactive. This reversible event of AMPylation ensures a rapid on/off regulation of BiP to maintain protein homeostasis in the ER.
DISCUSSION
AMPylation appears to be a promising posttranslational regulatory mechanism adopted by organisms of varying complexity due to the high conservation of Fic domains and its use of ATP as a substrate. Bacterial AMPylators have already been shown to play an essential role in hijacking host signaling pathways during pathogenesis, but the endogenous function of AMPylation in eukaryotes remained elusive. Our study presents BiP, a well known ER chaperone and a major regulator of UPR, as the first substrate of AMPylation by a eukaryotic protein. BiP from the whole S2 cell lysate was predominantly labeled by dFic enzyme in the presence of divalent cations Mg2+, Mn2+, and Ca2+ (Fig. 1A). Calcium appears to promote a catalytic reaction as shown by Fig. 2A. Indeed, ER is a major organelle for calcium storage with the concentration of calcium ions in the lumen in the millimolar range (48). Therefore, having calcium ions in the reaction may better mimic the chemical environment in the ER. It was also reported that Hsp70 chaperones bind to calcium ions, which may stabilize the protein structure (49). The stable conformation of BiP achieved by calcium binding may render it a better substrate for dFic.
The essential role of BiP in ER stress response and many cellular and pathological processes supports the notion that there may be multiple mechanisms for its regulation. Upon ER stress, BiP is transcriptionally activated to assist in the folding of high levels of unfolded proteins. However, the dynamic fluctuation of unfolded protein loads in the ER due to stress response or alternating rates of protein synthesis may require a more rapid regulation of BiP. This is also supported by the discrepancy between the long half-life of BiP, which is up to 48 h, and its excess activity being detrimental to cells, as maturation and secretion of critical proteins can be significantly delayed (50, 51). A post-translational modification provides an ideal mechanism to readily activate and deactivate BiP upon changes in unfolded protein loads in the ER. Previously, ADP-ribosylation has been suggested to inactivate BiP by attenuating substrate binding and interfering with the allosteric coupling between domains (35). In contrast to AMPylation, which we observed in the ATPase domain, ADP-ribosylation occurs in the substrate binding domain on arginine residues (Arg-470 and Arg-492). Therefore, we speculate that BiP undergoes different modifications on both domains, ensuring its tight regulation at multiple levels. The exact molecular event triggering such modifications or the order in which they occur remains to be explored. Furthermore, identifying the enzyme that catalyzes the rapid deAMPylation of BiP during ER stress and understanding its functional consequence will be of great interest.
AMPylation occurs on a conserved threonine residue located near the ATP binding pocket, which suggests that AMPylation may affect the ATPase activity of BiP by either blocking nucleotide binding or inhibiting efficient ATP hydrolysis. Unfortunately, the inverse relationship of BiP suitability for AMPylation and its ATPase activity makes the measurement of activity differences caused by AMPylation technically challenging.
Another possible inhibitory mechanism of AMPylation on BiP is uncoupling of two-domain allostery, similar to the effect of ADP-ribosylation (35). Upon ATP binding, Hsp70 proteins undergo a structural change wherein the two domains come in close contact and form a compact structure (52). The addition of the bulky AMP moiety can potentially hinder the contact between the domains and thereby disrupt the functional cycle of BiP. From an intermolecular perspective, AMPylation of BiP may also alter its interaction with binding partners such as DnaJ co-chaperone (53–55) or nucleotide exchange factors (56–58).
Induction of ER stress up-regulates not only BiP but also dFic, which was a surprising result considering that AMPylation of BiP decreases during ER stress. Nevertheless, it suggests that dFic is among many UPR genes that cells use to cope with the stress response. It is possible that dFic is induced along with other chaperones, but its activity or translocation is blocked until the unfolded protein load decreases and BiP has to be promptly inactivated. dFic could then be released from repression and subsequently inactivate BiP. Otherwise, excess levels of active BiP may prolong protein maturation and secretion, which would be deleterious to cells. How dFic is regulated upon ER stress is another interesting avenue to be explored. There might be another layer of posttranslational modification governing the function of dFic. It is also fascinating to speculate that autoAMPylation observed with dFic both in vitro and in vivo might be involved in its own regulation.
Previous studies have shown that flies without functional dFic in glial cells have impaired visual neurotransmission (15). This suggests that dFic substrate could be a component of visual signaling or a transporter of neurotransmitters. Alternatively, the blind phenotype could result from a loss of regulation on BiP. We can speculate that the protein responsible for the visual signaling is not properly matured or secreted in the absence of tight regulation of BiP in dfic null flies, which thereby results in visual defect. Indeed, imbalance of protein homeostasis is a cause of many pathological processes due to accumulation of aberrant protein or impaired protein secretion (59).
Interestingly, dFic is mainly localized to the cell surface on glial cells and is particularly enriched in capitate projections in contrast to the ER localization in S2 cells. Therefore, dFic may target a different molecule on the cell surface that could directly impact neurotransmission. It is possible that in glial cells there might be cell type-specific factors that can induce the secretion of dFic from the ER to cell surface.
We show that human Fic also AMPylates BiP in vitro and that it is transcriptionally activated by ER stress. This suggests that AMPylation is a conserved regulatory mechanism in multiple species possibly involved in UPR. As both Fic and BiP are highly conserved in many organisms, it will be worth investigating the role of AMPylation in other species.
Understanding the regulatory mechanism of BiP is of utmost importance, as misregulation of BiP is associated with numerous diseases including neurological disorders and various cancers (36–40). An increased level of BiP is a critical factor for tumor progression and has been shown to confer chemoresistance to a variety of cancer cell lines (38, 40, 60, 61). Accordingly, inhibition of BiP activity is emerging as an important cancer target. The discovery of BiP AMPylation and deAMPylation presents a new targetable avenue for drug development.
Acknowledgments
We thank members of the Orth laboratory and Lawrence Lum for helpful discussion and assistance. We acknowledge the services of the UT Southwestern Proteomics Core, supported by Cancer Prevention and Research Institute of Texas Grant RP120613 (to Drs. David Trudgian and Hamid Mirzaei).
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 EY021922 and R01-AI056404 (to K. O.) and Cell and Molecular Biology Training Grant 5T32GM008203 (to A. R. W.). This work was also supported by Welch Research Foundation Grant I-1561.
- GS
- glutamine synthetase
- dFic
- Drosophila Fic
- ConA
- concanavalin A
- UPR
- unfolded protein response
- hFic
- human Fic
- ER
- endoplasmic reticulum.
REFERENCES
- 1. Woolery A. R., Luong P., Broberg C. A., Orth K. (2010) AMPylation: something old is new again. Front Microbiol. 1, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shapiro B. M., Kingdon H. S., Stadtman E. R. (1967) Regulation of glutamine synthetase. VII. Adenylyl glutamine synthetase: a new form of the enzyme with altered regulatory and kinetic properties. Proc. Natl. Acad. Sci. U.S.A. 58, 642–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kingdon H. S., Shapiro B. M., Stadtman E. R. (1967) Regulation of glutamine synthetase. 8. ATP: glutamine synthetase adenylyltransferase, an enzyme that catalyzes alterations in the regulatory properties of glutamine synthetase. Proc. Natl. Acad. Sci. U.S.A. 58, 1703–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yarbrough M. L., Li Y., Kinch L. N., Grishin N. V., Ball H. L., Orth K. (2009) AMPylation of Rho GTPases by Vibrio VopS disrupts effector binding and downstream signaling. Science 323, 269–272 [DOI] [PubMed] [Google Scholar]
- 5. Woolery A. R., Yu X., LaBaer J., Orth K. (2014) AMPylation of Rho GTPases subverts multiple host signaling processes. J. Biol. Chem. 289, 32977–32988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Worby C. A., Mattoo S., Kruger R. P., Corbeil L. B., Koller A., Mendez J. C., Zekarias B., Lazar C., Dixon J. E. (2009) The fic domain: regulation of cell signaling by adenylylation. Mol. Cell 34, 93–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Müller M. P., Peters H., Blümer J., Blankenfeldt W., Goody R. S., Itzen A. (2010) The Legionella effector protein DrrA AMPylates the membrane traffic regulator Rab1b. Science 329, 946–949 [DOI] [PubMed] [Google Scholar]
- 8. Tan Y., Luo Z. Q. (2011) Legionella pneumophila SidD is a deAMPylase that modifies Rab1. Nature 475, 506–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mukherjee S., Liu X., Arasaki K., McDonough J., Galán J. E., Roy C. R. (2011) Modulation of Rab GTPase function by a protein phosphocholine transferase. Nature 477, 103–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Feng F., Yang F., Rong W., Wu X., Zhang J., Chen S., He C., Zhou J. M. (2012) A Xanthomonas uridine 5′-monophosphate transferase inhibits plant immune kinases. Nature 485, 114–118 [DOI] [PubMed] [Google Scholar]
- 11. Castro-Roa D., Garcia-Pino A., De Gieter S., van Nuland N. A., Loris R., Zenkin N. (2013) The Fic protein Doc uses an inverted substrate to phosphorylate and inactivate EF-Tu. Nat. Chem. Biol. 9, 811–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cruz J. W., Rothenbacher F. P., Maehigashi T., Lane W. S., Dunham C. M., Woychik N. A. (2014) Doc toxin is a kinase that inactivates elongation factor Tu. J. Biol. Chem. 289, 7788–7798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garcia-Pino A., Zenkin N., Loris R. (2014) The many faces of Fic: structural and functional aspects of Fic enzymes. Trends Biochem. Sci. 39, 121–129 [DOI] [PubMed] [Google Scholar]
- 14. Kinch L. N., Yarbrough M. L., Orth K., Grishin N. V. (2009) Fido, a novel AMPylation domain common to fic, doc, and AvrB. PLoS ONE 4, e5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rahman M., Ham H., Liu X., Sugiura Y., Orth K., Krämer H. (2012) Visual neurotransmission in Drosophila requires expression of Fic in glial capitate projections. Nat. Neurosci. 15, 871–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Munro S., Pelham H. R. (1986) An Hsp70-like protein in the ER: identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell 46, 291–300 [DOI] [PubMed] [Google Scholar]
- 17. Haas I. G. (1994) BiP (GRP78), an essential hsp70 resident protein in the endoplasmic reticulum. Experientia 50, 1012–1020 [DOI] [PubMed] [Google Scholar]
- 18. Gething M. J. (1999) Role and regulation of the ER chaperone BiP. Semin Cell Dev. Biol. 10, 465–472 [DOI] [PubMed] [Google Scholar]
- 19. Corsi A. K., Schekman R. (1997) The lumenal domain of Sec63p stimulates the ATPase activity of BiP and mediates BiP recruitment to the translocon in Saccharomyces cerevisiae. J. Cell Biol. 137, 1483–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McClellan A. J., Endres J. B., Vogel J. P., Palazzi D., Rose M. D., Brodsky J. L. (1998) Specific molecular chaperone interactions and an ATP-dependent conformational change are required during posttranslational protein translocation into the yeast ER. Mol. Biol. Cell 9, 3533–3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brodsky J. L., McCracken A. A. (1997) ER-associated and proteasome-mediated protein degradation: how two topologically restricted events came together. Trends Cell Biol. 7, 151–156 [DOI] [PubMed] [Google Scholar]
- 22. Vembar S. S., Brodsky J. L. (2008) One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ruggiano A., Foresti O., Carvalho P. (2014) Quality control: ER-associated degradation: protein quality control and beyond. J. Cell Biol. 204, 869–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Olzmann J. A., Kopito R. R., Christianson J. C. (2013) The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb. Perspect. Biol. 5, a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hiller M. M., Finger A., Schweiger M., Wolf D. H. (1996) ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science 273, 1725–1728 [DOI] [PubMed] [Google Scholar]
- 26. Gardner B. M., Pincus D., Gotthardt K., Gallagher C. M., Walter P. (2013) Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 5, a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mori K. (2009) Signalling pathways in the unfolded protein response: development from yeast to mammals. J. Biochem. 146, 743–750 [DOI] [PubMed] [Google Scholar]
- 28. Bukau B., Weissman J., Horwich A. (2006) Molecular chaperones and protein quality control. Cell 125, 443–451 [DOI] [PubMed] [Google Scholar]
- 29. Leustek T., Toledo H., Brot N., Weissbach H. (1991) Calcium-dependent autophosphorylation of the glucose-regulated protein, Grp78. Arch. Biochem. Biophys. 289, 256–261 [DOI] [PubMed] [Google Scholar]
- 30. Gaut J. R., Hendershot L. M. (1993) The immunoglobulin-binding protein in vitro autophosphorylation site maps to a threonine within the ATP binding cleft but is not a detectable site of in vivo phosphorylation. J. Biol. Chem. 268, 12691–12698 [PubMed] [Google Scholar]
- 31. Gaut J. R. (1997) In vivo threonine phosphorylation of immunoglobulin binding protein (BiP) maps to its protein binding domain. Cell Stress Chaperones 2, 252–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carlsson L., Lazarides E. (1983) ADP-ribosylation of the Mr 83,000 stress-inducible and glucose-regulated protein in avian and mammalian cells: modulation by heat shock and glucose starvation. Proc. Natl. Acad. Sci. U.S.A. 80, 4664–4668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leno G. H., Ledford B. E. (1990) Reversible ADP-ribosylation of the 78-kDa glucose-regulated protein. FEBS Lett. 276, 29–33 [DOI] [PubMed] [Google Scholar]
- 34. Ledford B. E., Leno G. H. (1994) ADP-ribosylation of the molecular chaperone GRP78/BiP. Mol. Cell Biochem. 138, 141–148 [DOI] [PubMed] [Google Scholar]
- 35. Chambers J. E., Petrova K., Tomba G., Vendruscolo M., Ron D. (2012) ADP ribosylation adapts an ER chaperone response to short-term fluctuations in unfolded protein load. J. Cell Biol. 198, 371–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pfaffenbach K. T., Lee A. S. (2011) The critical role of GRP78 in physiologic and pathologic stress. Curr. Opin. Cell Biol. 23, 150–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weng W. C., Lee W. T., Hsu W. M., Chang B. E., Lee H. (2011) Role of glucose-regulated Protein 78 in embryonic development and neurological disorders. J. Formos. Med. Assoc. 110, 428–437 [DOI] [PubMed] [Google Scholar]
- 38. Wang M., Wey S., Zhang Y., Ye R., Lee A. S. (2009) Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 11, 2307–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mei Y., Thompson M. D., Cohen R. A., Tong X. (2013) Endoplasmic reticulum stress and related pathological processes. J. Pharmacol. Biomed. Anal. 1, 1000107. [PMC free article] [PubMed] [Google Scholar]
- 40. Li J., Lee A. S. (2006) Stress induction of GRP78/BiP and its role in cancer. Curr. Mol. Med. 6, 45–54 [DOI] [PubMed] [Google Scholar]
- 41. Ceriani M. F. (2007) Basic protocols for Drosophila S2 cell line: maintenance and transfection. Methods Mol. Biol. 362, 415–422 [DOI] [PubMed] [Google Scholar]
- 42. Hao Y. H., Chuang T., Ball H. L., Luong P., Li Y., Flores-Saaib R. D., Orth K. (2011) Characterization of a rabbit polyclonal antibody against threonine-AMPylation. J. Biotechnol. 151, 251–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Engel P., Goepfert A., Stanger F. V., Harms A., Schmidt A., Schirmer T., Dehio C. (2012) Adenylylation control by intra- or intermolecular active-site obstruction in Fic proteins. Nature 482, 107–110 [DOI] [PubMed] [Google Scholar]
- 44. Pollard M. G., Travers K. J., Weissman J. S. (1998) Ero1p: a novel and ubiquitous protein with an essential role in oxidative protein folding in the endoplasmic reticulum. Mol. Cell 1, 171–182 [DOI] [PubMed] [Google Scholar]
- 45. Zinszner H., Kuroda M., Wang X., Batchvarova N., Lightfoot R. T., Remotti H., Stevens J. L., Ron D. (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Petrova K., Oyadomari S., Hendershot L. M., Ron D. (2008) Regulated association of misfolded endoplasmic reticulum lumenal proteins with P58/DNAJc3. EMBO J. 27, 2862–2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Swain J. F., Dinler G., Sivendran R., Montgomery D. L., Stotz M., Gierasch L. M. (2007) Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol. Cell 26, 27–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Koch G. L. (1990) The endoplasmic reticulum and calcium storage. Bioessays 12, 527–531 [DOI] [PubMed] [Google Scholar]
- 49. Sriram M., Osipiuk J., Freeman B., Morimoto R., Joachimiak A. (1997) Human Hsp70 molecular chaperone binds two calcium ions within the ATPase domain. Structure 5, 403–414 [DOI] [PubMed] [Google Scholar]
- 50. Dorner A. J., Wasley L. C., Kaufman R. J. (1992) Overexpression of GRP78 mitigates stress induction of glucose regulated proteins and blocks secretion of selective proteins in Chinese hamster ovary cells. EMBO J. 11, 1563–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Feder J. H., Rossi J. M., Solomon J., Solomon N., Lindquist S. (1992) The consequences of expressing hsp70 in Drosophila cells at normal temperatures. Genes Dev. 6, 1402–1413 [DOI] [PubMed] [Google Scholar]
- 52. Zhuravleva A., Clerico E. M., Gierasch L. M. (2012) An interdomain energetic tug-of-war creates the allosterically active state in Hsp70 molecular chaperones. Cell 151, 1296–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chevalier M., Rhee H., Elguindi E. C., Blond S. Y. (2000) Interaction of murine BiP/GRP78 with the DnaJ homologue MTJ1. J. Biol. Chem. 275, 19620–19627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shen Y., Meunier L., Hendershot L. M. (2002) Identification and characterization of a novel endoplasmic reticulum (ER) DnaJ homologue, which stimulates ATPase activity of BiP in vitro and is induced by ER stress. J. Biol. Chem. 277, 15947–15956 [DOI] [PubMed] [Google Scholar]
- 55. Qiu X. B., Shao Y. M., Miao S., Wang L. (2006) The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell. Mol. Life Sci. 63, 2560–2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chung K. T., Shen Y., Hendershot L. M. (2002) BAP, a mammalian BiP-associated protein, is a nucleotide exchange factor that regulates the ATPase activity of BiP. J. Biol. Chem. 277, 47557–47563 [DOI] [PubMed] [Google Scholar]
- 57. Dragovic Z., Broadley S. A., Shomura Y., Bracher A., Hartl F. U. (2006) Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. EMBO J. 25, 2519–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Andréasson C., Rampelt H., Fiaux J., Druffel-Augustin S., Bukau B. (2010) The endoplasmic reticulum Grp170 acts as a nucleotide exchange factor of Hsp70 via a mechanism similar to that of the cytosolic Hsp110. J. Biol. Chem. 285, 12445–12453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hipp M. S., Park S. H., Hartl F. U. (2014) Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 24, 506–514 [DOI] [PubMed] [Google Scholar]
- 60. Vandewynckel Y. P., Laukens D., Geerts A., Bogaerts E., Paridaens A., Verhelst X., Janssens S., Heindryckx F., Van Vlierberghe H. (2013) The paradox of the unfolded protein response in cancer. Anticancer Res. 33, 4683–4694 [PubMed] [Google Scholar]
- 61. Virrey J. J., Dong D., Stiles C., Patterson J. B., Pen L., Ni M., Schönthal A. H., Chen T. C., Hofman F. M., Lee A. S. (2008) Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Mol. Cancer Res. 6, 1268–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]