

Background: Mitochondrial glutaredoxin 2 is a member of the oxidoreductase family with disulfide reductase activity that repairs thiol oxidation-damaged proteins.
Results: Glutaredoxin 2 null mice develop early cataracts during aging.
Conclusion: Glutaredoxin 2 deletion causes mitochondrial malfunction and cataract formation and is a good model for studying the mechanism of human age-related cataracts.
Significance: Thiol oxidation repair system is essential for lens transparency.
Keywords: Age-related Cataract, Electron Transport System, Glutaredoxin 2, Oxidative Stress, α-Crystallin Protein
Abstract
Glutaredoxin 2 (Grx2) is an isozyme of glutaredoxin1 (thioltransferase) present in the mitochondria and nucleus with disulfide reductase and peroxidase activities, and it controls thiol/disulfide balance in cells. In this study, we investigated whether Grx2 gene deletion could induce faster age-related cataract formation and elucidated the biochemical changes effected by Grx2 gene deletion that may contribute to lens opacity. Slit lamp was used to examine the lenses in Grx2 knock-out (KO) mice and age-matched wild-type (WT) mice ages 1 to 16 months. In the Grx2 null mice, the lens nuclear opacity began at 5 months, 3 months sooner than that of the control mice, and the progression of cataracts was also much faster than the age-matched controls. Lenses of KO mice contained lower levels of protein thiols and GSH with a significant accumulation of S-glutathionylated proteins. Actin, αA-crystallin, and βB2-crystallin were identified by Western blot and mass spectroscopy as the major S-glutathionylated proteins in the lenses of 16-month-old Grx2 KO mice. Compared with the WT control, the lens of Grx2 KO mice had only 50% of the activity in complex I and complex IV and less than 10% of the ATP pool. It was concluded that Grx2 gene deletion altered the function of lens structural proteins through S-glutathionylation and also caused severe disturbance in mitochondrial function. These combined alterations affected lens transparency.
Introduction
Oxidative stress is considered to associate closely with aging (1) and many diseases, including Alzheimer (2), Parkinson (3), cancer (4), retina degeneration (5), and cataract of the eye (6, 7). Under oxidative stress, the thiol groups within proteins (PSH) are among the most oxidant-sensitive targets and can undergo a variety of reversible and irreversible redox modifications. Protein S-glutathionylation, the reversible formation of a mixed disulfide (PSSG)2 between protein thiols (PSH) and oxidized glutathione (GSSG), appears to be the most important mode of thiol oxidation. PSSG formation can potentially affect protein/enzyme structure, function, and activity or even disrupt cell signaling, depending on the importance of the cysteine residue in carrying out protein/enzyme function (7–10).
Because of the physical locale, the eye lens is vulnerable to oxidation, and cataract formation in the aging population is one of the leading causes of blindness in the world (11). The lens is a protein-rich avascular organ whose main function is to maintain transparency so that the incident light can be transmitted and focused on the retina for vision. Lens structural proteins are SH-rich and present in a reduced state with tight packing to provide the lens with the necessary refractive power and transparency (12). The lens has a single anterior layer of epithelial cells, which continuously differentiate into fiber cells that lie concentrically below the epithelium forming the major bulk of the lens organ. The structural proteins in these inactive organelle-free fiber cells once modified can accumulate with time and alter lens functions. Extensive research has shown that oxidative stress-induced protein modification is a major risk factor for lens transparency loss. In such cases, the lens proteins are cross-linked and aggregated, forming high molecular weight molecules that can scatter light leading to lens opacity and cataract development. Such morphological and functional changes of lens often occur during aging, as is the case of age-related cataracts in humans (7). Oxidatively modified proteins are found in animal and human lenses as a function of age (13), more so in an opaque lens where the level of accumulation is proportional to the severity of the opacity (14). The protein thiols are conjugated with GSSG to form PSSG or with oxidized free cysteine to form protein-S-S-cysteine (PSSC) (15). It is speculated (7, 16) that S-glutathionylated proteins may undergo a thiol transfer into protein-protein disulfides (PSSPs), which are elevated in the water-insoluble high molecular weight protein fractions of old or cataractous lenses. The above phenomenon is often accompanied by the loss of free glutathione both in old and in opaque lenses. Therefore, sulfhydryl homeostasis in the lens is critical for preserving its physiological function and maintaining its transparency. The lens is rich in GSH pools and other small antioxidant molecules such as ascorbate as well as antioxidant enzymes, including GSH peroxidase, catalase, and superoxide dismutase (12, 17), which can remove oxidants and protect the lens from extensive oxidative damage. However, during aging some of these antioxidants or antioxidant enzymes are either depleted or weakened. Therefore, the accumulated thiol-oxidized proteins require an efficient system to reverse or repair the damage.
In recent years, two thiol repair systems have been identified in the lens similar to other tissues. The first is the GSH-dependent glutaredoxin (Grx) system, which reduces the protein thiols that have been oxidized into protein-thiol mixed disulfides and favors PSSG over PSSC (18–20). The second repair system is the NADPH-dependent thioredoxin (Trx) system, which reduces the protein thiols that have been oxidized and formed PSSPs and restores their proper structure and original physiological function (21, 22). Both Grx and Trx belong to the oxidoreductase superfamily and are found in all living organisms. Grx has two isoforms in the human lens, glutaredoxin 1 (Grx1, also called thioltransferase) and glutaredoxin 2 (Grx2). Grx1 is a small 12-kDa cytosolic protein, which is well studied in the mammalian Grxs family, and its important roles in regulating cytosolic thiol/disulfide balance and various cellular functions have been established (23–26). The mitochondrial Grx2, however, was only discovered more recently (27, 28), with a molecular mass of 16 kDa and only 34% sequence identity with Grx1. Human Grx2 is characterized as an iron-sulfur protein and a redox sensor. It is present as an inactive dimer bridged by a [2Fe-2S]2+ cluster but is monomerized and activated under oxidative stress conditions (29). Grx2 is present in many tissues, including the brain, liver, heart, and the pancreas. In the eye, Grx2 has been found in almost all ocular tissues, including the cornea, iris, lens, retina, and optic nerve (30). Extensive studies have been carried out in the lens, demonstrating that the lens Grx2 shows disulfide reductase (27), peroxidase (31), ascorbate recycling activities, and anti-apoptotic properties (32). Both Grx1 and Grx2 can be up-regulated under oxidative stress conditions (32, 33) where, as a thiol repair enzymes, they are thought to provide a first line of defense against protein oxidation or misfolded protein aggregation.
Recently, the Grx system has received increasing attention because of its potential association with cardiovascular and neurological disorders, as well as age-related macular degeneration and cancer. Evidence from recent studies suggests that Grx2 may control mitochondrial redox balance and mitochondrial function, and it may also regulate cell apoptosis (34, 35). Grx2 has been shown to deglutathionylate the S-glutathionylated proteins and thereby regulate several mitochondrial targets, including complex I (36), and uncoupling protein-3 (37) of the respiratory chain. Grx2 is thus closely involved in energy homeostasis and ATP production in cells. Recently, we have found that Grx2 overexpression could protect human lens epithelial cells from H2O2-induced cell apoptosis via its ability to prevent S-glutathionylation in mitochondrial complex I (36). Similarly, primary lens epithelial cells isolated from the Grx2 knock-out (KO) mice also exhibited unusual sensitivity to oxidation. Imported recombinant Grx2 protein could rescue the cells from oxidative stress-induced cell apoptosis (38). Although these findings may imply the importance of Grx2 in mitochondrial function, little work has been done in vivo, and the roles of Grx2 in aging and in lens transparency are not well understood. We hypothesized previously (7) that unchecked PSSG formation under oxidative stress is the first major protein thiol modification that may trigger the cascading events in lens protein/enzyme function and activity loss, protein aggregation, and cataract formation. We further hypothesized that Grx is the key enzyme to prevent PSSG accumulation and preserve lens transparency. The purpose of this study is to test the hypothesis in vivo using the Grx2 knock-out mouse as a model. We found that Grx2 deletion accelerated cataract development during aging and that these lenses showed extensive PSSG accumulation in structural proteins and a severely compromised electron transport system and ATP production.
EXPERIMENTAL PROCEDURES
Materials
NADPH, GSH, dithiothreitol (DTT), 5′,5′-dithiobis(2-nitrobanzoic acid), β-hydroxyethyl disulfide, BSA, 2,6-dichloroindophenol (DCIP), decylubiquinone, antimycin-A, and all other chemicals were obtained from Sigma unless otherwise stated. Bicinchoninic acid (BCA) protein assay reagent and SuperSignal West Chemiluminescent substrate were from Pierce. Antibodies against actin, αA-crystallin, βB2 crystallin, β-actin, complex I 75-kDa subunit, and complex IV subunit IV were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Anti-glutathione monoclonal antibody (anti-PSSG antibody) was purchased from ViroGen (Watertown, MA). Anti-Grx2 antibody was purchased from Abcam (Cambridge, MA). Protein A/G magnetic beads were purchased from Thermo (Waltham, MA).
Generation of Grx2 Knock-out Mice
Custom screening of a bacterial artificial chromosome (BAC) library, which carries strain 129SV mouse genomic DNA, by hybridization with the mouse Grx2 cDNA was conducted by Invitrogen. The screening identified three positive clones. The DNA fragments containing the entire mouse Grx2 gene in the BAC clones were then subcloned into vector pKS (Stratagene, La Jolla, CA) for restriction mapping and DNA sequencing. A targeting vector, in which exon 2 and some of the flanking intron sequences were deleted, was constructed using the plasmid pPNT as backbone (Fig. 1A) (39). The targeting vector was linearized by digestion with restriction enzyme NotI and then transfected into R1 embryonic stem (ES) cells (40). Screening of 360 ES clones by Southern blot analysis of PstI-digested genomic DNA allowed identification of a total of 51 clones (14.2%) that contain the targeted mutation in the Grx2 gene (Fig. 1A). Clones 29, 91, 122 and 190 were then microinjected into mouse blastocysts harvested from C57BL/6 mice for generation of chimeric mice (41). All male chimeric mice derived from these ES clones showed germ line transmission of the 129SV chromosomes into their progeny, resulting in the generation of heterozygous Grx2 knock-out mice (Fig. 1B). Homozygous knock-out mice were then generated by breeding of heterozygous knock-out mice (Fig. 1B). The homozygous knock-out mice are healthy and fertile upon observation until 8 months of age. The heterozygous Grx2 knock-out mice in the 129SV and C57Bl/6 hybrid background were then backcrossed into C57BL/B mice to generate homozygous Grx2 knock-out mice and wild-type mice that were used in this study.
FIGURE 1.
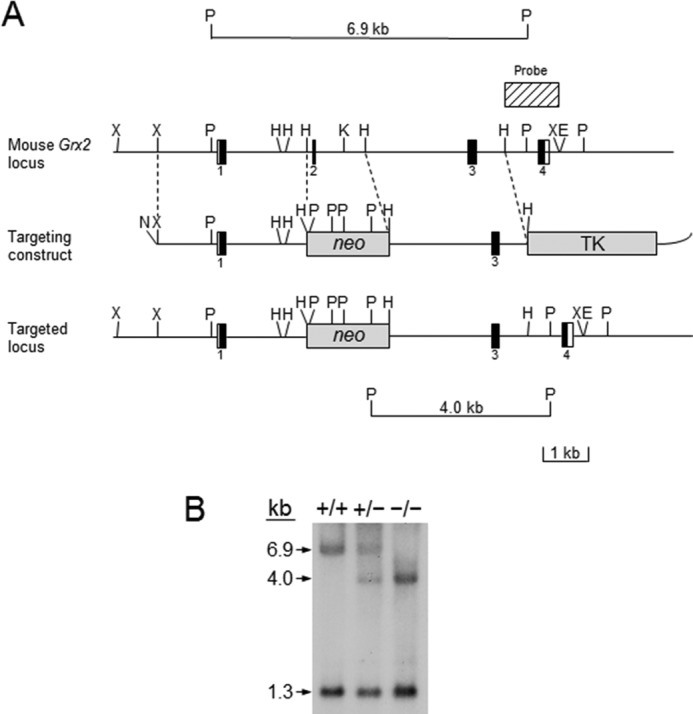
Targeted disruption of the mouse Grx2 gene. A, genomic structure and partial restriction map of the mouse Grx2 locus (top), the targeting vector (middle), and the targeted locus (bottom) are shown. The opened and black boxes represent the protein coding regions and noncoding regions in the exons, respectively. The number of exons is indicated below each exon. The striped box on top of the restriction map of the Grx2 locus represents the 3′ external sequence used for probing the DNA blot filters. B, BamHI; E, EcoRI; H, HindIII; N, NotI; X, XbaI; neo, neomycin resistance cassette; TK, herpes thymidine kinase gene under the control of a mouse promoter of the phosphoglycerate kinase-1 (Pgk-1) gene. Neo, neomycin resistance cassette. The sizes of the PstI restriction fragments from wild-type and targeted loci hybridized with the probe are shown at the top and bottom of the figure, respectively. B, Southern blot analysis of wild-type, heterozygous Grx2 knock-out, and homozygous Grx2 knock-out mice. The 6.9-kb hybridization band is derived from the wild-type Grx2 allele, and 4.0-kb hybridization band the mutated allele. The 1.3-kb hybridization fragment is derived from the PstI genomic fragment containing exon 4 from both the wild-type and the mutated alleles. +/+, +/−, and −/− represent wild-type, heterozygous Grx2 knock-out, and homozygous Grx2 knock-out mice, respectively.
Expression Studies of Various Tissues from Wild-type and Heterozygous and Homozygous Grx2 Knock-out Mice
The methods for isolation of RNA and proteins from mouse tissues and blot analysis have previously been described (42). A specific anti-Grx2 antibody used was from Abcam (catalog no. ab85267).
Reverse Transcription-Polymerase Chain Reaction (RT-PCR) of Grx2 mRNA from Hearts of Wild-type and Heterozygous and Homozygous Grx2 Knock-out Mice
One microgram of total heart RNA from mice of each genotype was used in reverse transcription using the iScript cDNA synthesis kit (Bio-Rad). The synthesized Grx2 cDNA was then PCR-amplified with forward primer GGGACCTTTGGCTATGTCCT (from exon 1) and reverse primer TCTTTGTGAAGCCTGTGAGTGT (from exon 4) using the GoTaq GreenMaster Mix (Promega, Madison, WI). The PCR-amplified DNA fragments were initially analyzed on an agarose gel and then purified for sequence determination.
Cataract Prevalence Rate and Opacity Scoring
For the present eye lens studies, we only use the homozygous Grx2 KO mouse (Grx2−/−) model. The lenses in Grx2 null mice and age-matched wild-type (WT) mice of the same background, both sexes at 3–16 months old were examined and imaged using a slit lamp ophthalmoscope (66 Instrument Co, Suzhou, China), equipped with a Sony digital camera, after the pupils were dilated with tropicamide phenylephrine eye drops (Santen Pharmaceutical Co., Ltd.) under nonanesthetized conditions. The eight age groups of mice were 3, 5, 7, 8, 9, 11, 13, and 16 months old. The number of Grx2 KO mouse eyes was 12 (3 months), 10 (5 months), 8 (7 months), 6 (8 months), 12 (9 months), 12 (11 months), 10 (13 months), and 10 (16 months), whereas the number of wild-type mouse eyes was 12 (3 months), 12 (5 months), 8 (7 months), 10 (8 months), 10 (9 months), 10 (11 months), 8 (13 months), and 8 (16 months). The LOCS II system (43) was used for cataract classification and grading (score). For the nucleus, score 0 illustrates a clear nucleus; score 1 is an early degree of nuclear opalescence; score 2 is a moderately advanced nuclear opacification, and score 3 represents an advanced nuclear opalescence. For the cortex, score 0 is clear; score 1 shows a small mini-spoke inside the pupillary margin; score 2 contains cortical spokes that obscure slightly more than two full quadrants; score 3 illustrates ∼50% opacification of the intra-pupillary zone, and score 4 means ∼90% opacification of the intra-pupillary zone. For the posterior subcapsular region, score 1 illustrates a cataract filling ∼3% of the area; score 2 stands for ∼30% opacification; score 3 illustrates about 50%, and score 4 shows >50% opacification of the area. The cataract score reported in this study is calculated based on the sum of the scores from nucleus, cortex, and posterior subcapsular opacity divided by the total eyes examined.
Isolation of Mitochondria from Mouse Lenses
The mitochondrial fraction was isolated according to the modified procedure of Rehncrona et al. (44). Briefly, each lens was homogenized in 1 ml of ice-cold buffer A (containing 225 mm mannitol, 65 mm sucrose, 1 mm EGTA, and 10 mm HEPES (pH 7.2)) using a glass-to-glass homogenizer, followed by centrifugation at 1,000 × g for 10 min at 4 °C. The supernatant was centrifuged again at 10,000 × g for 10 min at 4 °C. The pellet was resuspended with 1 ml of buffer B (containing 225 mm mannitol, 65 mm sucrose, and 10 mm HEPES (pH 7.2)) followed by centrifugation at 10,000 × g for 10 min at 4 °C. The final mitochondrial fraction was resuspended in 100 μl of buffer B and stored at −80 °C until used.
Western Blot Analysis
Equal amounts of proteins were subjected to SDS-PAGE on 12% polyacrylamide gel and transferred to a polyvinylidene difluoride (PVDF) membrane (GE Healthcare). Detection was done using the ECL Western blotting detection system (Thermo Scientific, Rockford, IL). The immunoblot was analyzed with an imaging system (VersaDoc 5000 MP Imaging System, Bio-Rad).
Grx2 Activity Assay
Grx2 activity was assayed according to a previously described method (27). The reaction mixture contained 0.2 mm NADPH, 0.5 mm GSH, 0.1 m potassium phosphate buffer (pH 7.4), 0.4 units of GSSG reductase, and an aliquot of mitochondrial fraction in a total volume of 0.5 ml. The reaction was carried out at 30 °C following a 5-min preincubation with a substrate of 2 mm hydroxyethyl disulfide. The decrease in absorbance of NADPH at 340 nm was monitored for 3 min using a Beckman DU 640 spectrophotometer (Beckman, Fullerton, CA). To determine the Grx2 activity, the slope of the linear portion of the time course for 340 nm of absorption loss in a control (Grx2-free) sample was subtracted from the slope of the samples containing Grx2.
GSH and Protein Sulfhydryl (PSH) Group Measurement
Each whole lens was homogenized in 1.0 ml of ice-cold buffer containing 50 mm Tris-HCl (pH 7.5), 100 mm NaCl, and 10 mm EDTA. The homogenate was centrifuged at 16,000 × g for 10 min at 4 °C. For total sulfhydryl group measurement, an aliquot of 50 μl of the fresh lens homogenate was mixed with 150 μl of 5′,5′-dithiobis(2-nitrobanzoic acid) reagent, and the absorbance was read at 412 nm. For GSH assay, lens homogenate was deproteinized by mixing with an equal volume of 20% trichloroacetic acid (TCA), followed by centrifugation for 10 min at 16,000 × g to remove proteins. The supernatant was used immediately for GSH analysis by 5′,5′-dithiobis(2-nitrobenzoic acid), following the published method (45). PSH level was calculated by subtracting the values of GSH from total sulfhydryl groups.
Quantitation of Protein-Thiol Mixed Disulfides in the Eye Lens
Protein-thiol mixed disulfides, protein-GSH (PSSG) and protein-cysteine (PSSC), were quantified following the method of Lou et al. (46) with modifications. Six mouse lenses were pooled and homogenized in 2 ml of 10% TCA followed by centrifugation at 1600 × g for 15 min. The pellet was thoroughly washed three times with 10% TCA to remove free GSH and GSSG. The final pellet was then washed with 0.5 ml of methanol/ether mixture (1:1, v/v) to remove any lipid components followed by drying on a heating block at 65 °C overnight. The dried protein pellet was weighed and ground into fine powder, which was suspended in 125 μl of 88% formic acid, with an addition of 0.5 ml of freshly made performic acid. The reaction tube was kept on ice for 2.5 h with occasional swirling to thoroughly oxidize and cleave the disulfide bonds of protein-thiol mixed disulfides of PSSG and PSSC to release glutathione sulfonic acid (GSO3H) and cysteic acid (CSO3H), respectively. The excess performic acid was neutralized using 90 ml of pre-chilled water followed by lyophilization. The lyophilized residue was washed into a 15-ml centrifuge tube with three 3-ml aliquots of ice-cold water. The suspension was centrifuged for 15 ml at 17,000 rpm to remove the oxidized insoluble proteins. The supernatant was transferred to a 10-ml pear-shaped flask and lyophilized twice. The resulting residue was subsequently suspended in 200 μl of H2O followed by centrifugation at 16,000 × g before using an aliquot for GSO3H and CSO3H quantification using a Dionex anion amino acid analyzer (DX500 model, Dionex, Sunnyvale, CA).
ATP Quantification
ATP level was measured with an ATP bioluminescence assay kit CLS II (Roche Applied Science) according to the manufacturer's recommendation. Briefly, lens homogenates were mixed with 9 volumes of boiling solution (PBS containing 100 mm Tris and 4 mm EDTA) and incubated for 2 min at 100 °C. The sample mix was then centrifuged at 1000 × g for 1 min, and 50 μl of the supernatant was mixed with 50 μl of luciferase reagent by automated injection. The luminescence intensity was detected by a Fluostar Optima microplate reader (BMG Labtech, Offenburg, Germany), and integrated for 1–10 s.
Detection of PSSG with Western Blot and Immunoprecipitation
WT or Grx2 KO mouse lenses were homogenized in 50 mm Tris (pH 7.4), containing 150 mm NaCl, 10 mm iodoacetic acid, and 1% protease inhibitor mixture. The homogenates were centrifuged at 13,000 × g for 10 min at 4 °C, followed by protein determination. The lens proteins (120 μg) were loaded onto SDS-polyacrylamide gels under nonreducing conditions and probed with a specific anti-GSH antibody. For immunoprecipitation, the glutathionylated proteins were pulled down with anti-GSH antibody, using the nonspecific mouse IgG as a negative control. The bound proteins were eluted by 0.2 m glycine (pH 2) containing 50 mm N-ethylmaleimide, separated by nonreducing SDS-PAGE, and visualized by silver staining or Coomassie Blue staining.
Mass Spectrometric Identification of S-Glutathionylated Proteins
The major bands immunoprecipitated with anti-GSH antibody were excised from a Coomassie Blue-stained SDS-polyacrylamide gel; proteins were digested overnight with trypsin, and peptides were analyzed by liquid chromatography/tandem mass spectrometry (LC-MS/MS) as described previously (47), except gel slices were not reduced. LTQ Velos ion trap mass spectrometer (Thermo Scientific, San Jose, CA) was used, and five data-dependent MS2 scans were collected following each MS survey scan. Peptide identification was performed by comparing observed MS2 spectra to theoretical fragmentation spectra of peptides generated from a protein database using Sequest (Version 27, revision 12, Thermo Scientific). Searches included a static modification of +57 on cysteines due to alkylation with iodoacetic acid, and a dynamic modification of +248 on cysteines to detect possible sites of glutathionylation. A mouse version of the Swiss-Prot database was used (Swiss Institute of Bioinformatics, Geneva, Switzerland) with added common contaminants added, and the resulting file amended by reversing the sequence of all 16,563 entries to estimate the protein false discovery rate. Peptide and protein identifications were then compiled using in-house software (48), using a two unique peptide minimum per entry and a protein false discovery rate of 5%.
Complex I Activity Assay
The enzymatic activity of complex I was assayed according to the method of Janssen et al. (49). In brief, 50 μg of mitochondria was mixed with 960 μl of reaction mixture containing 20 mm KH2PO4, 3.5 mg/ml BSA, 60 μm DCIP, 70 μm decylubiquinone (prepared in dimethyl sulfoxide, DMSO), and 1 μm antimycin-A at 30 °C. After 3 min, 20 μl of 10 mm NADH was added, and the absorbance was measured at 30-s interval for 4 min at 37 °C. After that, 1.0 μl of rotenone (1 mm, in DMSO) was added, and the absorbance was measured at 30-s intervals for an additional 4 min to confirm the measurement of rotenone-sensitive complex I activity. The enzyme activity was expressed as nanomoles of DCIP reduced per min/mg of protein. The substrate DCIP, which is a water-soluble final electron acceptor, is specific for complex I because DCIP receives electrons only from complex I and not from other nonmitochondrial NADH dehydrogenases. BSA is a necessary component in the assay mixture because it acts as a solubilization agent for rotenone and decylubiquinone.
Complex IV Enzyme Activity Assay
The complex IV rodent enzyme activity microplate assay kit (Abcam, Cambridge, MA) was used to determine the activity of the cytochrome c oxidase enzyme in the mouse lens, according to the manufacturer's recommendation. Briefly, 100 μg of mitochondrial proteins isolated from WT or Grx2 KO mouse lenses were loaded into a 96-well plate. Complex IV was immunocaptured in the 96-well plate, and the enzyme activity was determined colorimetrically by following the decrease in 550 nm absorbance during oxidation of reduced cytochrome c.
Statistics
Each experiment was performed at least three times, and statistical analyses were performed using one-way analysis of variance followed by a post hoc Tukey's test, using the SPSS software. All data were expressed as means ± S.D., and the differences were considered significant at p < 0.05.
RESULTS
General Characterization of Grx2 Knock-out Mice
Following the generation of Grx2 knock-out mice, we determined whether replacement of exon 2 with a neomycin resistance cassette disrupts expression of the mouse Grx2 gene. Surprisingly, as shown in Fig. 2A, tissues of homozygous Grx2 knock-out mice still express a slightly smaller species of Grx2 mRNA at a lower abundance than the transcripts present in tissues of WT mice. Because exon 2 is deleted in the disrupted Grx2 gene, the expressed Grx2 mRNA in homozygous knock-out mice is believed to derive from fusion of exons 1, 3, and 4 sequences. This assumption was confirmed by RT-PCR amplification of Grx2 mRNA isolated from mouse hearts. As shown in Fig. 2B, the expected size of the RT-PCR DNA fragment from WT Grx2 mRNA is 420 bp. However, the disrupted Grx2 allele in knock-out mice gives rise to a 356-bp RT-PCR fragment. The results of DNA sequencing confirmed that the 356-bp DNA fragment contains fusion of exons 1 and 3 sequences (Fig. 2C).
FIGURE 2.

General characterization of Grx2 knock-out mice. A, RNA blot analysis of Grx2 mRNA expression in tissues of wild-type and Grx2 knock-out mice. The RNA blot membrane was hybridized with a mouse Grx2 cDNA and then re-hybridized with a rat cDNA for glyceraldehyde-3-phosphate dehydrogenase (G3PD). B, RT-PCR amplification of wild-type and mutant Grx2 mRNA isolated from mouse hearts. C, sequence of the junction between exon 1 and exon 3 of the mutant Grx2 mRNA and the encoded amino acids. D, protein blot analysis of Grx2 in tissues of wild-type and Grx2 knock-out mice. The protein blot membrane initially reacted with a rabbit anti-mouse Grx2 antiserum. A duplicate protein blot membrane prepared from a second gel loaded with the same amount of samples reacted with rabbit anti-human copper-zinc superoxide dismutase (CuZnSOD) antibodies for confirming equal loading of proteins in each set of tissue samples from mice with three different genotypes. A, B, and D, +/+, +/−, and −/− represent wild-type, heterozygous Grx2 knock-out, and homozygous Grx2 knock-out mice, respectively.
As a consequence, translation of protein from this mRNA will stop in exon 3 due to the shifted reading frame (Fig. 2C). The resultant protein will only contain the mitochondrial translocation signal, which is encoded by exon 1, followed by eight amino acids translated from the out-of-frame exon 3 sequence. The aberrant Grx2 mRNA is likely susceptible to nonsense-mediated mRNA decay (50), resulting in a lower level of the mRNA in tissues of heterozygous and homozygous knock-out mice compared with that of WT Grx2 mRNA (Fig. 2A). Protein blot study showed an ∼50% decrease of Grx2 protein in tissues of heterozygous knock-out mice compared with those of WT mice, and no Grx2 protein was found in the same tissues of homozygous knock-out mice (Fig. 2D).
Characterization of the Eye Lens of Grx2 KO Mice
We analyzed the lens tissues from the Grx2 KO mice (Grx2−/−) and could not detect Grx2 protein or its activity. As shown in a Western blot of the mitochondrial fraction isolated from the lenses of Grx2 KO mice (Fig. 3A and B), the voltage-dependent anion channel (VDAC) protein (a mitochondrial marker used as loading control) is clearly present, but Grx2 was absent. In contrast, both proteins could be seen in the WT control mice. The mitochondrial fraction from the same lens showed no Grx2 activity in the mouse KO sample, but Grx2 activity of 6.7 ± 0.9 milliunits/mg protein was detected in the WT control (Fig. 3B).
FIGURE 3.

Characterization and validation of the eye lenses of Grx2 KO mice. The Grx2 KO mouse (5 months old) lens was evaluated for the presence of Grx2 expression and disulfide reductase activity, as well as the effects on other oxidoreductase enzymes, including the cytosolic glutaredoxin 1 (Grx1) and thioredoxin 1(Trx1), and the mitochondrial thioredoxin 2 (Trx2). The effect of Grx2 gene deletion on eye and lens development was evaluated by examining the eye and lens wet weight during aging with samples from 1-, 7-, and 16-month-old animals. A, mitochondrial fractions (40 μg) isolated from 5-month-old WT and Grx2 KO mice were analyzed for Grx2 expression by Western blot with anti-Grx2 antibody. A mitochondrion-specific protein, voltage-dependent anion channel (VDAC), was used as marker and loading control. B, Grx2 disulfide reductase activity in the lens of WT and Grx2 KO mice was measured using the same mitochondrial fraction (100 μg). Data are expressed as means ± S.D., n = 3, ***, p < 0.001 versus age-matched WT. C, aliquot of lens lysate (40 μg) was used to detect Grx1 and Trx1, whereas the mitochondrial fraction was used for Trx2 measurement by immunoblot analysis using anti-Grx1, anti-Trx1, or anti-Trx2 antibody, respectively. β-Actin was used as a loading control for Grx1 and Trx1 protein expression in the WT and KO samples (upper and middle panels), whereas VDAC was the loading control for Trx2 analysis of the WT and KO samples (bottom panel). Pixel densities of Grx1 and Trx1 were normalized to the β-actin band, and Trx2 was normalized to VDAC and is depicted in a bar graph. D, comparisons of the eye mass (mg) between WT and Grx2 KO mice in mg of wet weight. E, comparisons of lens in mg of wet weight between WT and Grx2 KO mice. Data are expressed as means ± S.D., n = 8.
To confirm that this Grx2 KO mouse line is Grx2-specific, we analyzed the lens tissues for other thiol repair enzymes in the oxidoreductase family, including the cytosolic Grx2 isozyme Grx1, the cytosolic thioredoxin 1 (Trx1), and mitochondrial thioredoxin 2 (Trx2) by immunoblotting analysis using specific antibodies. Fig. 3C indicates that Grx2 gene deletion does not affect other oxidoreductase enzymes, because levels of Grx1, Trx1, and Trx2 were equally reactive in the lenses of KO and WT mice.
To determine whether Grx2 KO mice have a phenotype associated with gross changes in eye or lens development, we compared the wet weights of the eye mass and the lens from ages 1, 7, and 16 months from WT and KO mice. Fig. 3, D and E, indicate that such gene deletion does not lead to significant differences in the growth of either the eye mass or lens in the three age groups between WT and Grx2 KO mice.
Accelerated Cataract Progression in Grx2 KO Mice
The potential effect of Grx2 deletion on lens transparency during aging was measured in Grx2 KO mice by comparison with age-matched WT mice. As shown in Fig. 4A, the lenses were clear in both KO and WT animals at 3 months of age, but KO animals exhibited a more rapid loss of transparency and became over 80% cataractous at 11 months, compared with WT mice which were only 20% cataractous at this age.
FIGURE 4.

Evaluation of the cataract prevalence rate and opacity score in the aging populations of Grx2 KO mice and age-matched WT mice. A, prevalence rate of cataract in Grx2 KO mice (●) and WT mice (■). Side panel shows the number of opacity eyes over the total eyes in respective Grx2 KO and WT mice, corresponding to the ages of the animals. B, opacity score in WT and Grx2 KO mice. Opacity was quantified using the LOCS II system, and the sum of the scores was used to analyze the severity of the cataract. The same ophthalmologist gave the cataract scores without prior knowledge of the mouse genotype. *, p < 0.05 versus age-matched WT. C, slit lamp images of representative eyes. Lenses of Grx2 KO and age-matched WT mice ranging from 3 to 16 months (m) were examined. Eyes were dilated under nonanesthetized conditions.
When examining the severity of cataracts using an opacity score from 0 to 5, the two animal populations showed extensive disparity, which corroborated with the degree of cataract prevalence. As shown in Fig. 4B, the KO group shows a lens opacity score of 0.1 at 5 months and steadily increases to 0.92 at 11 months, after which the score rises abruptly to 3.8 and 5.2 at 13 and 16 months, respectively. Similar to the cataract prevalence, the WT showed a slower formation of lens opacity with a score of 0.25 at 7 months, 0.4 at 9 months, and 1.38 and 1.63 at 13 and 16 months, respectively. In each age group, cataract development in the WT mice was far less than that in the KO animals. The opacity scores in the young animals increased predominantly due to increases in nuclear opacity, although opacity scores in 13–16-month-old mice increased due to development of additional cortical and post-subcapsular opacity.
The images of slip lamp observation in representative eyes of both KO and WT animals at 3, 5, 8, and 16 months old are shown in Fig. 4C. In general, the lens opacity in both animal groups first appeared in the center of the lens, or the nuclear region, and gradually spread out into the cortical, posterior, and posterior subcapsular areas as the animals aged. As shown in Fig. 4C, lower panel, lens opacity of the KO group was clearly seen in the nuclear region at 5 months and gradually expanded into an opaque ring at the outer cortical layers by 8 months, and eventually into full lens involvement in the 16-month-old mice. In contrast, lens opacity did not appear in the wild-type mouse (Fig. 4C, upper panel) until 8 months, when a light opacity appeared in the nuclear zone, and then a full cortical involvement was seen in the 16-month-old mouse.
Lens GSH and PSH Levels in Grx2 KO Mice
Because Grx2 reduces the disulfide linkages of S-glutathionylated proteins and thus may function as an essential repair enzyme to maintain the SH groups in lens structural proteins in a reduced state, we examined the levels of reduced glutathione (GSH) and free thiol in proteins (PSH) of the lenses in the population of WT and Grx2 KO mice. In general, both GSH and PSH levels were suppressed in the mouse lens during aging. Fig. 5A shows that the change in GSH was mild in WT mice, where GSH remained constant up to 7 months and only decreased by 25% at 16 months. However, the change was more drastic in the KO animals. Although the GSH level in the 1-month-old KO mice was the same as that of the age-matched WT mice, it dropped to 80 and 33% in the 7- and 16-month-old mice, respectively. A similar pattern was observed in the PSH levels of the lenses shown in Fig. 5B, in which the WT level decreased more slowly than that of the KO group. In the 1-month-old animals, the WT lost 10% of PSH levels of lenses in both the 7- and 16-month age groups, but the KO animals lost 25 and 30% at 7 and 16 months of age, respectively.
FIGURE 5.
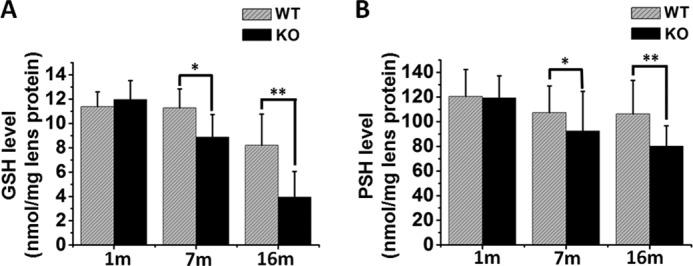
Quantification of free GSH, PSH, in the lenses of WT and Grx2 KO mice. Lenses from Grx2 KO and WT mice (1, 7,and 16 months old) were used for this study. Eight individual lens homogenates were used for the GSH and PSH analysis. A, free GSH levels in Grx2 KO and age-matched WT mice. Data are expressed as means ± S.D., n = 8, *, p < 0.05; **, p < 0.01. B, PSH levels in the lenses of WT and Grx2 KO mice were calculated from the difference between total SH groups and the GSH levels. Data are expressed as means ± S.D., n = 8, *, p < 0.05; **, p < 0.01 versus age-matched WT.
Effect of Grx2 Gene Deletion on the Level of Protein-Thiol Mixed Disulfides in the Mouse Lens
Formation and accumulation of protein-thiol mixed disulfides, either PSSG or PSSC in the lens, have been implicated to be part of the mechanism of protein misfolding and protein-protein aggregation in cataractogenesis (7). Because Grx2 deletion resulted in faster cataract development during aging (Fig. 4), we examined whether the PSSG and PSSC levels were elevated in the lenses of KO mice relative to the age-matched WT control. The method measured PSSG and PSSC in the lens by quantification of their in vitro-derived products glutathione sulfonic acid (GSO3H) and cysteic acid (CSO3H), respectively (40). Fig. 6A shows the chromatographic separation of CSO3H and GSO3H in lenses obtained from 1, 7, and 16-month-old WT and KO mice, with elution of CSO3H and GSO3H standards as the controls. The concentrations of PSSG are shown in Fig. 6B, in which PSSG in 1-month-old mice is low and similar for WT and Grx2 KO lenses (0.05 ± 0.02 versus 0.06 ± 0.03 μmol/g wet weight). By 7 and 16 months, the PSSG levels in WT mice were 0.09 ± 0.02 μmol/g wet weight and 0.15 ± 0.02, respectively, although PSSG levels in Grx2 KO mice were much higher at 0.13 ± 0.04 μmol/g wet weight (p < 0.05) and 0.2 ± 0.03 (p < 0.01), respectively.
FIGURE 6.

Quantification of protein-thiol mixed disulfides in the lenses of WT and Grx2 KO mice. Two duplicate samples processed from six pooled lenses of the KO or WT mice were used for the protein mixed disulfide quantification. Details are described under “Experimental Procedures.” A, typical chromatograms of PSSC and PSSG analyses. PSSG and PSSC standards at 5 nmol (panel a) and 10 nmol (panel b); WT samples from 1 month (panel c), 7 months (panel e), and 16 months (panel g); KO samples from 1 month (panel d), 7 months (panel f), and 16 months (panel h). B, quantified PSSG level expressed as μmol/g wet lens weight. C, quantified PSSC level expressed as μmol/g wet lens weight. Data are expressed as means ± S.D. based on triplicate experiments. *, p < 0.05 versus age-matched WT; **, p < 0.01 versus age-matched WT.
A similar pattern was observed in the PSSC levels. As shown in Fig. 6C, the PSSC levels in the 1-month-old lenses of WT and KO mice were 0.19 ± 0.03 and 0.21 ± 0.04 μmol/g wet weight, respectively. The lens PSSC accumulated with aging but much more so when Grx2 gene is deleted. By 7 months of age, PSSC in the KO group was 2-fold higher than that of the WT (0.51 ± 0.04 versus 0.29 ± 0.05 μmol/g wet weight). This trend continued to 16 months, with PSSC elevated to 0.82 ± 0.12 μmol/g wet weight in the KO lenses and 0.54 ± 0.04 μmol/g wet weight in the age-matched WT controls.
Accumulation and Identification of PSSG Proteins in Grx2 KO Mouse Lenses
To further examine the effect of Grx2 gene deletion on lens protein S-glutathionylation or PSSG formation, lens homogenates from WT and Grx2 KO mice were examined by immunoblotting with the anti-PSSG-specific antibody. As shown in Fig. 7A, aging progressively induces accumulation of S-glutathionylated proteins in the lenses of both WT and KO groups (see arrows). At 1 month of age, PSSG in both groups was equally low with one major band between 37 and 50 kDa and another at 25 kDa. In the 7-month-old lenses, the KO mice showed more PSSG in these regions compared with that of WT controls, with an additional band appearing at ∼15 kDa. Further elevation of PSSG was seen in the lenses of 16-month-old KO mice. The pixel density of the total GSH antibody-positive protein bands in Fig. 7A is depicted on the right. These results corroborate the PSSG analysis in Fig. 6B, indicating that the Grx2 null animal is more susceptible to age-related PSSG accumulation.
FIGURE 7.

Detection of glutathionylated proteins (PSSG) in the lenses of WT and Grx2 KO mice. A, comparison of protein glutathionylation in the lens of WT and Grx2 KO mice. Soluble fraction of lens homogenates from WT and Grx2 KO mice (1, 7, and 16 months old) were analyzed by Western blotting using anti-PSSG antibody under nonreducing conditions. GAPDH was used as a loading control. The graph on the right depicts the relative pixel density of all the PSSG bands (indicated by arrows) over GAPDH (with the 1-montg-old WT normalized to 1.0). B, S-glutathionylated protein identification. Whole lens lysate from 16-month-old Grx2 KO mice was immunoprecipitated for glutathionylated proteins using anti-PSSG antibody and then was separated with SDS-polyacrylamide gel followed by Coomassie Blue staining. Proteins showing positive immunoreactivity (indicated by arrows) with anti-PSSG antibody were cut and identified by liquid chromatography-tandem mass spectrometry as predominantly actin, αA-crystallin, and βB2-crystallin. For confirmation, glutathionylated proteins were immunoprecipitated (IP) by using anti-PSSG antibodies, followed by immunoblot (IB) detection of actin (C), βB2-crystallin (D), and αA-crystallin (E). Blots labeled “IB: actin,” “IB: αA-crystallin”, and “IB: βB2-crystallin” are control Western blots for actin, βB2-crystallin, and αA-crystalline, respectively. The right panels show the pixel density graphs of glutathionylated actin, βB2-crystallin, or αA-crystalline in comparison with the 1-month-old WT lens, respectively. Data represent the means ± S.D. of three independent experiments. *, p < 0.05 versus age-matched WT; **, p < 0.01 versus age-matched WT.
To identify the target proteins of Grx2, S-glutathionylated proteins were immunoprecipitated from the lenses of 16-month-old Grx2 KO mice using anti-PSSG antibody, separated by SDS-PAGE, followed by Coomassie Blue staining. The immunoreactive GSH-positive proteins (indicated by arrows in Fig. 7B) were excised, digested with trypsin, and subjected for identification by LC-MS/MS. This analysis identified several major lens structural proteins that immunoprecipitated with anti-PSSG antibody, including actin, βB2-crystallin, and αA-crystallin. However, sites of S-glutathionylation were not identified, because their relative abundance, compared with unmodified peptides, was low.
To confirm and quantify the presence of S-glutathionylated actin, βB2-crystallin, or αA-crystallin identified by LC-MS/MS analyses, immunoprecipitation and immunoblotting were carried out. Total PSSG proteins were first precipitated from whole lens homogenates of WT or Grx2 KO mice using anti-PSSG antibody, followed by probing with specific antibodies against actin, βB2-crystallin, or αA-crystallin. As shown in Fig. 7, C–E, and the respective pixel density graphs, in 1-month-old mice the level of glutathionylated actin or βB2-crystallin is low in both WT and Grx2 KO mice, although no glutathionylated αA-crystallin is found. However, all three proteins were increasingly glutathionylated in an age-dependent manner in both WT and Grx2 KO lenses. The PSSG levels in Grx2 null mice were consistently higher than those of the age-matched WT controls. As depicted in the pixel density graphs of Fig. 7, C–E, a 2.5-, 3.5-, or 3.5-fold elevation of glutathionylated actin, βB2-crystallin, or αA-crystallin was found, respectively, in the 16-month over the 1-month-old mouse.
ATP Levels in the Lenses of the WT and Grx2 Knock-out Mice
We have previously demonstrated in the lens epithelial cells (36, 38) that Grx2 protects the mitochondrial respiratory electron transport system and ATP production. Therefore, we examined the status of in vivo ATP formation in the lenses of Grx2 KO mice during aging. Fig. 8 summarizes the ATP production in the Grx2 KO and age-matched WT mice in three age groups. At 1 month, ATP levels in WT and Grx2 KO lens were the same (90.3 ± 10.2 versus 91.0 ± 20.3 μg/mg lens wet weight), but this basal level decreased with age, with a faster depletion in the Grx2 null mice than in the WT controls. By 7 months, the lenses in WT and Grx2 KO mice were 75.2 ± 8.9 and 60.3 ± 10.6 μg/mg lens wet weight, respectively. By 16 months old, the ATP level was further decreased to 20.3 ± 5.42 μg/mg lens wet weight (25% of the 1 month level) in the WT animals, and a barely detectable amount of 1.5 ± 0.5 μg/mg lens wet weight (<2% of 1-month level) in the lenses of Grx2 KO mice.
FIGURE 8.

ATP level in the lenses of WT and Grx2 KO mice. Whole lens homogenates from WT and Grx2 KO mice (1, 7, and 16 months old) were used for ATP measurement with an ATP assay kit. Total ATP levels in micrograms/lens wet weight are expressed as means ± S.D., n = 6, *, p < 0.05; **, p < 0.01.
S-Glutathionylated Complex I and Complex IV in the Mitochondrial Respiratory Chain of the Eye Lenses
Complex I, or NADH dehydrogenase, is the entry point and the first enzyme system of the mitochondrial respiratory chain for O2 reduction and ATP generation. We used purified mitochondria from the lenses of WT or KO mice to determine whether Grx2 gene deletion affects the function of mitochondrial complex I during aging. Fig. 9A shows that complex I activity is similar in the lenses of 1-month-old WT and Grx2 KO mice (278.4 ± 20.3 versus 261.5 ± 32.5 milliunits/g lens wet weight), and the activity was gradually lost during aging. However, Grx2 KO mice at 7 and 16 months displayed much lower activity compared with that of age-matched WT controls. The 7-month-old WT mice showed complex I activity of 243.2 ± 34.5 milliunits/g lens wet weight (87% of 1 month old) but Grx2 KO mice had only 172.5 ± 28.9 milliunits/g lens wet weight (66% of 1 month old). The 16-month-old WT mice displayed complex I activity at 182.5 ± 19.2 milliunits/g lens wet weight (65% of 1 month old) but only 130.8 ± 12.3 milliunits/g lens wet weight (50% of 1 month old) in the Grx2 null mice.
FIGURE 9.

Detection of glutathionylated complex I and complex IV in the mitochondria of lenses from WT and Grx2 KO mice. Mitochondrial fractions isolated from the lenses of WT and Grx2 KO mice (1, 7, and 16 months old) were used for enzymatic activity assays and Western blot analyses. A, complex I activity. B, comparison of the glutathionylated complex I protein (75-kDa subunit) from 16-month-old Grx2 KO and WT mice. Mitochondrial samples were immunoprecipitated, followed by Western blot detection of complex I 75-kDa subunit. The top panel shows the anti-GSH positive immunoprecipitated (IP) sample followed by identification by anti-complex I 75-kDa subunit antibody. The relative pixel density of the immunoblots (IB) of WT and Grx2 KO is shown in the bar graph after normalizing the WT value to 1.0. **, p < 0.01 versus WT. C, complex IV activity. Data are expressed as means ± S.D., n = 6. *, p < 0.05; **, p < 0.01 versus WT. D, comparison of the glutathionylated complex IV protein (16-kDa subunit) from 16-month-old Grx2 KO and WT mice. Mitochondrial samples were immunoprecipitated, followed by Western blot detection of complex IV 16-kDa subunit. Top panel shows the anti-GSH positive immunoprecipitated sample followed by identification of anti-complex IV 16-kDa subunit antibody. The relative pixel density of the immunoblots of WT and Grx2 KO is shown in the bar graph after normalized the WT value to 1.0. **, p < 0.01 versus WT.
We have recently reported (38) that under oxidative stressed conditions, complex I activity in the lens epithelial cells is suppressed due to S-glutathionylation of its 75-kDa catalytic subunit. Therefore, we explored the possibility that similar modification may occur in the lenses of aging Grx2 KO mice in vivo. The 75-kDa subunit of complex I was immunoprecipitated from the mitochondrial fraction obtained from either WT or Grx2 KO mouse lenses followed by immunoblotting with anti-PSSG antibody to detect the S-glutathionylated 75-kDa protein band. As shown in Fig. 9B, the 75-kDa band was immunoreactive with the anti-PSSG antibody in the 16-month-old Grx2 KO sample, and nearly 3-fold higher than that of the age-matched WT control (see the pixel density graph).
Because complex IV is an important downstream regulator for the mitochondrial respiratory chain, we also explored the potential effects of Grx2 gene deletion on complex IV. Fig. 9C shows that, similar to the results for complex I, complex IV activity was decreased during aging in both Grx2 KO and WT groups but more drastically in the KO animals. The complex IV activity in the 7- and 16-month-old WT animals was suppressed to 82 and 33%, respectively, but the activity at the same ages in the KO animals was suppressed more, to only 49 and 16%, respectively.
To examine whether S-glutathionylation may affect the activity of the 16-kDa catalytic subunit of complex IV in the absence of the Grx2 gene, we immunoprecipitated this subunit using the mitochondrial fractions isolated from the lenses of 16-month-old Grx2 KO and WT mice. The results shown in Fig. 9D confirm that more S-glutathionylation is found in this subunit from the Grx2 KO mice than that of the WT control. The increase of PSSG in the KO animal was nearly 2-fold over the WT control (see the pixel density graph in Fig. 9D).
DISCUSSION
Although Grx2 is believed to play a major role in regulating thiol/disulfide balance of mitochondria, its function in vivo in a whole animal remains elusive. We sought to evaluate the physiological role of Grx2 in the pathogenesis of cataract by producing a line of knock-out mice with global Grx2 deficiency. The Grx2 KO mouse model used in this study has been shown to lack Grx2 in the lens, in agreement with its absence in other tissues (see Figs. 2D and 3A). Interestingly, deletion of Grx2 did not affect the expression of other major oxidation repair enzymes in the oxidoreductase family. Therefore, it is likely that the phenotype displayed in this animal model is mostly, if not solely, due to Grx2 gene deletion.
Based on these results, it is of interest to note that Grx2 gene deletion produces a phenotype of early onset and faster progression in cataract formation during aging, in comparison with the age-matched wild-type mice. In this Grx2 KO mouse model, the opacity first appeared in the nuclear zone at the center of the lens, and it gradually spread outward into the outer cortical and posterior subcapsular regions. Such changes were very visible by the time the mice reached 13–16 months of age (Fig. 3).
Age-related cataract (ARC) or senile cataract is a very common health problem in the current human aging population and is the leading cause of blindness in the world. Extensive evidence has linked oxidative stress with the mechanism of ARC formation (7, 51, 52). Many attempts have been made to develop an oxidative stress-induced cataract model in animals, but few have shown any close correlation to ARC. Wolf et al. (53) demonstrated that deletion of glutathione peroxidase 1 gene (Gpx1), a major H2O2-degrading enzyme, resulted in a faster cataract progression during aging. However, based on the gross cataract scoring system and a hand-held slit lamp, they observed only a 20–30% higher opacity at the advanced age of 26 months in the Gpx1 null mice compared with the age-matched controls. In contrast, we used a clinical slit lamp and a quantitative LOCS II cataract grading system for this study. Our model showed an early onset of lens opacity at 5 months, 3 months earlier than that of the control mice. The cataract prevalence of the Grx2 KO mice was also consistently higher as a function of age in multiple aging populations. The Grx2 null mice also displayed a progressive and steady increase in lens opacity, faster than that of the WT mice, with a 20–30% higher score as early as 7–8 months of age, and a >100% higher score by the age of 13–16 months. Furthermore, Grx2 is a disulfide reductase, which has a specific catalytic function to reduce the PSSG target proteins accumulated in the lens during aging that is associated with oxidation. Reducing the accumulated PSSG in the early stage of protein oxidation can prevent the lens proteins from further modification into PSSP conjugates and the downstream high molecular weight aggregates that can lead to cataract formation. As far as we know, our current Grx2 null mouse model provides the first evidence that lacking this oxidative damage repair enzyme can lead to accelerated cataract formation during aging. Thus, we propose that the Grx2 KO mouse line can be a good model to study human ARC.
Previously, we (7) have proposed that the initiation of oxidative stress-induced cataract may, in part, involve the oxidation-induced PSSG formation in the lens enzymes/proteins, resulting in loss of enzyme activity, such as observed with glyceraldehyde-3-dehydrogenase (20) or protein function, as with the chaperone-like function of αA-crystallin (54). The extra charge and mass of the –SG group that S-conjugates onto a protein may cause conformational change (55) and/or destabilization of lens crystallin proteins (56, 57). These types of reactions may allow multiple modified lens proteins to aggregate and become water-insoluble. Progressive accumulation of such large particles during aging will eventually lead to cataract formation. The Grx2 KO mice showed similar alterations to those found in old human clear (13, 58) or opaque lenses (51, 55). In the Grx2 KO mice soon after lens opacification was initiated, the GSH pool was suppressed at 7 months and more so at 16 months of age. In the same lens, the PSH level was also correspondingly suppressed in these two age groups, indicating PSSP accumulation had taken place. Accompanying the loss in GSH and PSH, these lenses showed considerable elevation of PSSG and more so of PSSC, which is to be expected, as PSSG is a far better substrate than PSSC for Grx enzymes (19). In contrast, the wild-type mice with a normal rate of transparency loss during aging displayed much less change in GSH, PSH, and protein-thiol mixed disulfides.
Such discrepancies in the rate of age-related cataract progression between WT and KO mice is likely due to the presence of Grx2 in the WT mice. Reducing the accumulated PSSG in the early stage of protein oxidation can prevent the lens proteins from further modification into PSSP conjugates and the downstream high molecular weight aggregates that can lead to cataract formation. However, even the aging WT animals would succumb to the aging effect due to the chronic oxidative stress from the environment and the decline in antioxidants and oxidation damage repair enzymes (7, 12). Thus, the effect on lens transparency begins and progresses, albeit slower than that of the mice without a Grx2 presence.
Our mass spectrometry and immunoprecipitation studies have identified several lens structural proteins that have been in part modified into PSSG and accumulated in the lens. Of the three proteins shown in Fig. 7, αA-crystallin is an important and abundant structural protein. It possesses a chaperone-like function that stabilizes and maintains other lens proteins in a soluble form. However, on S-glutathionylation of one or both of its two SH groups, αA-crystallin's chaperone-like function can be drastically lost (54). Therefore, absence of Grx2 in the Grx2 KO mice likely contributes to the malfunction of αA-crystallin in the lens with a higher probability for compromised lens transparency.
Lens lacking Grx2 can affect the mitochondrial functions in the lens. Previous studies have indicated that Grx2 is a key regulator of redox homeostasis in the mitochondria, and it controls complex I activity in the electron transport system (ETS) for oxygen reduction and ATP production (37, 38). The results in this study as shown in Figs. 8 and 9 further support our earlier observation in the primary cell culture obtained from lenses of Grx2 KO mice that the 75-kDa catalytic subunit of complex I has been, in part, S-glutathionylated, resulting in lower complex I activity (38). In addition, this study showed that the downstream member of the ETS system, complex IV, has also suffered from activity loss, possibly due to S-glutathionylation at its 16-kDa subunit. The compromised ETS system in the Grx2 KO mice has resulted in lower ATP production. Furthermore, it is very likely that the oxygen free radical generated from incomplete oxygen reduction inside the mitochondria would leak out into cytosol, overwhelming the cytosolic capacity of Grx1 to protect the cytosolic proteins/enzymes from oxidative stress, leading to oxidation and malfunction of these proteins (see Fig. 10A). Although Grx2 in other mammalian tissues is known as an iron-sulfur protein and associated with iron metabolism, such property has never been explored in the lens and it worth the attention for future investigation.
FIGURE 10.

Potential mechanism for Grx2 gene deletion-induced cataract formation. A, proposed mechanism of ROS release into the cytosolic region of the cells. B, hypothesis for the thiol/disulfide imbalance-induced cataract formation in the lens. GR, glutathione reductase; Grx, glutaredoxin; Trx(R), reduced thioredoxin; Trx(O), oxidized thioredoxin; TR, thioredoxin reductase; PSSG, S-glutathionylated proteins; PSSP, protein-protein disulfides; ROS, reactive oxygen species.
Grx1 has recently been found to be present in the intermembrane space of the mitochondria (59). Our soluble mitochondrial preparation used for Grx2 disulfide reductase activity assay may likely contribute from mitochondrial Grx1 as both Grxs reduce the synthetic substrate (β-hydroxyethyl disulfide) used in this study, but we did not detect any Grx2 activity in the lens preparation of the Grx2 KO mice (Fig. 3B). However, as the mitochondrial Grx1 is located in the intermembrane space of the mitochondria, it should not interfere with or contribute to the function of Grx2 located in the mitochondrial matrix, where the ETS system is protected by this isozyme.
Besides the current evidence that mitochondrial Grx deletion may directly affect lens transparency, mice with global cytosolic Grx1 KO also displayed a phenotype of faster cataract progression with age,3 and its isolated lens epithelial cells became extra-sensitive to oxidation and were prone to apoptosis (60), parallel to its mitochondrial isoform. Thus, the Grx family is essential in protecting lens transparency.
In conclusion, Grx2 gene deletion induces a phenotype of faster progression of cataracts during aging, likely contributed from the combination of chaperone-like function loss and compromised mitochondrial functions. This is a first animal model that displays a close association to the absence of a single oxidative damage repair gene in the mitochondria with cataract progression that mimics the age-related cataract in humans. Results in the current studies provide further evidence for our previous hypothesis shown in Fig. 10B that oxidative stress-induced thiol/disulfide imbalance at the stage of PSSG accumulation is the first critical checkpoint to prevent the cascading events leading to cataract formation (7). The findings also suggest that Grx protein may be a potential anti-cataract agent for delaying or slowing down the age-related cataracts in humans.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 EY10595 (to M. F. L.) and NIEHS Pilot Grant P30 ES06639 (to Y. S. H.). This work was also supported by American Heart Association Greater Midwest Affiliate Grants 0455876Z and 0655631Z.
M. F. Lou, unpublished results.
- PSSG
- S-glutathionylated protein
- PSSC
- protein-S-S-cysteine
- DCIP
- 2,6-dichloroindophenol
- VDAC
- voltage-dependent anion channel
- PSSP
- protein-protein disulfide
- Trx
- thioredoxin
- Grx
- glutaredoxin
- ARC
- age-related cataract
- ETS
- electron transport system
- PSH
- protein sulfhydryl.
REFERENCES
- 1. Finkel T., Holbrook N. J. (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408, 239–247 [DOI] [PubMed] [Google Scholar]
- 2. Barnham K. J., Masters C. L., Bush A. I. (2004) Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 3, 205–214 [DOI] [PubMed] [Google Scholar]
- 3. Jenner P. (2003) Oxidative stress in Parkinson's disease. Ann. Neurol. 53, S26–S38 [DOI] [PubMed] [Google Scholar]
- 4. Bhat H. K., Calaf G., Hei T. K., Loya T., Vadgama J. V. (2003) Critical role of oxidative stress in estrogen-induced carcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 100, 3913–3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beatty S., Koh H., Phil M., Henson D., Boulton M. (2000) The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 45, 115–134 [DOI] [PubMed] [Google Scholar]
- 6. Spector A. (1995) Oxidative stress-induced cataract: mechanism of action. FASEB J. 9, 1173–1182 [PubMed] [Google Scholar]
- 7. Lou M. F. (2003) Redox regulation in the lens. Prog. Retin. Eye Res. 22, 657–682 [DOI] [PubMed] [Google Scholar]
- 8. Grek C. L., Zhang J., Manevich Y., Townsend D. M., Tew K. D. (2013) Causes and consequences of cysteine S-glutathionylation. J. Biol. Chem. 288, 26497–26504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mieyal J. J., Gallogly M. M., Qanungo S., Sabens E. A., Shelton M. D. (2008) Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 10, 1941–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gallogly M. M., Mieyal J. J. (2007) Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 7, 381–391 [DOI] [PubMed] [Google Scholar]
- 11. Taylor H. R., Keeffe J. E. (2001) World blindness: a 21st century perspective. Br. J. Ophthalmol. 85, 261–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harding J. J., Crabbe M. J. (1984) in The Lens: Development, Proteins, Metabolism and Cataract (Davson H., ed) Vol. 1, pp. 207–492, Academic Press, Orlando [Google Scholar]
- 13. Lou M. F., Dickerson J. E., Jr. (1992) Protein-thiol mixed disulfides in human lens. Exp. Eye Res. 55, 889–896 [DOI] [PubMed] [Google Scholar]
- 14. Lou M. F., Huang Q. L., Zigler J. S., Jr. (1989) Effect of opacification and pigmentation on human lens protein thiol/disulfide and solubility. Curr. Eye Res. 8, 883–890 [PubMed] [Google Scholar]
- 15. Lou M. F., Dickerson J. E., Jr., Tung W. H., Wolfe J. K., Chylack L. T., Jr. (1999) Correlation of nuclear color and opalescence with protein S-thiolation in human lenses. Exp. Eye Res. 68, 547–552 [DOI] [PubMed] [Google Scholar]
- 16. Lou M. (2004) in Redox-Genome Interactions in Health and Disease (Fuchs J., Podda M., Packer L., eds) pp. 325–350, Marcel Dekker, Inc., New York [Google Scholar]
- 17. Fecondo J. V., Augusteyn R. C. (1983) Superoxide dismutase, catalase and glutathione peroxidase in the human cataractous lens. Exp. Eye Res. 36, 15–23 [DOI] [PubMed] [Google Scholar]
- 18. Raghavachari N., Lou M. F. (1996) Evidence for the presence of thioltransferase in the lens. Exp. Eye Res. 63, 433–441 [DOI] [PubMed] [Google Scholar]
- 19. Qiao F., Xing K., Lou M. F. (2000) Modulation of lens glycolytic pathway by thioltransferase. Exp. Eye Res. 70, 745–753 [DOI] [PubMed] [Google Scholar]
- 20. Xing K. Y., Lou M. F. (2002) Effect of H2O2 on human lens epithelial cells and the possible mechanism for oxidative damage repair by thioltransferase. Exp. Eye Res. 74, 113–122 [DOI] [PubMed] [Google Scholar]
- 21. Yegorova S., Liu A., Lou M. F. (2003) Human lens thioredoxin: molecular cloning and functional characterization. Invest. Ophthalmol. Vis. Sci. 44, 3263–3271 [DOI] [PubMed] [Google Scholar]
- 22. Fernando M. R., Satake M., Monnier V. M., Lou M. F. (2004) Thioltransferase mediated ascorbate recycling in human lens epithelial cells. Invest. Ophthalmol. Vis. Sci. 45, 230–237 [DOI] [PubMed] [Google Scholar]
- 23. Mannervik B., Axelsson K. (1980) Role of cytoplasmic thioltransferase in cellular regulation by thiol-disulphide interchange. Biochem. J. 190, 125–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Axelsson K., Mannervik B. (1983) An essential role of cytosolic thioltransferase in protection of pyruvate kinase from rabbit liver against oxidative inactivation. FEBS Lett. 152, 114–118 [DOI] [PubMed] [Google Scholar]
- 25. Holmgren A. (1989) Thioredoxin and glutaredoxin systems. J. Biol. Chem. 264, 13963–13966 [PubMed] [Google Scholar]
- 26. Shelton M. D., Chock P. B., Mieyal J. J. (2005) Glutaredoxin: role in reversible protein S-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid. Redox Signal. 7, 348–366 [DOI] [PubMed] [Google Scholar]
- 27. Gladyshev V. N., Liu A., Novoselov S. V., Krysan K., Sun Q. A., Kryukov V. M., Kryukov G. V., Lou M. F. (2001) Identification and characterization of a new mammalian glutaredoxin (thioltransferase), Grx2. J. Biol. Chem. 276, 30374–30380 [DOI] [PubMed] [Google Scholar]
- 28. Lundberg M., Johansson C., Chandra J., Enoksson M., Jacobsson G., Ljung J., Johansson M., Holmgren A. (2001) Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. J. Biol. Chem. 276, 26269–26275 [DOI] [PubMed] [Google Scholar]
- 29. Lillig C. H., Berndt C., Vergnolle O., Lönn M. E., Hudemann C., Bill E., Holmgren A. (2005) Characterization of human glutaredoxin 2 as iron-sulfur protein: a possible role as redox sensor. Proc. Natl. Acad. Sci. U.S.A. 102, 8168–8173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tian X. L., Upadhyaya B., Wu H. L., Lou M. F. (2013) Expression and distribution of thiol-regulating enzyme glutaredoxin 2 (Grx2) in porcine ocular tissues. Invest. Ophthalmol. Vis. Sci. 54, (Abstr. 4936) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fernando M. R., Lechner J. M., Löfgren S., Gladyshev V. N., Lou M. F. (2006) Mitochondrial thioltransferase (glutaredoxin 2) has GSH-dependent and thioredoxin reductase-dependent peroxidase activities in vitro and in lens epithelial cells. FASEB J. 20, 2645–2647 [DOI] [PubMed] [Google Scholar]
- 32. Wu H. L., Xing K. Y., Lou M. F. (2009) Mitochondrial glutaredoxin (Grx2) protects human lens epithelial cells from H2O2-induced apoptosis via defending complex I from oxidation damage ARVO Meeting Abstracts (Abstr. 2541) [Google Scholar]
- 33. Moon S., Fernando M. R., Lou M. F. (2005) Induction of thioltransferase and thioredoxin/thioredoxin reductase systems in cultured porcine lenses under oxidative stress. Invest. Ophthalmol. Vis. Sci. 46, 3783–3789 [DOI] [PubMed] [Google Scholar]
- 34. Beer S. M., Taylor E. R., Brown S. E., Dahm C. C., Costa N. J., Runswick M. J., Murphy M. P. (2004) Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant defence. J. Biol. Chem. 279, 47939–47951 [DOI] [PubMed] [Google Scholar]
- 35. Enoksson M., Fernandes A. P., Prast S., Lillig C. H., Holmgren A., Orrenius S. (2005) Overexpression of glutaredoxin 2 attenuates apoptosis by preventing cytochrome c release. Biochem. Biophys. Res. Commun. 327, 774–779 [DOI] [PubMed] [Google Scholar]
- 36. Wu H., Xing K., Lou M. F. (2010) Glutaredoxin 2 prevents H2O2-induced cell apoptosis by protecting complex I activity in the mitochondria. Biochim. Biophys. Acta 1797, 1705–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mailloux R. J., Xuan J. Y., Beauchamp B., Jui L., Lou M., Harper M. E. (2013) Glutaredoxin-2 is required to control proton leak through uncoupling protein-3. J. Biol. Chem. 288, 8365–8379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu H., Lin L., Giblin F., Ho Y. S., Lou M. F. (2011) Glutaredoxin 2 knockout increases sensitivity to oxidative stress in mouse lens epithelial cells. Free Radic. Biol. Med. 51, 2108–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tybulewicz V. L., Crawford C. E., Jackson P. K., Bronson R. T., Mulligan R. C. (1991) Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell 65, 1153–1163 [DOI] [PubMed] [Google Scholar]
- 40. Nagy A., Rossant J., Nagy R., Abramow-Newerly W., Roder J. C. (1993) Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 90, 8424–8428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bradley A. (1987) in Tetratocarcinomas and Embryonic Stem Cells: A Practical Approach (Roberston E. J., ed) pp. 113–151, IRL Press at Oxford University Press, Oxford [Google Scholar]
- 42. Ho Y. S., Xiong Y., Ho D. S., Gao J., Chua B. H., Pai H., Mieyal J. J. (2007) Targeted disruption of the glutaredoxin 1 gene does not sensitize adult mice to tissue injury induced by ischemia/reperfusion and hyperoxia. Free Radic. Biol. Med. 43, 1299–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chylack L. T., Jr., Leske M. C., McCarthy D., Khu P., Kashiwagi T., Sperduto R. (1989) Lens opacities classification system II (LOCS II). Arch. Ophthalmol. 107, 991–997 [DOI] [PubMed] [Google Scholar]
- 44. Rehncrona S., Mela L., Siesjö B. K. (1979) Recovery of brain mitochondrial function in the rat after complete and incomplete cerebral ischemia. Stroke 10, 437–446 [DOI] [PubMed] [Google Scholar]
- 45. Lou M. F., Dickerson J. E., Jr., Garadi R., York B. M., Jr. (1988) Glutathione depletion in the lens of galactosemic and diabetic rats. Exp. Eye Res. 46, 517–530 [DOI] [PubMed] [Google Scholar]
- 46. Lou M. F., McKellar R., Chyan O. (1986) Quantitation of lens protein mixed disulfides by ion-exchange chromatography. Exp. Eye Res. 42, 607–616 [DOI] [PubMed] [Google Scholar]
- 47. Yan F. F., Pratt E. B., Chen P. C., Wang F., Skach W. R., David L. L., Shyng S. L. (2010) Role of Hsp90 in biogenesis of the beta-cell ATP-sensitive potassium channel complex. Mol. Biol. Cell 21, 1945–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wilmarth P. A., Riviere M. A., David L. L. (2009) Techniques for accurate protein identification in shotgun proteomic studies of human, mouse, bovine, and chicken lenses. J. Ocul. Biol. Dis. Infor. 2, 223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Janssen A. J., Trijbels F. J., Sengers R. C., Smeitink J. A., van den Heuvel L. P., Wintjes L. T., Stoltenborg-Hogenkamp B. J., Rodenburg R. J. (2007) Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin. Chem. 53, 729–734 [DOI] [PubMed] [Google Scholar]
- 50. Maquat L. E., Carmichael G. G. (2001) Quality control of mRNA function. Cell 104, 173–176 [DOI] [PubMed] [Google Scholar]
- 51. Spector A., Roy D. (1978) Disulfide-linked high molecular weight protein associated with human cataract. Proc. Natl. Acad. Sci. U.S.A. 75, 3244–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Augusteyn R. (1981) in Mechanisms of Cataract Formation in the Human Lens (Duncan G., ed) pp. 72–115, Academic Press, New York [Google Scholar]
- 53. Wolf N., Penn P., Pendergrass W., Van Remmen H., Bartke A., Rabinovitch P., Martin G. M. (2005) Age-related cataract progression in five mouse models for anti-oxidant protection or hormonal influence. Exp. Eye Res. 81, 276–285 [DOI] [PubMed] [Google Scholar]
- 54. Cherian M., Smith J. B., Jiang X. Y., Abraham E. C. (1997) Influence of protein-glutathione mixed disulfide on the chaperone-like function of α-crystallin. J. Biol. Chem. 272, 29099–29103 [DOI] [PubMed] [Google Scholar]
- 55. Hanson S. R., Chen A. A., Smith J. B., Lou M. F. (1999) Thiolation of the γB-crystallins in intact bovine lens exposed to hydrogen peroxide. J. Biol. Chem. 274, 4735–4742 [DOI] [PubMed] [Google Scholar]
- 56. Liang J. N., Pelletier M. R. (1988) Destabilization of lens protein conformation by glutathione mixed disulfide. Exp. Eye Res. 47, 17–25 [DOI] [PubMed] [Google Scholar]
- 57. Kono M., Sen A. C., Chakrabarti B. (1990) Thermodynamics of thermal and athermal denaturation of γ-crystallins: changes in conformational stability upon glutathione reaction. Biochemistry 29, 464–470 [DOI] [PubMed] [Google Scholar]
- 58. Truscott R. J., Augusteyn R. C. (1977) The state of sulphydryl groups in normal and cataractous human lenses. Exp. Eye Res. 25, 139–148 [DOI] [PubMed] [Google Scholar]
- 59. Pai H. V., Starke D. W., Lesnefsky E. J., Hoppel C. L., Mieyal J. J. (2007) What is the functional significance of the unique location of glutaredoxin 1 (GRx1) in the intermembrane space of mitochondria? Antioxid. Redox Signal. 9, 2027–2033 [DOI] [PubMed] [Google Scholar]
- 60. Löfgren S., Fernando M. R., Xing K. Y., Wang Y., Kuszynski C. A., Ho Y. S., Lou M. F. (2008) Effect of thioltransferase (glutaredoxin) deletion on cellular sensitivity to oxidative stress and cell proliferation in lens epithelial cells of thioltransferase knockout mouse. Invest. Ophthalmol. Vis. Sci. 49, 4497–4505 [DOI] [PubMed] [Google Scholar]