

Background: Carbon monoxide and connexin hemichannels are involved in several physiological and pathological processes.
Results: Carbon monoxide inhibits Cx43 and Cx46 hemichannel opening.
Conclusion: Connexin hemichannels are modulated by gaseous transmitters.
Significance: Our observations will help understand the effects of CO and CO donors in pathological conditions.
Keywords: Carbon Monoxide, Connexin, Ion Channel, Post-translational Modification (PTM), Redox Signaling, Carbonylation, Hemichannels
Abstract
Hemichannels (HCs) are hexamers of connexins that can form gap-junction channels at points of cell contacts or “free HCs” at non-contacting regions. HCs are involved in paracrine and autocrine cell signaling, and under pathological conditions may induce and/or accelerate cell death. Therefore, studies of HC regulation are of great significance. Nitric oxide affects the activity of Cx43 and Cx46 HCs, whereas carbon monoxide (CO), another gaseous transmitter, modulates the activity of several ion channels, but its effect on HCs has not been explored. We studied the effect of CO donors (CORMs) on Cx46 HCs expressed in Xenopus laevis oocytes using two-electrode voltage clamp and on Cx43 and Cx46 expressed in HeLa cells using a dye-uptake technique. CORM-2 inhibited Cx46 HC currents in a concentration-dependent manner. The C-terminal domain and intracellular Cys were not necessary for the inhibition. The effect of CORM-2 was not prevented by guanylyl-cyclase, protein kinase G, or thioredoxin inhibitors, and was not due to endocytosis of HCs. However, the effect of CORM-2 was reversed by reducing agents that act extracellularly. Additionally, CO inhibited dye uptake of HeLa cells expressing Cx43 or Cx46, and MCF-7 cells, which endogenously express Cx43 and Cx46. Because CORM-2 carbonylates Cx46 in vitro and induces conformational changes, a direct effect of that CO on Cx46 is possible. The inhibition of HCs could help to understand some of the biological actions of CO in physiological and pathological conditions.
Introduction
Hemichannels (HCs)2 are hexamers of connexins thought to be normally closed, preventing cell losses of important metabolites such as ATP and glutathione, as well as massive entry of Ca2+ (1, 2). However, recent studies show that HCs undergo brief openings under physiological conditions (1, 3); HC opening has been observed in ephaptic communication in the vertebrate retina (4), during the spread of Ca2+ waves (5), memory consolidation in the amygdala (6), and neurotransmitter release (7). It is also known that HCs with high activity (leaky HCs) are observed in several diseases where it contributes to cell damage or death (8–11). Overall, these studies support the idea that brief HC openings are compatible with life and are relevant in physiological conditions; however, uncontrolled HC opening is detrimental.
The mechanisms of HC gating vary among connexin isoforms. Phosphorylation (12), protease cleavage (13), and membrane potential (14) are well known factors. Recently, it has been proposed that changes in redox potential are also important modulators of Cx43 and Cx46 HCs (11, 15); for example, nitric oxide (NO) induces the S-nitrosylation of Cx43 intracellular Cys and causes HC opening (11). NO is probably the most studied of the gas transmitters, which also include carbon monoxide (CO) and hydrogen sulfide (H2S).
Under physiological conditions, CO is a subproduct of decomposition of heme groups that results from the work of heme oxygenases types I (HO-I) and II (HO-II); HO-I is inducible, whereas HO-II is constitutive (16). The actions of CO involve two pathways: (i) activation of guanylate cyclase with production of cGMP, and (ii) carbonylation of amino acids such as proline, threonine, lysine, and arginine (16). The physiological importance of CO is supported by the fact that cell cultures of HO-I knock-out mice show elevated concentrations of free radicals, and the few animals that reach birth (less than 5%) present serious disturbances in liver function and high mortality (17). These results suggest that CO is vital for the development and function of several tissues. Abnormal endogenous CO effects have been demonstrated in several diseases, and exogenous CO administration has potential value in the treatment of inflammation, sepsis, lung injury, cardiovascular diseases, transplantation, and cancer (18, 19). Despite its importance in health and disease, the molecular mechanism of action of CO is not well understood.
As mentioned above, connexin HCs are involved in a plethora of cellular functions, both under physiological and pathological conditions. Cx46 has gained considerable attention lately as a potential target for cancer treatment (20, 21) because it has been suggested that Cx46 helps cancer cells survive in hypoxic conditions (20). Because of its potential role of Cx46 in cancer biology and its sensitivity to gaseous transmitters such as NO (15), we decided to evaluate the possibility that Cx46 HCs are affected by HO-1 effectors (CO or guanylyl cyclase signaling pathways). To test this hypothesis, we determined the effects of the CO donor CORM-2 on HC activity. We studied Cx46 HC currents in Xenopus laevis oocytes using two-electrode voltage-clamp, and dye uptake through Cx43 and Cx46 HCs in HeLa cells. We found that CORM-2 produced a major reduction of Cx46 HC currents, an effect independent of the CO guanylyl cyclase/protein kinase G (PKG) signaling pathway, but associated to Cx46 carbonylation, suggesting that CO has a direct effect on the HCs.
MATERIALS AND METHODS
Chemicals
Tricarbonyldichlororuthenium(II) dimer (CORM-2), tricarbonylchloro(glycinato)ruthenium(II) (CORM-3), Na2[H3BCO2] (CORM-A1), β-mercaptoethanol (β-ME), dithiothreitol (DTT), KT5823, 1H-[1,2,4] oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), and MG132 were purchased from Sigma, and 4′,6-diamidino-2-phenylindole (DAPI) from Invitrogen. The protein carbonyl colorimetric assay kit was from Cayman Chemical Company (Ann Arbor, MI).
Plasmid Engineering
Details on the wild-type Cx46 and mutants were previously described (15). The rat Cx46 in pSP64T plasmid was obtained from Dr. Lisa Ebihara (Rosalind Franklin University of Medicine and Science), and site-directed mutagenesis was used to generate the Cx46-C3A and Cx46ΔCT mutants. In Cx46-C3A the intracellular Cys-218, Cys-283, and Cys-321 were substituted with Ala. Cx46ΔCT is a mutant in which the C-terminal domain was deleted after Gly-239. Human Cx43 and Cx46 were obtained from Promega and subcloned in to pF5A Flexi vector for stable transfection.
Cell Culture
HeLa cells and the human breast cancer cell line (MCF-7) were obtained from ATCC. HeLa cells were used to generate stable cell lines expressing wild-type Cx43 or Cx46. MCF-7 cells were used as an endogenous expression model for Cx43 and Cx46. Both cell types were cultured in DMEM with 10% fetal calf serum, at 37 °C, and in the presence of 5% CO2. G418 was used for selection of HeLa cells expressing wild-type Cx43 or Cx46.
cRNA Preparation and Injection into X. laevis Oocytes
Oocytes were injected with 12.5 ng of antisense Cx38 oligonucleotide alone or in combination with 25 ng of cRNA coding for Cx46, Cx46-C3A, or Cx46ΔCT. The antisense oligonucleotide is used to eliminate the endogenous expression of Cx38 that can form functional HCs (22). After cRNA injection, oocytes were maintained in Barth's solution (in mm: 88 NaCl, 1 KCl, 5 CaCl2, 0.8 MgCl2, 10 HEPES/NaOH, pH 7.4) supplemented with 0.1 mg/ml of gentamycin and 20 units/ml of penicillin and streptomycin for 24–48 h before the experiments.
Electrophysiological Recordings and Calculations
HC currents were measured as described by Retamal et al. (15). Each oocyte was placed in the 1-ml recording chamber and bathed at room temperature with ND96 solution (in mm: 96 NaCl, 2 KCl, 1.8 CaCl2, and 5 HEPES/NaOH, pH 7.4). Current-voltage (I-V) relationships were obtained from the current values at the end of the pulses. In some studies, the Boltzmann equation was fit to the data (15).
CORM Treatment
Oocytes were placed in the recording chamber and HC currents were measured under control conditions. Then, the oocytes were exposed to CORM-2, CORM-3, or CORM-A1 (100 μm) for 3 min, and HC currents were recorded once again. During this period, oocytes were held at −60 mV. Stock solutions (100 mm) were prepared by dissolving CORM-2 in pure ethanol, and CORM-3 or CORM-A1 in bath solution. CO-depleted CORM-2 solution was used as negative control. To prepare this solution, CORM-2 was dissolved in ND96 at the final concentration of 100 μm and the solution was placed at 37 °C overnight.
Dye Uptake
Dye uptake experiments were performed in ∼80% confluent cells grown on a glass coverslip. The cells were washed twice with recording solution (in mm: 148 NaCl, 5 KCl, 1.8 CaCl2, 1 MgCl2, 5 glucose, and 5 HEPES/NaOH, pH 7.4), and then each coverslip was placed in an inverted microscope (Nikon Ti-Eclipse) and exposed to 5 μm DAPI in recording solution. The DAPI emission fluorescence in each cell was recorded using a CCD monochrome camera (CFW-1310M; Scion, Frederick, MD). Images captured for 20 min at 30-s intervals were analyzed with an imaging software (NIS-Elements AR Analysis, Nikon), and the rate of dye uptake was calculated from the linear fit of the 15–20 min data.
Expression and Purification of Cx46
A synthetic gene coding for Cx46 followed by a tobacco etch virus protease cleavage sequence and a poly-His tag (His6) was expressed in Escherichia coli. This DNA, optimized for E. coli expression, was subcloned into the pQE60 plasmid, and expression was carried out in XL10-Gold cells grown in a modified M9 medium. The purification procedure was as previously described for Cx26, using a combination of metal affinity (Talon Co2+ column, Talon Superflow, Clontech) and gel-filtration chromatography (Superdex 200HR 10/300 GL column, GE Healthcare) (23). The tag was removed by incubation with tobacco etch virus protease for 12 h, at a Cx46/tobacco etch virus protease ratio of 1:15 (w/w). After removal of the His tag, purified Cx46 was isolated by gel-filtration chromatography on a Superdex 200HR column.
Protein Carbonyl Detection
Protein samples were derivatized by the reaction between 2,4-dinitrophenylhydrazine and protein carbonyls. Formation of a Schiff base produces the corresponding hydrazone, which was analyzed spectrophotometrically at 375 nm.
Tryptophan Fluorescence Measurements
Trp fluorescence was measured on a Jasco spectrofluorimeter model FP-6300. Samples containing purified Cx46 were excited at 295 nm and fluorescence emission was measured between the wavelengths of 310 and 500 nm. The measurements were carried out at room temperature.
Detection of CO Content in CO-depleted CORM-2 Solution
For the determination of the content of CO in solution, Hb was dissolved to a concentration of 1 mg/ml in ND96 (control) or in ND96 supplemented with 100 μm fresh CORM-2 or CO-depleted CORM-2 solutions. The solutions were supplemented with 0.1% sodium dithionite to deoxygenate them, to allow CO binding to Hb to form carboxyhemoglobin. The content of carboxyhemoglobin was assessed from the changes of the absorbance at 540 nm (24).
Immunofluorescence
MCF-7 cells grown in glass coverslips were fixed by incubation in 4% paraformaldehyde for 30 min, and incubated with PBS, 1% Triton for an additional 15 min. The cells were then incubated with normal goat serum (Santa Cruz, number SC-2043) for 1 h. After overnight incubation with the polyclonal primary antibodies against Cx43 (Invitrogen, number 710700) or Cx46 (Santa Cruz, number M-127) diluted in normal goat serum, the coverslips were washed with PBS and incubated with a secondary antibody (goat anti-rabbit, Thermo Scientific) for 1 h. Cx43 and Cx46 immunolocalization was determined in an epifluorescence-inverted microscope (Nikon, Eclipse Ti).
Statistical Analysis
Results are expressed as mean ± S.E. and n refers to the number of independent experiments. For statistical analyses, each treatment was compared with its respective control, and significance was determined using a one-way analysis of variance or paired Student's t test, as appropriate. Differences were considered significant at p < 0.05.
RESULTS
CORMs Inhibits Cx46 Hemichannel Currents
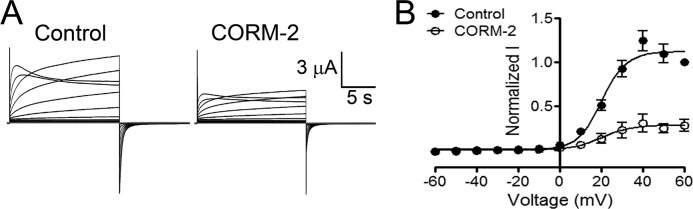
Because Cx46 is responsive to changes in redox potential (15) and CO modulates physiological and pathological processes by mechanisms that include effects on ion channels (25), we tested whether CO modulates rat Cx46 HC currents. With repeated depolarizing pulses from −60 to +60 mV for 2.5 s, with 2.5-s intervals, HC currents increase progressively until they reach a stable maximum. We previously described this phenomenon and called it “facilitation” (26). Application of 100 μm CORM-2 once the currents stabilized caused inhibition of Cx46 HCs (Fig. 1A) with a time constant (τ) of 29 s. This concentration of CORM-2 has been previously used in studies on other ion channels (27–30). To study the voltage dependence of the CORM-2 inhibition, oocytes were exposed to 100 μm CORM-2 for 3 min (to ensure a maximal effect) and HC currents were recorded using a protocol wherein oocytes were held at −60 mV and then exposed for 15 s to voltages ranging from −60 to +60 mV in 10 mV steps. After each pulse, the membrane potential was held for 10 s at −60 mV (15). Under control conditions, Cx46 HCs activated slowly at voltages over +10 mV, showing a clear tail current when the voltage returned to −60 mV (Fig. 1B, lower panel). At +60 mV the maximum current was 12.0 ± 1.1 μA. These results are consistent with our previous studies (15). After the 3-min exposure to 100 μm CORM-2, Cx46 HC currents were inhibited by 73 ± 4% (Fig. 1, B and D). Oocytes injected only with antisense for Cx38 show a small current (0.37 ± 0.17 μA) that did not change after CORM-2 application (0.39 ± 0.14 μA) (Fig. 1B, upper panel). Fig. 1C shows that increasing CORM-2 concentrations progressively decreased the HC currents with an IC50 of 3.4 μm. Unfortunately, the voltage clamp was difficult to maintain at higher concentrations of CORM-2. We do not know whether this is the result of alterations in plasma membrane properties or other effects. The V50 values of the whole-cell currents determined by fitting to the Boltzmann equation were 39 ± 1 mV under control conditions and 44 ± 10 mV after CORM-2. The lack of a statistically significant difference indicates that CORM-2 reduces either the number of Cx46 HCs opened by depolarization, and/or the single-channel conductance, without changing voltage sensitivity. Exposure to 100 μm CO-depleted CORM-2 solution (see “Materials and Methods”) increased Cx46 HC currents by 49 ± 17% (Fig. 1D). The CO-depleted CORM-2 solution did not react with Hb to form Hb-CO, whereas fresh CORM-2 did (Fig. 2). These results indicate that CORM-2 was effectively depleted of CO by overnight incubation at 37 °C. The absence of Cx46 HC inhibition by CO-depleted CORM-2 suggests that the inhibitory effect of CORM-2 is due to CO, and not to another CORM-2 product. We did not explore the origin of activation of Cx46 HCs by CO-depleted CORM-2; it may result from effects of products different from CO. In agreement with the results above, addition of 5.6 mg/ml of hemoglobin (a CO scavenger) to the bath solution before the addition of fresh CORM-2 prevented the current inhibition by CORM-2 (Δ = −6 ± 2%) (Fig. 1D). A clear inhibition of Cx46 HCs currents was also induced by CORM-3 and CORM-A1 (77 ± 3.9 and 39 ± 7.4% inhibition, respectively). (Fig. 1D). These data strongly support the conclusion that the HC current inhibition by CORM-2 is mediated by released CO.
FIGURE 1.

CORM-2 reduces Cx46 hemichannel currents. A, effect of CORM-2 on Cx46 HC currents. Cx46 was expressed in X. laevis oocytes and HC currents were measured with the two microelectrode voltage-clamp technique. Oocytes were depolarized to +60 mV for 2.5 s and then the membrane potential was held at −60 mV for an additional 2.5 s. This protocol was repeated at least 40 times, until the currents elicited were maximal and stable, and then CORM-2 (100 μm) was added. Time constant (τ) of the change in current amplitude was calculated by fitting a single exponential decay equation to the data (n = 5). B, typical records, at varying voltages, showing the effect of CORM-2 on oocytes injected only with Cx38 antisense (upper panel, n = 5) or expressing Cx46 HCs (lower panel, n = 23). Under control conditions, large outward currents were observed when Cx46 expressing oocytes were depolarized at voltages over +10 mV. Exposure of the same oocyte to 100 μm CORM-2 for 3 min substantially inhibited the Cx46 HC currents. During CORMs treatments oocytes were always held at −60 mV. C, Cx46 HC currents were recorded after 3 min of exposure to different concentrations of CORM-2. The percentage of current inhibition is shown in a semilogarithmic plot, and the solid line corresponds to the fit of the data to the Hill equation. The dose-response relationship for CORM-2 was studied at concentrations ranging from 0.01 to 100 μm (n = 7 for each concentration). D, dependence of the CORM-2 effect on CO. CO-2, 100 μm CORM-2 (n = 23); CO−, 100 μm CO-depleted CORM-2 solution (n = 10); Hb, 100 μm CORM-2 in the presence of 5.6 mg/ml of hemoglobin (n = 10); CO-3, 100 μm CORM-3 (n = 7); and CO-A1, 100 μm CORM-A1 (n = 7). Data are mean ± S.E. of currents measured at the end of +60 mV depolarization pulse, normalized to the control value; *, **, and *** denote p < 0.05, 0.01, and 0.001 compared with control, respectively; ### denotes p < 0.001 for CO-2 compared with CO- or Hb; n.s., denotes not statistically significant.
FIGURE 2.

Detection of CO release from CORM-2 using a Hb-based assay. Hb (1 mg) was dissolved in 1 ml of solution containing fresh CORM-2 (100 μm) (CO+), its vehicle (ethanol, 1:1000) (OH), or a CO-depleted CORM-2 solution (100 μm) (CO−). The solutions were supplemented with 0.1% sodium dithionite to deoxygenate them and allow CO binding to the Hb to form carboxyhemoglobin (CO-Hb). The resulting mixture was maintained at room temperature for 3 min and then the CO-Hb content was assessed from the changes in absorbance at 540 nm (n = 8 for each condition). Data are mean ± S.E.; ***, denote p < 0.001.
The C-terminal Domain of Cx46 Is Not Involved in Hemichannel Inhibition by CORM-2
Because the C-terminal domains of Cx46 and other connexin isoforms have a regulatory role, we studied the effect of CORM-2 on HCs formed by Cx46ΔCT, a mutant with deletion of the CT domain. Under control conditions, Cx46ΔCT HCs display outward currents activated at positive voltages and current inactivation at voltages over +50 mV (Fig. 3A, left record). After a 3-min exposure to 100 μm CORM-2 HC, the currents were reduced by 72 ± 7%, without changes in the activation voltage and current relaxation (Fig. 3A, right record) (n = 8). Similar to the effect of CORM-2 over wild-type Cx46 HCs, the V50 of Cx46ΔCT HC did not change (18 ± 2 versus 17 ± 7 mV before and after CORM-2, respectively) (Fig. 3B). These results show that the C-terminal domain of Cx46 is not necessary for the reduction of HC current by CORM-2.
FIGURE 3.
The C-terminal domain of Cx46 is not involved in CORM-2 hemichannel inhibition. A, representative Cx46ΔCT (C-terminal domain deletion) HC current records under control conditions (left-hand records) and after exposure to 100 μm CORM-2 (right-hand records) (n = 8 for each condition). B, I/V relationship. Currents at the end of each pulse were normalized to the maximal current obtained at +60 mV under control conditions, and the I/V relationship was fitted with a Bolzmann equation. Data are presented as mean ± S.E. (n = 8).
The Reversibility of the Inhibition by CORM-2 Is Greatly Enhanced by Reducing Agents
Because Cys residues have a critical role in the CO sensitivity of BK(Ca) channels (31) and CO modulates the redox balance in several cell types (16), we studied the role of Cx46 Cys on HC inhibition by CO. Cx46 has 9 Cys, but only the 3 intracellular Cys appear to be affected by free radicals (15). Therefore, we decided to test the effect of 100 μm CORM-2 on HCs formed by a mutant where these three Cys were replaced with Ala (Cx46-C3A). Exposure to 100 μm CORM-2 for 3 min reduced Cx46-C3A HC currents by 70 ± 6% (Fig. 4A), an inhibition similar to that produced on wild-type Cx46 HCs (73 ± 4%), with no statistically significant differences between V50 values (control: 40 ± 7 mV; CORM-2: 45 ± 12 mV; Fig. 4A). These results indicate that intracellular Cys are not required for the inhibitory effect of CORM-2.
FIGURE 4.

Cx46 hemichannel inhibition by CORM-2 is modulated by reducing agents. A, effect of CORM-2 on HCs formed by Cx46-C3A (Cx46 without intracellular Cys). Currents at the end of each pulse were normalized to the maximal current obtained at +60 mV under control conditions, and the I/V relationship was fitted with a Bolzmann equation. Exposure to 100 μm CORM-2 was for 3 min. Data are mean ± S.E. (n = 8). B, reversal by reducing agents of the Cx46 HC inhibition by CORM-2. Oocytes were exposed to 100 μm CORM-2 for 3 min and then to 5 mm β-ME (n = 6), 1 mm DTT (n = 6), or 5 mm extracellular reduced glutathione (GSHe) (n = 8) for 1 min. GSH was injected to a calculated final concentration of 5 mm (intracellular reduced glutathione, GSHi) (n = 6). Currents were normalized to the control value and are expressed as mean ± S.E. *** indicates p < 0.001 compared with CORM-2. C, effect of CORM-2 on HCs formed by Cys-less Cx46 (Cx46-CL). Currents at the end of each pulse were normalized to the maximal current obtained at +60 mV under control conditions, and the I/V relationship was fitted with a Bolzmann equation. Exposure to 100 μm CORM-2 was for 3 min. Data are mean ± S.E. (n = 11). D, oocytes expressing Cx46 or Cx46ΔCT were exposed to 100 μm CORM-2 for 3 min, followed by washing with ND96 (1 ml/min for 3 min). Currents measured at +60 mV were normalized to the control value in the absence of CORM-2. Data are mean ± S.E. (n = 5 for each time point).
Fig. 4D shows that the inhibition of Cx46 HC by exposure to 100 μm CORM-2 for 3 min is reversible. Following extensive washing with ND96, Cx46 HCs currents displayed a slow rate of recovery over time. Linear regression analysis showed a recovery of 0.007 ± 0.002 nA/min for the Cx46 HC currents, whereas Cx46ΔCT HC currents recovered twice as fast (0.014 ± 0.003 nA/min). This recovery is very slow, and may be the result of insertion of non-oxidized Cx46 at the plasma membrane and/or decarbonylation of Cx46 HCs that occurs naturally on oocytes.
Carbonylation is a potential mechanism for the inhibitory effect of CORM-2 (32). Carbonylation induced by CO can be reversed by reducing agents and CO may induce secondary Cys carbonylation in response to lipid peroxidation products, a process that can be sensitive to reducing agents (33, 34). Therefore, we tested whether the inhibition of Cx46 HCs currents by exposure to 100 μm CORM-2 for 3 min was reversed by β-ME or dithiothreithol (DTT). After 1-min exposure to 5 mm β-ME or 1 mm DTT (reducing agents that permeate the plasma membrane), the inhibition of Cx46 HC currents by CORM-2 was reversed to 98 ± 0.1 and 102 ± 1.4% of the control value, respectively. Addition of 5 mm reduced glutathione (GSH, a reducing agent that does not permeate the plasma membrane) also recovered the Cx46 HCs currents inhibited by CORM-2 (to 98 ± 1.9% of the control value). However, when GSH was injected into the oocytes (5 mm estimated final concentration), the recovery of Cx46 HCs currents was minimal (Fig. 4B). Because CO-inhibition present in Cx46-C3A HCs was strongly affected by extracellular reducing agents, and secondary carbonylation of Cys can occur in response to lipid peroxidation products (34), we tested the role of the 6 extracellular Cys in the inhibition of HCs by CORM-2. In HCs formed by a Cx46 where all Cys were replaced with Ala (Cx46-CL) CORM-2 produced only a 25 ± 3.9% inhibition (Fig. 4C). Together, these data indicate that the HC inhibition by CO requires the presence of one or more extracellular Cys.
Guanylyl Cyclase, PKG, and Proteasome Pathways Are Not Involved in the CORM-2 Effect
It has been reported that CO activates guanylyl cyclase (18), which elevates the cGMP intracellular concentration with the consequent PKG activation. Because Cx46 is a phosphoprotein, an involvement of guanylyl cyclase and PKG in the effect of CORM-2 is possible. However, pre-incubation for 45 min with 2 μm KT5823 (a specific PKG inhibitor) or 50 μm ODQ (1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one, a specific guanylyl cyclase inhibitor) (27, 28) did not affect the current inhibition induced by 100 μm CORM-2 (Fig. 5A). It is also known that oxidized proteins can traffic to proteasomes and be degraded (35), which could account for the decrease in Cx46 HC currents by CORM-2 as a result of a loss of HCs from the plasma membrane. Fig. 5A shows that the inhibition of Cx46 HC currents by CORM-2 in oocytes treated with 10 μm MG132 (a proteasome inhibitor) (32) was 78 ± 10%, a value indistinguishable from that in oocytes treated with CORM-2 alone. Because decarbonylation depends on reducing agents acting by a redox-thioredoxin-dependent mechanism (33, 34), we explored the effects of the thioredoxin reductase inhibitor auranofin on the inhibition of Cx46 HC currents by CORM-2. Oocytes were preincubated with 60 μm auranofin for 30 min and then exposed to 100 μm CORM-2 for 3 min in the continued presence of auranofin. Under these conditions, CORM-2 inhibited Cx46 HC currents by ∼60%, indicating that thioredoxin reductase is not directly involved in the HC current recovery induced by reducing agents (Fig. 5A). These data suggest that CO-induced inhibition is not due to HC internalization or activation of a cGMP-dependent pathway.
FIGURE 5.

Guanylyl cyclase, PKG, and proteasome pathways are not involved in the inhibitory effect of CORM-2 on Cx46 hemichannels. Oocytes expressing Cx46 were exposed to 100 μm CORM-2 for 3 min and HC currents were measured at +60 mV in oocytes under control conditions (solid bar), and after a 45-min pre-treatment with the PKG inhibitor KT5823 (2 μm), the guanylyl cyclase inhibitor ODQ (50 μm), the proteasome inhibitor MG132 (10 μm), or the thioredoxin reductase inhibitor auranofin (60 μm) (n = 7 for each inhibitor). Data are mean ± S.E.
CORM-2 Inhibits Human Cx43 and Cx46 Hemichannels in Mammalian Cultured Cells
Because signaling and protein-protein interactions are different in frog oocytes and mammalian cells, it is important to demonstrate that the response of HCs to CO is preserved in mammalian cells. To this end, we studied the effect of CORM-2 on HeLa and MCF-7 cells expressing human Cx43 and/or Cx46 using a dye-uptake technique. Under control conditions, Cx46-transfected HeLa cells present a prominent DAPI uptake when exposed to 5 μm of the dye (rate of uptake = 64 ± 8 AU/min) (Fig. 6A). After adding 1 μm CORM-2 (a concentration that shows the maximum effect in Cx43 and Cx46, Fig. 6, B and C), the uptake was reduced drastically (to 23 ± 5 AU/min). Comparable results were obtained in HeLa cells expressing Cx43, where CORM-2 reduced the rate of dye uptake from 48 ± 3 to 25 ± 2 AU/min (Fig. 6C). As negative control we used non-transfected HeLa cells (parental cells); in these, 1 μm CORM-2 had no effect on the rate of dye uptake (6.3 ± 0.7 before versus 6.8 ± 0.4 AU/min after CORM-2) (Fig. 6D). These data demonstrate that the effect of CORM-2 on Cx46 HCs is independent of the cell type and that CORM-2 not only inhibits Cx46 HCs, but also Cx43 HCs. Because regulation of HCs can be altered in overexpressing HeLa cells, we also tested the effect of CORM-2 in cells with native HC expression. In MCF-7 cells, a human breast cancer cell line that expresses Cx43 and Cx46 (21) (Fig. 6E), a decrease of dye uptake by CORM-2 was also observed; uptakes were 30 ± 3 before and 10 ± 1 AU/min after 10 μm CORM-2 (Fig. 6D). The basal dye uptake was inhibited by 200 μm La3+ (85.4% inhibition) and 100 μm Gap27 (65% inhibition), suggesting that the basal rate of uptake is mainly mediated by connexin HCs (Fig. 6F). Also, dye uptake was partially sensitive to 5 μm carbenoxolone (33% inhibition). Because at this concentration carbenoxolone inhibits Panx, but not connexin HCs (36), the results suggest that ∼⅓ of the dye uptake could be mediated by Panx hemichannels.
FIGURE 6.

CORM-2 induces hemichannel inhibition in human cell lines. A, under control conditions dye uptake was observed in HeLa cells transfected with human Cx46 (filled circles, n = 8), which was inhibited by 1 μm CORM-2 (open circles, n = 8). For comparison the dye uptake in parental cells under control conditions is also shown (filled triangles, n = 8). B, dependence of inhibition of the rate of DAPI uptake on CORM-2 concentration (0.1–10 μm) in HeLa cells expressing Cx46 HCs (n = 3 for each condition). C, dependence of the inhibition of the rate of DAPI uptake on CORM-2 concentration (0.1–10 μm) in HeLa cells expressing Cx43 HCs (n = 3 for each condition). D, average of CORM-2-induced inhibition (1 μm) on the rate of dye uptake in HeLa cells expressing Cx43 (n = 8), Cx46 (n = 8), parental HeLa cells (n = 6), and MCF-7 cells (n = 7). At least 30 cells were analyzed in each experiment. Data were normalized as the rate of dye uptake before CORM-2 addition. Data are mean ± S.E.; *, **, and *** denote p < 0.05, 0.01, and 0.001 compared with the corresponding control; n.s. denotes no significant difference. For all panels, cells where exposed to 5 μm DAPI and images were acquired every 30 s for 20 min. MCF-7 cells express functional hemichannels in their plasma membrane. E, expression of Cx43 and Cx46. The proteins are expressed, but do not form clearly distinguishable gap-junction plaques. The green signal corresponds to Cx43 or Cx46 immunolocalization, and the blue signal corresponds to staining of the nucleus by DAPI. F, functional HCs are present in the plasma membrane of MCF-7 cells. MCF-7 cells where exposed to 5 μm DAPI and pictures were taken every 30 s for 20 min. The basal dye uptake was inhibited by the HC blockers La3+ (200 μm) and Gap27 (100 μm). The uptake was also inhibited by carbenoxolone (Cbx, 5 μm), which selectively inhibits Panx HCs over connexin HCs at this concentration. Data are mean ± S.E. from >90 cells in 6 independent experiments; *** denote p < 0.001 compared with basal dye uptake; ### denote p < 0.001 when compared with the La3+ and Gap27 effect; and &&& denotes p < 0.001 when compared with the Gap27 and Cbx effect.
CORM-2 Induces Cx46 Carbonylation in Vitro
If the effect of CO on the HCs is direct, it could be mediated by carbonylation of Cx46. Protein carbonyls were estimated from derivatization with 2,4-dinitrophenylhydrazine coupled to a reaction that forms hydrazine, which is measured by its absorbance at 370 nm. Fig. 7A shows a statistically significant increase in the absorbance after exposure to 100 μm CORM-2 for 3 min at room temperature in both bovine serum albumin (BSA) (from 0.0243 ± 0.0015 to 0.0425 ± 0.0015) and Cx46 (from 0.0119 ± 0.0025 to 0.0275 ± 0.0047). No changes in absorbance were observed when 2,4-dinitrophenylhydrazine was omitted. Co-incubation with CORM-2 and 5 mm DTT did not affect the CORM-2 effect (0.029 ± 0.0017). These results show that Cx46 is carbonylated by CO and that this effect is not directly reversed by DTT. If CO decreases HC activity by carbonylation, it may be possible to determine conformational changes by the effect on Trp fluorescence in response to CORM-2 treatment. Under control conditions Trp fluorescence emission shows a bell shape (Fig. 7B) with maximum emission of 14.8 ± 0.7 AU at 337 ± 0.7 nm. Exposure to 100 μm CORM-2 for 1 min reduced the maximum fluorescence to 3.40 ± 0.18 AU, with a red shift of the emission maximum to 343 ± 0.4 nm. These results indicate important conformational changes of purified Cx46 in response to CO. Exposure to 5 mm β-ME applied 1 min after CORM-2 did not recover Trp fluorescence. The latter result confirms that reducing agents cannot decarbonylate Cx46 in vitro, which is in line with previous findings (33, 34). The changes in Trp fluorescence suggest that one or more Trp residues move to a more hydrophilic environment in response to carbonylation. Unfortunately, Cx46 has many Trp residues distributed throughout the protein, and it is not possible to ascribe the structural change to a specific domain.
FIGURE 7.

CORM-2 induces Cx46 carbonylation and conformational changes in vitro. A, purified BSA and Cx46 (both at 1.5 mg/ml) were exposed to CORM-2 for 3 min at room temperature. Then, the proteins were assayed for carbonylation (see “Materials and Methods” for details). Data are presented as absorbance at 370 nm, which is proportional to carbonylation (n = 3 for BSA and n = 6 for Cx46). In some experiments purified Cx46 was simultaneously exposed to 5 mm DTT and 100 μm CORM-2 (n = 3). B, representative Trp spectra of purified Cx46 under control conditions (solid black line) (n = 4), and after exposure to CORM-2 vehicle ethanol (1:500) (dotted black line) (n = 2), 200 μm CORM-2 (solid gray line) (n = 4), or 5 mm β-ME (dotted gray line) (n = 2). Excitation was at 295 nm and emission was measured between 310 and 420 nm. Data are mean ± S.E.; * and ** denote p < 0.05 and 0.01, respectively.
DISCUSSION
We found that CORM-2 causes a large reduction in currents through HCs formed by Cx46 expressed in X. laevis oocytes. We conclude that the effect of CORMs on Cx46 HCs is produced by CO, because CO-depleted CORM-2 solution had no inhibitory effect, the CO scavenger hemoglobin prevented the CORM-2 effect, and other CO releasing molecules (CORM-3 and CORM-A1) have the same inhibitory effect. Interestingly, with the CO-depleted CORM-2 solution we observed an increase in HC activity that suggests that unidentified CORM-2 subproducts activate Cx46 HCs. Consistent with this interpretation; CORM-2 subproducts activate non-selective cation currents in human endothelial cells (37).
It is well known that the signaling effects of CO may involve two mechanisms (i) direct carbonylation of proline, threonine, lysine, and/or arginine residues (16) and (ii) activation of guanylyl cyclase, with elevation of cGMP levels and activation of PKG (18). The lack of effects of guanylyl cyclase/PKG specific inhibitors on the response of Cx46 HCs to CORM-2 strongly suggests that the effect of CO does not involve the guanylyl cyclase/PKG pathways. The Cx46 HC current reduction could also result from internalization and proteasomal degradation of carbonylated Cx46. However, our results do not support this hypothesis because the effect of CO was fully and rapidly reversed by GSHe, β-ME, and DTT and the inhibition of Cx46 HC currents by CO was not prevented by a proteasome inhibitor. Therefore, Cx46 carbonylation seems more likely.
The inhibition of Cx46 HCs by CO was fully reversed by extracellular reducing agents, depended on Cx46 extracellular Cys, and did not involve the regulatory C-terminal domain. We also found that CORM-2 inhibits connexin HCs expressed in human cell lines, indicating that the effect of CO is independent of the experimental system and that CO affect both human and rat Cx46 HCs. However, the loss of effect at 10 μm was unexpected and denotes that the CO effect in mammalian cells could be more complex. We also demonstrated that CORM-2 induces Cx46 carbonylation in vitro, which induce Cx46 conformational changes detected by Trp fluorescence. Thus, this work suggests that CO inhibits Cx46 HCs through carbonylation of extracellular residues and that inhibition is reversible in a Cys-dependent mechanism.
CO may induce secondary Cys-carbonylation in response to lipid peroxidation products, a process that can be sensitive to reducing agents (34). Because the inhibition of Cx46 HCs by CO is reversed by cell-membrane impermeable reducing agents, the possibility of a secondary Cys carbonylation cannot be ruled out. This possibility is supported by the fact that the inhibition by CO was much reduced in HC formed by Cys-less Cx46. As mentioned above, the effect of CORM-2 was reversed by membrane-impermeable reducing agents. This, in combination with our results in vitro, point to a complex enzyme-mediated decarbonylation mechanism. We found that carbonylation of purified Cx46 by CO was not reversed by DTT and that the conformational changes induced by CO were not reverted by β-ME. Thioredoxin- and peroxiredoxin-dependent decarbonylation mechanisms have been proposed (33, 34). Interestingly these two enzymes can act at the extracellular face of the plasma membrane (38), suggesting that this enzymatic mechanism may be involved in our system.
Most of our experiments were done at 100 μm CORM-2. At this concentration and exposure time of usually 3 min, CORM-2 should release about ∼75 μmol of CO/liter of solution (39, 40). The physiological concentration of CO is in the range of a few micromolar (41) and we estimated the IC50 at 3.4 μm CORM-2. Therefore, Cx43 and Cx46 HCs can be good sensors of physiological and pathological changes in CO concentration, confirming the regulation of connexin HCs by redox potential (42). Epithelial lens cells in culture express HO-1 in response to an oxidative stress (43). Because lens fibers express Cx46, it is possible that the CO generated in response to cellular stress keeps the HCs closed, preventing cell damage. Because HCs can play important roles in the progression of certain forms of cancer (44), our results may help to understand the molecular mechanism of CO as an anticancer agent (18). The present results suggest the possibility of targeting the CO signaling pathway to treat disorders such as cancer, where activation of Cx46 HCs is involved in the resistance of tumors to hypoxia.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM79629 and 3R01GM079629-03S1 and American Heart Association, Texas Affiliate Inc. Grant 14GRNT18750014 (to G. A. A.), Fondecyt Grant 1120214 and Anillo Grant ACT1104 (to M. A. R.), Centro de costo Grant 23400077 (to C. G. L.), and Fondecyt Grant 3130577 (to V. F.).
- HC
- hemichannel
- Cx
- connexin
- β-ME
- β-mercaptoethanol
- ODQ
- 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- AU
- arbitrary unit
- PKG
- protein kinase G
- CORM-2
- tricarbonyldichlororuthenium(II) dimer
- CORM-3
- tricarbonylchloro(glycinato)ruthenium(II)
- CORM-A1
- Na2[H3BCO2]
- Hb
- hemoglobin.
REFERENCES
- 1. Sáez J. C., Schalper K. A., Retamal M. A., Orellana J. A., Shoji K. F., Bennett M. V. (2010) Cell membrane permeabilization via connexin hemichannels in living and dying cells. Exp. Cell Res. 316, 2377–2389 [DOI] [PubMed] [Google Scholar]
- 2. Sánchez H. A., Mese G., Srinivas M., White T. W., Verselis V. K. (2010) Differentially altered Ca2+ regulation and Ca2+ permeability in Cx26 hemichannels formed by the A40V and G45E mutations that cause keratitis ichthyosis deafness syndrome. J. Gen. Physiol. 136, 47–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kar R., Batra N., Riquelme M. A., Jiang J. X. (2012) Biological role of connexin intercellular channels and hemichannels. Arch. Biochem. Biophys. 524, 2–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Klaassen L. J., Fahrenfort I., Kamermans M. (2012) Connexin hemichannel mediated ephaptic inhibition in the retina. Brain Res. 1487, 25–38 [DOI] [PubMed] [Google Scholar]
- 5. Orellana J. A., Sánchez H. A., Schalper K. A., Figueroa V., Sáez J. C. (2012) Regulation of intercellular calcium signaling through calcium interactions with connexin-based channels. Adv. Exp. Med. Biol. 740, 777–794 [DOI] [PubMed] [Google Scholar]
- 6. Stehberg J., Moraga-Amaro R., Salazar C., Becerra A., Echeverría C., Orellana J. A., Bultynck G., Ponsaerts R., Leybaert L., Simon F., Sáez J. C., Retamal M. A. (2012) Release of gliotransmitters through astroglial connexin 43 hemichannels is necessary for fear memory consolidation in the basolateral amygdala. FASEB J. 26, 3649–3657 [DOI] [PubMed] [Google Scholar]
- 7. Wang N., De Bock M., Decrock E., Bol M., Gadicherla A., Vinken M., Rogiers V., Bukauskas F. F., Bultynck G., Leybaert L. (2013) Paracrine signaling through plasma membrane hemichannels. Biochim. Biophys. Acta 1828, 35–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chi J., Li L., Liu M., Tan J., Tang C., Pan Q., Wang D., Zhang Z. (2012) Pathogenic connexin-31 forms constitutively active hemichannels to promote necrotic cell death. PLoS One 7, e32531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liang G. S., de Miguel M., Gómez-Hernández J. M., Glass J. D., Scherer S. S., Mintz M., Barrio L. C., Fischbeck K. H. (2005) Severe neuropathy with leaky connexin32 hemichannels. Ann. Neurol. 57, 749–754 [DOI] [PubMed] [Google Scholar]
- 10. Mese G., Sellitto C., Li L., Wang H. Z., Valiunas V., Richard G., Brink P. R., White T. W. (2011) The Cx26-G45E mutation displays increased hemichannel activity in a mouse model of the lethal form of keratitis-ichthyosis-deafness syndrome. Mol. Biol. Cell 22, 4776–4786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Retamal M. A., Cortés C. J., Reuss L., Bennett M. V., Sáez J. C. (2006) S-Nitrosylation and permeation through connexin 43 hemichannels in astrocytes: induction by oxidant stress and reversal by reducing agents. Proc. Natl. Acad. Sci. U.S.A. 103, 4475–4480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnstone S. R., Billaud M., Lohman A. W., Taddeo E. P., Isakson B. E. (2012) Posttranslational modifications in connexins and pannexins. J. Membr. Biol. 245, 319–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang K., Gu S., Yin X., Weintraub S. T., Hua Z., Jiang J. X. (2012) Developmental truncations of connexin 50 by caspases adaptively regulate gap junctions/hemichannels and protect lens cells against ultraviolet radiation. J. Biol. Chem. 287, 15786–15797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kronengold J., Srinivas M., Verselis V. K. (2012) The N-terminal half of the connexin protein contains the core elements of the pore and voltage gates. J. Membr. Biol. 245, 453–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Retamal M. A., Yin S., Altenberg G. A., Reuss L. (2009) Modulation of Cx46 hemichannels by nitric oxide. Am. J. Physiol. Cell Physiol. 296, C1356–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cattaruzza M., Hecker M. (2008) Protein carbonylation and decarboylation: a new twist to the complex response of vascular cells to oxidative stress. Circ. Res. 102, 273–274 [DOI] [PubMed] [Google Scholar]
- 17. Poss K. D., Tonegawa S. (1997) Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. U.S.A. 94, 10925–10930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Motterlini R., Otterbein L. E. (2010) The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 9, 728–743 [DOI] [PubMed] [Google Scholar]
- 19. Gullotta F., di Masi A., Ascenzi P. (2012) Carbon monoxide: an unusual drug. IUBMB Life 64, 378–386 [DOI] [PubMed] [Google Scholar]
- 20. Banerjee D., Gakhar G., Madgwick D., Hurt A., Takemoto D., Nguyen T. A. (2010) A novel role of gap junction connexin46 protein to protect breast tumors from hypoxia. Int. J. Cancer 127, 839–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burr D. B., Molina S. A., Banerjee D., Low D. M., Takemoto D. J. (2011) Treatment with connexin 46 siRNA suppresses the growth of human Y79 retinoblastoma cell xenografts in vivo. Exp. Eye Res. 92, 251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ripps H., Qian H., Zakevicius J. (2002) Pharmacological enhancement of hemi-gap-junctional currents in Xenopus oocytes. J. Neurosci. Methods 121, 81–92 [DOI] [PubMed] [Google Scholar]
- 23. Fiori M. C., Figueroa V., Zoghbi M. E., Saéz J. C., Reuss L., Altenberg G. A. (2012) Permeation of calcium through purified connexin 26 hemichannels. J. Biol. Chem. 287, 40826–40834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adachi K., Asakura T. (1982) Effect of liganded hemoglobin S and hemoglobin A on the aggregation of deoxyhemoglobin. J. Biol. Chem. 257, 5738–5744 [PubMed] [Google Scholar]
- 25. Peers C. (2011) Ion channels as target effectors for carbon monoxide. Exp. Physiol. 96, 836–839 [DOI] [PubMed] [Google Scholar]
- 26. Retamal M. A., Yin S., Altenberg G. A., Reuss L. (2010) Voltage-dependent facilitation of Cx46 hemichannels. Am. J. Physiol. Cell Physiol. 298, C132–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Althaus M., Fronius M., Buchäckert Y., Vadász I., Clauss W. G., Seeger W., Motterlini R., Morty R. E. (2009) Carbon monoxide rapidly impairs alveolar fluid clearance by inhibiting epithelial sodium channels. Am. J. Respir. Cell Mol. Biol. 41, 639–650 [DOI] [PubMed] [Google Scholar]
- 28. Dong D. L., Zhang Y., Lin D. H., Chen J., Patschan S., Goligorsky M. S., Nasjletti A., Yang B. F., Wang W. H. (2007) Carbon monoxide stimulates the Ca2+-activated big conductance K channels in cultured human endothelial cells. Hypertension 50, 643–651 [DOI] [PubMed] [Google Scholar]
- 29. Wilkinson W. J., Gadeberg H. C., Harrison A. W., Allen N. D., Riccardi D., Kemp P. J. (2009) Carbon monoxide is a rapid modulator of recombinant and native P2X(2) ligand-gated ion channels. Br. J. Pharmacol. 158, 862–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wilkinson W. J., Kemp P. J. (2011) Carbon monoxide: an emerging regulator of ion channels. J. Physiol. 589, 3055–3062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Telezhkin V., Brazier S. P., Mears R., Müller C. T., Riccardi D., Kemp P. J. (2011) Cysteine residue 911 in C-terminal tail of human BK(Ca)α channel subunit is crucial for its activation by carbon monoxide. Pflugers Arch. 461, 665–675 [DOI] [PubMed] [Google Scholar]
- 32. Wong C. M., Cheema A. K., Zhang L., Suzuki Y. J. (2008) Protein carbonylation as a novel mechanism in redox signaling. Circ. Res. 102, 310–318 [DOI] [PubMed] [Google Scholar]
- 33. Wong C. M., Marcocci L., Liu L., Suzuki Y. J. (2010) Cell signaling by protein carbonylation and decarbonylation. Antioxid. Redox Signal. 12, 393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wong C. M., Marcocci L., Das D., Wang X., Luo H., Zungu-Edmondson M., Suzuki Y. J. (2013) Mechanism of protein decarbonylation. Free Radic. Biol. Med. 65, 1126–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grune T., Reinheckel T., Davies K. J. (1997) Degradation of oxidized proteins in mammalian cells. FASEB J. 11, 526–534 [PubMed] [Google Scholar]
- 36. Dahl G., Qiu F., Wang J. (2013) The bizarre pharmacology of the ATP release channel pannexin1. Neuropharmacology 75, 583–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dong D. L., Chen C., Huang W., Chen Y., Zhang X. L., Li Z., Li Y., Yang B. F. (2008) Tricarbonyldichlororuthenium(II) dimer (CORM2) activates non-selective cation current in human endothelial cells independently of carbon monoxide releasing. Eur. J. Pharmacol. 590, 99–104 [DOI] [PubMed] [Google Scholar]
- 38. Griffiths H. R., Dias I. H., Willetts R. S., Devitt A. (2014) Redox regulation of protein damage in plasma. Redox. Biol. 2, 430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Desmard M., Foresti R., Morin D., Dagouassat M., Dagoussat M., Berdeaux A., Denamur E., Crook S. H., Mann B. E., Scapens D., Montravers P., Boczkowski J., Motterlini R. (2012) Differential antibacterial activity against Pseudomonas aeruginosa by carbon monoxide-releasing molecules. Antioxid. Redox Signal. 16, 153–163 [DOI] [PubMed] [Google Scholar]
- 40. Motterlini R., Clark J. E., Foresti R., Sarathchandra P., Mann B. E., Green C. J. (2002) Carbon monoxide-releasing molecules: characterization of biochemical and vascular activities. Circ. Res. 90, E17–E24 [DOI] [PubMed] [Google Scholar]
- 41. Kajimura M., Fukuda R., Bateman R. M., Yamamoto T., Suematsu M. (2010) Interactions of multiple gas-transducing systems: hallmarks and uncertainties of CO, NO, and H2S gas biology. Antioxid. Redox Signal. 13, 157–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Retamal M. A. (2014) Connexin and Pannexin hemichannels are regulated by redox potential. Front. Physiol. 5, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Padgaonkar V. A., Giblin F. J., Fowler K., Leverenz V. R., Reddan J. R., Dziedzic D. C. (1997) Heme oxygenase synthesis is induced in cultured lens epithelium by hyperbaric oxygen or puromycin. Exp. Eye Res. 65, 435–443 [DOI] [PubMed] [Google Scholar]
- 44. Schalper K. A., Carvajal-Hausdorf D., Oyarzo M. P. (2014) Possible role of hemichannels in cancer. Front. Physiol. 5, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]