

Background: ROS and iron availability influence [2Fe-2S] cluster occupancy in IscR.
Results: Prx3 is a Grx3/glutathione-dependent 1-Cys peroxiredoxin essential for survival under oxidative stress and pathogenesis of Vibrio vulnificus, and IscR directly activates prx3 by sensing ROS and iron starvation.
Conclusion: IscR-dependent prx3 expression contributes to the pathogenesis of V. vulnificus.
Significance: This study elucidated the IscR-mediated regulation of an antioxidant enzyme.
Keywords: Bacterial Pathogenesis, Gene Regulation, Oxidative Stress, Peroxiredoxin, Reactive Oxygen Species (ROS), IscR, Vibrio vulnificus, Iron Starvation
Abstract
Peroxiredoxins (Prxs) are ubiquitous antioxidant enzymes that reduce toxic peroxides. A new Vibrio vulnificus Prx, named Prx3, was identified and characterized in this study. Biochemical and mutational analyses revealed that Prx3 reduces H2O2, utilizing glutaredoxin 3 (Grx3) and glutathione (GSH) as reductants, and requires only N-terminal peroxidatic cysteine for its catalysis. These results, combined with the monomeric size of Prx3 observed under non-reducing conditions, suggested that Prx3 is a Grx3/GSH-dependent 1-Cys Prx and oxidized without forming intermolecular disulfide bonds. The prx3 mutation impaired growth in the medium containing peroxides and reduced virulence in mice, indicating that Prx3 is essential for survival under oxidative stress and pathogenesis of V. vulnificus. The Fe-S cluster regulator IscR activates prx3 by direct binding to a specific binding sequence centered at −44 from the transcription start site. The binding sequence was homologous to the Type 2 IscR-binding sequence, most likely recognized by the Fe-S clusterless apo-IscR in Escherichia coli. The iscR3CA mutant, chromosomally encoding the apo-locked IscR, exhibited 3-fold higher levels of activation of prx3 than the wild type and accumulated more IscR3CA protein in cells. The IscR-dependent activation of prx3 by aerobic growth and iron starvation was also associated with the increase in cellular levels of IscR protein. Taken together, the results suggested that IscR senses iron starvation as well as reactive oxygen species and shifts to the apo-form, which leads to the increase of cellular IscR and in turn prx3 expression, contributing to the survival and virulence of V. vulnificus during pathogenesis.
Introduction
Bacteria continually encounter toxic reactive oxygen species (ROS),2 such as hydrogen peroxide (H2O2) and peroxynitrite (ONOO−), in their growth environments (1, 2). In addition, pathogenic bacteria are inevitably exposed to ROS that are crucial for the optimal microcidal activity of neutrophils and other phagocytes of the host (3–5). Oxidative stress caused by increased levels of ROS can lead to the damage of cellular components, including metal centers, protein, DNA, and membrane lipid (4, 5). Therefore, pathogens have evolved sophisticated mechanisms to overcome oxidative stress, and the mechanisms are closely linked to their virulence (4, 5). The mechanisms of bacterial defense against oxidative stress include highly specific and effective antioxidant enzymes (4–7). Among them, peroxiredoxins (Prxs) are a ubiquitous family of cysteine-based peroxidases widely distributed across all kingdoms of life and catalyze the reduction of peroxides, such as H2O2, organic hydroperoxide, and ONOO− (6, 8).
Based on catalytic mechanisms, Prxs are divided into three classes referred to as typical 2-Cys, atypical 2-Cys, and 1-Cys Prxs (9). 2-Cys Prxs have two conserved catalytic cysteines, peroxidatic (CP) and resolving (CR) cysteines. CP reacts with peroxides and forms a cysteine sulfenic acid intermediate (CP-SOH), which is followed by the formation of an intermolecular disulfide bond with CR from another subunit (typical 2-Cys Prxs) or with CR located within the same subunit (atypical 2-Cys Prxs) (10, 11). Disulfide-bonded 2-Cys Prxs are subsequently reduced and reactivated by thiol-containing reductants, such as thioredoxin (Trx) and alkyl hydroperoxidase subunit F (AhpF) (10, 11). Finally, 1-Cys Prxs contain only CP and not CR, and the CP-SOH of 1-Cys Prxs is reduced mostly, if not exclusively, by glutaredoxin (Grx) and reduced glutathione (GSH) (12–15). In numerous bacteria, the catalytic mechanisms, physiological functions, and expression patterns of the 2-Cys Prxs, including alkyl hydroperoxidase subunit C (AhpC) have been extensively studied (for a recent review, see Ref. 6). In this context, two 2-Cys Prxs, Prx1 and Prx2, in which the expression patterns and kinetic properties are differentially optimized to decompose distinct levels of H2O2 were previously identified in Vibrio vulnificus, a facultative aerobic pathogen (16, 17). However, studies on the bacterial 1-Cys Prxs are presently limited (13, 18, 19); therefore, the regulatory mechanisms used by bacteria to sense environmental signals and modulate their expression have not yet been addressed at the molecular levels.
IscR, an iron-sulfur (Fe-S) cluster-containing transcription factor, functions as a sensor of the cellular Fe-S cluster status and homeostatically regulates Fe-S cluster biogenesis (20, 21). Under conditions favoring stabilization of the Fe-S cluster (mostly in anaerobic growth), sufficient Fe-S cluster occupies IscR to result in [2Fe-2S]-IscR (holo-IscR). The holo-IscR represses the isc operon (iscRSUA-hscBA-fdx) encoding IscR and all of the proteins required for the biogenesis of the Fe-S cluster. When the [2Fe-2S] cluster in IscR is disrupted under conditions such as oxidative stress or iron starvation, the resulting clusterless IscR (apo-IscR) relieves the repression of the isc operon; increases the cellular level of IscR, most likely in its apo-form; and, accordingly, promotes Fe-S cluster biogenesis (20–24). IscR is a global regulator that controls the expression of more than 40 genes in Escherichia coli (25). IscR binds two distinct DNA sequences, Type 1 and Type 2, depending on the [2Fe-2S] occupancy of the protein (26, 27). Most, if not all, promoters containing a Type 2 sequence are regulated by apo-IscR, whereas promoters containing a Type 1 sequence are regulated exclusively by holo-IscR (21, 26–29).
A transcriptome analysis previously predicted that V. vulnificus IscR is a global regulator that controls the expression of many genes involved in important cellular processes in addition to Fe-S cluster biogenesis (30). Among the genes up-regulated by IscR, a gene was predicted to encode a putative Prx (here called Prx3) (30). However, the catalytic mechanism of Prx3 and IscR regulation of its expression have not yet been experimentally verified. In the present study, biochemical analyses supported that Prx3 is indeed a Grx3/GSH-dependent 1-Cys Prx. A V. vulnificus null mutant, in which the prx3 gene was inactivated, was constructed by allelic exchanges, and the possible roles of Prx3 in the survival and pathogenesis of the pathogen were explored. Here, the results provided molecular genetic evidence that Prx3 is essential for survival under oxidative stress and virulence in mice and that IscR activates prx3 by directly binding to its promoter in response to endogenously produced ROS and diminishing iron levels.
EXPERIMENTAL PROCEDURES
Strains, Plasmids, and Culture Conditions
The strains and plasmids used in this study are listed in Table 1. Unless noted otherwise, the V. vulnificus strains were grown aerobically in Luria-Bertani (LB) medium supplemented with 2.0% (w/v) NaCl (LBS) at 30 °C. Anerobic conditions were obtained by using an anaerobic chamber with an atmosphere of 90% N2, 5% CO2, and 5% H2 (Coy Laboratory Products, Grass Lake, MI). For anaerobic culture, the medium was preincubated to remove resolved O2 in the anaerobic chamber, which was verified by adding 0.00001% (w/v) resazurin salt (Sigma) to the medium as described previously (31).
TABLE 1.
Plasmids and bacterial strains used in this study
| Strain or plasmid | Relevant characteristicsa | Reference or source |
|---|---|---|
| Bacterial strains | ||
| V. vulnificus | ||
| M06-24/O | Clinical isolate; virulent | Laboratory collection |
| JK134 | M06-24/O with Δprx3 | This study |
| JK093 | M06-24/O with ΔiscR | Ref. 30 |
| JK128 | M06-24/O with iscR3CA encoding the apo-locked IscR, IscR3CA | This study |
| E. coli | ||
| DH5α | λ− φ80dlacZΔM15Δ (lacZYA-argF)U169 recA1 endA1 hsdR17 (rK− mK−) supE44 thi-1 gyrA relA1; plasmid replication | Laboratory collection |
| S17–1 λ pir | λ-pir lysogen; thi pro hsdR hsdM+ recA RP4–2 Tc::Mu-Km::Tn7;Tpr Smr; host for π-requiring plasmids; conjugal donor | Ref. 35 |
| BL21(DE3) | F− ompT hsdSB (rB− mB−) gal dcm (DE3) | Laboratory collection |
| Plasmids | ||
| pHIS-parallel1 | His6 tag fusion expression vector; Apr | Ref. 32 |
| pJK1321 | pHIS-parallel1 with the wild-type prx3; Apr | This study |
| pBANG1304 | pHIS-parallel1 with grxC; Apr | This study |
| pBANG1301 | pHIS-parallel1 with gor; Apr | This study |
| pBANG1104 | pHIS-parallel1 with trxA; Apr | Ref. 17 |
| pOH053 | pRSET A with trxB; Apr | Ref. 17 |
| pJK0928 | pET22b (+) with iscR; Apr | Ref. 30 |
| pJK1322 | pHIS-parallel1 with the mutant prx3 encoding Prx3-C48S; Apr | This study |
| pJK1323 | pHIS-parallel1 with the mutant prx3 encoding Prx3-C73S; Apr | This study |
| pDM4 | R6K γ ori sacB; suicide vector; oriT of RP4; Cmr | Ref. 34 |
| pJK1127 | pDM4 with Δprx3; Cmr | This study |
| pKS1101 | pBAD24 with oriT of RP4; Apr | Ref. 37 |
| pJK1113 | pKS1101 with nptI; Apr, Kmr | This study |
| pJK1303 | pJK1113 with prx3; Apr, Kmr | This study |
| pGEM-T Easy | PCR product cloning vector; Apr | Promega |
| pJK1402 | pGEM-T Easy with a 309-bp fragment of the putative promoter region of prx3; Apr | This study |
| pJK0927 | pGEM-T Easy with iscR; Apr | This study |
| pJK0929 | pGEM-T Easy with iscR3CA; Apr | This study |
| pJK1250 | pDM4 with iscR3CA; Cmr | This study |
a Tpr, trimethoprim-resistant; Smr, streptomycin-resistant; Apr, ampicillin-resistant; Cmr, chloramphenicol-resistant; Kmr, kanamycin-resistant.
Overexpression and Purification of Recombinant Protein
Each open reading frame (ORF) of the genes encoding Prx3 (NCBI accession number ADV89163), glutaredoxin 3 (Grx3; NCBI accession, ADV89460), and glutathione reductase (GR; NCBI accession, ADV85104) was amplified by PCR using a pair of oligonucleotide primers as listed in Table 2. The amplified PCR products were cloned into a His6 tag expression vector, pHIS-parallel1 (32), to result in pJK1321 (for prx3), pBANG1304 (for grx3), and pBANG1301 (for gor encoding GR) as described in Table 1. His-tagged proteins were expressed in E. coli BL21(DE3) and purified by affinity chromatography according to the manufacturer's procedure (Qiagen, Valencia, CA). Similarly, the expression and purification of the His-tagged thioredoxin A (TrxA), thioredoxin reductase (TrR), and IscR were carried out using pBANG1104, pOH053, and pJK0928 (Table 1), respectively, as described previously (17, 30).
TABLE 2.
Oligonucleotides used in this study
| Name | Oligonucleotide sequence (5′ → 3′)a | Use |
|---|---|---|
| For expression of proteins | ||
| PRX301-F | CCATGGTGATCGCTCAAGGCCAAACT | Prx3 overexpression |
| PRX301-R | ACTAGTCGTTAGCTACCGATGAGCAAC | |
| GRX301-F | CACGGGACCATGGGCATGCCAAAGAT | GrxC overexpression |
| GRX301-R | CAATAAGTGTCGACTTAGTGACGGATTGC | |
| GR01-F | GAGAAAGCCATGGCAACGCATTTTGAC | GR overexpression |
| GR01-R | GTGCTTGTCGACTTAGCGCATCGTGACG | |
| For mutagenesis | ||
| PRX3C48S-F | GGCGTTCACGCCAACCAGCTCAGAAGCACATTTACCG | Construction of Prx3-C48S mutant |
| PRX3C48S-R | CGGTAAATGTGCTTCTGAGCTGGTTGGCGTGAACGCC | |
| PRX3C73S-F | GCGTCGATCTGATTGCCAGCGTTTCTGTCAACGATGC | Construction of Prx3-C73S mutant |
| PRX3C73S-R | GCATCGTTGACAGAAACGCTGGCAATCAGATCGACGC | |
| ISCR3CA1-F | CGCAACTAAGGCGCAGGGCAAAGGAGATGCGCAAGGCGGCAC | Construction of IscR3CA mutant |
| ISCR3CA1-R | GTGCCGCCTTGCGCATCTCCTTTGCCCTGCGCCTTAGTTGCG | |
| ISCR3CA2-F | CAAGGCGGCACTCGCGCGCTTACTCATACACTTTGGCGTGACC | |
| ISCR3CA2-R | GGTCACGCCAAAGTGTATGAGTAAGCGCGCGAGTGCCGCCTTG | |
| For mutant construction | ||
| PRX302-F | GTGGTTGATAGACCAGCCATTCGTC | Construction of prx3 mutant |
| PRX302-R | GATGGATCCTTTTTTGCCAGCAAA | |
| PRX303-F | AAAGGATCCATCGACAATGGCGTA | |
| PRX303-R | CGCTTTCGTGACCTTAATTGGTTTG | |
| For amplification of gene ORF | ||
| PRX304-F | CCATGGTGATCGCTCAAGGCCAAACT | Amplification of prx3 ORF |
| PRX304-R | GCATGCTTAGCTACCGATGAGCAACCA | |
| For primer extension analysis, EMSA, and DNase I protection assay | ||
| PRX305-F | ATATTTGGACATAAAAAGACCCCC | Amplification of prx3 upstream region |
| PRX305-R | CTAAAACAGGGTGGTGGACCA | Amplification of prx3 upstream region, Extension of prx3 transcript |
| For qRT-PCR | ||
| PRX3_qRT-F | TGAAAGCCTGGGGTGAAGCA | Expression of prx3 |
| PRX3_qRT-R | ATCGCGTAGCGTTGAGAGCG | |
Site-directed Mutagenesis of prx3 and Purification of the Mutant Prx3 Proteins
Each of the two cysteine residues in Prx3 (Cys-48 and -73) was replaced with serine by using a QuikChange® site-directed mutagenesis kit (Agilent Technologies, Loveland, CO) (17). The complementary mutagenic primers listed in Table 2 were used in conjunction with the plasmid pJK1321 (as a template DNA) to create pJK1322 (for Prx3-C48S) and pJK1323 (for Prx3-C73S) (Table 1). The mutations were confirmed by DNA sequencing, and the mutant proteins were expressed and purified as described above.
NADPH Consumption Assay
Peroxidase activities of the wild-type and mutant Prx3 were determined by monitoring the decrease in A340 due to NADPH oxidation as described previously (17) with minor modification. The reaction mixture (200 μl in a final volume) contained 2 μm TrxA and 0.3 μm TrR (for the TrxA-dependent peroxidase activity) or 2 μm Grx3, 0.3 μm GR, and 1 mm GSH (for the Grx3/GSH-dependent peroxidase activity) in addition to 50 mm HEPES-NaOH (pH 7.0), 200 μm NADPH, and 4 μm Prx3 (or the mutant Prx3). The reaction was initiated by adding 0.1 mm H2O2 to the reaction mixture preincubated at room temperature for 5 min, and then the decrease in A340 was scored (Infinite M200 reader, Tecan, Männedorf, Switzerland). To determine the initial rates of H2O2 reduction, the decrease in A340 of the reaction mixture containing various concentrations of Grx3 (0, 1, 2, 3, and 4 μm), GSH (0, 0.25, 0.5, 1.0, and 1.5 mm), or Prx3 (0, 1, 2, 3, 4, and 5 μm) was scored for the first 1 min, and the amounts of NADPH oxidized per min were calculated using the molar extinction coefficient of 6,220 m−1 cm−1 for NADPH (33). The data were fitted by nonlinear regression analysis using SigmaPlot version 10.0 (SPSS Inc., Chicago, IL), and the best fits are shown as lines.
Western Blot Analysis
The purified His-tagged Prx3 was used to raise rabbit anti-Prx3 polyclonal antibodies (AB Frontier, Seoul, South Korea). The V. vulnificus strains grown aerobically or anaerobically to an A600 of 0.3 were harvested to isolate total proteins. Proteins (10 μg) were resolved on SDS-PAGE and immunoblotted as described previously (30). For immunoblotting of IscR, the anti-IscR polyclonal antibody prepared previously was used (30). When necessary, the wild type grown aerobically to an A600 of 0.3 was exposed to various concentrations of an iron chelator, 2,2′-dipyridyl (DP) (Sigma) for 10 min and then harvested.
Generation and Complementation of prx3 Mutant
The prx3 gene was inactivated in vitro by deletion of the prx3 ORF (282 of 474 bp) using the PCR-mediated linker-scanning mutation method as described previously (30). Pairs of primers PRX302-F and -R (for amplification of the 5′ amplicon) or PRX303-F and -R (for amplification of the 3′ amplicon) were designed (Table 2). The 282-bp deleted prx3 was amplified by PCR using the mixture of both amplicons as the template and PRX302-F and PRX303-R as primers. The resulting DNA fragment containing the deleted prx3 was ligated with SpeI-SphI-digested pDM4 (34) to generate pJK1127 (Table 1). The E. coli S17-1 λ pir, tra strain (35) containing pJK1127 was used as a conjugal donor to V. vulnificus MO6-24/O to generate the prx3 mutant JK134 (Table 1). The conjugation and isolation of the transconjugants were conducted as described previously (30). For complementation, a pair of primers PRX304-F and -R was designed and used to amplify the prx3 ORF (Table 2). A plasmid pJK1113 was constructed by cloning a 1.2-kb nptI DNA conferring resistance to kanamycin (36) into a broad host range vector pKS1101 (37). The amplified prx3 ORF was cloned into pJK1113 under an arabinose-inducible promoter, PBAD to create pJK1303 (Table 1). pJK1303 was transferred into JK134 by conjugation as described above.
Survival under Oxidative Stress
The V. vulnificus strains were grown to an A600 of 0.5 in the LBS broth and then challenged with 250 μm H2O2 or 200 μm ONOO− (Millipore Corp., Bedford, MA) at final concentrations. The cultures were further incubated at 30 °C with shaking for 6 h, and their growth was monitored spectrophotometrically at 600 nm (A600). l-Arabinose was added to the medium at a final concentration of 0.02% (w/v) to induce the expression of recombinant prx3 on pJK1303.
Mouse Mortality Test
Mouse mortalities of the wild type and prx3 mutant were compared as described previously (30). Groups of (n = 20) 7-week-old ICR female mice (specific pathogen-free, Seoul National University) were starved without food and water for 12 h until infection. Then the mice, without iron-dextran pretreatment, were intragastrically administered with 100 μl of the inoculum, representing ∼109 cells of either the wild type or the prx3 mutant. Mouse mortalities were recorded for 24 h. All manipulations of mice were approved by the Animal Care and Use Committee at Seoul National University.
RNA Purification and Transcript Analysis
Total cellular RNAs from the V. vulnificus strains grown aerobically or anaerobically to an A600 of 0.3 were isolated using an RNeasy® minikit (Qiagen) (30). When necessary, the strains were exposed to DP and then harvested as described above for Western blot analysis. For primer extension analysis, a 21-base oligonucleotide primer PRX305-R (Table 2) complementary to the coding region of prx3 was end-labeled with [γ-32P]ATP and added to the RNA. The primer was then extended with SuperScript II RNase H− reverse transcriptase (Invitrogen). The cDNA products were purified and resolved on a sequencing gel alongside sequencing ladders generated from pJK1402 with the same primer. The plasmid pJK1402 was constructed by cloning the 309-bp prx3 upstream region extending from −198 to +111, amplified by PCR using a pair of primers PRX305-F and -R (Table 2), into pGEM-T Easy (Promega, Madison, WI). The primer extension products were visualized using a phosphor image analyzer (BAS1500, Fuji Photo Film Co. Ltd., Tokyo, Japan).
For quantitative real-time PCR (qRT-PCR), cDNA was synthesized using the iScriptTM cDNA synthesis kit (Bio-Rad), and real-time PCR amplification of the cDNA was performed by using the Chromo 4 real-time PCR detection system (Bio-Rad) with a pair of primers, PRX3_qRT-F and -R (Table 2), as described previously (30). Relative expression levels of the specific transcripts were calculated by using the 16 S rRNA expression level as the internal reference for normalization.
Iron Content Analysis of the Purified IscR
The purified IscR was digested with concentrated HNO3 and diluted with distilled water. The diluted IscR solution was nebulized into the plasma using MiraMist nebulizer (Burgener Research Inc., Mississauga, Canada), and the concentrations of iron ions were measured using inductively coupled plasma-atomic emission spectrometry (Optima-4300 DV, PerkinElmer). The operating conditions developed by Cubadda and Raggi (38) were adopted, and calibration was performed using the reference iron solution as external standards.
Electrophoretic Mobility Shift Assay (EMSA) and DNase I Protection Assay
The 309-bp upstream region of the prx3 promoter was amplified by PCR using [γ-32P]ATP-labeled PRX305-F and unlabeled PRX305-R as primers (Table 2). The labeled 309-bp DNA (5 nm) probe was incubated with various concentrations of purified His-tagged IscR for 30 min at 30 °C in a 20-μl reaction mixture containing 1× binding buffer (25) and 0.1 μg of poly(dI-dC) (Sigma). Electrophoretic analysis of the DNA-protein complexes was performed as described previously (39).
The same labeled 309-bp upstream region was used for DNase I protection assays. The binding of IscR to the labeled DNA was performed as described above, and DNase I digestion of the DNA-protein complexes followed the procedures described previously (39). After precipitation with ethanol, the digested DNA products were resolved on a sequencing gel alongside sequencing ladders of pJK1402 generated with the primer PRX305-F (for the coding strand) or PRX305-R (for the noncoding strand). The gels were visualized as described above for the primer extension analysis.
Generation of the iscR3CA Mutant Expressing Apo-locked IscR
The three cysteine residues (Cys-92, -98, and -104) essential for [2F-2S] cluster ligation are highly conserved in IscR homologues in bacteria (26). Therefore, the three cysteines in IscR were substituted with alanine by using a QuikChange® site-directed mutagenesis kit (Agilent Technologies) (17). The complementary mutagenic primers listed in Table 2 were used in conjunction with the plasmid pJK0927 (iscR on pGEM-T Easy as a template DNA) to create pJK0929 containing iscR3CA (Table 1). The iscR3CA gene was subcloned into the SpeI-SphI-digested pDM4 to result in pJK1250 (Table 1). The E. coli S17-1 λ pir, tra strain containing pJK1250 was used as a conjugal donor to V. vulnificus MO6-24/O. The iscR3CA mutant JK128, of which the iscR coding region on the chromosome was replaced with iscR3CA encoding an apo-locked IscR3CA, was isolated and confirmed by DNA sequencing (Table 1).
Data Analyses
Averages and S.E. values were calculated from at least three independent experiments. Mouse mortality was evaluated using the log rank test program. All other data were analyzed by Student's t tests with the SAS program (SAS software; SAS Institute Inc.). Significance of differences between experimental groups was accepted at a p value of < 0.05.
RESULTS
Sequence Analysis of Prx3
The deduced amino acid sequence revealed that Prx3 is composed of 157 amino acids with a theoretical molecular mass of 16,579 Da and a pI of 4.80. The predicted profile of the hydrophobicity revealed that Prx3 is a cytosolic soluble protein as observed from other Prxs (9). The alignment of the amino acid sequence of Prx3 to those of Prx1 and Prx2 (16, 17) exhibited no significant identities (data not shown). Rather, a database search for homology to the amino acid sequence of Prx3 singled out a protein, NCBI accession number NP_230994, of Vibrio cholerae, a species closely related to V. vulnificus (Fig. 1). Although the putative protein NP_230994 exhibits a high level of identity (78% in amino acid sequences) with Prx3, its functions have not yet been addressed. Prx3 is also homologous to three peroxidases, plant Populus trichocarpa PrxD (40), parasite Plasmodium falciparum AOP (41), and human Prx5 (42), with about 35–52% identity in amino acid sequences (Fig. 1). These proteins belong to the Prx5 subfamily, which is classified based on the conservation of sequences proximal to the active site (8). Prx3 contains N-terminal cysteine (Cys-48) within the PXXXTXXC motif at the active site and second cysteine (Cys-73), which are highly conserved in members of the Prx5 subfamily (Fig. 1) (15). The combined information suggested that Prx3 is a peroxidase belonging to the Prx5 subfamily.
FIGURE 1.

Amino acid sequence relatedness of V. vulnificus Prx3 (VvPrx3) and members of the Prx5 subfamily. The amino acid sequences retrieved from the NCBI protein database (accession numbers: ADV89163 for V. vulnificus Prx3, NP_230994 for the V. vulnificus Prx3 homologue in V. cholerae, 1TP9_A for P. trichocarpa PrxD (PtPrxD), XP_002808799 for P. falciparum AOP (PfAOP), NM_012094 for human Prx5 (HsPrx5)) were aligned using the ClustalW2 program. Identical (asterisks), highly conserved (double dots), conserved (dots), and missing (dashes) sequences are indicated. All cysteine residues are shaded in gray. The positions of the peroxidatic (CP), second (C2), and resolving cysteines (CR) are indicated above the amino acid sequences. The active site PXXXTXXC motifs are indicated by an open box.
Prx3 Is a Grx3/GSH-dependent Peroxidase
To define the catalytic mechanism of Prx3, the peroxidase activity of Prx3 reducing H2O2 was determined by measuring NADPH consumption. When reacted with 0.1 mm H2O2, Prx3 actively consumed NADPH only when Grx3, GSH, and GR are present together as a reducing system (Fig. 2A). In contrast, no apparent peroxidase activity was observed when Grx3/GSH/GR were replaced by TrxA/TrR, indicating that TrxA is not able to reduce oxidized Prx3 to reactivate its peroxidase activity (Fig. 2A). The peroxidase activities of Prx3 determined from the initial rate of NADPH oxidation were proportional to the concentrations of Grx3 and GSH and saturated at about 4 μm and 1.5 mm, respectively (Fig. 2, B and C). These results indicated that Prx3 obtains electrons from NADPH through Grx3/GSH/GR to complete a catalytic cycle. Thus, hereafter, Grx3/GSH/GR was used as a reducing system to further characterize the peroxidase activity of Prx3. When reacted with 0.1 mm tert-butyl hydroperoxide or ONOO−, Prx3 also actively consumed NADPH (data not shown). The combined results suggested that Prx3 is a Grx3/GSH-dependent peroxidase and catalyzes the reduction of not only H2O2 but also tert-butyl hydroperoxide and ONOO−, as observed in other Prxs (43).
FIGURE 2.
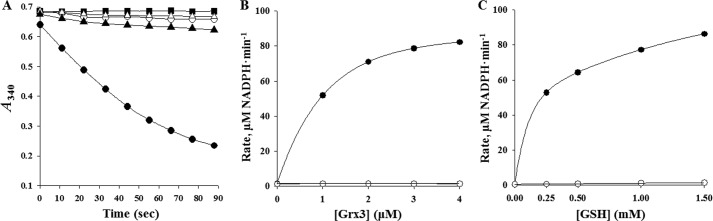
Peroxidase activities of Prx3 with H2O2. A, the peroxidase activities of Prx3 were determined by measuring NADPH oxidation in the reaction mixtures. The reaction mixture contained Prx3 (■), Grx3/GSH/GR (○), Prx3 and Grx3/GSH/GR (●), TrxA/TrR (▵), or Prx3 and TrxA/TrR (▴) in addition to NADPH. B and C, the Grx3/GSH-dependent peroxidase activities of Prx3 were determined by measuring the initial rates of NADPH oxidation (μm NADPH·min−1). B, the reaction mixture contained GSH/GR with (●) or without (○) Prx3 in addition to NADPH and various concentrations of Grx3 as indicated. C, the reaction mixture contained Grx3/GR with (●) or without (○) Prx3 in addition to NADPH and various concentrations of GSH as indicated. All of the reactions were initiated by adding 0.1 mm H2O2 to the reaction mixture. The S.E. values were too small to be shown by error bars. B and C, each solid line represents a fit of the data to a curve of exponential rise to maximum.
Prx3 Is a 1-Cys Prx Requiring Only N-terminal CP for Catalytic Ability
To further understand the catalytic mechanism of Prx3 at the molecular level, the mutants Prx3-C48S and Prx3-C73S, in which each of the two conserved cysteines was substituted with a serine, respectively, were constructed and subjected to the NADPH consumption assay. When reacted with 0.1 mm H2O2, the activity of the wild-type Prx3, determined from the initial rate of NADPH oxidation, increased in a concentration-dependent manner (Fig. 3). In contrast, Prx3-C48S did not exhibit any activity at any concentrations tested. These results indicated that Cys-48 is CP of Prx3, crucial for the peroxidase activity. The mutation of Cys-73 did not affect the activity of Prx3 (Fig. 3), suggesting that Cys-73 is not crucial for its catalysis, similar to the other members of the Prx5 subfamily (11, 15, 44). The results, combined with the amino acid sequence analysis (Fig. 1), indicated that Prx3 is a 1-Cys Prx requiring only N-terminal CP for its catalytic ability.
FIGURE 3.
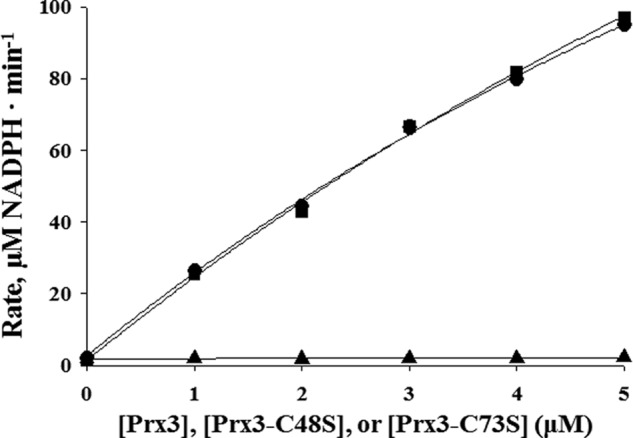
Peroxidase activities of wild-type and mutant Prx3. The peroxidase activities of wild-type Prx3, Prx3-C48S, and Prx3-C73S were determined by measuring the initial rates of NADPH oxidation (μm NADPH·min−1). The reaction mixture contained Grx3/GSH/GR and either Prx3 (●), Prx3-C48S (▴), or Prx3-C73S (■) in addition to NADPH. The reaction was initiated by adding 0.1 mm H2O2 to the reaction mixture. The S.E. values were too small to be shown by error bars. Each solid line represents a fit of the data to a curve of exponential rise to maximum.
The molecular size of the purified Prx3 under different conditions was determined by SDS-PAGE (Fig. 4A). In the presence of DTT (i.e. under reducing conditions), the reduced Prx3 band was detected at a molecular size corresponding to the monomeric form (Fig. 4A). Besides this band, an extra Prx3 band migrating slightly faster than the reduced Prx3 was also detected under non-reducing conditions. Double bands might represent the simultaneous presence of the Prx3 monomers with different conformations. The upper band might represent reduced or oxidized Prx3 without any disulfide linkages, whereas the lower band represents oxidized Prx3 with an intramolecular linkage between Cys-48 and Cys-73 that increases the compactness and thereby electrophoretic mobility of the protein, as observed elsewhere (15). When cellular proteins on non-reducing SDS-PAGE were immunoblotted using rabbit anti-Prx3, the two bands of monomeric Prx3 were also detected (Fig. 4B). Taken together, Prx3 is a 1-Cys Prx and does not form dimers linked by intermolecular disulfide bonds upon oxidation that was observed in Prx1 and Prx2 (17).
FIGURE 4.
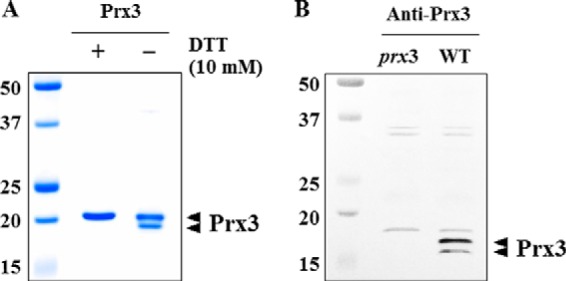
SDS-PAGE and Western blot analyses of Prx3. A, Purified His-tagged Prx3 proteins were resolved on SDS-PAGE with (+) or without (−) 10 mm DTT as indicated. B, total proteins were isolated from the WT and prx3 mutant grown to an A600 of 0.3 and then resolved by non-reducing SDS-PAGE and immunoblotted using a rabbit anti-Prx3 antiserum. The positions of protein size markers (in kDa; Bio-Rad) are shown on the left of the gel. Arrowheads, two conformations of monomeric Prx3s.
Prx3 Is Essential for Survival under Oxidative Stress and Virulence in Mice
In an effort to understand the role of Prx3 in resistance to ROS, the growth of an isogenic mutant, JK134, which lacked a functional prx3 gene (Table 1), was evaluated in the presence of peroxides. Compared with the wild type, the prx3 mutant exhibited substantially impaired growth in LBS medium containing H2O2 (Fig. 5B) or ONOO− (Fig. 5C). However, neither defective nor advantageous growth was observed for the strains in LBS without oxidant (Fig. 5A). These results suggested that the prx3 mutant is more sensitive to H2O2- and ONOO−-induced oxidative stress than its parental wild type. Complementation with a functional prx3 gene (pJK1303) restored the impaired growth of the prx3 mutant to the growth of the wild type (Fig. 5, B and C). Therefore, the impaired growth of JK134 apparently resulted from inactivation of prx3 rather than any polar effects on genes downstream of prx3. Noticeably, Prx3 manages to detoxify high levels of peroxides, 250 μm for H2O2 and 200 μm for ONOO− (Fig. 5, B and C). These results indicated that Prx3 plays a crucial role in the survival of V. vulnificus under oxidative stress by scavenging high levels of peroxides provided exogenously.
FIGURE 5.
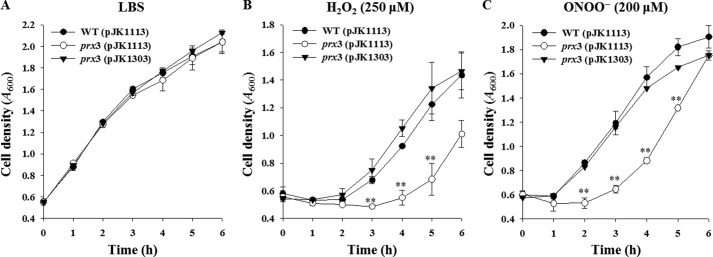
Growth of the V. vulnificus strains under oxidative stress. The V. vulnificus strains were compared for their ability to grow in LBS broth supplemented without oxidant (A) or with 250 μm H2O2 (B) or 200 μm ONOO− (C) at final concentrations. Error bars, S.E. **, p < 0.005 relative to the wild type at the indicated time. WT (pJK1113), wild type; prx3 (pJK1113), prx3 mutant; prx3 (pJK1303), complemented strain.
To further investigate the role of Prx3 in pathogenesis, the virulence of the wild type and prx3 mutant was compared in mice. For this purpose, mice were infected intragastrically with the wild type or prx3 mutant, and the numbers of dead mice were counted. As shown in Fig. 6, the death of mice infected with the prx3 mutant was consistently and significantly delayed (p = 0.0287, log rank test) compared with that of mice infected with the parental wild type. At 24 h postinfection, the percentages of mice that survived after challenge with the prx3 mutant or the wild type were 90 and 60%, respectively (Fig. 6). This attenuated virulence of the prx3 mutant suggested that Prx3 is essential for the pathogenesis of V. vulnificus. Recent work also has shown that the cells of mouse intestine infected with V. vulnificus generate ROS (45). Therefore, it is reasonable to assume that Prx3 could contribute to the pathogenesis by protecting the pathogen from the ROS imposed by host cells to ensure its survival and multiplication during infection.
FIGURE 6.
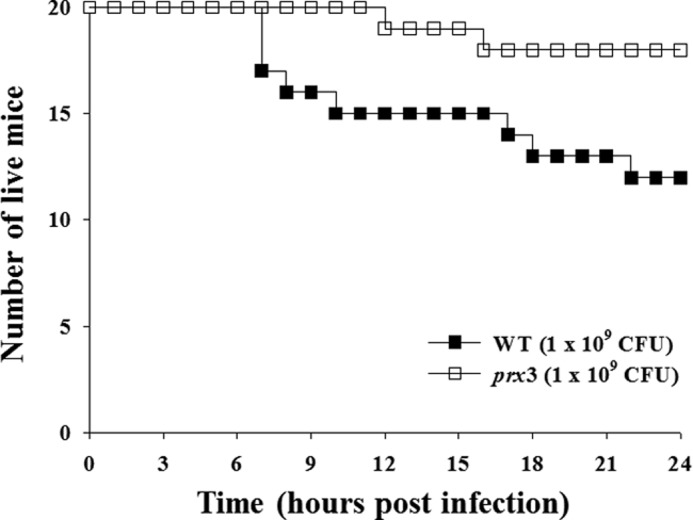
Effect of prx3 mutation on mouse mortality. Seven-week-old specific pathogen-free female ICR mice were intragastrically infected with the WT or prx3 mutant at doses of 109 cfu.
IscR Positively Regulates prx3 Transcription
Although prx3 was previously predicted as a member of the V. vulnificus IscR regulon by a microarray analysis (30), no IscR regulation of any prx genes has been reported yet in other bacteria to our knowledge. Therefore, in order to experimentally verify this prediction, the prx3 promoter activity of the iscR mutant, which was previously constructed by deletion of the iscR ORF (30) (Table 1), was compared with that of the parental wild type by primer extension analyses. A single reverse transcript was produced from primer extension of RNA isolated from the wild type grown aerobically (Fig. 7A). The 5′-end of the prx3 transcript was located 30-bp upstream of the translational initiation codon of prx3 and subsequently designated +1 (Fig. 7B). The putative promoter constituting this transcription start site was named Pprx3. Using different sets of primers, no other transcription start sites were identified by primer extension analyses (data not shown). As shown in Fig. 7A, the Pprx3 activity was abolished in the iscR mutant, as determined based on the intensity of the prx3 reverse transcript band. This result confirmed that V. vulnificus IscR positively regulates the expression of prx3 via activation of Pprx3 during aerobic growth. The sequences for the −10 and −35 regions of Pprx3 were assigned on the basis of similarity to consensus sequences of the E. coli σ70 promoter (Fig. 7B).
FIGURE 7.
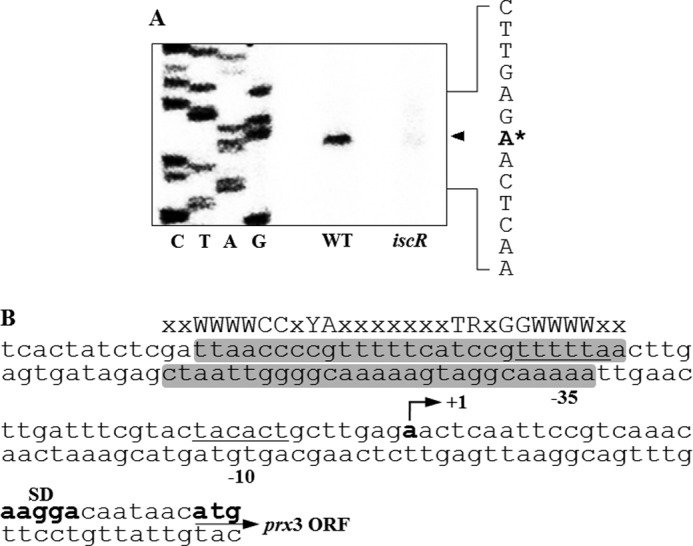
Effect of IscR on the Pprx3 activity and sequence analysis of the prx3 regulatory region. A, Pprx3 activities were determined by primer extension of the RNAs isolated from the WT and the iscR mutant grown aerobically to an A600 of 0.3. Lanes C, T, A, and G, nucleotide sequencing ladders of pJK1402. The asterisk indicates the transcription start site of Pprx3. B, the transcription start site of Pprx3 is indicated by a bent arrow, and the positions of the putative −10 and −35 regions are underlined. The sequences for binding of IscR determined later in this study (Fig. 9) are presented as shaded boxes. The consensus sequences of the Type 2 IscR-binding site are indicated above the V. vulnificus DNA sequence. The ATG translation initiation codons and the putative ribosome-binding site (SD) are also indicated in boldface. W, A or T; Y, C or T; R, A or G; x, any nucleotide.
IscR Directly Binds to a Specific Binding Site in Pprx3 in Vitro
There are still several possible ways for IscR to affect the Pprx3 activity. One is by binding directly to the prx3 promoter region to stimulate the Pprx3 activity, whereas another is by modulating the cellular level of another trans-acting factor(s), which in turn binds directly to the prx3 promoter region. To distinguish these two possibilities, the 309-bp labeled DNA probe encompassing the prx3 promoter region was incubated with increasing amounts of the purified IscR and then subjected to electrophoresis. Inductively coupled plasma-atomic emission spectrometry analysis revealed that the purified IscR does not contain any detectable levels of iron (data not shown), indicating that most of the IscR protein was in the Fe-S clusterless apo-form. As seen in Fig. 8, the addition of IscR resulted in a shift of the 309-bp DNA fragment to a single band with slower mobility. The binding of IscR was also specific, because assays were performed in the presence of 0.1 μg of poly(dI-dC) as a nonspecific competitor. In a second EMSA, the same but unlabeled 309-bp DNA fragment was used as a self-competitor to confirm the specific binding of IscR. The unlabeled 309-bp DNA competed for the binding of IscR in a dose-dependent manner (Fig. 8), confirming that IscR binds specifically to the prx3 promoter region.
FIGURE 8.
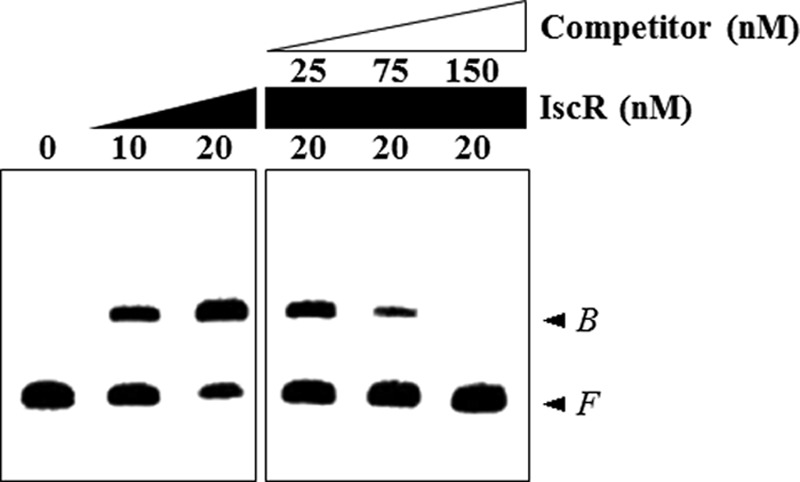
Specific binding of IscR to Pprx3. A 309-bp DNA fragment of the upstream region of Pprx3 was radioactively labeled and then used as a DNA probe. The radiolabeled probe DNA (5 nm) was mixed with increasing amounts of IscR, as indicated. For competition analysis, the same but unlabeled DNA fragment was used as a self-competitor DNA. Various amounts of the self-competitor DNA were added to a reaction mixture containing the 5 nm labeled DNA prior to the addition of 20 nm IscR. B, bound DNA; F, free DNA.
To determine the precise location of the IscR binding to the prx3 promoter region, DNase I protection assays were performed using the same 309-bp DNA fragment used for the EMSA. As shown in Fig. 9, regions extending from −56 to −30 (in the coding strand) and from −58 to −32 (in the noncoding strand) relative to the transcription start site of Pprx3 were clearly protected by IscR. Several nucleotides also showed enhanced cleavages, which have been frequently observed in DNase I protection analyses of the binding sites of IscR (25, 28). The predicted IscR-binding site, centered at −44, indicated that IscR is a class II activator interacting with domain 4 of the RNA polymerase σ subunit (46). This location of IscR binding was similar to those of IscR binding on the promoter of E. coli sufA and ydiU, whose expression is activated by apo-IscR (25, 26, 28). This result suggested that apo-IscR activates Pprx3 by directly binding to a specific site of the prx3 promoter region. Inspection of the sequences extending from −58 to −30 revealed a 29-bp imperfect palindrome (Fig. 7B). The sequence of the palindrome scored about 90% similarity to a consensus sequence of the Type 2 IscR-binding site, to which the apo-form of IscR most likely binds in E. coli grown aerobically (26).
FIGURE 9.

Sequences for binding of IscR to Pprx3. A 309-bp DNA fragment of the upstream region of Pprx3 was radioactively labeled and then used to as a DNA probe. The radiolabeled probe DNA (25 nm) was mixed with increasing amounts of IscR as indicated and then digested with DNase I. The regions protected by IscR are indicated by open boxes, and the nucleotides showing enhanced cleavage are indicated by black boxes. Lanes C, T, A, and G, nucleotide sequencing ladders of pJK1402. The transcription start site of Pprx3 (+1) and the putative −10 and −35 regions are indicated on the left of the nucleotide sequencing ladders.
Apo-IscR Plays a Role in Activating Pprx3 in Vivo
To ascertain whether the apo-form of IscR indeed activates Pprx3 in vivo, the prx3 transcripts in the wild type, iscR mutant, and iscR3CA mutant grown aerobically were quantified by qRT-PCR analyses. As shown in Fig. 10A, the level of prx3 transcript in the iscR3CA mutant, which chromosomally encodes the apo-locked IscR, IscR3CA, was almost 30-fold greater than that in the iscR mutant, indicating that apo-IscR is able to activate Pprx3 in vivo. In addition, the prx3 expression of the iscR3CA mutant was almost 3-fold greater than that of the wild type, in which both holo- and apo-IscR coexist (Fig. 10A). Because both holo- and apo-IscR bind to the Type 2 sequence with similar affinity in E. coli (26), the increased activity of Pprx3 in the iscR3CA mutant was possibly attributable to the elevated cellular level of IscR3CA. To examine this possibility, the IscR levels were compared in the wild type and iscR3CA mutant. The IscR3CA level in the iscR3CA mutant was almost 3-fold greater than the IscR level in the wild type, as determined based on the intensity of the IscR (or IscR3CA) protein band of Western blot analysis (Fig. 10B). This elevated IscR3CA level in the iscR3CA mutant was perhaps not surprising because apo-IscR derepresses its own expression (20, 21). The combined results confirmed that IscR in its apo-form activates prx3 in vivo.
FIGURE 10.
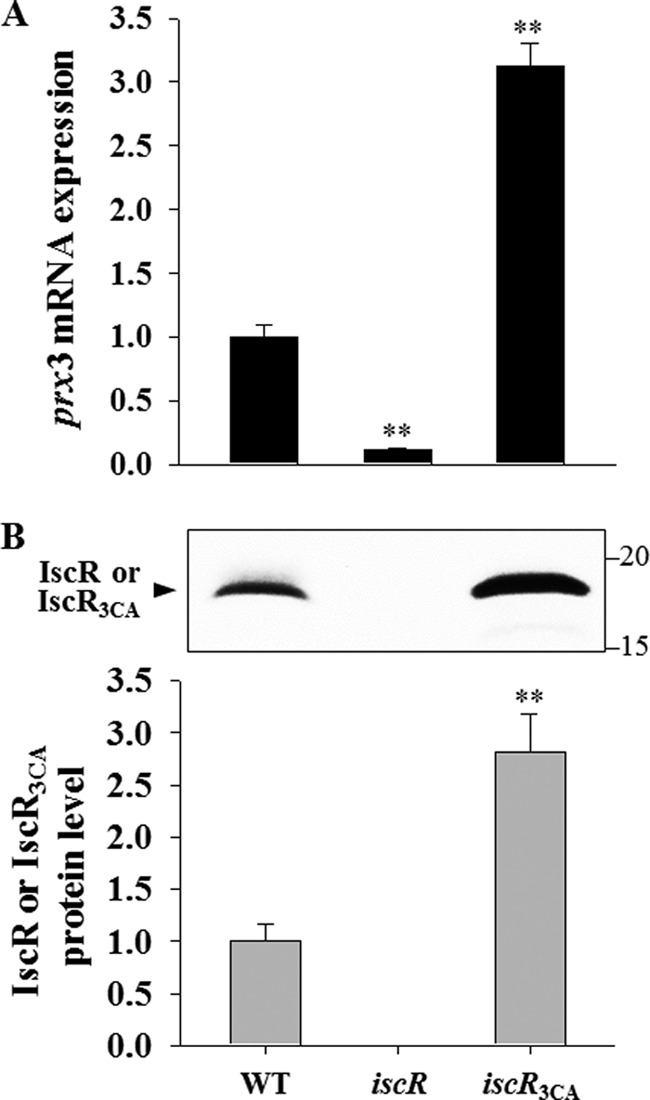
Effects of mutations of three cysteines on the IscR activity. Total RNAs and proteins were isolated from the strains grown aerobically to an A600 of 0.3. A, the prx3 mRNA levels in the total RNA were determined by qRT-PCR analyses, and the prx3 mRNA level in the wild type was set to 1. B, protein samples were resolved by SDS-PAGE, and IscR or IscR3CA was detected by Western blotting using a rabbit anti-IscR antiserum. The positions of protein size markers (in kDa; Bio-Rad) are shown on the right of the gel. In the graph, the IscR protein levels were calculated based on the band intensities, and the IscR protein level in the wild type was set to 1. Error bars, S.E. **, p < 0.005 relative to WT. iscR, iscR mutant; iscR3CA, a strain expressing apo-locked IscR3CA.
IscR Activates prx3 Expression by Sensing ROS and Iron Starvation
Recently, it was discovered that the [2Fe-2S] cluster in IscR is disrupted to shift to the apo-form by oxidative stress and iron starvation in E. coli (20–24). This prompted us to examine the effects of aerobic growth and iron starvation on the IscR-dependent expression of prx3 using qRT-PCR analyses. The level of the prx3 transcript in the wild type grown aerobically increased and was almost 6-fold greater compared with that reached by the bacteria grown anaerobically (Fig. 11A). In contrast, this increase in the prx3 transcript under aerobic conditions was not apparent in the iscR mutant. In conjunction with an 11-fold increased cellular level of IscR, as determined by Western blot analyses (Fig. 11B), the results suggested that IscR shifts to the apo-form by sensing trace amounts of ROS generated endogenously during aerobic metabolism, and the activation of prx3 is primarily due to the increased amounts of apo-IscR. Consistent with this, the IscR-dependent activation of prx3 was not substantial during anaerobic growth when the holo-form of IscR predominates (21), albeit the wild type produces slightly more prx3 transcript than the iscR mutant (Fig. 11A).
FIGURE 11.

Effects of oxidative stress and iron starvation on the IscR activity. Total RNAs and proteins were isolated from the wild type and iscR mutant grown anaerobically or aerobically to an A600 of 0.3 (A and B) or from the strains grown aerobically to an A600 of 0.3 after being exposed to various concentrations of DP for 10 min as indicated (C and D). A and C, the prx3 mRNA levels were determined by qRT-PCR analyses, and the prx3 mRNA level in the wild type grown aerobically was set to 1. B and D, protein samples were resolved by SDS-PAGE, IscR was detected by Western blotting, and results are presented as described in the legend to Fig. 10. In the graph, the IscR protein levels were calculated based on the band intensities, and the IscR protein level in the wild type grown aerobically was set to 1. Error bars, S.E. **, p < 0.005 relative to the strains grown anaerobically (A and B) or to the strains unexposed to DP (C and D). −, grown anaerobically; +, grown aerobically.
To examine the effect of iron availability on the expression of prx3, the strains were grown aerobically and exposed to a range of iron chelator DP. The level of the prx3 transcript in the wild type was gradually elevated along with increasing concentrations of DP in the medium (Fig. 11C). The result implied that the DP-mediated iron starvation decreases the Fe-S cluster occupancy of IscR to result in the apo-form that activates prx3. The cellular levels of IscR also exhibited a similar dependence on the iron availability and increased in response to iron starvation (Fig. 11D). Again, the expression of prx3 in the iscR mutant was low and not further induced under iron-depleted conditions (Fig. 11C). The results indicated that the increase in the apo-IscR protein levels also accounts for the activation of prx3 by iron starvation. Taken together, the results led us to propose a model in which IscR senses iron starvation as well as ROS and shifts to the clusterless apo-form, leading to derepression of the isc operon, elevated apo-IscR protein levels, and, accordingly, the activation of prx3.
DISCUSSION
In earlier studies, we identified two distinct 2-Cys Prxs, Prx1 and Prx2, in V. vulnificus (16, 17). Prx1 (formerly V. vulnificus AhpC) is an AhpF-dependent peroxidase (16). Prx1 is expressed only in cells exposed to high levels of exogenous H2O2 and is effective at decomposing large amounts of peroxides rapidly (17). In contrast to Prx1, Prx2 is a TrxA/TrR-dependent peroxidase, and its expression is induced during aerobic growth. Prx2 is more effective at scavenging low levels of H2O2, such as those generated endogenously during aerobic metabolism. In this study, molecular genetic analyses demonstrated that Prx3 is a 1-Cys Prx (Figs. 1 and 3). Prx3 is not only induced in cells grown aerobically without exposure to exogenous ROS (Fig. 11, A and B) but also essential for survival of the pathogen under high levels of exogenous ROS (250 μm H2O2 or 200 μm ONOO−) (Fig. 5). The results indicated that Prx3 is probably able to decompose both low endogenous and high exogenous levels of ROS. This ability of Prx3 to detoxify an extended range of ROS appears to be a costly overlap with that of Prx1 and Prx2 (16, 17). However, Prx3 adopts Grx3/GSH/GR, instead of AhpF or TrxA/TrR, as a reducing system to reactivate its oxidized form (Fig. 2). Therefore, having multiple Prxs that utilize different thiol-reduction systems and thereby dividing the burden of reactivating oxidized Prxs could provide an evolutionary advantage to V. vulnificus that must cope continually and consistently with the prevalence of ROS.
OxyR is a central regulator of the oxidative stress response in a number of bacteria, and its regulon includes many genes, primarily encoding antioxidants, including most, if not all, bacterial Prxs (AhpC) and catalases (47, 48). OxyR senses high levels of ROS and is activated via the oxidation of specific cysteine residues but is normally inactive during routine aerobic growth (22, 31, 48, 49). This study demonstrated for the first time that IscR, rather than OxyR, activates the expression of a Prx (Prx3) (Figs. 7–9). Data of this study suggested that IscR senses ROS by means of the decrease of its [2Fe-2S] occupancy, and the resulting apo-IscR activates the expression of Prx3 (Fig. 11, A and B). Consistent with this, IscR3CA, locked apo-IscR, is more efficient in the activation of Prx3 than is IscR (Fig. 10). IscR has been extensively studied for its role in maintaining Fe-S cluster homeostasis in E. coli (21, 50). In response to ROS that destabilizes the Fe-S cluster and increases the cellular demands for Fe-S cluster biogenesis, IscR shifts to the apo-form, derepresses the isc operon, and up-regulates the alternative Suf pathway, leading to increased Fe-S biogenesis (28, 50). Conceivably, IscR-dependent activation of prx3 could be beneficial to V. vulnificus because Prx3 is able to directly decompose ROS, maintain intracellular concentrations of ROS within the ranges favoring stabilization of the Fe-S cluster, and circumvent the cost-expensive Fe-S cluster biogenesis.
In addition to this, IscR could have considerable benefits in the regulation of prx3. First, the expression of prx3 during aerobic growth (Fig. 11, A and B) indicates that sensing of ROS via the decrease of the Fe-S cluster occupancy in IscR is more sensitive than that via the oxidation of cysteines in OxyR. Therefore, V. vulnifcus could use IscR to activate prx3 upon exposure to low levels of ROS that are insufficient to oxidize OxyR. Second, the IscR regulon includes many genes seemingly related to the pathogenesis of bacteria (30, 51). Accordingly, the iscR mutant of V. vulnificus is defective in motility, hemolytic activity, and adhesion to host cells, leading to attenuated virulence (30). Therefore, the regulation of these genes and prx3 coordinately by a common regulator, IscR, could facilitate the cooperation of their gene products, which could be crucial for the overall success of the pathogen during pathogenesis.
It is highly intriguing that IscR senses iron starvation in addition to ROS to induce the expression of prx3 (Fig. 11, C and D). ROS restrains iron availability in cells by oxidizing ferrous ion to insoluble ferric ion (24). Furthermore, ROS induces the expression of Fur and Dps, which decrease cellular levels of iron by hindering iron transport into cells and by sequestering the transported iron, respectively (22, 52). Therefore, it is perhaps not surprising that IscR induces Prx3, a decomposer of ROS, upon sensing iron starvation to maintain iron availability within safe limits. Consistent with this, under anaerobic conditions in which ROS is hardly present, iron starvation did not induce the expression of prx3 (data not shown). Additionally, pathogenic bacteria encounter a period of iron starvation upon entering their hosts and accordingly have evolved to sense iron starvation as a signal indicating that they are within a host (53). In this context, therefore, we postulate that V. vulnificus senses iron starvation as a marker of host environment via destabilization of the Fe-S cluster in IscR and in turn activates the expression of prx3, which is required for its successful pathogenesis (Fig. 6).
In summary, this study characterized a 1-Cys Prx of V. vulnificus, Prx3, that catalyzes the reduction of peroxides utilizing Grx3 and GSH as reductants. The prx3 mutant showed reduced survival under oxidative stress and virulence in mice, indicating that Prx3 plays an important role in V. vulnificus pathogenesis by protecting the pathogen from oxidative stress during infection. IscR controls the prx3 expression by directly binding to the Type 2 IscR-binding site centered at position −44 from the transcription start site. Biochemical and molecular genetic evidence demonstrated that IscR senses iron starvation as well as ROS and shifts to the clusterless apo-form, which leads to the increase in its own levels in cells and accordingly, the activation of prx3.
This work was supported by Mid-career Researcher Program Grant 2012R1A2A1A03009679 through the National Research Foundation funded by the Ministry of Science, ICT, and Future Planning; the R&D Convergence Center Support Program of the Ministry of Agriculture, Food, and Rural Affairs; and Ministry of Food and Drug Safety, Republic of Korea, Grant 14162MFDS972 (to S. H. C).
- ROS
- reactive oxygen species
- Prx
- peroxiredoxin
- CP
- peroxidatic cysteine
- CR
- resolving cysteine
- Trx
- thioredoxin
- AhpF
- alkyl hydroperoxidase subunit F
- Grx
- glutaredoxin
- AhpC
- alkyl hydroperoxidase subunit C
- GR
- glutathione reductase
- TrR
- thioredoxin reductase
- DP
- 2,2′-dipyridyl
- qRT-PCR
- quantitative real-time PCR.
REFERENCES
- 1. Storz G., Imlay J. A. (1999) Oxidative stress. Curr. Opin. Microbiol. 2, 188–194 [DOI] [PubMed] [Google Scholar]
- 2. Storz G., Zheng M. (2000) in Bacterial Stress Responses (Gisela Storz, Hengge-Aronis R., eds) pp. 47–59, ASM Press, Washington, D. C. [Google Scholar]
- 3. Miller R. A., Britigan B. E. (1997) Role of oxidants in microbial pathophysiology. Clin. Microbiol. Rev. 10, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nathan C., Shiloh M. U. (2000) Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. U.S.A. 97, 8841–8848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fang F. C. (2004) Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat. Rev. Microbiol. 2, 820–832 [DOI] [PubMed] [Google Scholar]
- 6. Dubbs J. M., Mongkolsuk S. (2007) Peroxiredoxins in bacterial antioxidant defense. Subcell. Biochem. 44, 143–193 [DOI] [PubMed] [Google Scholar]
- 7. Imlay J. A. (2008) Cellular defenses against superoxide and hydrogen peroxide. Annu. Rev. Biochem. 77, 755–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Poole L. B., Hall A., Nelson K. J. (2011) Overview of peroxiredoxins in oxidant defense and redox regulation. Curr. Protoc. Toxicol. 49, 7.9.1–7.9.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Knoops B., Loumaye E., Van Der Eecken V. (2007) Evolution of the peroxiredoxins. Subcell. Biochem. 44, 27–40 [DOI] [PubMed] [Google Scholar]
- 10. Hall A., Karplus P. A., Poole L. B. (2009) Typical 2-Cys peroxiredoxins-structures, mechanisms and functions. FEBS J. 276, 2469–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Seo M. S., Kang S. W., Kim K., Baines I. C., Lee T. H., Rhee S. G. (2000) Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J. Biol. Chem. 275, 20346–20354 [DOI] [PubMed] [Google Scholar]
- 12. Noguera-Mazon V., Lemoine J., Walker O., Rouhier N., Salvador A., Jacquot J. P., Lancelin J. M., Krimm I. (2006) Glutathionylation induces the dissociation of 1-Cys d-peroxiredoxin non-covalent homodimer. J. Biol. Chem. 281, 31736–31742 [DOI] [PubMed] [Google Scholar]
- 13. Clarke D. J., Ortega X. P., Mackay C. L., Valvano M. A., Govan J. R., Campopiano D. J., Langridge-Smith P., Brown A. R. (2010) Subdivision of the bacterioferritin comigratory protein family of bacterial peroxiredoxins based on catalytic activity. Biochemistry 49, 1319–1330 [DOI] [PubMed] [Google Scholar]
- 14. Pedrajas J. R., Padilla C. A., McDonagh B., Bárcena J. A. (2010) Glutaredoxin participates in the reduction of peroxides by the mitochondrial 1-CYS peroxiredoxin in Saccharomyces cerevisiae. Antioxid. Redox Signal. 13, 249–258 [DOI] [PubMed] [Google Scholar]
- 15. Djuika C. F., Fiedler S., Schnölzer M., Sanchez C., Lanzer M., Deponte M. (2013) Plasmodium falciparum antioxidant protein as a model enzyme for a special class of glutaredoxin/glutathione-dependent peroxiredoxins. Biochim. Biophys. Acta 1830, 4073–4090 [DOI] [PubMed] [Google Scholar]
- 16. Baek W. K., Lee H. S., Oh M. H., Koh M. J., Kim K. S., Choi S. H. (2009) Identification of the Vibrio vulnificus ahpCl gene and its influence on survival under oxidative stress and virulence. J. Microbiol. 47, 624–632 [DOI] [PubMed] [Google Scholar]
- 17. Bang Y. J., Oh M. H., Choi S. H. (2012) Distinct characteristics of two 2-Cys peroxiredoxins of Vibrio vulnificus suggesting differential roles in detoxifying oxidative stress. J. Biol. Chem. 287, 42516–42524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jeong W., Cha M. K., Kim I. H. (2000) Thioredoxin-dependent hydroperoxide peroxidase activity of bacterioferritin comigratory protein (BCP) as a new member of the thiol-specific antioxidant protein (TSA)/alkyl hydroperoxide peroxidase C (AhpC) family. J. Biol. Chem. 275, 2924–2930 [DOI] [PubMed] [Google Scholar]
- 19. Hugo M., Turell L., Manta B., Botti H., Monteiro G., Netto L. E., Alvarez B., Radi R., Trujillo M. (2009) Thiol and sulfenic acid oxidation of AhpE, the one-cysteine peroxiredoxin from Mycobacterium tuberculosis: kinetics, acidity constants, and conformational dynamics. Biochemistry 48, 9416–9426 [DOI] [PubMed] [Google Scholar]
- 20. Schwartz C. J., Giel J. L., Patschkowski T., Luther C., Ruzicka F. J., Beinert H., Kiley P. J. (2001) IscR, an Fe-S cluster-containing transcription factor, represses expression of Escherichia coli genes encoding Fe-S cluster assembly proteins. Proc. Natl. Acad. Sci. U.S.A. 98, 14895–14900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Giel J. L., Nesbit A. D., Mettert E. L., Fleischhacker A. S., Wanta B. T., Kiley P. J. (2013) Regulation of iron-sulphur cluster homeostasis through transcriptional control of the Isc pathway by [2Fe–2S]–IscR in Escherichia coli. Mol. Microbiol. 87, 478–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zheng M., Wang X., Templeton L. J., Smulski D. R., LaRossa R. A., Storz G. (2001) DNA microarray-mediated transcriptional profiling of the Escherichia coli response to hydrogen peroxide. J. Bacteriol. 183, 4562–4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Outten F. W., Djaman O., Storz G. (2004) A suf operon requirement for Fe–S cluster assembly during iron starvation in Escherichia coli. Mol. Microbiol. 52, 861–872 [DOI] [PubMed] [Google Scholar]
- 24. Imlay J. A. (2006) Iron-sulphur clusters and the problem with oxygen. Mol. Microbiol. 59, 1073–1082 [DOI] [PubMed] [Google Scholar]
- 25. Giel J. L., Rodionov D., Liu M., Blattner F. R., Kiley P. J. (2006) IscR-dependent gene expression links iron-sulphur cluster assembly to the control of O2-regulated genes in Escherichia coli. Mol. Microbiol. 60, 1058–1075 [DOI] [PubMed] [Google Scholar]
- 26. Nesbit A. D., Giel J. L., Rose J. C., Kiley P. J. (2009) Sequence-specific binding to a subset of IscR-regulated promoters does not require IscR Fe–S cluster ligation. J. Mol. Biol. 387, 28–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rajagopalan S., Teter S. J., Zwart P. H., Brennan R. G., Phillips K. J., Kiley P. J. (2013) Studies of IscR reveal a unique mechanism for metal-dependent regulation of DNA binding specificity. Nat. Struct. Mol. Biol. 20, 740–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yeo W. S., Lee J. H., Lee K. C., Roe J. H. (2006) IscR acts as an activator in response to oxidative stress for the suf operon encoding Fe-S assembly proteins. Mol. Microbiol. 61, 206–218 [DOI] [PubMed] [Google Scholar]
- 29. Wu Y., Outten F. W. (2009) IscR controls iron-dependent biofilm formation in Escherichia coli by regulating type I fimbria expression. J. Bacteriol. 191, 1248–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lim J. G., Choi S. H. (2014) IscR is a global regulator essential for pathogenesis of Vibrio vulnificus and induced by host cells. Infect. Immun. 82, 569–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim S., Bang Y. J., Kim D., Lim J. G., Oh M. H., Choi S. H. (2014) Distinct characteristics of OxyR2, a new OxyR-type regulator, ensuring expression of Peroxiredoxin 2 detoxifying low levels of hydrogen peroxide in Vibrio vulnificus. Mol. Microbiol. 93, 992–1009 [DOI] [PubMed] [Google Scholar]
- 32. Sheffield P., Garrard S., Derewenda Z. (1999) Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr. Purif. 15, 34–39 [DOI] [PubMed] [Google Scholar]
- 33. Pauwels F., Vergauwen B., Vanrobaeys F., Devreese B., Van Beeumen J. J. (2003) Purification and characterization of a chimeric enzyme from Haemophilus influenzae Rd that exhibits glutathione-dependent peroxidase activity. J. Biol. Chem. 278, 16658–16666 [DOI] [PubMed] [Google Scholar]
- 34. Milton D. L., O'Toole R., Horstedt P., Wolf-Watz H. (1996) Flagellin A is essential for the virulence of Vibrio anguillarum. J. Bacteriol. 178, 1310–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Simon R., Priefer U., Pühler A. (1983) A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat. Biotechnol. 1, 784–791 [Google Scholar]
- 36. Oka A., Sugisaki H., Takanami M. (1981) Nucleotide sequence of the kanamycin resistance transposon Tn903. J. Mol. Biol. 147, 217–226 [DOI] [PubMed] [Google Scholar]
- 37. Kim S. M., Lee D. H., Choi S. H. (2012) Evidence that the Vibrio vulnificus flagellar regulator FlhF is regulated by a quorum sensing master regulator SmcR. Microbiology 158, 2017–2025 [DOI] [PubMed] [Google Scholar]
- 38. Cubadda F., Raggi A. (2005) Determination of cadmium, lead, iron, nickel and chromium in selected food matrices by plasma spectrometric techniques. Microchem. J. 79, 91–96 [Google Scholar]
- 39. Lim J. G., Park J. H., Choi S. H. (2014) Low cell density regulator AphA upregulates the expression of Vibrio vulnificus iscR gene encoding the Fe-S cluster regulator IscR. J. Microbiol. 52, 413–421 [DOI] [PubMed] [Google Scholar]
- 40. Rouhier N., Gelhaye E., Sautiere P. E., Brun A., Laurent P., Tagu D., Gerard J., de Faÿ E., Meyer Y., Jacquot J. P. (2001) Isolation and characterization of a new peroxiredoxin from poplar sieve tubes that uses either glutaredoxin or thioredoxin as a proton donor. Plant Physiol. 127, 1299–1309 [PMC free article] [PubMed] [Google Scholar]
- 41. Sarma G. N., Nickel C., Rahlfs S., Fischer M., Becker K., Karplus P. A. (2005) Crystal Structure of a Novel Plasmodium falciparum 1-Cys Peroxiredoxin. J. Mol. Biol. 346, 1021–1034 [DOI] [PubMed] [Google Scholar]
- 42. Knoops B., Clippe A., Bogard C., Arsalane K., Wattiez R., Hermans C., Duconseille E., Falmagne P., Bernard A. (1999) Cloning and characterization of AOEB166, a novel mammalian antioxidant enzyme of the peroxiredoxin family. J. Biol. Chem. 274, 30451–30458 [DOI] [PubMed] [Google Scholar]
- 43. Poole L. B. (2007) The catalytic mechanism of peroxiredoxins. Subcell. Biochem. 44, 61–81 [DOI] [PubMed] [Google Scholar]
- 44. Rouhier N., Gelhaye E., Jacquot J. P. (2002) Glutaredoxin-dependent peroxiredoxin from poplar protein-protein interaction and catalytic mechanism. J. Biol. Chem. 277, 13609–13614 [DOI] [PubMed] [Google Scholar]
- 45. Chung K. J., Cho E. J., Kim M. K., Kim Y. R., Kim S. H., Yang H. Y., Chung K. C., Lee S. E., Rhee J. H., Choy H. E., Lee T. H. (2010) RtxA1-induced expression of the small GTPase Rac2 plays a key role in the pathogenicity of Vibrio vulnificus. J. Infect. Dis. 201, 97–105 [DOI] [PubMed] [Google Scholar]
- 46. Browning D. F., Busby S. J. (2004) The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2, 57–65 [DOI] [PubMed] [Google Scholar]
- 47. Chiang S. M., Schellhorn H. E. (2012) Regulators of oxidative stress response genes in Escherichia coli and their functional conservation in bacteria. Arch. Biochem. Biophys. 525, 161–169 [DOI] [PubMed] [Google Scholar]
- 48. Dubbs J. M., Mongkolsuk S. (2012) Peroxide-sensing transcriptional regulators in bacteria. J. Bacteriol. 194, 5495–5503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Imlay J. A. (2013) The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat. Rev. Microbiol. 11, 443–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Py B., Barras F. (2010) Building Fe-S proteins: bacterial strategies. Nat. Rev. Microbiol. 8, 436–446 [DOI] [PubMed] [Google Scholar]
- 51. Miller H. K., Kwuan L., Schwiesow L., Bernick D. L., Mettert E., Ramirez H. A., Ragle J. M., Chan P. P., Kiley P. J., Lowe T. M., Auerbuch V. (2014) IscR is essential for Yersinia pseudotuberculosis type III secretion and virulence. PLoS Pathog. 10, e1004194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Antelmann H., Helmann J. D. (2011) Thiol-based redox switches and gene regulation. Antioxid. Redox Signal. 14, 1049–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Skaar E. P. (2010) The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 6, e1000949. [DOI] [PMC free article] [PubMed] [Google Scholar]