

Background: TRPM2 channels play an essential role in cell death following oxidative stress.
Results: Dominant negative TRPM2-S decreases growth of neuroblastoma xenografts and increases doxorubicin sensitivity through modulation of HIF-1/2α expression, mitophagy, and mitochondrial function.
Conclusion: TRPM2 is important for neuroblastoma growth and viability through modulation of HIF-1/2α.
Significance: Modulation of TRPM2 may be a novel approach in cancer therapeutics.
Keywords: Hypoxia-inducible Factor (HIF), Mitochondria, Mitophagy, Neuroblastoma, Oxidative Stress, TRP, TRPM
Abstract
The calcium-permeable ion channel TRPM2 is highly expressed in a number of cancers. In neuroblastoma, full-length TRPM2 (TRPM2-L) protected cells from moderate oxidative stress through increased levels of forkhead box transcription factor 3a (FOXO3a) and superoxide dismutase 2. Cells expressing the dominant negative short isoform (TRPM2-S) had reduced FOXO3a and superoxide dismutase 2 levels, reduced calcium influx in response to oxidative stress, and enhanced reactive oxygen species, leading to decreased cell viability. Here, in xenografts generated with SH-SY5Y neuroblastoma cells stably expressing TRPM2 isoforms, growth of tumors expressing TRPM2-S was significantly reduced compared with tumors expressing TRPM2-L. Expression of hypoxia-inducible factor (HIF)-1/2α was significantly reduced in TRPM2-S-expressing tumor cells as was expression of target proteins regulated by HIF-1/2α including those involved in glycolysis (lactate dehydrogenase A and enolase 2), oxidant stress (FOXO3a), angiogenesis (VEGF), mitophagy and mitochondrial function (BNIP3 and NDUFA4L2), and mitochondrial electron transport chain activity (cytochrome oxidase 4.1/4.2 in complex IV). The reduction in HIF-1/2α was mediated through both significantly reduced HIF-1/2α mRNA levels and increased levels of von Hippel-Lindau E3 ligase in TRPM2-S-expressing cells. Inhibition of TRPM2-L by pretreatment with clotrimazole or expression of TRPM2-S significantly increased sensitivity of cells to doxorubicin. Reduced survival of TRPM2-S-expressing cells after doxorubicin treatment was rescued by gain of HIF-1 or -2α function. These data suggest that TRPM2 activity is important for tumor growth and for cell viability and survival following doxorubicin treatment and that interference with TRPM2-L function may be a novel approach to reduce tumor growth through modulation of HIF-1/2α, mitochondrial function, and mitophagy.
Introduction
Transient receptor potential (TRP)3 channels are a superfamily of monovalent and divalent cation-permeable ion channels with six transmembrane domains. They are homologs of the Ca2+-permeable Drosophila melanogaster TRP channel, which is essential for phototransduction (1). TRPM channels, members of the melastatin subfamily of TRP channels, have important roles in cell proliferation and survival (2–5). The second member of this subfamily that was cloned, TRPM2 (previously LTRPC2), consists of 32 exons encoding a protein of 1503 amino acids with a predicted molecular mass of ∼170 kDa (6). TRPM2 channels are permeable to Ca2+, Na+, and K+ and are widely expressed in many cell types including brain, hematopoietic cells, heart, vascular smooth muscle, and endothelial cells (7, 8). The role of TRPM2 in numerous cell types including brain, heart, endothelial cells, and inflammatory cells under physiological conditions as well as oxidative stress is actively being investigated (9). Extracellular signals that activate TRPM2 include oxidative stress, TNFα, and amyloid β-peptide (10–13). Stimulation with these extracellular signals results in production of ADP-ribose (ADPR), which plays an important role in channel activation by binding to the TRPM2 C-terminal NUDT9-H domain (7, 13–16). ADPR is produced in the mitochondria (15) or through activation of poly(ADPR) polymerase (PARP) (17, 18). TRPM2 is also positively regulated by intracellular Ca2+ and calmodulin (19–21) and is inhibited by acidification (22–24), providing a mechanism for limiting Ca2+ entry during ischemia or inflammation. At least four physiological splice variants of full-length TRPM2 (TRPM2-L) have been reported (12, 25, 26). Similar to other TRP channels, TRPM2 functions as a tetramer, and the association of splice variants with TRPM2-L may modulate its function (27–29). One of these splice variants, the short isoform (TRPM2-S; 845 residues), is missing the four C-terminal transmembrane domains and the putative Ca2+ pore. When TRPM2-S (S) and TRPM2-L (L) are coexpressed, S can inhibit calcium influx through L to function as a dominant negative (26).
We recently demonstrated that TRPM2 channel isoforms are highly expressed in neuroblastoma (30). TRPM2-L protected neuroblastoma cells from low or moderate oxidative stress through increased levels of forkhead box transcription factor 3a (FOXO3a) and superoxide dismutase 2 (30), whereas cells expressing the dominant negative S isoform had reduced FOXO3a and superoxide dismutase 2 levels and enhanced reactive oxygen species (ROS) with increased susceptibility to oxidant death. This finding is consistent with studies in pyramidal neurons subjected to oxidant injury that showed that inhibition of TRPM2 enhanced cellular damage (31). It is also consistent with recent in vivo experiments with knock-out mice that show that L function is protective, rather than deleterious, in a number of pathological conditions. In wild type mice subjected to intraperitoneal injection of endotoxin, survival was 5 times greater than in the TRPM2 knock-out due to reduced NADPH oxidase-mediated ROS production in phagocytes (32). Using a different KO mouse model, we recently demonstrated that cardiac function after ischemia/reperfusion was also worse in the TRPM2 KO and that ROS levels were significantly higher in TRPM2 KO cardiac myocytes subjected to hypoxia/reperfusion (33). ROS are produced naturally during respiration by the mitochondrial electron transport chain and play a major pathological role in tissue injury through protein oxidation, lipid peroxidation, and DNA oxidation and mutagenesis (34–37).
To examine the influence of TRPM2 isoforms on tumor growth, we examined mice with xenograft tumors genetically modified for gain or loss of TRPM2 function. In xenografts, tumor growth was significantly greater in neuroblastoma SH-SY5Y cells exogenously expressing L compared with cells expressing dominant negative S. The determinant mechanisms for the reduction of tumor growth in S-expressing cells were explored. Hypoxia-inducible factors (HIFs) are transcription factors that are often up-regulated in cancer and play an important role in tumor progression through modulation of expression of genes involved in glycolysis (lactate dehydrogenase A (LDHA)), angiogenesis (vascular endothelial growth factor (VEGF)), ROS production, mitochondrial function (BNIP3 and NDUFA4L2), growth, and survival (38–42). This study was undertaken to determine how TRPM2 splice variants determine tumor survival or death. We evaluated TRPM2 current in neuroblastoma cells expressing TRPM2-L and TRPM2-S isoforms, mitochondrial function, and relevant transcription factors. We found that in TRPM2-S-expressing cells there was a reduction of HIF-1α and HIF-2α expression, mitophagy, and mitochondrial function. TRPM2-L-expressing cells treated with the inhibitor clotrimazole or cells expressing TRPM2-S compared with TRPM2-L were significantly more sensitive to doxorubicin.
EXPERIMENTAL PROCEDURES
Tissues and Generation of Stably Transfected Neuroblastoma Cell Lines
Human adrenal gland and neuroblastoma tissues were obtained from the National Cancer Institute Cooperative Human Tissue Network, Pediatric Division, and The Research Institute at Nationwide Children's Hospital (Columbus, OH) under protocols approved by the Institutional Review Board of the Pennsylvania State University College of Medicine. The neuroblastoma cell line SH-SY5Y was purchased from the American Type Culture Collection (ATCC, Manassas, VA). SH-SY5Y cells were cultured in 50% DMEM and 50% Ham's F-12 supplemented with 10% heat-inactivated FBS. SH-SY5Y cells at 90% confluence were transfected with pcDNA3.1/V5-His TOPO expressing L, S, both L and S, or empty vector for 48 h using Lipofectamine 2000 (Invitrogen) (26). Stably transfected cell lines were selected using 600 μg/ml G418 (Geneticin; an analog of neomycin) (Gemini Bio-Products, West Sacramento, CA), and cell cultures were maintained in the presence of 250 μg/ml G418 for a minimum of 2 months before cells were used for experiments. For doxorubicin studies, plates were treated when cells were 70–80% confluent and harvested at the time points noted. For exogenous expression of HIF-1α or HIF-2α, human HIF-1α or HIF-2α constructs with mutations at ubiquitination sites to prevent degradation were obtained from Addgene (Cambridge, MA). For knockdown of HIF-1α or HIF-2α expression, shRNA plasmids targeted to HIF-1α or HIF-2α were purchased from OriGene (Rockville, MD). All constructs were transfected into SH-SY5Y cells stably expressing TRPM2-L or -S using the Neon Transfection System (Invitrogen) following the manufacturer's instructions, and doubled stably transfected cells were further selected by 0.5 μg/ml puromycin and 250 μg/ml G418.
RT-PCR of TRPM2 in Human Neuroblastoma and Adrenal Tissues
RNA was prepared from normal adrenal and neuroblastoma samples using the mirVana isolation kit (Life Technologies, Foster City, CA) and quality-controlled by BioAnalyzer RNA 6000 nanochip (Agilent Technologies, Santa Clara, CA). cDNA was synthesized using a High Capacity cDNA reverse transcription kit (Life Technologies), and 2.5 ng of each cDNA was applied to PCR. The following primer pairs based on the TRPM2 coding sequence were designed to specifically recognize TRPM2-L or TRPM2-S: TRPM2-L: forward primer, 5′-ACGTGCTCATGGTGGACTTC-3′; reverse primer, 5′-AGGGTCATAGAAGAGCTGCC-3′; TRPM2-S: forward primer, 5′-CTACTTCGCCTTCCTCTGCC-3′; reverse primer, 5′-GTCAGGGTCATAGAAGAGCTAC-3′. Semiquantitative PCR was performed for 35 and 40 cycles (initially 94 °C for 2 min, denaturation at 94 °C for 30 s, annealing at 65 °C for 30 s and 72 °C for 30 s, and then 72 °C for 30 s). The 110-bp product for TRPM2-L and 145-bp product for TRPM2-S were quantified by the BioAnalyzer High Sensitivity chip (Agilent Technologies), respectively.
Measurement of [Ca2+]i
Changes in [Ca2+]i were measured with fluorescence microscopy-coupled digital video imaging as described previously (43, 44). Stably transfected SH-SY5Y cells expressing TRPM2-L, TRPM2-S, or empty vector were adhered to fibronectin-coated glass coverslips and loaded for 20 min with 0.1 μm Fura-2 AM (Molecular Probes, Inc., Eugene, OR). Fura-2-loaded cells were excited alternately at 360 and 380 nm, and fluorescence emissions (510 nm) were captured. The ratio of F360/F380 was measured at baseline and over 20 min following H2O2 treatment.
TRPM2 Current and Estimation of the Ratio of the Conductance for Ca2+ and Na+ Ions (GCa/GNa) in Neuroblastoma Cells
TRPM2 currents were measured in neuroblastoma cells (30 °C) stably expressing L, S, or L and S with whole cell patch clamp (45). Fire-polished pipettes (tip diameter, 2 μm) with resistances of 2–4 megaohms when filled with pipette solutions were used. Pipette solution contained 110 mm CsCl, 20 mm triethanolamine chloride, 10 mm HEPES, 10 mm EGTA, and 5 mm MgATP, pH 7.2, and bathing solution contained 127 mm NaCl, 5.4 mm CsCl, 2 mm CaCl2, 1.3 mm MgSO4, 4 mm 4-aminopyridine, 10 mm HEPES, 10 mm Na-HEPES, 15 mm glucose, and 0.001 mm verapamil, pH 7.4. Solutions were designed to minimize L-type Ca2+ current (ICa), Na+-K+-ATPase current (INaKATPase), Na+/Ca+ exchange current (INaCa), and potassium currents (IK). Neuroblastoma cells were held at −70 mV. To inactivate fast inward Na+ current, the holding potential was switched to −40 mV before application of voltage ramp (+100 to −100 mV; 500 mV/s). In some experiments, ADPR (300 μm) was included in the pipette solutions to activate TRPM2 channels (14). In other experiments, after full activation of TRPM2 channels by ADPR, flufenamic acid (0.5 mm) or clotrimazole (50 μm) was added to the extracellular medium to inhibit TRPM2 currents (46, 47).
To estimate TRPM2 GCa and GNa, neuroblastoma cells were voltage-clamped at −80 mV. Pipette solution was identical to that used above and contained ADPR. Extracellular solution contained 140 mm NaCl, 10 mm HEPES, and 15 mm glucose, pH 7.4. After break-in, steady-state inward Na+ currents were obtained at Em of −80, −90, and −100 mV. Extracellular solution was then changed to one containing 110 mm CaCl2, 10 mm HEPES, and 15 mm glucose, pH 7.4. Steady-state inward Ca2+ currents were obtained at Em of −80, −90, and −100 mV from the same cell. By Ohm's law, Em − Erev = I/G. Taking derivatives, ΔI/ΔEm = G. GCa/GNa is given by the ratio of the slopes of I versus Em plots with Ca2+ or Na+ as the permeant ion.
Subcellular Fractionation and TRPM2 Localization
The Qproteome mitochondria isolation kit (Qiagen, Inc., Valencia, CA) was utilized to separate cytoplasmic, plasma membrane/endoplasmic reticulum (ER), and mitochondrial fractions from wild type SH-SY5Y cells to determine endogenous TRPM2-L localization. To further examine subcellular localization of TRPM2 isoforms, SH-SY5Y cells were transduced with lentiviruses encoding DsRed-ER together with GFP-tagged TRPM2-L or TRPM2-S using the ViraPower lentiviral expression system (Invitrogen) as described previously (48) and stained with rabbit antibody to Tom20 (Santa Cruz Biotechnology, Dallas, TX) followed by secondary antibody conjugated with Alexa Fluor 405 (Invitrogen). Confocal images of fluorescently labeled cells were acquired with a Leica AOBS SP8 laser-scanning confocal microscope (Leica, Heidelberg, Germany) using a high resolution Leica 40×/1.3 or 63×/1.4 numerical aperture Plan-Apochromat oil immersion objectives. The laser lines used for excitation were continuous wave 405 (for Alexa Fluor 405), 80-MHz pulsed 489 nm (for GFP), and 80-MHz pulsed 558 nm (for DsRed). The respective emission signals were collected sequentially to avoid cross-excitation, and spectral scanning was performed on all dyes to confirm signal specificity. Images (z-sections) were compiled, and finally three-dimensional image restoration was performed using Imaris software (Bitplane). An appropriate threshold was set for each voxel intensity to exclude possible contributions from background, and the commonly used Pearson coefficient (RP) was determined (49).
Xenograft Tumors Expressing TRPM2 Isoforms
To determine the role of TRPM2 isoforms in tumor growth, athymic Nude-FOXn1nu female mice (Harlan Laboratories, Inc., Indianapolis, IN) were injected in one flank with 1.5 × 107 SH-SY5Y cells stably expressing L, S, or empty vector. Approximately 8–10 mice per group were used in each of five experiments. Tumor length and width were measured twice weekly with a caliper to determine tumor growth over 6–7 weeks, and tumor area was calculated. At the completion of each experiment, tumors were harvested, weighed, and frozen for analysis. All protocols and procedures applied to the mice in this study were approved by the Institutional Animal Care and Use Committee of The Pennsylvania State University College of Medicine.
Immunoblot Analysis
For cell culture or tumors from neuroblastoma xenografts, whole cell lysates were prepared with Triton lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1% Triton X-100, 1 mm EDTA, 10 mm NaF, protease inhibitor, and phosphatase inhibitor) followed by 10-min centrifugation at 10,000 rpm at 4 °C. The supernatants were collected and subjected to 8–15% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) as described previously (30). All gels then were transblotted onto nitrocellulose membranes. Blots were probed with anti-TRPM2-C (1:300; Bethyl Laboratories, Montgomery, TX) (26) or anti-V5-HRP (1:2000; Invitrogen) antibody to confirm expression of endogenous or transfected TRPM2 channels. Anti-TRPM2-C recognizes endogenous TRPM2-L but not TRPM2-S. Blots were also probed with antibodies to BNIP3 (1:1000; Abcam, Cambridge, MA), caspase 3 (1:500; Cell Signaling Technology Inc., Boston, MA), caspase 7 (1:750; Cell Signaling Technology Inc.), COX4.1 and COX4.2 (1:1000; Abcam), enolase 2 (1:5000; Abnova, Taipei, Taiwan), FOXO3a (1:400; Cell Signaling Technology Inc.), HIF-1α (1:250; Cell Signaling Technology Inc.), HIF-2α (1:750; Novus, Littleton, CO), LDHA (1:15,000; Cell Signaling Technology Inc.), NDUFA4L2 (1:1000; Abcam), Hsp60 (1:5000; Cell Signaling Technology Inc.), p62 (1:4000; American Research Products, Inc., Waltham, MA), PARP (1:750; Cell Signaling Technology Inc.), mitochondrial transcription factor A (1:2000; Cell Signaling), Tom20 (1:5000, Santa Cruz Biotechnology), VEGF (1:30,000; Abcam); VHL (1:250; Cell Signaling Technology Inc.), actin (1:10,000; Sigma), tubulin (1:20,000), and C1ORF43 (1:1000; Abcam). Blots were washed and incubated with appropriate horseradish peroxidase (HRP)-conjugated antibodies (1:2000). Enhanced chemiluminescence (ECL) was used for detection of signal. The intensity of bands was quantitated with densitometry and normalized with loading controls.
Neuroblastoma Proteomic Analysis
Label-free proteomic analysis was performed using GeLC-MS/MS technology. Extracted proteins in radioimmune precipitation assay buffer from pooled xenograft tumors stably transfected with TRPM2-L (n = 5) or TRPM2-S (n = 5) and harvested at 6 weeks after cell injection were processed for GeLC-MS/MS analysis as described previously (50). Briefly, electrospray ionization tandem MS was performed using a Bruker HCT Ultra ion trap mass spectrometer. Mass spectra processing was performed using Bruker Daltonics Esquire 6.1-DataAnalysis (Version 3.4). The generated deisotoped peak list was submitted to in-house Mascot server 2.2.07 for searching against the Swiss-Prot database (Release 2011_06) (Version 56.6, 536,029 sequences). Mascot search parameters were set as follows: species, Homo sapiens (20,413 sequences); enzyme, trypsin with maximal one missed cleavage; fixed modification, cysteine carboxymethylation; variable modification, methionine oxidation; 0.45-Da mass tolerance for precursor peptide ions; and 0.6-Da mass tolerance for MS/MS fragment ions. All peptides matches were filtered using an ion score. Label-free quantitation was performed in Mascot Distiller using the average protein ratio, a minimum of one peptide with unique sequence, and a 0.05 significant threshold, and the results were analyzed with MS Data Miner (51). Label-free quantified proteins with greater than a 2-fold increase were selected and clustered by biological functions. To identify biological networks of differentially expressed proteins, we performed a standard network analysis using IPA software and the Ingenuity knowledge database for physical and functional interactions as described previously (52).
Quantitation of HIF-1/2α mRNA
Total RNA was isolated from xenograft samples harvested at 6–7 weeks using an RNeasy mini kit (Qiagen, Germantown, MD). cDNA then was prepared using the SuperScript first strand synthesis system for reverse transcription-PCR (Invitrogen). HIF-1α (forward, 5′-ACGTTCCTTCGATCAGTTGTCA; reverse, 5′-TTTGAGGACTTGCGCTTTCA) and HIF-2α mRNA (forward, 5′-CATGCGCTAGACTCCGAGAAC; reverse, 5′-CCTGACCCTTGGTGCACAA) were quantitated by real time PCR with SYBR Green FastMix followed by standardization to 18 S RNA expression.
Mitochondrial DNA Quantitation
Genomic DNA and mitochondrial DNA (mtDNA) from xenograft tumors were isolated using a Wizard Genomic DNA purification kit (Promega, Madison, WI) and quantitated by real time quantitative PCR amplification on a StepOnePlus PCR machine (Applied Biosystems, Carlsbad, CA) with primers that recognize mitochondrial 12 S DNA (forward, 5′-TAGCCTAAACCTCAACAGT; reverse, 5′-TGCGCTTACTTTGTAGCCTTCAT) and nuclear 18 S DNA (forward, 5′-CCCTGCCCTTTGTACACACC; reverse, 5′-GATCCGAGGGCCTCACTA) (53) and PerfeCTa SYBR Green FastMix (Quanta Bioscience, Gaithersburg, MD). The ratios (12 S/18 S) obtained for measurements (five xenografts/group transfected with vector, TRPM2-L, or TRPM2-S) were calculated and analyzed.
Electron Microscopy Analysis of Tumors from Xenografts Expressing TRPM2-L or TRPM2-S
Tumor xenografts were excised and weighed. Tumors were bisected, and a central 3–5-mm tumor central slice was fixed in 10% buffered formalin for paraffin sectioning. A small portion of the central slice was diced and fixed in fresh, ice-cold Karnovsky's fixative (Electron Microscopy Sciences, Hatfield, PA) for EM. After a 45-h fixation at 4 °C, EM samples were processed and embedded in EMbed 812 resin (Electron Microscopy Sciences). Diamond-cut 90-nm sections were mounted on 200-mesh copper grids and stained with 2% aqueous uranyl acetate and lead citrate. Sections were imaged at 1200× and 3000× on a JEOL JEM-1400 transmission electron microscope (JEOL, Peabody, MA).
Measurement of Mitochondrial Membrane Potential (ΔΨm) and Mitochondrial Ca2+ Uptake
To evaluate the integrity of mitochondrial function, we measured Δψm and mitochondrial Ca2+ uptake. To assess Δψm, neuroblastoma cells were permeabilized and resuspended in intracellular-like medium containing 120 mm KCl, 10 mm NaCl, 1 mm KH2PO4, 20 mm HEPES-Tris, 2 μg/ml thapsigargin, 80 μg/ml digitonin, pH 7.2, and protease inhibitors (EDTA-free Complete tablets, Roche Applied Science). Permeabilized cells were loaded with Fura-FF (0.5 μm) at 0 s and JC-1 (800 nm; Molecular Probes) at 20 s to measure extramitochondrial Ca2+ and Δψm, respectively. Fluorescence signals were monitored in a temperature-controlled (37 °C) multiwavelength excitation and dual wavelength emission spectrofluorometer (Delta RAM, Photon Technology International, Birmingham, NJ) using 490-nm excitation/535-nm emission for the monomer, 570-nm excitation/595-nm emission for the J-aggregate of JC-1, and 340- and 380-nm excitation/510-nm emission for Fura-FF (54, 55). The ratiometric dye Fura-FF was calibrated as described previously (54). At 450 s, a 10 μm Ca2+ pulse was added, and Δψm and extramitochondrial Ca2+ were monitored simultaneously at six measurements per second. Δψm was calculated as the ratio of the fluorescence of the JC-1 oligomeric to monomeric forms. The cytosolic Ca2+ clearance rate was taken to represent mitochondrial Ca2+ uptake.
Measurement of Mitochondrial O2 Consumption and ATP Levels
The oxygen consumption rate was measured in neuroblastoma SH-SY5Ycells at 37 °C in an XF96 extracellular flux analyzer (Seahorse Bioscience). Neuroblastoma cells were sequentially exposed to oligomycin, carbonyl cyanide p-trifluoromethoxyphenylhydrazone, 2-deoxyglucose, and rotenone using the XF Cell Mito Stress kit (Seahorse Bioscience) according to the manufacturer's instructions. Preliminary experiments were performed to select optimal seeding density (105 cells/well) and compound concentrations according to the manufacturer's instructions. To measure ATP levels, neuroblastoma SH-SY5Y cells or cells isolated from neuroblastoma xenografts were lysed, and ATP (luminescence) levels were measured using a Cell Titer-Glo luminescent cell viability assay kit (Promega) according to the manufacturer's instructions.
Cell Proliferation Assay
Cells from stably transfected cell lines were seeded on 96-well plates and cultured in medium with 250 μg/ml G418 for 96 h. Cell proliferation was assessed by measurement at A490 nm/690 nm using an XTT (2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide)cell proliferation assay (Trevigen Inc., Gaithersburg, MD) following the manufacturer's instructions (56). In some experiments, cells were treated with doxorubicin (0.1–0.3 μm) or clotrimazole (10 μm) for specified durations during cell culture. Viability after doxorubicin treatment was quantitated by trypan blue exclusion and XTT assay.
Statistics
All results are expressed as mean ± S.E. For analysis of TRPM2 current as a function of group and voltage, two-way analysis of variance was used. For analysis of protein expression levels, Δψm, mitochondrial Ca2+ uptake, and O2 consumption, one-way analysis of variance or Student's t test was used. In all analyses, p ≤ 0.05 was taken to be statistically significant.
RESULTS
Expression of TRPM2 Isoforms in Adrenal Gland and Neuroblastoma
TRPM2 has been shown to be highly expressed in a number of malignancies including neuroblastoma (25, 30). To quantitate expression of TRPM2-L and TRPM2-S isoforms in neuroblastoma with a different approach, RNA was prepared from 11 adrenal glands and 17 neuroblastoma tumor samples, and RT-PCR was performed with primers specific for TRPM2-L or TRPM2-S. TRPM2-L mRNA expression was significantly greater in neuroblastoma tumor samples than in adrenal gland (Fig. 1, A and C; p < 0.003). TRPM2-S mRNA required RT-PCR at 40 cycles for detection in the majority of samples but was also statistically greater in neuroblastoma compared with adrenal gland (Fig. 1, B and D; p < 0.05). These results show that neuroblastoma tumors have statistically greater expression of TRPM2-L, that this increase is at least partially on a transcriptional basis, and that these tumors have relatively little TRPM2-S.
FIGURE 1.
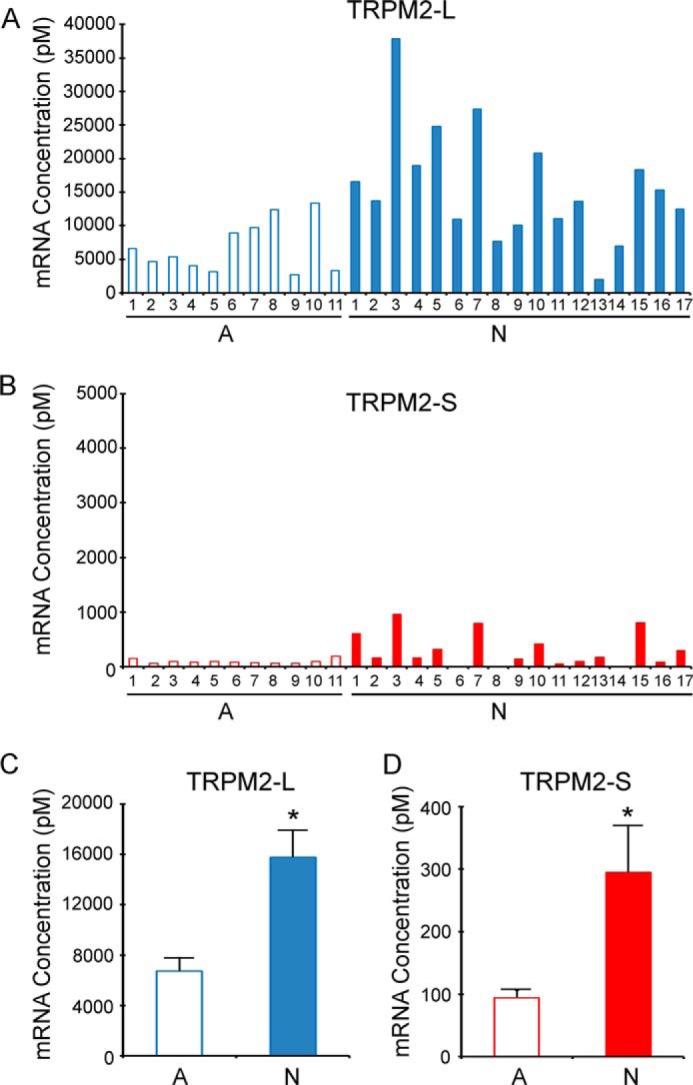
RT-PCR quantitation of endogenous TRPM2 expressed in adrenal glands and neuroblastoma. RNA was prepared from 11 age-matched normal adrenal gland (A) and 17 primary neuroblastoma samples (N). RT-PCR was performed with primers specific for TRPM2-L (A) or TRPM2-S (B) and quantitated as described under “Experimental Procedures.” C and D, statistical differences in TRPM2-L and TRPM2-S mRNA between adrenal gland tissue (11 samples) and neuroblastoma (17 tumor samples) were analyzed with Student's t test. *, p < 0.003 for TRPM2-L and p < 0.05 for TRPM2-S. Error bars represent ±S.E.
Generation and Characterization of Neuroblastoma SH-SY5Y Cell Lines Stably Expressing TRPM2-L or TRPM2-S
To study the role of TRPM2 isoforms in neuroblastoma, we generated neuroblastoma SH-SY5Y cells stably expressing V5-tagged L, S, or empty vector (V). Expression of transfected TRPM2-L or TRPM2-S was confirmed by Western blotting of lysates with anti-V5 antibody (Fig. 2A). On long exposures, the parent cell line (wild type (WT)), empty vector, and TRPM2-S stably transfected cells were found to express low levels of endogenous TRPM2-L (Fig. 2B) or TRPM2-S (30).
FIGURE 2.

TRPM2 isoform expression and function in SH-SY5Y neuroblastoma cells. SH-SY5Y cells were stably transfected with V (pcDNA3.1/V5-His TOPO), L, or S. The parent WT cell line was also studied. Western blots using whole cell lysates from these cells were probed with anti-V5-HRP (A) or anti-TRPM2-C (B) antibody. Blots were also probed with anti-tubulin antibody as a control for equivalent loading. C, SH-SY5Y cells stably transfected with V or expressing L or S were loaded with Fura-2 AM as described under “Experimental Procedures.” Cells were treated with 250 μm H2O2 for 20 min, and F360/F380 was measured in single cells with digital video imaging at baseline and at 2–5-min intervals after treatment. Peak mean percent increase of F360/F380 above baseline ±S.E. is shown at 20 min. Two experiments were performed, and a total of 27 (V), 27 (S), or 31 (L) single cells were studied in each group. *, p ≤ 0.002. Error bars represent ±S.E.
To show that transfected channels are functional, Fura-2-loaded cells were stimulated with 250 μm H2O2. [Ca2+]i (quantitated as percent increase in F360/F380) was measured with digital video imaging over 20 min. The greatest increase in [Ca2+]i was observed in TRPM2-L-expressing cells, and the lowest increase was observed in cells expressing dominant negative TRPM2-S compared with empty vector (Fig. 2C). This is consistent with previous data that show that S inhibits calcium influx through TRPM2-L (26).
TRPM2 Current in Neuroblastoma Cells
We next characterized cation current in SH-SY5Y cells expressing TRPM2-L, TRPM2-S, or both. Under our experimental conditions, intracellular application of ADPR (300 μm) elicited large inward and outward cation currents in TRPM2-L- but not TRPM2-S-expressing cells (Fig. 3A; p < 0.001). Omission of ADPR in pipette solutions resulted in much smaller currents in L- and S-expressing cells. ADPR-activated currents displayed the characteristic TRPM2 linear I-V relationship with reversal potential close to 0 mV (57, 58). Either extracellular flufenamic acid (0.5 mm) or clotrimazole (50 μm) abolished the current elicited by ADPR in L-expressing cells (data not shown). Coexpression of S with L significantly inhibited TRPM2 current (Fig. 3A; p < 0.001). These characteristics indicate that (i) the ADPR-activated current was mediated by TRPM2-L, (ii) cells expressing TRPM2-S exhibited only leakage currents, and (iii) TRPM2-S significantly inhibited TRPM2-L current. These observations are consistent with the lower increase in [Ca2+]i (nonspecific leak) in cells expressing TRPM2-S exposed to low dose H2O2 (Fig. 2C).
FIGURE 3.
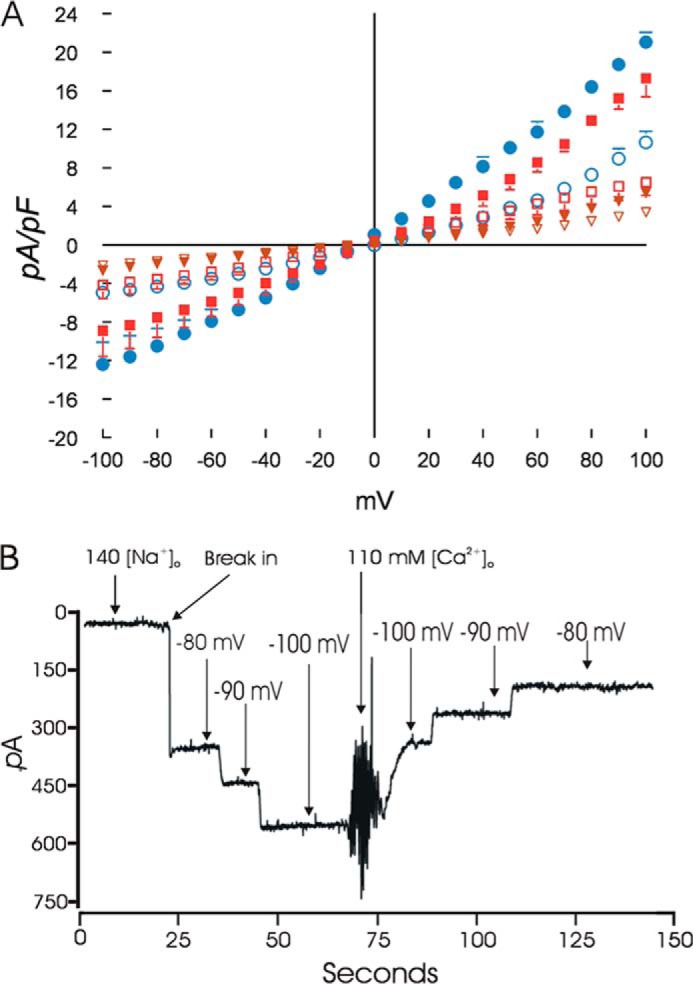
ADPR activates cationic currents in L- but not S-expressing neuroblastoma cells. A, SH-SY5Y cells stably expressing L, S, or both were studied with the standard patch clamp whole cell configuration. The composition of pipette and external solutions and voltage ramp protocols are given under “Experimental Procedures.” I-V relationships of cationic current (means ± S.E.) from L- (●; n = 5), S- (▾; n = 4), or L + S (■; n = 3)-expressing cells stimulated with 300 μm ADPR are shown. Error bars are not shown if they fell within the boundaries of the symbol. Two-way analysis of variance indicated p < 0.0001 for L versus S and L versus L + S. Omission of ADPR in pipette solutions resulted in much smaller currents in L- (○; n = 6), S- (▿; n = 6), and L + S (□; n = 4)-expressing cells. B, TRPM2 currents do not undergo inactivation, and GCa and GNa estimates are shown. SH-SY5Y cells expressing L were held at −80 mV. ADPR was included in the pipette solution to activate TRPM2 channels. With 140 mm [Na+]o, after break-in, a large inward current that did not undergo inactivation was observed. The current showed linear increase with increasing hyperpolarization. Following a change of the medium to one containing 110 mm [Ca2+]o, current became smaller and demonstrated a linear decrease with depolarization from −100 to −80 mV. GCa and GNa were estimated from the slope of the I-V relationship (“Experimental Procedures”). Error bars represent ±S.E. pF, picofarad.
To estimate GCa/GNa, neuroblastoma cells expressing L were held at −80 mV in 140 mm [Na+]o pipette solution containing 300 μm ADPR. After break-in, a large inward current (Na+ ions) was evident (Fig. 3B). There was no inactivation of WT TRPM2 currents. Stepping the voltage from −80 to −100 mV resulted in linear increases in Na+ current (slope, 8.86 ± 0.74 pA/mV; n = 6). Changing to medium containing 110 mm [Ca2+]o decreased inward current (Ca2+ ions; slope, 6.36 ± 0.39 pA/mV; n = 6). GCa/GNa for TRPM2-L in neuroblastoma cells is 0.76 ± 0.12 (n = 6). This compares favorably with GCa/GNa for TRPM2 channels in WT mouse cardiac ventricular myocytes (59).
In SH-SY5Y cells expressing both L and S, both Na+ conductance (4.36 ± 1.25 pA/mV; n = 4) and Ca2+ conductance (3.06 ± 1.06 pA/mV; n = 4) were smaller compared with cells expressing L alone, consistent with S inhibiting cationic currents mediated by L channels. However, the relative conductance (GCa/GNa, 0.74 ± 0.13) was not affected by co-expression of S.
TRPM2 Subcellular Localization
Because of the recent report that TRPC3 is located on the mitochondrial membrane (60), subcellular localization of TRPM2 was examined. Using subcellular fractionation, we determined that endogenous TRPM2-L was primarily located in the plasma membrane/ER fraction compared with the cytoplasmic or mitochondrial fraction (data not shown). To further distinguish plasma membrane/ER from mitochondrial localization, we performed three-dimensional confocal microscopy with SH-SY5Y cells transduced with lentivirus expressing GFP-tagged TRPM2-L or TRPM2-S isoform with endoplasmic reticulum identified by DsRed-ER and mitochondria identified by staining with antibody to Tom20 (Fig. 4). Results were consistent with fractionation studies and showed that in these cells although TRPM2-L and TRPM2-S are expressed on the plasma membrane both are also found in the cytoplasm. They strongly associate with ER compared with their association with mitochondria (Fig. 4). RP values for coexpression of both TRPM2-L and TRPM2-S with ER were found to be 0.80 ± 0.05 and 0.71 ± 0.14, respectively, whereas RP values with mitochondria were found to be 0.53 ± 0.09 and 0.4 ± 0.17, respectively. A closer look at their three-dimensionally rendered sectional views (Fig. 4, M–P and Q–T) of cells expressing GFP-TRPM2 also show predominately colocalized TRPM2/ER voxels (yellow color), whereas non-colocalized mitochondrial voxels (blue color) are still visible. Thus both three-dimensionally rendered visualization and subsequent quantitative measures are the direct indication of the preferential association of TRPM2 with ER regardless of the isoform.
FIGURE 4.

Subcellular localization of TRPM2 was determined with three-dimensional confocal microscopy. Confocal microscopy was performed with SH-SY5Y cells transduced with lentivirus expressing GFP-tagged TRPM2-L or TRPM2-S isoforms (green) and DsRed-ER to identify endoplasmic reticulum (red). Mitochondria were detected with antibody to Tom20 and appropriate secondary antibody (blue). A–F, sectional views (both xy and xz planes) of TRPM2-L (A), ER (B), mitochondria (C), and the overlays of TRPM2-L with ER (D), TRPM2-L with mitochondria (E), and TRPM2-L with ER and mitochondria (F). G–L, sectional views (both xy and xz planes) of TRPM2-S (G), ER (H), mitochondria (I), and the overlays of TRPM2-S with ER (J), TRPM2-S with mitochondria (K), and TRPM2-S with ER and mitochondria (L). M–P, three-dimensionally rendered views of the overlays of TRPM2-L with ER (M), TRPM2-L with mitochondria (N), TRPM2-L with ER and mitochondria (O), and a colocalization plot of the overlay of TRPM2-L with ER (P). Q–T, three-dimensionally rendered views of the overlays of TRPM2-S with ER (Q), TRPM2-S with mitochondria (R), TRPM2-S with ER and mitochondria (S), and three-dimensionally rendered sectional xz views of the respective overlays (T). The broken line in S shows the xz position of overlays shown in T. The measured RP, a measure of colocalized fluorescence signal intensities, is shown in the graph (U). The overlays of three-dimensional images as well as the quantitatively determined RP indicated that although TRPM2-L and TRPM2-S are expressed in the plasma membrane both are also expressed in the cytoplasm and are relatively strongly associated with the ER compared with their association with mitochondria (MITO) (p < 0.05). Scale bars, 10 μm. Error bars represent ±S.E.
TRPM2-S Inhibits Growth of Neuroblastoma Xenografts
Expression of endogenous TRPM2-L and -S in SH-SY5Y cells is low in SH-SY5Y cells. To evaluate effects of TRPM2 on tumor growth, athymic female mice were injected in one flank with SH-SY5Y cells stably expressing L, S, or empty vector, and tumor size was measured twice weekly for 6–7 weeks. In five experiments, tumors from cells expressing dominant negative S demonstrated significantly reduced tumor volume (Fig. 5A) and tumor weights (Fig. 5B) compared with cells expressing L or V. Differences in tumor sizes were visually apparent (Fig. 5C). Reduced PARP and procaspase 3 protein and increased caspase 3 cleavage products were detected in tumors expressing TRPM2-S (Fig. 5D). These results demonstrate that TRPM2-S-expressing cells have reduced cell growth and viability in vivo.
FIGURE 5.

TRPM2-S expression significantly reduces growth of SH-SY5Y xenografts. Athymic female mice were injected with 1.5 × 107 SH-SY5Y cells stably transfected with L, S, or V, and tumor volume (A) and weight (B) were measured at 6 weeks (Wk). Five experiments were performed, and a representative experiment (n = 8 in each group) is shown. *, p ≤ 0.01. Photographs of representative tumors in all groups (V, L, and S) are shown in C for visual comparison. D, Western blotting was performed on two experiments to examine PARP and caspase 3 expression and cleavage products (Clvd). A representative blot from one experiment is shown. Scale bars, 1 cm. Error bars represent ±S.E.
HIF-1α Is Down-regulated in TRPM2-S-expressing Xenografts
To determine the mechanisms responsible for the reduced growth of TRPM2-S-expressing tumors, we performed global label-free proteomic analysis using GeLC-MS/MS technology. Comparing xenograft tumors expressing TRPM2-L with those expressing TRPM2-S, a number of proteins transcriptionally regulated by HIF-1α were differentially expressed between these two groups. Mitochondrial transcription factor A, which is inhibited by HIF-1α, was up-regulated in TRPM2-S-expressing cells in proteomic analysis, whereas a number of proteins whose transcription is increased by HIF-1α were down-regulated (Fig. 6, A and B). HIF-1α expression was predicted to be reduced in TRPM2-S-expressing cells but could not be directly measured with MS/MS.
FIGURE 6.
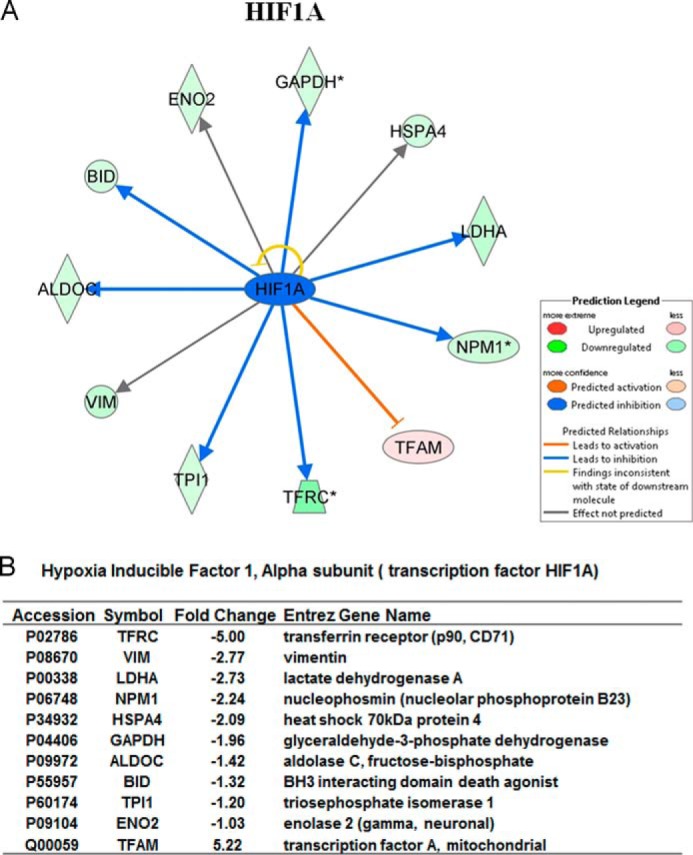
Reduced HIF-1α expression in TRPM2-S-expressing cells determined by proteomic analysis. Differentially expressed proteins between TRPM2-L- and TRPM2-S-expressing xenografts (n = 5 each) were determined using GeLC-MS/MS. A, the HIF-1α network was generated according to Ingenuity pathway knowledge criteria. Pink, significantly up-regulated proteins; green, significantly down-regulated proteins; blue, predicted reduction in expression. B, analysis of -fold change and accession numbers of differentially expressed proteins in the HIF-1α signaling pathway. * indicates that proteins were identified multiple times in different sample fractions. Only the highest expression value was included in ingenuity pathway analysis.
To validate the results of proteomic analysis, Western blotting was performed on lysates of xenograft tumors harvested 6–7 weeks after SH-SY5Y cell injection. Western blotting was performed in two experiments with similar results (Fig. 7). Transfected V5-tagged TRPM2-L and TRPM2-S expression was confirmed by probing with anti-V5 antibody. As predicted by proteomic analysis, a statistically significant increase in expression of the transcriptional regulators HIF-1α and HIF-2α was observed in tumors expressing endogenous (V) or transfected L compared with tumors from cells transfected with S. The cells expressing endogenous or transfected TRPM2-L and more HIF-1α and -2α formed larger tumors (Fig. 5), consistent with reports that HIF-2α promotes a more aggressive tumor phenotype in patients with neuroblastoma (61, 62). Western blotting also confirmed modulation of expression of proteins downstream of HIF-1α identified by proteomic analysis. This included increased expression of mitochondrial transcription factor A and reduced expression of LDHA and enolase 2. Reduced expression of several target genes downstream of HIF-1/2α was also demonstrated in tumors expressing TRPM2-S including antioxidants (FOXO3a) (63, 64), VEGF (angiogenesis), proteins involved in inhibition of mitochondrial O2 consumption and ROS production (BNIP3 and NDUFA4L2), and mitochondrial electron transport chain activity (cytochrome oxidase 4.1/4.2 in complex IV) (38–41, 65, 66). The expression level of these proteins was not measured by proteomic analysis because they were not detectable by MS/MS. These data suggest that the decrease in HIF-1/2α expression plays a role in reduced survival in cells expressing TRPM2-S.
FIGURE 7.

Tumors expressing TRPM2-L demonstrate higher expression of HIF-1α and HIF-2α and downstream signaling proteins. Lysates were prepared from tumors xenografts harvested 6 weeks after injection with SH-SY5Y cells stably expressing V, L, or S. Western blotting was performed, and blots were probed with anti-V5-HRP, anti-HIF-1α, anti-HIF-2α, and antibodies to the downstream target genes enolase 2 (ENO2), LDHA, FOXO3a, VEGF, BNIP3, NDUFA4L2, COX4.1/4.2, and mitochondrial transcription factor A (TFAM). C1ORF43 was probed to confirm equivalent loading. Western blotting was performed on two xenograft experiments (n = 6 for each TRPM2 isoform in each experiment), and results were similar. Representative Western blots from one xenograft experiment are shown. Protein expression was quantitated by densitometry, and the mean ± S.E. of protein/normalizing control for V, L, and S was calculated. *, p ≤ 0.05. Error bars represent ±S.E.
HIF-1/2α Transcription Is Decreased and VHL Levels Are Increased in TRPM2-S- Versus TRPM2-L-expressing SH-SY5Y Cells
To determine whether decreased HIF-1/2α expression in TRPM2-S resulted from reduced transcription, HIF-1/2α mRNA levels in SH-SY5Y cells expressing different TRPM2 isoforms were quantitated by RT-PCR. HIF-1 and -2α mRNA levels were significantly reduced in TRPM2-S-expressing cells compared with cells expressing TRPM2-L (Fig. 8A). HIF-2α mRNA levels were also significantly reduced in TRPM2-S-expressing cells compared with empty vector. von Hippel-Lindau E3 ligase expression was quantitated with Western blotting of tumor xenografts. von Hippel-Lindau E3 ligase levels were significantly increased in TRPM2-S-expressing cells (Fig. 8B). These data suggest that reduced expression of HIF-1/2α following TRPM2-S expression is mediated through both reduced transcription and enhanced degradation through increased levels of von Hippel-Lindau E3 ligase.
FIGURE 8.

HIF-1/2α transcription is reduced and VHL expression is increased in TRPM2-S-expressing cells. A, HIF-1 or -2α mRNA expression was quantitated by RT-PCR in five xenograft tumors generated from SH-SY5Y cells expressing empty vector, TRPM2-L, or TRPM2-S. Expression of each mRNA was analyzed in each tumor in six replicates, and the mean ± S.E. for each group (n = 30) is shown. *, p ≤ 0.001. B, Western blotting was performed with anti-VHL and anti-C1ORF43 antibodies in one xenograft experiment (V, n = 4; L and S, n = 5), and results of densitometry analysis are shown (*, p < 0.05). Error bars represent ±S.E.
TRPM2-S-expressing Cells Have Decreased Autophagy/Mitophagy
In xenograft tumors harvested 6 weeks after injection of SH-SY5Y cells expressing vector, TRPM2-L, or TRPM2-S, mtDNA levels were determined using PCR. Mitochondrial DNA (12 S/18 S ratio) was significantly greater in tumors expressing TRPM2-S compared with tumors expressing TRPM2-L or empty vector (Fig. 9A). To examine differences in autophagy/mitophagy in cells expressing different TRPM2 isoforms, we performed Western blotting of Hsp60 and Tom20 in xenograft tumors from SH-SY5Y cells expressing different TRPM2 isoforms. Hsp60 accumulates in mitochondria if not removed by mitophagy, and Tom20 is a mitochondrial translocase receptor that is reduced in mitophagy. Both Hsp60 and Tom20 were significantly increased in TRPM2-S-expressing cells (Fig. 9B). These data suggest that autophagy/mitophagy is reduced in TRPM2-S-expressing cells, contributing to accumulation of mtDNA (Fig. 9A).
FIGURE 9.

Mitophagy is reduced in TRPM2-S-expressing cells. A, mtDNA (Mito DNA) analysis was performed on five xenograft tumors from cells stably expressing TRPM2-L, TRPM2-S, or empty vector. Each sample was studied in triplicate, and results of 12 S DNA quantification for each analysis/mean 18 S genomic DNA analysis for each tumor sample were determined. The mean ± S.E. for each tumor group is shown. *, p < 0.01. B, Western blotting was performed on lysates from xenograft tumors harvested 6 weeks after injection with SH-SY5Y cells stably expressing V, L, or S. Two xenograft experiments were studied with similar results, and representative Western blot results of one xenograft are shown. Results of densitometry analysis of one of the experiments (V, n = 4; L and S, n = 5) are shown (*, p < 0.03). C and D, loss or gain of HIF-1/2α function. SH-SY5Y cells stably expressing TRPM2-L or -S and scrambled (SCR) shRNA or shRNA targeted to HIF-1α or HIF-2α (C) or expressing pBabe vector or HIF-1 or -2α with mutations inhibiting degradation in pBabe (D) were harvested in exponential culture. Western blotting was performed, and blots were probed with antibodies to HIF-1α, HIF-2α, BNIP3, and C1ORF43. E, ultrastructural studies of neuroblastoma xenografts expressing different TRPM2 isoforms. Tumor sections from mice injected with WT SH-SY5Y cells or cells expressing TRPM2-L examined with electron microscopy (1200× and 3000×) show normal elongated mitochondria and cristae. Tumors expressing TRPM2-S show a high percentage of dysmorphic mitochondria with a round, swollen appearance and degenerated cristae. A representative picture of 40 (WT) or 70 fields (L and S) photographed at 3000× for xenograft tumors from each isoform (WT, n = 4; L and S, n = 7 tumors) is shown. Error bars represent ±S.E. Scale bars, 1 μm.
The role of HIF-1/2α in modulation of expression of the mitochondrial target protein BNIP3, which has a role in a mitophagy, was then examined in gain and loss of function studies. Reduction in HIF-1/2α with targeted shRNA resulted in a decrease in BNIP3 expression in TRPM2-L- and TRPM2-S-expressing cells (Fig. 9C). Overexpression of HIF-1/2α resulted in increased expression of BNIP3. These data suggest that reduced HIF-1/2α expression in TRPM2-S-expressing cells has an important role in modulating mitophagy.
We assessed mitochondrial morphology with electron microscopy in tumors from wild type SH-SY5Y cells or cells expressing L or S. Compared with WT cells or cells expressing TRPM2-L, which had typical elongated mitochondria with normal cristae, cells expressing TRPM2-S showed an abundance of dysmorphic mitochondria with a swollen appearance and degenerated cristae (Fig. 9E).
Reduced Mitochondrial Membrane Potential and Ca2+ Uptake in Neuroblastoma Cells Expressing TRPM2-S
Because cells expressing S produce more ROS (30) and NDUFA4L2 expression was reduced in TRPM2-S-expressing cells (Fig. 7), mitochondrial function was evaluated in SH-SY5Y cells expressing different TRPM2 isoforms. Cells stably expressing empty vector, TRPM2-L, TRPM2-S, or both L and S were studied. We measured Δψm and mitochondrial Ca2+ uptake in a permeabilized cell system that mimics the cytosolic milieu while inhibiting ER clearance with the addition of thapsigargin. Compared with cells expressing either L or vector, after six Ca2+ pulses (orange arrows), Δψm was severely impaired in cells expressing S or both L and S (Fig. 10, A and B). Because Δψm is vulnerable to Ca2+ challenge in S- and L + S-expressing cells, total mitochondrial [Ca2+] uptake (measured as the amount of mitochondrial Ca2+ released with the uncoupler carbonyl cyanide m-chlorophenylhydrazone) after six pulses of Ca2+ was assessed. Compared with L-expressing cells, mitochondrial Ca2+ uptake was almost absent in neuroblastoma cells expressing either S or both L and S (Fig. 10, C and D). Fig. 10D shows the lack of mitochondrial calcium uptake in TRPM2-S- and TRPM2-S plus TRPM2-L-expressing cells, and Fig. 10C shows the increase in extramitochondrial Ca2+ in these cells. These results demonstrate that in TRPM2-S-expressing cells in which NDUFA4L2 is decreased, Δψm is compromised, and mitochondrial Ca2+ uptake is decreased.
FIGURE 10.

Δψm and mitochondrial Ca2+ uptake are lower in neuroblastoma cells expressing TRPM2-S. SH-SY5Y cells stably expressing TRPM2-L, TRPM2-S, both TRPM2-L and TRPM2-S, or empty vector were studied. Cells were permeabilized with digitonin and supplemented with succinate. A, the ratiometric indicator JC-1 was added at 20 s and used to monitor Δψm. Units of Δψm represent the ratio of the fluorescence intensity of the JC-1 oligomeric form to that of the monomeric form. Black arrows indicate addition of digitonin or the mitochondrial uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP) (2 μm), and orange arrows indicate addition of Ca2+ (10 μm), respectively. Tg, thapsigargin. B, normalized Δψm after Ca2+ pulses but before carbonyl cyanide m-chlorophenylhydrazone addition. C, the ratiometric dye Fura-FF was added at 0 s and used to monitor extramitochondrial Ca2+. Repeated pulses of Ca2+ (10 μm) were added as indicated (orange arrows). The cytosolic Ca2+ clearance rate after the first Ca2+ pulse was measured. D, summary of cytosolic Ca2+ clearance (mitochondrial Ca2+ uptake) rates. **, p < 0.01; ***, p < 0.001; n = 3–4. Error bars represent ±S.E.
Mitochondrial O2 Consumption Is Lower in Neuroblastoma Cells Expressing TRPM2-S
After observing that mitochondrial Δψm and Ca2+ uptake were impaired in SH-SY5Y neuroblastoma cells transfected with TRPM2-S (Fig. 10), we examined whether mitochondrial complex activities were altered in these cells. Both basal and maximal oxygen consumption rates (OCRs) were significantly reduced in S-expressing cells compared with cells expressing L or vector (Fig. 11). These results show that when TRPM2-S is expressed and NDUFA4L2 is depleted mitochondrial bioenergetics are significantly impaired in neuroblastoma.
FIGURE 11.
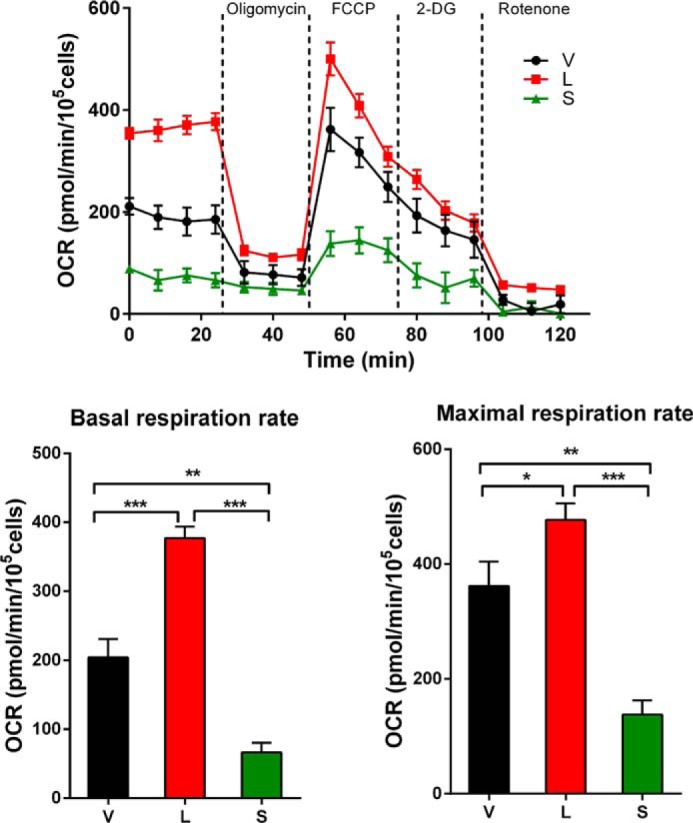
O2 consumption is lowest in neuroblastoma cells expressing TRPM2-S. The O2 consumption rate was measured in SH-SY5Y cells transfected to stably express L, S, or V. Top, after basal OCR was obtained, oligomycin (1 μm) was added to inhibit F0F1-ATPase (complex V). The uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (1 μm) was then added, and maximal OCR was measured. 2-Deoxyglucose (2-DG) (100 μm) was added to inhibit the glycolytic pathway, and then rotenone (1 μm) was added to inhibit NADH dehydrogenase (complex I), respectively. Three experiments were done, and a representative experiment is shown. Each point in the traces represents the average of five different wells. A summary of basal (bottom left) and maximal (bottom right) OCR of three groups of cells (n = 5 each) is shown. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Error bars represent ±S.E.
Inhibition of TRPM2-L Decreases Survival after Doxorubicin Treatment in Vitro
Because doxorubicin modulates cell viability through ROS production (67) and TRPM2-S-expressing cells were shown to generate increased ROS (30), we examined whether cells expressing TRPM2-S have increased susceptibility to doxorubicin-induced cell death. SH-SY5Y neuroblastoma cells stably expressing empty vector, TRPM2-L, or TRPM2-S were exposed to 0.033, 0.1, or 0.3 μm doxorubicin for 24 h. Cell viability was reduced in all three cell lines in a dose- and time-dependent manner. Cells expressing TRPM2-S had significantly reduced viability after doxorubicin treatment compared with cells expressing TRPM2-L or empty vector (Fig. 12, A and B). Consistent with viability assays, TRPM2-S-expressing cells showed increased cleavage of caspase 3, caspase 7, and PARP compared with TRPM2-L- or empty vector-expressing cells (Fig. 12C). As an alternative approach, TRPM2 function was inhibited with the TRPM2 inhibitor clotrimazole. Cells were pretreated with vehicle (0.1% DMSO) or clotrimazole (10 μm) (Fig. 12D) and exposed to 0.1 or 0.3 μm doxorubicin. These experiments demonstrated that inhibition of TRPM2 (exogenous or endogenous) with clotrimazole also significantly enhances sensitivity to doxorubicin.
FIGURE 12.

TRPM2-S expression or inhibition of TRPM2-L with clotrimazole reduces cell viability after treatment with doxorubicin. A and B, SH-SY5Y cells stably transfected with V, L, or S were treated with 0.033, 0.1, or 0.3 μm doxorubicin (Doxo) for 24 h. Cell viability was determined by trypan blue exclusion (A) or XTT assay (B). Mean ± S.E. of two experiments done in triplicate (A) or four experiments each done in six replicates (B) is shown. *, p ≤ 0.05. C, increased cell death in TRPM2-S-expressing cells was confirmed by examining cleavage (Clvd) of PARP, caspase 3, and caspase 7 on Western blots. Representative results of three Western blots are shown. D, SH-SY5Y cells stably expressing V or L were pretreated with or without 10 μm clotrimazole (Clo) for 10 min and then treated with doxorubicin (0.1 or 0.3 μm) or vehicle with or without clotrimazole for 24 h. Cell viability assessed with the XTT assay was significantly reduced in cells pretreated with clotrimazole. A representative result of four experiments is shown. *, p < 0.05. Error bars represent ±S.E.
TRPM2-S Influences Cell Viability through Modulation of HIF-1α and HIF-2α
To determine whether modulation of HIF-1α or HIF-2α expression has a functional role in regulating proliferation of cells expressing different TRPM2 isoforms or their susceptibility to doxorubicin, we created cell lines with loss or gain of HIF-1α or -2α function. Reduction of HIF-1α or -2α levels with shRNAs (Fig. 13, A and C) or enhancement of HIF-1α or -2α with constructs (Fig. 14, A and C) was demonstrated by Western blotting (insets). Proliferation of SH-SY5Y cells expressing TRPM2-L was significantly reduced following HIF-1α (Fig. 13A) or HIF-2α (Fig. 13C) depletion.
FIGURE 13.

Loss of HIF-1α or HIF-2α function modulates survival of TRPM2-L- and TRPM2-S-expressing cells. A and B, SH-SY5Y cells stably expressing TRPM2-L or TRPM2-S were transfected with shRNA targeted to HIF-1α or scrambled shRNAs as described under “Experimental Procedures”. A, cell proliferation was measured by XTT assay, and results are expressed as percentage of XTT at 24 h after cell seeding. Two experiments were performed with six replicates each, and the mean ± S.E. of one representative experiment is shown. *, p < 0.05. The inset shows a Western blot for cells transfected with scrambled shRNA (Scr) or shRNA targeted to HIF-1α. B, these cells were treated with 0.1 or 0.3 μm doxorubicin (Doxo) for 48 h, and viability was determined by XTT assay. Three experiments were performed with four replicates each, and the mean ± S.E. of one representative experiment is shown. *, p < 0.05. C and D, SH-SY5Y cells stably expressing TRPM2-L or TRPM2-S were transfected with shRNA targeted to HIF-2α or scrambled shRNAs as described under “Experimental Procedures.” C, cell proliferation was measured by XTT assay, and results are expressed as percentage of XTT after cell seeding. Three experiments were performed with six replicates each, and the mean ± S.E. of one representative experiment is shown. *, p < 0.05. The inset for TRPM2-L shows a Western blot for cells transfected with scrambled shRNA (Scr) or shRNAs targeted to HIF-2α. D, these cells were also treated with 0.1 or 0.3 μm doxorubicin for 48 h, and viability was determined by XTT assay. Three experiments were performed with four replicates each, and the mean ± S.E. of one representative experiment is shown. *, p < 0.05. Error bars represent ±S.E.
FIGURE 14.

Gain of HIF-1α or HIF-2α function modulates survival of TRPM2-L- and TRPM2-S-expressing cells. SH-SY5Y cells stably expressing TRPM2-L or TRPM2-S were transfected with pBabe (empty vector), HIF-1α (A and B), or HIF-2α (C and D) mutants that are nondegradable as described under “Experimental Procedures.” A and C, cell proliferation was measured by XTT assay, and results are expressed as percentage of XTT after cell seeding. Three experiments were performed with six replicates each, and the mean ± S.E. of one representative experiment is shown. *, p < 0.05. Representative Western blots demonstrating increased expression of HIF-1α (A) or HIF-2α (C) compared with transfection control are shown in insets. B and D, these cells were treated with 0.1 or 0.3 μm doxorubicin (Doxo) for 24 h, and viability was determined by XTT assay. Three experiments were performed, and representative results are shown. *, p < 0.05. Error bars represent ±S.E.
Reduction of HIF-1α had no significant effect on proliferation of TRPM2-S-expressing cells, and the decrease in HIF-2α had a modest effect likely because levels were already reduced. When cells depleted of HIF-1α or HIF-2α were treated with 0.1 or 0.3 μm doxorubicin, the viability of TRPM2-L-expressing cells was significantly reduced (Fig. 13, B and D). For TRPM2-L-expressing cells depleted of HIF-2α, the greatest reduction was observed at longer time periods after treatment (72 h; data not shown). Viability of TRPM2-S-expressing cells after doxorubicin treatment was significantly further reduced by both HIF-1 and -2α depletion.
SH-SY5Y cells expressing TRPM2-L or TRPM2-S were transfected with HIF-1α or HIF-2α constructs mutated to reduce their degradation. Increased expression of HIF-1α resulted in significantly increased proliferation in both TRPM2-L- and TRPM2-S-expressing cells (Fig. 14A). Increased expression of HIF-2α also resulted in a significant increase in proliferation of TRPM2-S-expressing cells, but enhanced levels of HIF-2α in TRPM2-L-expressing cells consistently resulted in decreased cell proliferation (Fig. 14C). These data suggest that an optimal range of HIF-2α levels may play an important role in regulating cell proliferation and that very high levels are inhibitory. Increased expression of HIF-1α or HIF-2α significantly enhanced the viability of TRPM2-S-expressing cells after treatment with 0.1 or 0.3 μm doxorubicin but had minimal effect on the viability of TRPM2-L-expressing cells (Fig. 14, B and D). These data demonstrate that HIF-1α and HIF-2α modulation following TRPM2-L and TRPM2-S expression plays an important role in regulation of proliferation and viability.
Neuroblastoma Cells Expressing TRPM2-S Have Lower ATP Levels
Because TRPM2-L-expressing cells demonstrated increased tumor growth, current function, and OCR compared with cells transfected to express TRPM2-S, we examined whether cells expressing different TRPM2 isoforms differ in bioenergetic capacity. ATP levels were measured in SH-SY5Y cells stably expressing vector, S, or L at 8–24 h after doxorubicin (0.5 μm) treatment. ATP levels progressively declined with time after doxorubicin treatment in neuroblastoma cells expressing S or vector (Fig. 15A). ATP levels in cells expressing TRPM2-S were significantly less at 24 h than in cells transfected with empty vector or TRPM2-L. By sharp contrast, ATP levels remained stable after doxorubicin exposure in cells expressing L. These findings show that enhanced expression of TRPM2-L helps maintain energy production after doxorubicin treatment. ATP levels were also measured in lysates from neuroblastoma xenografts expressing vector, S, or L 24 h after doxorubicin treatment in vivo. The lowest ATP levels were measured in xenografts expressing TRPM2-S (Fig. 15B) that had low levels of NDUFA4L2. Lower ATP production in neuroblastoma cells expressing S correlates with the significantly smaller size of tumors expressing S.
FIGURE 15.
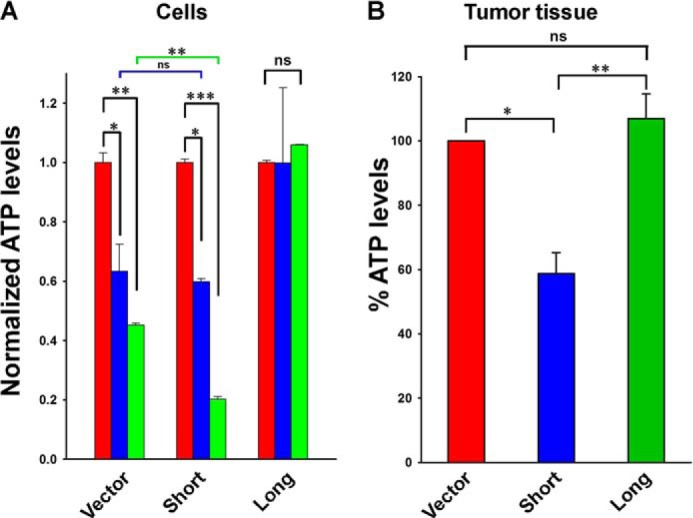
ATP production is reduced in TRPM2-S-expressing cells. A, SH-SY5Y cells stably expressing empty vector, TRPM2-L, or TRPM2-S were treated for 8 (blue) or 24 h (green) with 0.5 μm doxorubicin. ATP levels were measured with the Cell Titer-Glo assay, and results standardized to baseline measurements in each group are shown. B, samples from xenograft tumors expressing empty vector, TRPM2-L, or TRPM2-S were harvested 24 h after mice were treated with 3 mg/kg doxorubicin. Samples were homogenized, and ATP levels were measured. Mean ± S.E. is shown. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant; n = 3. Error bars represent ±S.E.
DISCUSSION
TRPM2 is widely recognized as an ion channel with an important role in cell survival in a number of physiological and pathological conditions including ischemic injury and oxidative stress (30, 32, 33, 59). TRPM2 isoforms are also highly expressed in a number of malignancies including neuroblastoma (30), melanoma, lung (25), and breast cancer.4 TRPM2 has been shown to have an important role in survival of neuroblastoma cells following oxidative stress, and TRPM2-L confers protection of neuroblastoma from oxidative stress-induced cell death through FOXO3a (30). Here, using a mouse xenograft model, we showed that neuroblastoma tumor growth in vivo is significantly diminished by expression of dominant negative TRPM2-S and that the mechanism involves reduced HIF-1α and HIF-2α expression, mitochondrial protein expression, and mitophagy. Furthermore, expression of TRPM2-S enhanced susceptibility to doxorubicin. These studies have high significance and clinical impact because they demonstrate that TRPM2 channels may be a novel target to modulate tumor growth and enhance sensitivity to cancer therapeutics.
The first important finding of this study is the characterization of TRPM2 current in neuroblastoma cells. In these cells, ADPR activates current through TRPM2-L but not TRPM2-S, and TRPM2-S inhibits current through TRPM2-L, consistent with previous reports utilizing measurement of the intracellular calcium concentration (26). Flufenamic acid and clotrimazole also inhibited current through TRPM2-L. We determined that TRPM2 conductance to Ca2+ is ≅76% that of Na+ in neuroblastoma cells (Fig. 3B). Co-expression of TRPM2-S did not affect the relative Ca2+/Na+ conductance of TRPM2-L. These data demonstrate that decreased tumor growth in cells expressing TRPM2-S is associated with decreased calcium current in these cells.
Here, we showed with RT-PCR that the increased expression of TRPM2 isoforms in neuroblastoma involved transcriptional regulation; large increases in TRPM2-L mRNA were demonstrated. The TRPM2 promoter region has a number of methylation sites that can be transcriptionally regulated in malignancies (68). TRPM2 is activated by oxidative stress (10, 30). However, although early reports supported the paradigm that increased calcium entry following TRPM2 activation contributes to cell death (69–71), more recent publications demonstrate that TRPM2 protects the lung from endotoxin-induced injury (32) and the heart from ischemia-reperfusion injury (33). In addition, TRPM2 is implicated to be beneficial to Guamanian patients with amyotrophic lateral sclerosis and Parkinsonism dementia (58) and protects pyramidal neurons (31) and neuroblastoma cell lines (30) from oxidant injury. The finding that TRPM2 is highly expressed in a number of cancers suggests that it may have a role in tumor growth and chemotherapy sensitivity.
To examine the function of TRPM2 isoforms in tumor formation, we utilized the neuroblastoma cell line SH-SY5Y, which expresses low levels of endogenous TRPM2-L and TRPM2-S. The second major finding of this report is that tumor growth was significantly reduced in cells expressing TRPM2-S compared with cells expressing TRPM2-L or vector (Fig. 5). To identify the molecular pathways involved in modulation of tumor growth, we utilized GeLC-MS/MS to assess TRPM2 isoform-induced differential expression of the whole proteome of xenograft tumors transfected to express TRPM2-S or TRPM2-L. Multiple members of the HIF-1α signaling pathway showed significant down-regulation in TRPM2-S-expressing cells (Fig. 6). Down-regulation of HIF-1α and HIF-2α in TRPM2-S-expressing cells compared with cells expressing TRPM2-L or empty vector was confirmed by Western blotting of xenografts (Fig. 7).
HIF-1/2α are up-regulated in a number of cancers and are important in tumor progression. In neuroblastoma, increased expression of HIF-2α is associated with disseminated disease and a poor outcome (61, 62, 72). The third major finding of this report is that expression of HIF-1/2α and downstream targets were modulated by TRPM2 isoforms. This suggests that modulation of TRPM2 function may be a mechanism through which HIF-1/2α expression may be targeted in malignancy. The mechanism of HIF-1/2α modulation in SH-SY5Y cells involved both reduced HIF-1/2α transcription and increased VHL expression (Fig. 8). The role of calcium in regulation of HIF-1/2α expression is complex, but the finding here of increased HIF-1/2α mRNA in cells expressing TRPM2-L compared with TRPM2-S is consistent with reports that HIF-1α transcription can be induced by Ca2+ ionophores (73). Hypoxia-inducible transcription factors regulate expression of genes involved in glycolysis (LDHA), oxidative stress (FOXO3a), and angiogenesis (VEGF) (38, 39, 42), and this was observed here in TRPM2-expressing cells. Increased expression of the downstream target FOXO3a was observed previously in TRPM2-L-expressing neuroblastoma cells, and findings here confirmed that this is a pathway through which TRPM2-L may protect against moderate oxidative stress-induced death by increasing superoxide dismutase 2 (30). In tumors expressing TRPM2-S, decreased expression of HIF-1α and HIF-2α and downstream target genes FOXO3a, LDHA, and VEGF provides one mechanism for the reduced growth of xenografts expressing TRPM2-S and is consistent with the report that HIF-2α promotes a more aggressive tumor phenotype in patients with neuroblastoma.
The fourth major finding of this study is that proteins expressed in the mitochondria and regulated by HIF-1α and -2α including BNIP3 (65), NDUFA4L2 (74), COX4.1, and COX4.2 (40) were significantly down-regulated in these TRPM2-S-expressing cells. BNIP3 has been reported to function either as a proapoptotic or a prosurvival factor (41, 65), and recent evidence shows that ablation of BNIP3 triggers cell death (41). In cardiac cells, inhibition of BNIP3 leads to accumulation of dysfunctional mitochondria and induction of cardiac dysfunction (65). O2 consumption and ATP levels were significantly decreased in mouse embryonic fibroblasts in which BNIP3 was ablated (40), similar to the decrease in mitochondrial function and in tumor growth observed here in neuroblastoma cells expressing TRPM2-S. BNIP3 levels can be regulated both by HIF-1α and FOXO3a, and FOXO3a was shown previously to be down-regulated in TRPM2-S-expressing cells (30). The combined deficiency of transcription factors HIF-1/2α and FOXO3a may contribute to decreased expression of BNIP3. Mitochondrial homeostasis is dynamically regulated by the processes of mitophagy and mitochondrial biogenesis. Although it is possible that the increased levels of mitochondrial DNA and proteins Tom20 and Hsp60 in TRPM2-S-expressing cells could be related to increased mitochondrial biogenesis, the observation of decreased BNIP3 protein levels and accumulation of damaged mitochondria suggests that mitophagy is impaired in TRPM2-S-expressing cells and that BNIP3 has an important role. The finding that HIF-1/2α has an important role in BNIP3 modulation suggests a pathway through which TRPM2 is involved.
NDUFA4L2 is a component of NADH dehydrogenase, which is involved in regulation of complex I activity (39). NDUFA4L2 is reduced in these TRPM2-S-expressing cells and in xenografts, implicating complex I dysfunction in these cells and consistent with the report that hypoxic NDUFA4L2-silenced HeLa cells demonstrated profound inhibition of cell proliferation and significantly higher ROS accumulation compared with control cells (39). We generated three SH-SY5Y clones stably expressing TRPM2-S with G418 selection, and in all three, expression of NDUFA4L2 was significantly reduced. Decreased NDUFA4L2 may be a key component of the decrease in mitochondrial function and cell viability observed in these cells. HIF-1α coordinates a switch in expression of the mitochondrial electron transport chain complex IV cytochrome oxidase subunit from COX4.1 to COX4.2, which is more efficient in hypoxia (40). Interestingly, in neuroblastoma xenografts studied here, both subunits were decreased in TRPM2-S-expressing cells (Fig. 7). Decreased expression of COX4.1 and COX4.2 may contribute to reduced ATP production and increased production of H2O2 (66), and lower ATP production may contribute to the reduced growth of tumors expressing TRPM2-S.
We examined the role of TRPM2 in chemosensitivity by inhibiting TRPM2-L function through expression of dominant negative splice variant TRPM2-S or treatment with the TRPM2 inhibitor clotrimazole. Expression of TRPM2-S or clotrimazole treatment significantly increased the sensitivity of SH-SY5Y cells to doxorubicin, suggesting that inhibition of TRPM2-L in cancer may be a therapeutic approach to enhance the response to doxorubicin when used in combination with chemotherapy. We modulated HIF-1α and -2α expression in TRPM2-L- and TRPM2-S-expressing cells to determine their role in cell proliferation and the sensitivity of cells expressing TRPM2 isoforms to doxorubicin. TRPM2-S-expressing cells with gain of HIF-1α or -2α function showed enhanced proliferation, and TRPM2-L-expressing cells with loss of HIF-1α or -2α function showed reduced proliferation. These data suggest that the changes in HIF-1α or -2α expression observed in cells expressing different TRPM2 isoforms may play an important role in cell proliferation and viability after doxorubicin treatment.
The mechanisms through which TRPM2 isoforms regulate HIF-1/2α here are not known. Pathways involving Ca2+ influx through TRPM2-L may be involved in modulation of HIF-1/2α expression, leading to enhanced cell survival and mitochondrial function. For example, one mechanism through which Ca2+ influx through TRPM2 could modulate HIF expression is by stabilization of HIF-1α. Calcium influx through TRPM2 can activate calcineurin, which dephosphorylates RACK1, blocking RACK1 dimerization and increasing HIF-α levels by impeding its ubiquitination and degradation (75). HIF-1α translation can also be calcium-regulated by protein kinase C-α (76), and HIF-1α transcriptional activity can be modulated by increased Ca2+ influx (73) or Ca2+/calmodulin kinase-mediated phosphorylation of p300 (77). Other pathways in TRPM2-S-expressing cells not restricted to those mediating calcium entry may be involved in mediating HIF-1/2α expression but have not yet been explored.
Although effects on tumor growth and doxorubicin sensitivity in TRPM2-S-expressing cells may result from involvement of a number of signaling mechanisms, our data support an essential role for the pathway shown in Fig. 16. A critical role for low level mitochondrial Ca2+ uptake in regulation of bioenergetics has been demonstrated recently (78, 79). We have shown that in cells that express TRPM2-S both calcium entry (Figs. 2C and 3A) and mitochondrial calcium uptake (Fig. 10D) are significantly reduced. Although our confocal studies showed that TRPM2 and mitochondria are in close proximity, they could not distinguish whether TRPM2 localization on ER sites is in close contact with mitochondria or represents direct mitochondria association. In this context, recent studies have demonstrated a close spatial relationship between ER and mitochondria (ER-mitochondrial tethering) (80, 81). It is possible that through this mechanism TRPM2 in the ER can influence mitochondrial Ca2+ uptake. Ca2+ entry through TRPM2-L may also provide reducing equivalents for optimal oxidative phosphorylation. Diminished Ca2+ uptake in mitochondria can metabolically compromise cells (79, 82), inhibiting the function of Ca2+-dependent enzymes responsible for and thus reducing ATP production (Fig. 15). In TRPM2-S-expressing neuroblastoma cells, HIF-l and 2α expression are reduced. Reduced HIF-1/2α expression results in decreased expression of mitochondrial genes that are downstream including BNIP3 and NDUFA4L2 (Figs. 7 and 9), further compromising mitochondrial function. The decrease in BNIP3 contributes to reduced mitophagy (Fig. 9), leading to accumulation of dysfunctional mitochondria and increased ROS (30), reducing cell viability and tumor growth. The importance of cytosolic Ca2+ in mitophagy, demonstrated here in cells expressing TRPM2-S, is consistent with previous reports that buffering of intracellular Ca2+ with 1,2bis(2aminophenoxy)ethaneN,N,N′,N′-tetraacetic acid or inhibition of plasma membrane Ca2+ channels can prevent autophagy, whereas the Ca2+ ionophore ionomycin can induce it (83–85). The finding that TRPM2-L is important in mitochondrial function in tumor growth as well as in protecting cells from cardiac ischemia (59) suggests that it may have a basic role in mitochondrial bioenergetics that may apply to a number of physiological systems.
FIGURE 16.

Proposed schema of TRPM2-S inhibition of tumor growth. Expression of TRPM2-S on the plasma membrane results in reduced intracellular calcium entry and mitochondrial calcium uptake in response to TRPM2 activation. Decreased intracellular calcium results in reduced expression of HIF-1/2α and genes transcriptionally regulated by HIF-1/2α including BNIP3 and NDUFA4L2. Decreased mitochondrial calcium uptake together with decreased expression of mitochondrial genes downstream of HIF impairs mitochondrial bioenergetics and increases dysfunctional mitochondria. The decrease in BNIP3 contributes to decreased mitophagy and accumulation of dysfunctional/damaged mitochondria, which result in increased ROS accumulation, contributing to cell death and reduced tumor growth.
A dramatic effect on tumor growth and cell viability after doxorubicin treatment was demonstrated here by blocking TRPM2 function with dominant negative TRPM2-S or clotrimazole. Inhibition of TRPM2-L may have significant implications in therapy of cancers including neuroblastoma. Another approach could be manipulation of alternative splicing mechanisms to alter isoform expression, utilizing the different physiological and pathological functions of TRPM2-L and -S; but at present, little is known about the mechanisms responsible for differential splicing of TRPM2.
Acknowledgment
Confocal images were generated using Leica SP8 (S10OD010756-01A1) located in The Pennsylvania State University Microscopy Imaging Core Facility.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1-DK46778 (to B. A. M.), RO1-HL86699 (to M. M.), and RO1-HL58672 and RO1-HL74854 (to J. Y. C.). This work was also supported by grants from the Hyundai Hope-on-Wheels Foundation (to B. A. M.) and by the Four Diamonds Fund of the Pennsylvania State University College of Medicine.
S.-j. Chen and B. A. Miller, unpublished observations.
- TRP
- transient receptor potential
- ADPR
- ADP-ribose
- BNIP3
- Bcl2/adenovirus E1B 19 kDa-interacting protein 3
- Δψm
- mitochondrial membrane potential
- Em
- membrane potential
- FOXO3a
- forkhead box transcription factor 3a
- GCa
- conductance for Ca2+
- GNa
- conductance for Na+
- HIF
- hypoxia-inducible factor
- NDUFA4L2
- NADH dehydrogenase (ubiquinone) 1α subcomplex 4-like 2
- OCR
- oxygen consumption rate
- ROS
- reactive oxygen species
- TRPC
- TRP-canonical
- TRPM
- TRP-melastatin
- TRPM2-L
- full-length TRPM2
- TRPM2-S
- short isoform of TRPM2
- PARP
- poly(ADPR) polymerase
- L
- TRPM2-L
- S
- TRPM2-S
- LDHA
- lactate dehydrogenase A
- RP
- Pearson coefficient
- VHL
- von Hippel-Lindau E3 ligase
- GeLC-MS/MS
- in-gel tryptic digestion followed by liquid chromatography-tandem mass spectrometry
- XTT
- 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
- V
- empty vector
- COX
- cytochrome oxidase.
REFERENCES
- 1. Ramsey I. S., Delling M., Clapham D. E. (2006) An introduction to TRP channels. Annu. Rev. Physiol. 68, 619–647 [DOI] [PubMed] [Google Scholar]
- 2. Duncan L. M., Deeds J., Hunter J., Shao J., Holmgren L. M., Woolf E. A., Tepper R. I., Shyjan A. W. (1998) Down-regulation of the novel gene melastatin correlates with potential for melanoma metastasis. Cancer Res. 58, 1515–1520 [PubMed] [Google Scholar]
- 3. Prawitt D., Enklaar T., Klemm G., Gärtner B., Spangenberg C., Winterpacht A., Higgins M., Pelletier J., Zabel B. (2000) Identification and characterization of MTR1, a novel gene with homology to melastatin (MLSN1) and the trp gene family located in the BWS-WT2 critical region on chromosome 11p15.5 and showing allele-specific expression. Hum. Mol. Genet. 9, 203–216 [DOI] [PubMed] [Google Scholar]
- 4. Aarts M., Iihara K., Wei W. L., Xiong Z. G., Arundine M., Cerwinski W., MacDonald J. F., Tymianski M. (2003) A key role for TRPM7 channels in anoxic neuronal death. Cell 115, 863–877 [DOI] [PubMed] [Google Scholar]
- 5. Tsavaler L., Shapero M. H., Morkowski S., Laus R. (2001) Trp-p8, a novel prostate-specific gene, is up-regulated in prostate cancer and other malignancies and shares high homology with transient receptor potential calcium channel proteins. Cancer Res. 61, 3760–3769 [PubMed] [Google Scholar]
- 6. Nagamine K., Kudoh J., Minoshima S., Kawasaki K., Asakawa S., Ito F., Shimizu N. (1998) Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics 54, 124–131 [DOI] [PubMed] [Google Scholar]
- 7. Miller B. A., Zhang W. (2011) TRP channels as mediators of oxidative stress. Adv. Exp. Med. Biol. 704, 531–544 [DOI] [PubMed] [Google Scholar]
- 8. Hecquet C. M., Ahmmed G. U., Vogel S. M., Malik A. B. (2008) Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ. Res. 102, 347–355 [DOI] [PubMed] [Google Scholar]
- 9. Faouzi M., Penner R. (2014) Trpm2. Handb. Exp. Pharmacol. 222, 403–426 [DOI] [PubMed] [Google Scholar]
- 10. Hara Y., Wakamori M., Ishii M., Maeno E., Nishida M., Yoshida T., Yamada H., Shimizu S., Mori E., Kudoh J., Shimizu N., Kurose H., Okada Y., Imoto K., Mori Y. (2002) LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell 9, 163–173 [DOI] [PubMed] [Google Scholar]
- 11. Fonfria E., Marshall I. C., Boyfield I., Skaper S. D., Hughes J. P., Owen D. E., Zhang W., Miller B. A., Benham C. D., McNulty S. (2005) Amyloid β-peptide(1–42) and hydrogen peroxide-induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J. Neurochem. 95, 715–723 [DOI] [PubMed] [Google Scholar]
- 12. Wehage E., Eisfeld J., Heiner I., Jüngling E., Zitt C., Lückhoff A. (2002) Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J. Biol. Chem. 277, 23150–23156 [DOI] [PubMed] [Google Scholar]
- 13. Gasser A., Glassmeier G., Fliegert R., Langhorst M. F., Meinke S., Hein D., Krüger S., Weber K., Heiner I., Oppenheimer N., Schwarz J. R., Guse A. H. (2006) Activation of T cell calcium influx by the second messenger ADP-ribose. J. Biol. Chem. 281, 2489–2496 [DOI] [PubMed] [Google Scholar]
- 14. Kolisek M., Beck A., Fleig A., Penner R. (2005) Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol. Cell 18, 61–69 [DOI] [PubMed] [Google Scholar]
- 15. Perraud A. L., Takanishi C. L., Shen B., Kang S., Smith M. K., Schmitz C., Knowles H. M., Ferraris D., Li W., Zhang J., Stoddard B. L., Scharenberg A. M. (2005) Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J. Biol. Chem. 280, 6138–6148 [DOI] [PubMed] [Google Scholar]
- 16. Tóth B., Csanády L. (2010) Identification of direct and indirect effectors of the transient receptor potential melastatin 2 (TRPM2) cation channel. J. Biol. Chem. 285, 30091–30102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fonfria E., Marshall I. C., Benham C. D., Boyfield I., Brown J. D., Hill K., Hughes J. P., Skaper S. D., McNulty S. (2004) TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br. J. Pharmacol. 143, 186–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Buelow B., Song Y., Scharenberg A. M. (2008) The poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J. Biol. Chem. 283, 24571–24583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McHugh D., Flemming R., Xu S. Z., Perraud A. L., Beech D. J. (2003) Critical intracellular Ca2+ dependence of transient receptor potential melastatin 2 (TRPM2) cation channel activation. J. Biol. Chem. 278, 11002–11006 [DOI] [PubMed] [Google Scholar]
- 20. Du J., Xie J., Yue L. (2009) Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proc. Natl. Acad. Sci. U.S.A. 106, 7239–7244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tong Q., Zhang W., Conrad K., Mostoller K., Cheung J. Y., Peterson B. Z., Miller B. A. (2006) Regulation of the transient receptor potential channel TRPM2 by the Ca2+ sensor calmodulin. J. Biol. Chem. 281, 9076–9085 [DOI] [PubMed] [Google Scholar]
- 22. Du J., Xie J., Yue L. (2009) Modulation of TRPM2 by acidic pH and the underlying mechanisms for pH sensitivity. J. Gen. Physiol. 134, 471–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Starkus J. G., Fleig A., Penner R. (2010) The calcium-permeable non-selective cation channel TRPM2 is modulated by cellular acidification. J. Physiol. 588, 1227–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Csanády L. (2010) Permeating proton found guilty in compromising TRPM2 channel activity. J. Physiol. 588, 1661–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Orfanelli U., Wenke A. K., Doglioni C., Russo V., Bosserhoff A. K., Lavorgna G. (2008) Identification of novel sense and antisense transcription at the TRPM2 locus in cancer. Cell Res. 18, 1128–1140 [DOI] [PubMed] [Google Scholar]
- 26. Zhang W., Chu X., Tong Q., Cheung J. Y., Conrad K., Masker K., Miller B. A. (2003) A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. J. Biol. Chem. 278, 16222–16229 [DOI] [PubMed] [Google Scholar]
- 27. Goel M., Sinkins W. G., Schilling W. P. (2002) Selective association of TRPC channel subunits in rat brain synaptosomes. J. Biol. Chem. 277, 48303–48310 [DOI] [PubMed] [Google Scholar]
- 28. Hofmann T., Schaefer M., Schultz G., Gudermann T. (2002) Subunit composition of mammalian transient receptor potential channels in living cells. Proc. Natl. Acad. Sci. U.S.A. 99, 7461–7466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li M., Jiang J., Yue L. (2006) Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J. Gen. Physiol. 127, 525–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen S. J., Zhang W., Tong Q., Conrad K., Hirschler-Laszkiewicz I., Bayerl M., Kim J. K., Cheung J. Y., Miller B. A. (2013) Role of TRPM2 in cell proliferation and susceptibility to oxidative stress. Am. J. Physiol. Cell Physiol. 304, C548–C560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bai J. Z., Lipski J. (2010) Differential expression of TRPM2 and TRPV4 channels and their potential role in oxidative stress-induced cell death in organotypic hippocampal culture. Neurotoxicology 31, 204–214 [DOI] [PubMed] [Google Scholar]
- 32. Di A., Gao X. P., Qian F., Kawamura T., Han J., Hecquet C., Ye R. D., Vogel S. M., Malik A. B. (2012) The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat. Immunol. 13, 29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miller B. A., Wang J., Hirschler-Laszkiewicz I., Gao E., Song J., Zhang X. Q., Koch W. J., Madesh M., Mallilankaraman K., Gu T., Chen S. J., Keefer K., Conrad K., Feldman A. M., Cheung J. Y. (2013) The second member of transient receptor potential-melastatin channel family protects hearts from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 304, H1010–H1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schumacker P. T. (2006) Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell 10, 175–176 [DOI] [PubMed] [Google Scholar]
- 35. Trachootham D., Alexandre J., Huang P. (2009) Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug Discov. 8, 579–591 [DOI] [PubMed] [Google Scholar]
- 36. Kang Y. J., Chen Y., Epstein P. N. (1996) Suppression of doxorubicin cardiotoxicity by overexpression of catalase in the heart of transgenic mice. J. Biol. Chem. 271, 12610–12616 [DOI] [PubMed] [Google Scholar]
- 37. Ewer M. S., Ewer S. M. (2010) Cardiotoxicity of anticancer treatments: what the cardiologist needs to know. Nat. Rev. Cardiol. 7, 564–575 [DOI] [PubMed] [Google Scholar]
- 38. Semenza G. L. (2009) Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin. Cancer Biol. 19, 12–16 [DOI] [PubMed] [Google Scholar]
- 39. Tello D., Balsa E., Acosta-Iborra B., Fuertes-Yebra E., Elorza A., Ordóñez Á., Corral-Escariz M., Soro I., López-Bernardo E., Perales-Clemente E., Martínez-Ruiz A., Enríquez J. A., Aragonés J., Cadenas S., Landázuri M. O. (2011) Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 14, 768–779 [DOI] [PubMed] [Google Scholar]
- 40. Zhang H., Bosch-Marce M., Shimoda L. A., Tan Y. S., Baek J. H., Wesley J. B., Gonzalez F. J., Semenza G. L. (2008) Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 283, 10892–10903 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41. Bellot G., Garcia-Medina R., Gounon P., Chiche J., Roux D., Pouysségur J., Mazure N. M. (2009) Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 29, 2570–2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Maynard M. A., Ohh M. (2007) The role of hypoxia-inducible factors in cancer. Cell. Mol. Life Sci. 64, 2170–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miller B. A., Barber D. L., Bell L. L., Beattie B. K., Zhang M. Y., Neel B. G., Yoakim M., Rothblum L. I., Cheung J. Y. (1999) Identification of the erythropoietin receptor domain required for calcium channel activation. J. Biol. Chem. 274, 20465–20472 [DOI] [PubMed] [Google Scholar]
- 44. Cheung J. Y., Zhang X. Q., Bokvist K., Tillotson D. L., Miller B. A. (1997) Modulation of calcium channels in human erythroblasts by erythropoietin. Blood 89, 92–100 [PubMed] [Google Scholar]
- 45. Wang J., Gao E., Rabinowitz J., Song J., Zhang X. Q., Koch W. J., Tucker A. L., Chan T. O., Feldman A. M., Cheung J. Y. (2011) Regulation of in vivo cardiac contractility by phospholemman: role of Na+/Ca2+ exchange. Am. J. Physiol. Heart Circ. Physiol. 300, H859–H868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hill K., McNulty S., Randall A. D. (2004) Inhibition of TRPM2 channels by the antifungal agents clotrimazole and econazole. Naunyn Schmiedebergs Arch. Pharmacol. 370, 227–237 [DOI] [PubMed] [Google Scholar]
- 47. Hill K., Benham C. D., McNulty S., Randall A. D. (2004) Flufenamic acid is a pH-dependent antagonist of TRPM2 channels. Neuropharmacology 47, 450–460 [DOI] [PubMed] [Google Scholar]
- 48. Young M. M., Takahashi Y., Khan O., Park S., Hori T., Yun J., Sharma A. K., Amin S., Hu C. D., Zhang J., Kester M., Wang H. G. (2012) Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 287, 12455–12468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Barlow A. L., Macleod A., Noppen S., Sanderson J., Guérin C. J. (2010) Colocalization analysis in fluorescence micrographs: verification of a more accurate calculation of Pearson's correlation coefficient. Microsc. Microanal. 16, 710–724 [DOI] [PubMed] [Google Scholar]
- 50. Barrero C. A., Perez-Leal O., Aksoy M., Moncada C., Ji R., Lopez Y., Mallilankaraman K., Madesh M., Criner G. J., Kelsen S. G., Merali S. (2013) Histone 3.3 participates in a self-sustaining cascade of apoptosis that contributes to the progression of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 188, 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dyrlund T. F., Poulsen E. T., Scavenius C., Sanggaard K. W., Enghild J. J. (2012) MS data miner: a web-based software tool to analyze, compare, and share mass spectrometry protein identifications. Proteomics 12, 2792–2796 [DOI] [PubMed] [Google Scholar]
- 52. Barrero C. A., Datta P. K., Sen S., Deshmane S., Amini S., Khalili K., Merali S. (2013) HIV-1 Vpr modulates macrophage metabolic pathways: a SILAC-based quantitative analysis. PLoS One 8, e68376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vadrot N., Ghanem S., Braut F., Gavrilescu L., Pilard N., Mansouri A., Moreau R., Reyl-Desmars F. (2012) Mitochondrial DNA maintenance is regulated in human hepatoma cells by glycogen synthase kinase 3β and p53 in response to tumor necrosis factor α. PLoS One 7, e40879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mallilankaraman K., Doonan P., Cárdenas C., Chandramoorthy H. C., Müller M., Miller R., Hoffman N. E., Gandhirajan R. K., Molgó J., Birnbaum M. J., Rothberg B. S., Mak D. O., Foskett J. K., Madesh M. (2012) MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 151, 630–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mallilankaraman K., Cárdenas C., Doonan P. J., Chandramoorthy H. C., Irrinki K. M., Golenár T., Csordás G., Madireddi P., Yang J., Müller M., Miller R., Kolesar J. E., Molgó J., Kaufman B., Hajnóczky G., Foskett J. K., Madesh M. (2012) MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 14, 1336–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Roehm N. W., Rodgers G. H., Hatfield S. M., Glasebrook A. L. (1991) An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J. Immunol. Methods 142, 257–265 [DOI] [PubMed] [Google Scholar]
- 57. Perraud A. L., Fleig A., Dunn C. A., Bagley L. A., Launay P., Schmitz C., Stokes A. J., Zhu Q., Bessman M. J., Penner R., Kinet J. P., Scharenberg A. M. (2001) ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 411, 595–599 [DOI] [PubMed] [Google Scholar]
- 58. Hermosura M. C., Cui A. M., Go R. C., Davenport B., Shetler C. M., Heizer J. W., Schmitz C., Mocz G., Garruto R. M., Perraud A. L. (2008) Altered functional properties of a TRPM2 variant in Guamanian ALS and PD. Proc. Natl. Acad. Sci. U.S.A. 105, 18029–18034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Miller B. A., Hoffman N. E., Merali S., Zhang X.-Q., Wang J., Rajan S., Shanmughapriya S., Gao E., Barrero C. A., Mallilankaraman K., Song J., Gu T., Hirschler-Laszkiewicz I., Koch W. J., Feldman A. M., Madesh M., Cheung J. Y. (2014) TRPM2 channels protect against cardiac ischemia-reperfusion injury: role of mitochondria. J. Biol. Chem. 289, 7615–7629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Feng S., Li H., Tai Y., Huang J., Su Y., Abramowitz J., Zhu M. X., Birnbaumer L., Wang Y. (2013) Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc. Natl. Acad. Sci. U.S.A. 110, 11011–11016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Holmquist-Mengelbier L., Fredlund E., Löfstedt T., Noguera R., Navarro S., Nilsson H., Pietras A., Vallon-Christersson J., Borg A., Gradin K., Poellinger L., Påhlman S. (2006) Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 10, 413–423 [DOI] [PubMed] [Google Scholar]
- 62. Noguera R., Fredlund E., Piqueras M., Pietras A., Beckman S., Navarro S., Påhlman S. (2009) HIF-1α and HIF-2α are differentially regulated in vivo in neuroblastoma: high HIF-1α correlates negatively to advanced clinical stage and tumor vascularization. Clin. Cancer Res. 15, 7130–7136 [DOI] [PubMed] [Google Scholar]
- 63. Kops G. J., Dansen T. B., Polderman P. E., Saarloos I., Wirtz K. W., Coffer P. J., Huang T. T., Bos J. L., Medema R. H., Burgering B. M. (2002) Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419, 316–321 [DOI] [PubMed] [Google Scholar]
- 64. Li M., Chiu J. F., Mossman B. T., Fukagawa N. K. (2006) Down-regulation of manganese-superoxide dismutase through phosphorylation of FOXO3a by Akt in explanted vascular smooth muscle cells from old rats. J. Biol. Chem. 281, 40429–40439 [DOI] [PubMed] [Google Scholar]
- 65. Gustafsson A. B. (2011) Bnip3 as a dual regulator of mitochondrial turnover and cell death in the myocardium. Pediatr. Cardiol. 32, 267–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fukuda R., Zhang H., Kim J. W., Shimoda L., Dang C. V., Semenza G. L. (2007) HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129, 111–122 [DOI] [PubMed] [Google Scholar]
- 67. Gilliam L. A., Moylan J. S., Patterson E. W., Smith J. D., Wilson A. S., Rabbani Z., Reid M. B. (2012) Doxorubicin acts via mitochondrial ROS to stimulate catabolism in C2C12 myotubes. Am. J. Physiol. Cell Physiol. 302, C195–C202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Uemura T., Kudoh J., Noda S., Kanba S., Shimizu N. (2005) Characterization of human and mouse TRPM2 genes: identification of a novel N-terminal truncated protein specifically expressed in human striatum. Biochem. Biophys. Res. Commun. 328, 1232–1243 [DOI] [PubMed] [Google Scholar]
- 69. Sumoza-Toledo A., Penner R. (2011) TRPM2: a multifunctional ion channel for calcium signalling. J. Physiol. 589, 1515–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yamamoto S., Shimizu S., Kiyonaka S., Takahashi N., Wajima T., Hara Y., Negoro T., Hiroi T., Kiuchi Y., Okada T., Kaneko S., Lange I., Fleig A., Penner R., Nishi M., Takeshima H., Mori Y. (2008) TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 14, 738–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sano Y., Inamura K., Miyake A., Mochizuki S., Yokoi H., Matsushime H., Furuichi K. (2001) Immunocyte Ca2+ influx system mediated by LTRPC2. Science 293, 1327–1330 [DOI] [PubMed] [Google Scholar]
- 72. Mohlin S., Hamidian A., Påhlman S. (2013) HIF2A and IGF2 expression correlates in human neuroblastoma cells and normal immature sympathetic neuroblasts. Neoplasia 15, 328–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu Q., Möller U., Flügel D., Kietzmann T. (2004) Induction of plasminogen activator inhibitor I gene expression by intracellular calcium via hypoxia-inducible factor-1. Blood 104, 3993–4001 [DOI] [PubMed] [Google Scholar]
- 74. Fredlund E., Ovenberger M., Borg K., Påhlman S. (2008) Transcriptional adaptation of neuroblastoma cells to hypoxia. Biochem. Biophys. Res. Commun. 366, 1054–1060 [DOI] [PubMed] [Google Scholar]
- 75. Liu Y. V., Hubbi M. E., Pan F., McDonald K. R., Mansharamani M., Cole R. N., Liu J. O., Semenza G. L. (2007) Calcineurin promotes hypoxia-inducible factor 1α expression by dephosphorylating RACK1 and blocking RACK1 dimerization. J. Biol. Chem. 282, 37064–37073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hui A. S., Bauer A. L., Striet J. B., Schnell P. O., Czyzyk-Krzeska M. F. (2006) Calcium signaling stimulates translation of HIF-α during hypoxia. FASEB J. 20, 466–475 [DOI] [PubMed] [Google Scholar]
- 77. Yuan G., Nanduri J., Bhasker C. R., Semenza G. L., Prabhakar N. R. (2005) Ca2+/calmodulin kinase-dependent activation of hypoxia inducible factor 1 transcriptional activity in cells subjected to intermittent hypoxia. J. Biol. Chem. 280, 4321–4328 [DOI] [PubMed] [Google Scholar]
- 78. Wallace D. C. (2012) Mitochondria and cancer. Nat. Rev. Cancer 12, 685–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cárdenas C., Miller R. A., Smith I., Bui T., Molgó J., Müller M., Vais H., Cheung K. H., Yang J., Parker I., Thompson C. B., Birnbaum M. J., Hallows K. R., Foskett J. K. (2010) Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kornmann B. (2013) The molecular hug between the ER and the mitochondria. Curr. Opin. Cell Biol. 25, 443–448 [DOI] [PubMed] [Google Scholar]
- 81. Lackner L. L., Ping H., Graef M., Murley A., Nunnari J. (2013) Endoplasmic reticulum-associated mitochondria-cortex tether functions in the distribution and inheritance of mitochondria. Proc. Natl. Acad. Sci. U.S.A. 110, E458–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rizzuto R., De Stefani D., Raffaello A., Mammucari C. (2012) Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578 [DOI] [PubMed] [Google Scholar]
- 83. Høyer-Hansen M., Bastholm L., Szyniarowski P., Campanella M., Szabadkai G., Farkas T., Bianchi K., Fehrenbacher N., Elling F., Rizzuto R., Mathiasen I. S., Jäättelä M. (2007) Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 25, 193–205 [DOI] [PubMed] [Google Scholar]
- 84. Knöferle J., Koch J. C., Ostendorf T., Michel U., Planchamp V., Vutova P., Tönges L., Stadelmann C., Brück W., Bähr M., Lingor P. (2010) Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proc. Natl. Acad. Sci. U.S.A. 107, 6064–6069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Decuypere J. P., Bultynck G., Parys J. B. (2011) A dual role for Ca2+ in autophagy regulation. Cell Calcium 50, 242–250 [DOI] [PubMed] [Google Scholar]