

Background: Group A Streptococcus responds to the human antimicrobial peptide LL-37 by up-regulating virulence factors controlled by the CsrRS system.
Results: We provide genetic and biochemical evidence for direct binding between LL-37 and the CsrS receptor.
Conclusion: LL-37 signaling through CsrS is mediated by a direct binding interaction.
Significance: These findings advance our understanding of streptococcal virulence regulation in response to a host signal.
Keywords: Antimicrobial Peptide (AMP), Cell Surface Receptor, Histidine Kinase, Host-Pathogen Interaction, Streptococcus pyogenes (S. pyogenes)
Abstract
Group A Streptococcus (GAS) responds to subinhibitory concentrations of LL-37 by up-regulation of virulence factors through the CsrRS (CovRS) two-component system. The signaling mechanism, however, is unclear. To determine whether LL-37 signaling reflects specific binding to CsrS or rather a nonspecific response to LL-37-mediated membrane damage, we tested LL-37 fragments for CsrRS signaling and for GAS antimicrobial activity. We identified a 10-residue fragment (RI-10) of LL-37 as the minimal peptide that retains the ability to signal increased expression of GAS virulence factors, yet it has no detectable antimicrobial activity against GAS. Substitution of individual key amino acids in RI-10 reduced or abrogated signaling. These data do not support the hypothesis that CsrS detects LL-37-induced damage to the bacterial cell membrane but rather suggest that LL-37 signaling is mediated by a direct interaction with CsrS. To test whether LL-37 binds to CsrS, we used the purified CsrS extracellular domain to pull down LL-37 in vitro, a result that provides further evidence that LL-37 binds to CsrS. The dissociation of CsrS-mediated signaling from membrane damage by LL-37 fragments together with in vitro evidence for a direct LL-37-CsrS binding interaction constitute compelling evidence that signal transduction by LL-37 through CsrS reflects a direct ligand/receptor interaction.
Introduction
Group A Streptococcus (GAS),2 the etiologic agent of streptococcal pharyngitis, is also capable of causing invasive infections including streptococcal toxic shock and necrotizing fasciitis that are associated with high mortality rates (1). Invasive infections are thought to be preceded by local oropharyngeal infection or colonization (1). However, the mechanisms by which a local infection is established and progresses to an invasive infection are not completely understood.
GAS harbors multiple regulatory systems that enable the organism to respond to environmental signals. Among these are the two-component systems, typically characterized by a sensor histidine kinase, which transduces an extracellular signal by phosphotransfer to a second intracellular component, the response regulator. Phosphorylation of the response regulator modulates its binding to target promoters where regulator binding acts to repress or activate gene transcription (2). GAS has up to 13 two-component systems, among which the CsrRS (also known as CovRS) system is the best studied (3–7). The CsrRS system has recently been hypothesized to play a role in oropharyngeal colonization and persistence by GAS (8–10). Up-regulation of virulence factors under regulatory control of this system results in increased resistance to opsonophagocytosis and intracellular survival in epithelial cells, neutrophils, and macrophages, which may facilitate oropharyngeal colonization (8, 11, 12). The CsrRS system also plays a role in the transition of a local infection to a more invasive process. Bacteria recovered from sterile sites both in a mouse model of infection and in patients with invasive GAS disease frequently have inactivating mutations of this two-component system (6, 7, 13–15). For most genes that encode CsrRS-regulated virulence factors, inactivation of CsrR or CsrS results in constitutive up-regulation of gene expression (4, 15).
LL-37 is a 37-residue amphipathic antimicrobial peptide (AMP) that is produced by leukocytes and epithelial cells of the skin, respiratory tract, and gastrointestinal tract (16, 17). A member of the cathelicidin class of AMPs, LL-37 has antimicrobial activity against many bacteria, as well as immunomodulatory effects (16–18). The structure of the peptide is well described as a helix, and its antimicrobial properties have been localized to the C-terminal domain consisting of amino acids 17–32 (FK-16) (19–22). The smallest fragment that maintains any antimicrobial properties for Escherichia coli is 12 residues in length and corresponds to amino acids 18–29 (KR-12) of the LL-37 sequence (22, 23).
Gryllos et al. (24) first described the signaling relationship between LL-37 and the CsrS sensor. They found that exposure of GAS to subinhibitory concentrations of LL-37 resulted in increased expression of multiple virulence factors including the has operon, responsible for capsular polysaccharide biosynthesis, and that CsrS was essential for this effect. The CsrRS system regulates 10–15% of the GAS genome (7); the effects of LL-37 signaling confer an “invasive phenotype” to the pathogen, reflected in a marked increase in resistance to opsonophagocytic killing by human blood leukocytes (11). Multiple additional genes, including sda1, slo, ska, and spyCEP/scpC, which encode virulence factors streptodornase D/DNase, streptolysin O, streptokinase, and the IL-8 protease SpyCEP/ScpC, respectively, respond to LL-37 through the CsrRS system (1, 11, 24). Testing of multiple AMPs for CsrRS-mediated signaling revealed that only LL-37 and to a lesser degree the rhesus macaque cathelicidin RL-37 (68% identity with LL-37) had the observed effect (24). Human alpha and beta defensins and cathelicidins from mouse (mCRAMP) and sheep (SMAP 29) were inactive in signaling through CsrS. Tran-Winkler et al. (11) subsequently demonstrated that replacement of three acidic amino acids in the extracellular domain of CsrS with uncharged residues abrogated the signaling effects of LL-37. These results suggested the targeted acidic residues might be part of an LL-37 binding site on CsrS.
In this study, we investigated the mechanism by which LL-37 interacts with CsrS and results in up-regulation of virulence factors. We considered two hypotheses. The first was that CsrS transduces a stress response, as has been demonstrated for other two-component receptors, because of sufficient disruption of the bacterial membrane to cause a conformational change that results in signal transduction by modulating the autokinase and/or phosphatase activity of CsrS (25). The second hypothesis we entertained was that LL-37 binds directly and specifically to CsrS to induce a conformational change and downstream signaling in a classic ligand/receptor interaction. We favored the latter hypothesis because CsrS signaling seemed to be specific to LL-37 and was not observed for other cationic AMPs (24). In this study, we identified a 10-residue fragment of LL-37 (RI-10) that has similar activity to full-length LL-37 with respect to its ability to signal through CsrS. This short peptide lacks all antimicrobial activity against GAS and exhibits specific amino acid and chirality requirements for its signaling effect. We also demonstrate a physical interaction of LL-37 with CsrS in pulldown experiments. These data provide strong evidence that LL-37 signaling through CsrS reflects a specific binding interaction between LL-37 and the extracellular domain of CsrS.
EXPERIMENTAL PROCEDURES
Bacterial Strains
GAS strain 854 (24) was used for all experiments involving pathogenic GAS. This is an M1T1 strain originally isolated from a retroperitoneal abscess. Bacterial growth, unless otherwise indicated, was in Todd Hewitt broth supplemented with 0.5% yeast extract (THY; BD Biosciences) at 37 °C under static conditions or on THY agar supplemented with 5% defibrinated sheep blood (Northeast Laboratory Services, Portland, ME).
LL-37, SMAP 29, mCRAMP, and LL-37 Fragments
LL-37, mCRAMP, SMAP 29, and peptides corresponding to portions of the LL-37 molecule, including RI-10 with specific amino acid substitutions and D-RI-10, were synthesized by Anaspec (Fremont, CA), and purity was confirmed by HPLC and mass spectrometry. Additional SMAP 29, a kind gift from Robert I. Lehrer (UCLA, Los Angeles, CA), was synthesized as previously described, and purity was confirmed by HPLC and mass spectrometry (11, 26). All peptides were dissolved in deionized water at 1 mg/ml and stored at −20 °C until use.
Virulence Factor Expression Analysis by qRT-PCR
qRT-PCR analyses were performed essentially as previously described (3, 8). Briefly, GAS 854 was grown in THY broth. When indicated, the growth medium was supplemented with LL-37 or a peptide fragment at a concentration of 300 nm. Cells were grown to midexponential phase (A600 nm 0.3–0.4), and 4.5 ml of culture were harvested for total RNA isolation using standard and previously described protocols (Qiagen RNeasyTM) (4). The quality and concentration of RNA were determined spectrophotometrically using a Nanodrop ND-1000 instrument. Five ng of total RNA were used for qRT-PCR using the SYBR Green RT-PCR system (Qiagen) in an ABI Prism 7300 real time PCR instrument (Applied Biosystems/Invitrogen). Previously described primers were utilized for quantification of hasB, recA, sda1, slo, ska, and spyCEP/scpC transcripts (4, 11). The change in gene expression during growth in the presence of peptide compared with that in unsupplemented THY was calculated by the ΔΔCt method and normalized to the housekeeping gene recA. Experiments were performed three or four times in triplicate. The data shown represent mean values ± standard deviation. Statistical significance of differences in gene expression was evaluated by unpaired two-tailed Student's t test of log10 normalized values. The data are depicted as fold increase in gene expression on a log2 scale as compared with GAS grown in the absence of added peptide.
Measurement of GAS Hyaluronic Acid Capsule
Capsular polysaccharide was measured as previously described (27, 28). Briefly, GAS 854 was grown in 10 ml of THY or the same medium supplemented with 300 nm LL-37, RI-10, or scrambled LL-37. Bacterial cells were harvested at midexponential phase (A600 nm 0.3–0.4) by centrifugation and resuspended in 500 μl of 10 mm Tris, pH 7.5. An equal volume of chloroform was added, and the mixture was vortexed vigorously. The quantity of hyaluronic acid in the aqueous phase was determined by adding 1-ethyl-2-[3-(1-ethylnaphtho[1,2-d]thiazolin-2-ylidene)-2-methylpropenyl]naphtho[1,2-d]thiazolium bromide (Stains-All; Sigma-Aldrich) to 100 μl, measuring absorbance at 640 nm, and comparing against a standard curve. The values were corrected for background absorbance determined from an acapsular derivative of GAS 854 (8). The experiment was repeated three times. The results are expressed as fg/cfu.
Live/Dead Staining, Fluorescence Microscopy, and Microplate Live/Dead Quantification
GAS 854 was grown to midexponential phase (A600 nm 0.3–0.4) in THY broth. One-ml aliquots of broth culture were collected by centrifugation, washed in PBS, and resuspended in 0.5× PBS. AMP was added to the desired concentration, and suspensions were incubated with occasional mixing for 1 h at room temperature. Cells were collected by centrifugation and resuspended in 1 ml of PBS. Aliquots (100 μl) were added in triplicate to a 96-well microassay clear-bottomed plate. Propidium iodide and SYTO 9 (100 μl) were added according to the manufacturer's instructions (Invitrogen) to achieve final concentrations of 6 μm SYTO 9 and 30 μm propidium iodide. The bacteria/dye mixture was protected from light and incubated at room temperature for 15 min. Fluorescence measurements were then obtained on a Tecan Infinite 200 Pro Microplate Reader (Tecan, Mannedorf, Switzerland) with excitation wavelength centered at 485 nm and emission wavelengths centered at 530 and 630 nm. The intensity ratio of λ530/λ630 for each sample was determined and compared with a standard curve constructed by measuring the intensity ratios of mixtures of known quantities of live and ethanol-killed GAS cells. Experiments were performed three times in triplicate and are reported as mean values of percent live cells ± standard deviation. After the microplate assay, 5 μl of reaction mixture were placed on a slide for fluorescence microscopy of the stained bacteria. Microscopy was performed in the Harvard Digestive Diseases Center core facility on a Nikon Eclipse TE2000-E inverted microscope with a kypton/argon laser capable of excitation at wavelengths of 488, 568, and 647 nm. The data were collected and analyzed with Slidebook 5 (3i, Denver, CO) using standard protocols.
Minimum Inhibitory Concentration and Minimum Bactericidal Concentration Assays
Minimum inhibitory concentration (MIC) assays and minimum bactericidal concentration (MBC) assays were adapted from previously published protocols (29–31). For MIC determination, GAS 854 was grown in THY broth to midexponential phase (A600 nm 0.4–0.5) and diluted 1:200 into 100 μl of 0.5× Todd-Hewitt broth containing the desired concentration of AMP (0–200 μm) in a 96-well plate. A650 nm was measured on a VERSAmax microplate reader (Molecular Devices, Sunnyvale, CA) immediately after setting up the experiment and after incubation at 37 °C in 5% CO2 for 20 h. MIC was defined as the minimum peptide concentration that prevented a detectable increase in A650 nm at 20 h. The assay was repeated for each peptide, and the values are reported as a range of concentrations if different results were obtained in the two experiments.
MBC assays were designed to detect the minimal concentration of peptide that results in 99.9% killing of a defined density of bacteria (31). GAS 854 was grown in THY at 37 °C to an A600 nm of ∼0.16. Cells were diluted to a concentration of 104–105 cfu/ml in 0.5× PBS containing AMP at desired concentration up to a maximum of 200 μm. Initial cfu were determined by culturing dilutions on solid medium just prior to addition of AMP. Reaction mixtures were incubated at 37 °C for 1 h at which point the cells were once again plated for cfu counts. The lowest peptide concentration that achieved 99.9% killing of the initial inoculum was considered to be the MBC. The assay was performed three times, and the values are reported as a range if different results were obtained in replicate experiments.
DNA Manipulation and Cloning
The extracellular domain of CsrS from amino acids 40–183 of the full-length CsrS coding region was cloned into a pCold vector (Takara, Otsu, Japan) that included sequences encoding a His6 affinity tag and maltose-binding protein at the N terminus of the multiple cloning site (pColdMBP) (32). The gene had initially been cloned after PCR amplification from GAS 854 chromosomal DNA and cloning into pORI23 (33) with a C-terminal His6 (pORI23CsrSHis). Primers used for this amplification were CsrHisF (5′-AACCGGGATCCGGCATGGGATACGTTATTCGTGAG-3′) and CsrSHisR (5′-AACCGCTGCAGCTAGTGATGGTGATGGTGATGGCTTCCTGATCCACTCTCTTTAGACTGGGCCAAAGG-3′). The selected CsrS coding region was then amplified by PCR from plasmid pORI23CsrSHis with primers pColdECDF (5′-AACCGACTAGTAGTTCAACAAACTATTTCTTATTGAAG-3′) and CsrSECDN183R (5′-AACCGCTGCAGCTAATTGCCTAAATCATGAAAGACTTGAAC-3′). The product was cloned into restriction sites SpeI and PstI of the pColdMBP multiple cloning site to create plasmid pCold MBP-ECD. For expression of MBP, pCold MBP-ECD was digested with XbaI and SpeI and religated to remove the CsrS gene and to introduce a translation termination codon at the multiple cloning site (pColdMBPstop). Finally, for construction of an expression vector harboring the CsrS ECD with a triple amino acid substitution as previously described (11), the full-length CsrS was introduced into the pGEM-T vector system (Promega, Madison, WI) after amplification from pORI23CsrSHis with Easy-A Hi-Fi Polymerase (Agilent Technologies, Santa Clara, CA) using primers CsrSHisF and CsrSHisR. Three acidic residues clustered in the extracellular domain (Asp148, Glu151, and Asp152) were changed to polar uncharged residues (Asn148, Gln151, and Asn152) by QuikChange mutagenesis (Agilent Technologies, Santa Clara, CA) using previously described primers csrS418-F(muNHIQN) and csrS480-R(muNHIQN) (11) to create pGemCsrSHis3×mut. The CsrS extracellular domain was then amplified from this plasmid, using primers pColdECDF and CsrSECDN183R, and cloned into pColdMBP, as described for the wild-type extracellular domain, to form pColdMBP-ECD3×mut. Fidelity of all cloned sequences was confirmed by DNA sequencing (Genewiz, South Plainfield, NJ).
Protein Expression and Purification
To purify MBP-ECD, pCold MBP-ECD was expressed from E. coli BL21 DE3-T1 (Sigma). Cells were grown in an Applikon (Foster City, CA) 5-liter Bioreactor to A600 nm 5–7, at which point expression was induced by decreasing the temperature to 15 °C and adding isopropyl β-d-thiogalactopyranoside to a final concentration of 0.8 mm. Cells were allowed to express for 2–3 h, harvested, and frozen (−80 °C) for later use. For purification, bacterial pellets were thawed and resuspended in buffer A (50 mm sodium phosphate, 500 mm NaCl, 20 mm imidazole, pH 8.0) with 0.1% Tween 20 and EDTA-free protease inhibitors (EMD Millipore, Darmstadt, Germany). Resuspended bacteria were exposed to lysozyme at 1 mg/ml for 30 min and DNase I at 0.05 mg/ml for an additional 30 min on ice. Bacterial cells were then lysed with a Sonic Dismembrator (Thermo Fisher Scientific, Waltham, MA) using a half-inch probe at amplitudes of 40–50% for a total time of 30 min, including cooling for 1 min between 30-s bursts, all on ice. Cleared lysate was filtered and loaded onto a 5-ml nickel-nitrilotriacetic acid-agarose (Invitrogen) column equilibrated with buffer A on an AKTApurifier FPLC system (GE Healthcare). The column was washed with 20 column volumes of buffer A at 1 ml/min. Protein was then eluted with a gradient of 20–250 mm imidazole over 20 column volumes. The eluted protein was then loaded onto 5 ml of amylose resin (New England Biolabs, Ipswich, MA), equilibrated with buffer B (50 mm sodium phosphate, 500 mm NaCl, pH 8.0), washed with 20 column volumes of the same buffer, and finally eluted with buffer B containing 10 mm maltose, all by gravity at 4 °C. For pulldown assays, ∼2 mg of purified MBP-ECD were loaded onto 0.5 ml of nickel-nitrilotriacetic acid-agarose equilibrated in buffer A, washed in the same buffer with 40 column volumes, and eluted with buffer A containing 250 mm imidazole. Both MBP alone and MBP-ECD 3× mutant were expressed from vectors pColdMBPstop and pColdMBP-ECD3×mut, respectively, and purified using an identical protocol as described above for pCold MBP-ECD. Protein concentrations were determined by A280 on a Nanodrop ND-1000 (Thermo Fisher Scientific).
Pulldown Experiments
LL-37 (10 μm) was mixed with either MBP-ECD, MBP-ECD 3×mutant, or MBP (each at 1 μm) in 1 ml of buffer B. The mixtures were kept on ice for 45 min. A 200-μl volume of amylose resin equilibrated in buffer B was added to the protein solution and allowed to bind, rotating at 4 °C for 1 h. The resin was washed 7–10 times in batch format with 1 ml of the same buffer, and then bound fusion protein was eluted with two 200-μl amounts of buffer B containing 10 mm maltose. Samples were separated by 12.5% (w/v) SDS-PAGE and transferred to nitrocellulose. Rabbit anti-sera to LL-37 (46545 and 46546) were a kind gift of Robert Lehrer (UCLA, Los Angeles, CA). Western analysis was carried out using standard protocols with anti-LL-37 rabbit serum (46546) at a 1:1000 dilution and secondary HRP-conjugated anti-rabbit antibodies (GE Healthcare) at a 1:10,000 dilution. The blot was developed using ECL reagents (Thermo Fisher Scientific) and exposed to x-ray film. The same procedure was repeated with MBP-ECD, substituting RK-19 for LL-37 as a negative control. The anti-LL-37 rabbit serum used (46545) for Western analysis in this experiment was able to recognize the smaller fragment (RK-19) with sensitivity comparable with that of LL-37 (not shown).
In separate experiments, biotinylation of LL-37 or SMAP 29 was performed in 100 μl of 50 mm sodium phosphate, 500 mm sodium chloride, pH 8.0, with 100 μm peptide and 200 μm sulfo-NHS-biotin (Thermo Fisher Scientific). The reaction was incubated on ice for 2 h and then added to 900 μl of 20 mm Tris, 500 mm sodium chloride, pH 8.0 (Tris buffer). The pulldown experiment then proceeded as described above with MBP-ECD at 1 μm and a final peptide concentration of 10 μm. The amylose resin was washed with 1 ml of Tris buffer five times, and MBP-ECD was eluted with two 200-μl amounts of Tris buffer containing 10 mm maltose. The samples were separated by SDS-PAGE in 12.5% acrylamide, and biotinylated protein was detected in the gel using previously described methods (34) by incubating the gels with streptavidin DyLight at a 1:1000 dilution (Thermo Fisher Scientific) in PBS containing 0.01% Tween and 5% bovine serum albumin for 1 h after fixing in 50% isopropanol/5% acetic acid. The gels were visualized and analyzed using an Odyssey Infrared Imaging System (LI-COR, Lincoln, NE).
RESULTS
The Sheep Antimicrobial Peptide SMAP 29 Has Antimicrobial Activity against GAS Strain 854 but Does Not Signal through CsrRS
Our laboratory has previously demonstrated that GAS 854, when exposed to 100–300 nm LL-37, responds through the CsrRS two-component system to up-regulate expression of multiple virulence factors including the has operon responsible for capsular polysaccharide synthesis (24). Using chloramphenicol acetyltransferase to report activity of the has operon promoter, Gryllos et al. (24) also showed that the mouse (mCRAMP) and sheep (SMAP 29) cathelicidins had little or no CsrRS signaling activity in GAS strain DLS003. In the current study, we used the M1T1 strain 854, an invasive clinical isolate that produces larger quantities of hyaluronic acid capsule in response to LL-37 as compared with GAS DLS003. Using qRT-PCR, we found that exposure of GAS 854 to 300 nm mCRAMP resulted in increased expression of the CsrRS-regulated genes hasB and sda1 (Fig. 1). The increase in gene expression, however, was only 50% of that seen in response to human LL-37. Exposure to the sheep SMAP-29 had no effect on transcription of either hasB or sda1 in GAS 854. However, assays of MIC and MBC showed that mCRAMP and SMAP 29 had similar or more potent antimicrobial activity against GAS as LL-37 (Table 1). These results indicate that the toxicity of the peptides, particularly SMAP-29, does not directly correlate with signaling through CsrS.
FIGURE 1.

Stimulation of CsrRS-regulated gene expression by LL-37, mCRAMP, and SMAP 29. The data represent fold increases in transcript abundance for hasB or sda1 in GAS grown in the presence of 300 nm peptide compared with that for GAS grown in the absence of peptide.
TABLE 1.
MIC and MBC of LL-37, mCRAMP, and SMAP29
Antimicrobial activity for GAS was assayed by determination of the minimum concentration of peptide that prevented growth during overnight incubation in 0.5× Todd-Hewitt broth (MIC) and the minimum concentration of peptide that resulted in 99.9% killing after 1 h of incubation in 0.5× phosphate-buffered saline (MBC).
| Peptide | MIC | MBC |
|---|---|---|
| μm | μm | |
| LL-37 | 50 | 20–30 |
| mCRAMP | 12.5 | 20–30 |
| SMAP 29 | 12.5–25 | 0.5–1 |
A 10-Amino Acid Fragment of LL-37, RI-10, Signals through CsrS
The fact that AMPs with similar antimicrobial activity against GAS had vastly different CsrRS signaling activity suggested that these two properties are not necessarily linked. Accordingly, we sought to identify the core region of LL-37 responsible for CsrS signaling and to determine whether there was a correlation between CsrS signaling capacity and antimicrobial activity for GAS. We tested a series of 13 synthetic peptides designed to correspond to fragments of LL-37 (Table 2). The peptide sequences spanned the region of LL-37 shown previously to be most active in damaging bacterial cell membranes (16, 19). The peptides were tested for CsrS signaling activity by exposing GAS to 300 nm peptide and then quantifying CsrS signaling by qRT-PCR analysis of expression of hasB. As expected, full-length LL-37 resulted in a 30-fold increase in hasB transcription, whereas a 37-amino acid peptide containing the same amino acids in random order (scrambled LL-37) had no activity. We identified several shorter peptides that retained CsrS signaling activity, as reflected in up-regulation of hasB transcription (Fig. 2A). The smallest fragment of LL-37 that retained signaling activity was 10 residues in length (RI-10). Signaling effects of RI-10 were confirmed for additional genes known to be regulated by CsrRS (sda1, spyCEP/scpC, slo, and ska; Fig. 2B) (4, 11, 27). We also tested the quantity of hyaluronic acid expressed by GAS 854 in the presence of LL-37 and RI-10 and found increases that were consistent with the qRT-PCR results (Fig. 2C). RI-10 had no effect on transcription of hasB or sda1 in a GAS 854 strain with a deletion of CsrS (not shown). Signaling was not solely dependent on peptide length. KR-10 and IV-10, both also 10 residues in length, had no significant CsrS signaling activity (Fig. 2A). These peptides differ from RI-10 by a shift of one residue toward the N terminus (KR-10) or C terminus (IV-10) of the LL-37 parent sequence (Table 2). Together, these results identify the LL-37 domain represented by RI-10 as critical for LL-37 signaling. Of note, we observed that the entire RI-10 sequence is present in each of the LL-37 fragments that had CsrS signaling activity (Table 2).
TABLE 2.
Amino acid sequences of synthetic peptides corresponding to fragments of mature, fulllength LL-37
Fragments are named by the first two amino acids followed by the total number of residues in the peptide. All peptides found to signal through CsrRS include the 10-amino acid sequence represented by RI-10 (highlighted in blue).
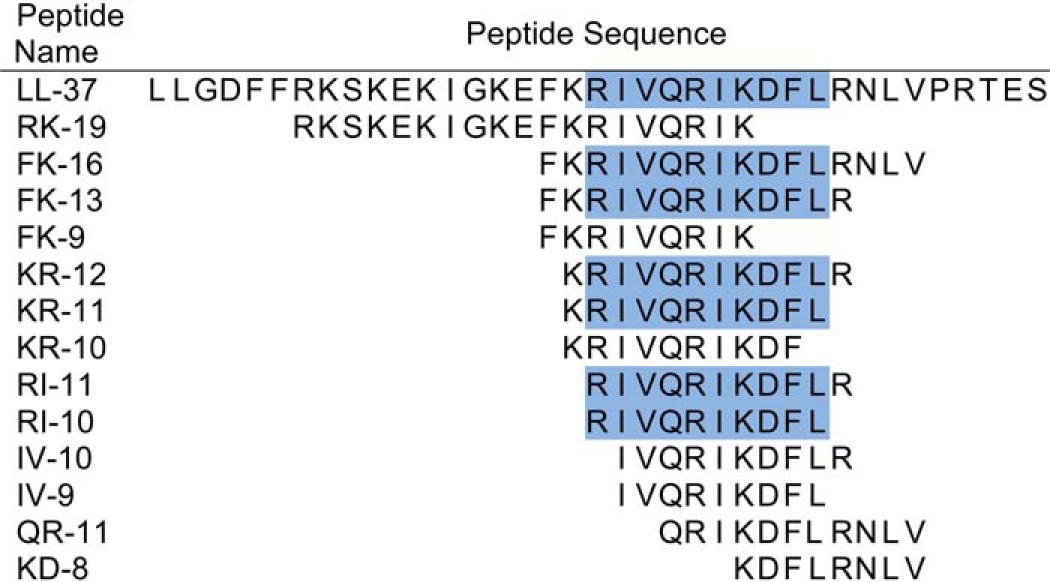
FIGURE 2.

Stimulation of CsrRS-regulated gene expression by LL-37 fragments. A, fold increase in transcript abundance for hasB in GAS grown in the presence of 300 nm peptide (Table 2) compared with that for GAS grown in the absence of peptide. *, p < 0.05 for comparison with the value for scrambled LL-37. B, fold increase in transcript abundance for selected CsrRS-regulated genes in GAS grown in the presence of 300 nm LL-37 or RI-10 compared with that for GAS grown in the absence of peptide. C, cell-associated hyaluronic acid produced by GAS 854 grown in the absence or presence of 300 nm LL-37, RI-10, or scrambled LL-37. *, p < 0.05 for comparison to the value for GAS 854 grown in the absence of peptide.
Neither LL-37 nor Any of Its Fragments Cause Significant GAS Membrane Damage at a Subinhibitory Concentration of 300 nm
We next sought to investigate whether LL-37 and its fragments signaled through CsrS by damaging the GAS cell membrane. We tested the effect of select peptides to induce membrane damage at a concentration of 300 nm, the concentration at which LL-37 and some of its fragments were shown to signal through CsrS, described above. GAS membrane injury was assessed by live/dead staining with SYTO 9 (green) and propidium iodide (red). At this concentration of peptide, bacteria treated with each of the peptides appeared viable by SYTO 9 staining, and none of the tested peptides resulted in bacterial uptake of propidium iodide, which only occurs if the cell membrane is damaged (Fig. 3). GAS treated with a higher concentration of 50 μm LL-37 were positive for propidium iodide staining, consistent with extensive membrane damage, whereas bacteria treated with 50 μm RI-10 did not stain with propidium iodide, and all appeared viable (Fig. 3A). The microscopy findings were confirmed and quantified in a fluorescence microplate assay with similar results (Fig. 3B). The fact that RI-10 signals through CsrS with similar potency to LL-37 but does not induce GAS membrane injury serves to dissociate CsrS signaling from membrane damage.
FIGURE 3.
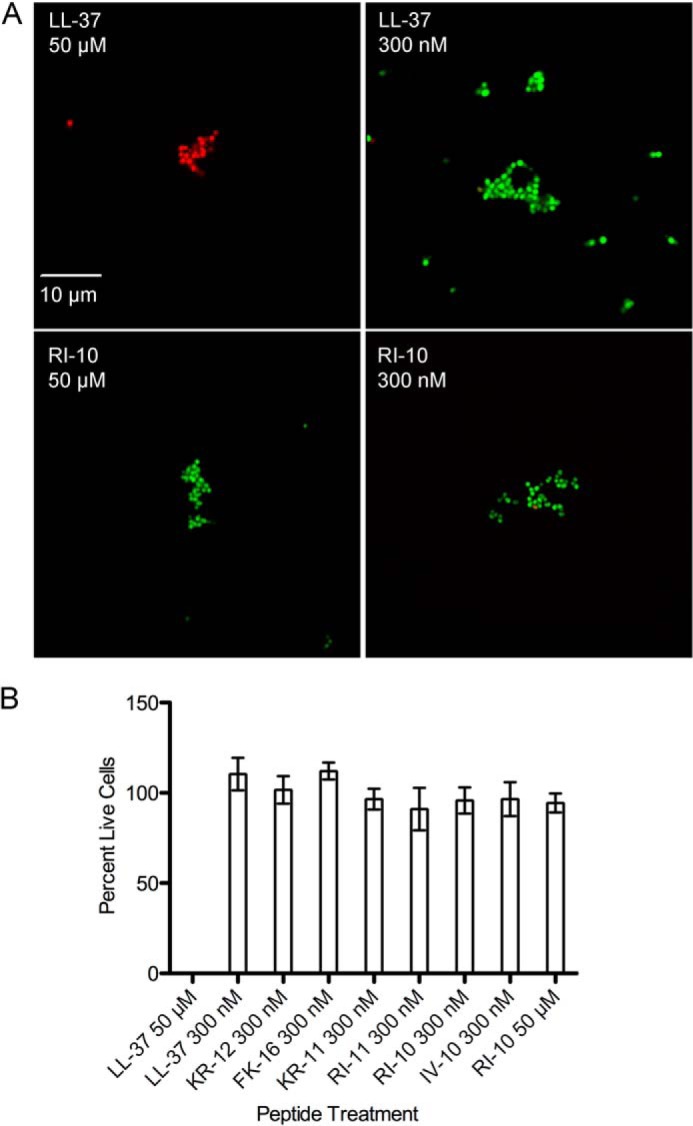
GAS membrane damage by antimicrobial peptides. A, fluorescence microscopy of GAS after exposure to antimicrobial peptides in 0.5× PBS for 1 h followed by staining with SYTO 9 (green, penetrates intact cell membranes) and propidium iodide (red, stains cells with membrane damage). B, quantification of cell viability by fluorescence measurement in a microplate fluorometer and comparison with a standard curve of different ratios of live/dead bacteria.
MICs and MBCs of LL-37 and Its Fragments
We used two additional methods to assess the GAS antimicrobial activity of LL-37 and its fragments: 1) determination of the MIC of each peptide, defined as the concentration of peptide that did not allow growth of GAS 854 in overnight culture, and 2) determination of the MBC, defined as the minimal peptide concentration that achieved 99.9% killing of GAS 854 in 0.5× PBS (Table 3). Consistent with previous reports for other GAS strains (30, 35), we found that LL-37 had antimicrobial activity for GAS strain 854, with an MIC of 50 μm and an MBC of 20–30 μm. The slightly lower concentration of peptide required for killing compared with that for inhibition of growth reflects the different conditions of the two assays: the killing assay was performed under hypotonic conditions that enhance antimicrobial activity. FK-16, which corresponds to the region considered to encompass the core of LL-37 antimicrobial activity (19, 20), demonstrated more potent antimicrobial activity than full-length LL-37, with lower MIC and MBC of 1 and 0.75–1 μm, respectively. KR-12 and RI-11, both of which are competent for CsrS signaling, had less potent antimicrobial activity than LL-37. RI-10, which is also signaling-competent, did not exhibit detectable antimicrobial activity against GAS at the concentrations and under the conditions we tested. In contrast, RK-19, which lacks CsrS signaling activity, had inhibitory activity in the MIC assay at a concentration of 200 μm. These data indicate that GAS antimicrobial activity by LL-37 fragments is neither sufficient nor necessary for signaling through CsrS and demonstrate a dissociation between the signaling and antimicrobial properties of the peptides.
TABLE 3.
MIC and MBC of LL-37 fragments
Antimicrobial activity for GAS was assayed as described in Table I.
| Peptide | MIC | MBC |
|---|---|---|
| μm | μm | |
| LL-37 | 50 | 20–30 |
| RK-19 | 200 | >200 |
| FK-16 | 1 | 0.75–1.0 |
| FK-13 | 25 | 30 |
| FK-9 | >200 | >200 |
| KR-12 | 100 | 200 |
| KR-11 | >200 | >200 |
| KR-10 | >200 | >200 |
| RI-11 | 200 | >200 |
| RI-10 | >200 | >200 |
| IV-10 | >200 | >200 |
| IV-9 | >200 | >200 |
| QR-11 | >200 | >200 |
| KD-8 | >200 | >200 |
Signaling by RI-10 Exhibits Amino Acid Specificity
In light of our findings, we hypothesized that if there is a direct ligand/receptor interaction between RI-10 and CsrS, we would be able to identify some amino acid specificity for signaling. We selected specific amino acids of RI-10, made both conservative and nonconservative replacements (Table 4), and then tested these peptides by qRT-PCR for their effect on transcription of CsrRS-regulated genes as described above (Fig. 4). We found that a conservative change from arginine to lysine at the first amino acid position did not change the signaling activity of RI-10 as reflected in transcription of hasB, sda1, slo, or spyCEP/scpC. However, a change to alanine at the same position, removing the positive charge, decreased signaling (statistically significant for three of four genes tested). Changing the C-terminal leucine to either alanine or phenylalanine, removing the large hydrophobic side chain or increasing its bulk, respectively, both resulted in complete abrogation of signaling by these peptides. Signaling by RI-10, therefore, has specific amino acid requirements and is not simply dependent on nonspecific properties such as hydrophobicity. Of interest, the mouse cathelicidin mCRAMP has phenylalanine rather than leucine at this position. Because mCRAMP exhibits CsrS signaling activity, albeit less than LL-37, it appears that the substitution of phenylalanine for leucine does not completely abrogate signaling in the context of the full-length cathelicidin. We also tested an all-D enantiomer of RI-10 (D-RI-10), i.e. a mirror image of RI-10 that is predicted to have a similar amphipathic nature and charge distribution. We hypothesized that if the binding and signaling capacity of RI-10 were due to nonspecific properties such as charge or amphipathicity, then an all-D enantiomer should behave similarly. However, if RI-10 signaling depends on specific binding to CsrS, then the corresponding D peptide is likely to have reduced activity in signaling through CsrS. We found that D-RI-10 had no signaling activity (Fig. 4). These data support specific binding of RI-10 (and, by inference, LL-37) to CsrS and a critical role for particular amino acid residues within this signaling core region of LL-37.
TABLE 4.
RI-10 amino acid substitutions used in this study
| Peptide | Peptide sequence | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| RI-10 | R | I | V | Q | R | I | K | D | F | L |
| R1A | A | I | V | Q | R | I | K | D | F | L |
| R1K | K | I | V | Q | R | I | K | D | F | L |
| L10A | R | I | V | Q | R | I | K | D | F | A |
| L10F | R | I | V | Q | R | I | K | D | F | F |
| D8N | R | I | V | Q | R | I | K | N | F | L |
| D-RI10a | r | i | v | q | r | i | k | d | f | l |
a All d-amino acids.
FIGURE 4.
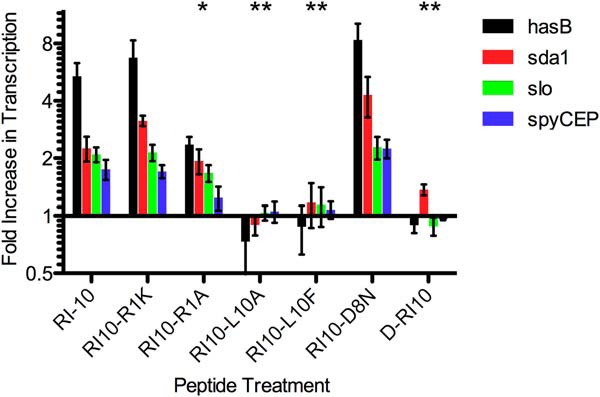
Amino acid specificity of RI-10 binding to the extracellular domain of CsrS. The data represent fold increase in transcript abundance for four CsrRS-regulated genes in GAS grown in the presence of 300 nm peptide (RI-10 amino acid substitutions; Table 4) compared with that for GAS grown in the absence of peptide. * denotes three of four and ** denotes four of four virulence factors for which expression in the presence of peptide is significantly different (p < 0.05) from that for GAS grown in the presence of RI-10.
LL-37 Binds the Extracellular Domain of CsrS
The data described above suggested that signal transduction through CsrS is unlikely to be caused by a stress response to membrane damage. Rather, these new data, together with previously published studies showing specificity of signaling for LL-37 and not other AMPs, suggest strongly that LL-37 interacts directly with the CsrS extracellular domain (24). To test this hypothesis, we expressed and purified a recombinant fusion protein representing the predicted CsrS extracellular domain (amino acids 40–183) fused at the N terminus to MBP and a His6 tag (MBP-ECD) for enhancement of solubility and affinity purification, respectively. As a control, we expressed and purified His6-tagged MBP alone from the same expression vector in the same manner as MBP-ECD. We then proceeded with pulldown experiments using amylose resin after mixing LL-37 with purified MBP-ECD or MBP (Fig. 5A). Western blot analysis demonstrated a marked increase in LL-37 recovered for the sample containing MBP-ECD compared with MBP alone, supporting a binding interaction between the CsrS ECD and LL-37. Tran-Winkler et al. (11) previously demonstrated that changing a cluster of three acidic amino acids in the CsrS ECD to uncharged polar residues abrogated the signaling effects of LL-37 in vivo. To test whether the affinity of the CsrS ECD for LL-37 was reduced by these amino acid substitutions, we expressed and purified an MBP-ECD construct harboring this triple amino acid substitution (MBP-ECD 3× mutant). LL-37 bound to the mutant construct to a greater extent than to MBP alone, but far less than to the wild-type MBP-ECD construct (Fig. 5A). Thus, the impaired responsiveness to LL-37 signaling of a GAS strain expressing a CsrS ECD harboring amino acid substitutions D148N, E151Q, and D152N correlates with decreased affinity of LL-37 binding. To address the possibility of a nonspecific charge interaction between LL-37 and the CsrS ECD, we tested whether MBP-ECD could pull down the LL-37 fragment RK-19 (Fig. 5B) or the sheep cathelicidin SMAP 29 (Fig. 5C), both cationic peptides and neither able to signal an increase in transcription of virulence factors through CsrS. Neither control peptide was detected in the eluate, indicating that they were not able to bind to MBP-ECD and that the effect seen with LL-37 is unlikely to be a nonspecific electrostatic interaction. Taken together, these data provide evidence for a direct binding interaction between LL-37 and the extracellular domain of CsrS.
FIGURE 5.

LL-37 binds to the CsrS extracellular domain. A, the CsrS extracellular domain fused to maltose-binding protein (MBP-ECD), the CsrS extracellular domain with three point mutations at acidic residues fused to maltose-binding protein (MBP-ECD 3× mutant), or MBP alone was mixed with LL-37 and then pulled down with amylose resin. Left panel, Coomassie-stained SDS-PAGE of proteins eluted from the amylose resin demonstrating similar amounts of MBP-ECD, MBP-ECD 3× mutant, and MBP alone as a negative control. Right panel, anti-LL-37 Western blot of the amylose eluate. B, MBP-ECD was mixed with LL-37 or with RK-19 and then pulled down with amylose resin. Left panel, Coomassie-stained SDS-PAGE of proteins eluted from the amylose resin demonstrating similar amounts of MBP-ECD. Right panel, Western blot of eluates with anti-LL-37 antibodies that recognize both LL-37 and RK-19. C, MBP-ECD was mixed with biotinylated LL-37 or biotinylated SMAP 29 and then pulled down with amylose resin. Left panel, Coomassie-stained SDS-PAGE of proteins eluted from the amylose resin demonstrating equal loading of MBP-ECD. Right panel, in-gel detection of biotin-labeled LL-37 and SMAP 29 with streptavidin DyLight.
DISCUSSION
GAS exposure to subinhibitory concentrations of LL-37 stimulates the coordinated up-regulation of expression of multiple virulence determinants controlled by the CsrRS two-component system (11, 24). A critical consequence of this regulatory program is a marked and multifaceted increase in GAS resistance to effective killing by host immune effectors, notably phagocytic leukocytes (8, 11). LL-37 is produced by neutrophils where it is found as a component of granules. Production by epithelial cells is induced by injury, inflammation, and exposure to microbial products. LL-37 has been detected in mucosal secretions in human airways and has been found in saliva of healthy individuals at concentrations ranging from 30 to 700 nm (36, 37). LL-37 is not produced by skin keratinocytes under resting conditions but can reach concentrations as high as 300 μm in inflamed skin, e.g. in psoriasis (38). At concentrations of 100–300 nm, LL-37 signals through CsrRS to increase expression of GAS genes that control synthesis of the hyaluronic acid capsule, an important antiphagocytic factor; of SpyCEP, an IL-8 protease that inactivates the chemotactic cytokine, IL-8, to reduce neutrophil influx to the site of infection; of SLO and its co-toxin NADase that damage phagocytes and reduce their bacterial killing activity; and of DNase that degrades neutrophil extracellular traps, permitting bacterial escape (11, 24). Together, these and other changes in gene expression triggered by LL-37 confer a virulence phenotype that is strongly associated with invasive infection in animal models and that mimics the behavior of naturally occurring CsrRS mutants in human infection (6, 7, 11, 13–15). LL-37-induced changes in virulence factor expression, like inactivating mutations in CsrS or CsrR, result in a striking increase in GAS resistance to opsonophagocytic killing in human blood in vitro (11). The virulence phenotype induced by LL-37 has been suggested to mediate the transition from asymptomatic colonization to invasive GAS infection (11). Therefore, defining the mechanism through which LL-37 signaling occurs is of central importance to development of new strategies for preventing or treating invasive GAS disease.
The goal of the present investigation was to study the nature of the interaction of LL-37 with GAS to distinguish more clearly whether CsrRS signaling is a consequence of LL-37-induced membrane damage or rather a direct effect of specific binding of LL-37 to CsrS. Our results strongly support the latter hypothesis. We initially made the observation that nonspecific membrane damage did not lead to signaling through CsrS by testing the sheep cathelicidin SMAP 29. We then were able to extend this observation to LL-37 by testing the CsrRS signaling and antibacterial activities of a series of LL-37 fragments. We found that these two activities could be dissociated in RI-10, a 10-amino acid internal fragment of LL-37 that exhibited CsrRS signaling activity that was only slightly reduced compared with full-length LL-37 but had no detectable antimicrobial activity against GAS as assessed by assays of bacterial killing, growth inhibition, and cell membrane injury. Similarly, the closely related peptides RI-11, KR-11, and KR-12 had reduced antimicrobial activity compared with LL-37 but similar CsrRS signaling activity. This dissociation of CsrRS signaling from antimicrobial activity provides compelling evidence that LL-37 signaling is not simply a consequence of membrane injury but rather reflects a specific interaction with CsrS, an interaction that appears to depend on a 10-amino acid core sequence of LL-37 represented by the RI-10 peptide. Indeed, all signaling-competent peptides in the qRT-PCR signaling assays harbored all 10 of these residues (Table 2). Additional evidence for binding specificity is presented from amino acid substitutions of RI-10. Both conservative and nonconservative changes (RI-10 L10A, L10F, and R1A) resulted in decreased signaling ability, suggesting that binding and/or signaling was affected by specific amino acids in the peptide. An all-D enantiomer (D-RI-10) also was unable to signal through CsrS, suggesting that the interaction between the two is not simply because of the amphipathic nature of the peptide, further supporting the specific nature of LL-37 binding to CsrS. These data all support the hypothesis of a direct ligand/receptor interaction between LL-37 and CsrS.
If CsrRS signaling by LL-37 reflects a specific interaction rather than a response to membrane damage, the interaction is expected to occur with the ECD of the cell surface-associated histidine kinase, CsrS. Previous work showed that CsrS has a surface-exposed domain that can be labeled by biotinylation of surface proteins on intact GAS cells (11). We found that a fusion protein representing the predicted CsrS ECD fused to MBP could pull down LL-37 from solution, whereas a control construct of MBP could not. We also tested an analogous construct in which a cluster of three acidic amino acids in the ECD were substituted with their uncharged analogs, a change shown previously to abrogate LL-37 signaling through CsrS (11). The triple-mutant construct showed detectable binding to LL-37, but binding was much reduced compared with the native ECD. These results indicate that LL-37 binds to the CsrS ECD and that binding is in part dependent on a cluster of three acidic residues shown previously to be required for LL-37-induced signaling through CsrS.
The GAS proteases SpeB and SIC have been shown to cleave LL-37, reducing its antimicrobial activity (39–41). In addition, LL-37 may be degraded by the proteolytic action of human plasmin associated with the GAS cell surface upon exposure of the bacteria to human blood plasma (35). It is therefore likely that GAS is exposed to fragments of LL-37 during the process of establishing infections. However, the antimicrobial and signaling effects of LL-37 fragments for GAS had not previously been investigated. Signaling-competent LL-37 fragments generated by proteolysis could induce an invasive phenotype during infection, even as the antimicrobial activity of LL-37 is inactivated by GAS and/or host proteases.
The observation of direct binding of LL-37 to the CsrS ECD, together with the finding that CsrRS signaling can be dissociated from GAS antimicrobial activity, provides new and direct support for specific binding to CsrS as the mechanism for LL-37-mediated signaling through CsrRS. The striking specificity of CsrRS signaling by LL-37 is consistent with a role of this two-component system in adaptation of GAS to survival in the human host in the face of attack by innate immune effectors. The ability of GAS to detect and respond to LL-37 as an indicator of the host innate immune response enables rapid deployment of virulence factors that enhance bacterial resistance to host control of GAS infection.
Acknowledgments
We thank Maghnus O'Seaghdha and Onkar Sharma for advice and technical assistance. We thank Meredith Benson and John Love for helpful advice. We thank James Chou and the Chou laboratory for helpful discussions, advice, and materials. We are also grateful to the New England Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Biomolecule Production Core Laboratory for assistance with protein production and the Harvard Digestive Diseases Imaging Core for technical assistance.
This work was supported, in whole or in part, by NICHD, National Institutes of Health Grant K12-HD000850 (to J. J. V.) and Public Health Service Grant AI29952 from the National Institutes of Health.
- GAS
- group A Streptococcus
- AMP
- antimicrobial peptide
- qRT-PCR
- quantitative RT-PCR
- MIC
- minimum inhibitory concentration
- MBC
- minimum bactericidal concentration
- MBP
- maltose-binding protein
- cfu
- colony-forming units.
REFERENCES
- 1. Cole J. N., Barnett T. C., Nizet V., Walker M. J. (2011) Molecular insight into invasive group A streptococcal disease. Nat. Rev. Microbiol. 9, 724–736 [DOI] [PubMed] [Google Scholar]
- 2. Stock A. M., Robinson V. L., Goudreau P. N. (2000) Two-component signal transduction. Annu. Rev. Biochem. 69, 183–215 [DOI] [PubMed] [Google Scholar]
- 3. Churchward G. (2007) The two faces of Janus: virulence gene regulation by CovR/S in group A streptococci. Mol. Microbiol. 64, 34–41 [DOI] [PubMed] [Google Scholar]
- 4. Gryllos I., Grifantini R., Colaprico A., Jiang S., Deforce E., Hakansson A., Telford J. L., Grandi G., Wessels M. R. (2007) Mg2+ signalling defines the group A streptococcal CsrRS (CovRS) regulon. Mol. Microbiol. 65, 671–683 [DOI] [PubMed] [Google Scholar]
- 5. Levin J. C., Wessels M. R. (1998) Identification of csrR/csrS, a genetic locus that regulates hyaluronic acid capsule synthesis in group A Streptococcus. Mol. Microbiol. 30, 209–219 [DOI] [PubMed] [Google Scholar]
- 6. Sumby P., Whitney A. R., Graviss E. A., DeLeo F. R., Musser J. M. (2006) Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog. 2, e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Graham M. R., Smoot L. M., Migliaccio C. A., Virtaneva K., Sturdevant D. E., Porcella S. F., Federle M. J., Adams G. J., Scott J. R., Musser J. M. (2002) Virulence control in group A Streptococcus by a two-component gene regulatory system: global expression profiling and in vivo infection modeling. Proc. Natl. Acad. Sci. U.S.A. 99, 13855–13860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Love J. F., Tran-Winkler H. J., Wessels M. R. (2012) Vitamin D and the human antimicrobial peptide LL-37 enhance group A Streptococcus resistance to killing by human cells. MBio 3, e00394–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Treviño J., Perez N., Ramirez-Peña E., Liu Z., Shelburne S. A., 3rd, Musser J. M., Sumby P. (2009) CovS simultaneously activates and inhibits the CovR-mediated repression of distinct subsets of group A Streptococcus virulence factor-encoding genes. Infect. Immun. 77, 3141–3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alam F. M., Turner C. E., Smith K., Wiles S., Sriskandan S. (2013) Inactivation of the CovR/S virulence regulator impairs infection in an improved murine model of Streptococcus pyogenes naso-pharyngeal infection. PLoS One 8, e61655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tran-Winkler H. J., Love J. F., Gryllos I., Wessels M. R. (2011) Signal transduction through CsrRS confers an invasive phenotype in group A Streptococcus. PLoS Pathog. 7, e1002361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. O'Seaghdha M., Wessels M. R. (2013) Streptolysin O and its co-toxin NAD-glycohydrolase protect group A Streptococcus from xenophagic killing. PLoS Pathog. 9, e1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Engleberg N. C., Heath A., Miller A., Rivera C., DiRita V. J. (2001) Spontaneous mutations in the CsrRS two-component regulatory system of Streptococcus pyogenes result in enhanced virulence in a murine model of skin and soft tissue infection. J. Infect. Dis. 183, 1043–1054 [DOI] [PubMed] [Google Scholar]
- 14. Engleberg N. C., Heath A., Vardaman K., DiRita V. J. (2004) Contribution of CsrR-regulated virulence factors to the progress and outcome of murine skin infections by Streptococcus pyogenes. Infect. Immun. 72, 623–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ikebe T., Ato M., Matsumura T., Hasegawa H., Sata T., Kobayashi K., Watanabe H. (2010) Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoS Pathog. 6, e1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burton M. F., Steel P. G. (2009) The chemistry and biology of LL-37. Nat. Prod. Rep. 26, 1572–1584 [DOI] [PubMed] [Google Scholar]
- 17. Dürr U. H., Sudheendra U. S., Ramamoorthy A. (2006) LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta 1758, 1408–1425 [DOI] [PubMed] [Google Scholar]
- 18. Bowdish D. M., Davidson D. J., Lau Y. E., Lee K., Scott M. G., Hancock R. E. (2005) Impact of LL-37 on anti-infective immunity. J. Leukoc. Biol. 77, 451–459 [DOI] [PubMed] [Google Scholar]
- 19. Li X., Li Y., Han H., Miller D. W., Wang G. (2006) Solution structures of human LL-37 fragments and NMR-based identification of a minimal membrane-targeting antimicrobial and anticancer region. J. Am. Chem. Soc. 128, 5776–5785 [DOI] [PubMed] [Google Scholar]
- 20. Vandamme D., Landuyt B., Luyten W., Schoofs L. (2012) A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cell. Immunol. 280, 22–35 [DOI] [PubMed] [Google Scholar]
- 21. Porcelli F., Verardi R., Shi L., Henzler-Wildman K. A., Ramamoorthy A., Veglia G. (2008) NMR structure of the cathelicidin-derived human antimicrobial peptide LL-37 in dodecylphosphocholine micelles. Biochemistry. 47, 5565–5572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang G. (2008) Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J. Biol. Chem. 283, 32637–32643 [DOI] [PubMed] [Google Scholar]
- 23. Epand R. F., Wang G., Berno B., Epand R. M. (2009) Lipid segregation explains selective toxicity of a series of fragments derived from the human cathelicidin LL-37. Antimicrob. Agents Chemother. 53, 3705–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gryllos I., Tran-Winkler H. J., Cheng M. F., Chung H., Bolcome R., 3rd, Lu W., Lehrer R. I., Wessels M. R. (2008) Induction of group A Streptococcus virulence by a human antimicrobial peptide. Proc. Natl. Acad. Sci. U.S.A. 105, 16755–16760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ruiz N., Silhavy T. J. (2005) Sensing external stress: watchdogs of the Escherichia coli cell envelope. Curr. Opin. Microbiol. 8, 122–126 [DOI] [PubMed] [Google Scholar]
- 26. Sawai M. V., Waring A. J., Kearney W. R., McCray P. B., Jr., Forsyth W. R., Lehrer R. I., Tack B. F. (2002) Impact of single-residue mutations on the structure and function of ovispirin/novispirin antimicrobial peptides. Protein Eng. 15, 225–232 [DOI] [PubMed] [Google Scholar]
- 27. Gryllos I., Levin J. C., Wessels M. R. (2003) The CsrR/CsrS two-component system of group A Streptococcus responds to environmental Mg2+. Proc. Natl. Acad. Sci. U.S.A. 100, 4227–4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schrager H. M., Rheinwald J. G., Wessels M. R. (1996) Hyaluronic acid capsule and the role of streptococcal entry into keratinocytes in invasive skin infection. J. Clin. Invest. 98, 1954–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nizet V., Ohtake T., Lauth X., Trowbridge J., Rudisill J., Dorschner R. A., Pestonjamasp V., Piraino J., Huttner K., Gallo R. L. (2001) Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 414, 454–457 [DOI] [PubMed] [Google Scholar]
- 30. Lauth X., von Köckritz-Blickwede M., McNamara C. W., Myskowski S., Zinkernagel A. S., Beall B., Ghosh P., Gallo R. L., Nizet V. (2009) M1 protein allows group A streptococcal survival in phagocyte extracellular traps through cathelicidin inhibition. J. Innate Immun. 1, 202–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Feng X., Sambanthamoorthy K., Palys T., Paranavitana C. (2013) The human antimicrobial peptide LL-37 and its fragments possess both antimicrobial and antibiofilm activities against multidrug-resistant Acinetobacter baumannii. Peptides 49, 131–137 [DOI] [PubMed] [Google Scholar]
- 32. Hayashi K., Kojima C. (2010) Efficient protein production method for NMR using soluble protein tags with cold shock expression vector. J. Biomol. NMR 48, 147–155 [DOI] [PubMed] [Google Scholar]
- 33. Que Y. A., Haefliger J. A., Francioli P., Moreillon P. (2000) Expression of Staphylococcus aureus clumping factor A in Lactococcus lactis subsp. cremoris using a new shuttle vector. Infect. Immun. 68, 3516–3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Benson M. A., Lilo S., Nygaard T., Voyich J. M., Torres V. J. (2012) Rot and SaeRS cooperate to activate expression of the staphylococcal superantigen-like exoproteins. J. Bacteriol. 194, 4355–4365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hollands A., Gonzalez D., Leire E., Donald C., Gallo R. L., Sanderson-Smith M., Dorrestein P. C., Nizet V. (2012) A bacterial pathogen co-opts host plasmin to resist killing by cathelicidin antimicrobial peptides. J. Biol. Chem. 287, 40891–40897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mumcu G., Cimilli H., Karacayli U., Inanc N., Ture-Ozdemir F., Eksioglu-Demiralp E., Ergun T., Direskeneli H. (2012) Salivary levels of antimicrobial peptides Hnp 1–3, LL-37 and S100 in Behcet's disease. Arch. Oral Biol. 57, 642–646 [DOI] [PubMed] [Google Scholar]
- 37. Tao R., Jurevic R. J., Coulton K. K., Tsutsui M. T., Roberts M. C., Kimball J. R., Wells N., Berndt J., Dale B. A. (2005) Salivary antimicrobial peptide expression and dental caries experience in children. Antimicrob. Agents Chemother. 49, 3883–3888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim J. E., Kim B. J., Jeong M. S., Seo S. J., Kim M. N., Hong C. K., Ro B. I. (2005) Expression and modulation of LL-37 in normal human keratinocytes, HaCaT cells, and inflammatory skin diseases. J. Korean Med. Sci. 20, 649–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Frick I. M., Akesson P., Rasmussen M., Schmidtchen A., Björck L. (2003) SIC, a secreted protein of Streptococcus pyogenes that inactivates antibacterial peptides. J. Biol. Chem. 278, 16561–16566 [DOI] [PubMed] [Google Scholar]
- 40. Schmidtchen A., Frick I. M., Andersson E., Tapper H., Björck L. (2002) Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol. Microbiol. 46, 157–168 [DOI] [PubMed] [Google Scholar]
- 41. Johansson L., Thulin P., Sendi P., Hertzén E., Linder A., Akesson P., Low D. E., Agerberth B., Norrby-Teglund A. (2008) Cathelicidin LL-37 in severe Streptococcus pyogenes soft tissue infections in humans. Infect. Immun. 76, 3399–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]