

Abstract
Uncontrolled proliferation is the hallmark of cancer and other proliferative disorders and abnormal cell cycle regulation is, therefore, common in these diseases. Cyclin-dependent kinases (CDKs) play a crucial role in the control of the cell cycle and proliferation. These kinases are frequently deregulated in various cancers, viral infections, neurodegenerative diseases, ischemia and some proliferative disorders. This led to a rigorous pursuit for small-molecule CDK inhibitors for therapeutic uses. Early efforts to block CDKs with nonselective CDK inhibitors led to little specificity and efficacy but apparent toxicity, but the recent advance of selective CDK inhibitors allowed the first successful efforts to target these kinases for the therapies of several diseases. Major ongoing efforts are to develop CDK inhibitors as monotherapies and rational combinations with chemotherapy and other targeted drugs.
Keywords: cancer, kinase, CDK, small molecule inhibitors
1. Overview of CDKs and CDK Inhibitors
The cell cycle is a period between the successive divisions of a cell. During this period, the contents of the cell must be accurately replicated. The processes that permit the cell to divide are very precisely controlled by a multitude of enzymatic reactions amongst which the protein kinase-triggered protein phosphorylation plays a major role. In eukaryotes, there are four main stages/phases of cell cycle namely the Gap-1 (G1) phase, Synthesis (S) phase, Gap-2 (G2) and Mitosis (M) phases. An extended phase of Gap-1 phase is coined as Gap-0 (G0) phase or Resting phase [1].
Cyclin-dependent kinases (CDKs) are members of subfamily of serine/threonine kinases which are found in both unicellular organisms such as yeast and multicellular organisms such as plants, humans and other mammals [1,2,3,4,5]. These kinases are dependent on cyclin-binding activity for their activation, thereby resulting in specific complex formation. In fission yeast Schizosaccharomyces pombe, the CDK1 gene encodes a 34 kDa protein serine/threonine kinase. The mutations in CDK1 cause the cell to halt at one of two distinct points: G1 or M phases [3]. In human genome, there are 21 genes that encode CDKs (Figure 1) and five genes encode more distant protein kinases termed as “CDK like kinases (CDKLs)” [6].
Figure 1.

Phylogenetic tree of human CDKs (created using UniProt multiple alignment).
According to Malumbres and Barbacid [5], various CDKs activate the different stages of the cell cycle from G1 to mitosis. Each CDK/cyclin complex is responsible for transition or progression of a given phase within the cell cycle. Complexes of cyclin D in combination with CDKs 4 and 6; cyclin E in combination with CDK2 regulate the G0–G1 transition and the early phases of G1 via phosphorylation of Rb. The cyclin E/CDK2 complex is also involved in G1–S transition. CDK2 can also bind cyclin A for the entire progression of S-phase. The complexes of cyclin A in combination with CDK1 participate in the S–G2 and cyclin B in combination with CDK1 participates in the G2–M. CDK3 also seems to participate in the phosphorylation of Rb as it is highly related to CDK’s 1 and 2 and interacts with both cyclins A and E [7]. CDK5 is expressed in brain and are activated by proteins p35 and p39 which are not considered to be cyclins. CDK5 mostly acts in neural cells and is associated with activities such as cell survival, transcription, migration and membrane transportation [8]. CDK7, a constituent of both CDK-activating kinase (CAK) and general transcription factor TFIIH, acts upstream of cell cycle-regulatory CDKs and affects transcription [9]. The regulation of transcription is also performed by CDKs 8 [10] and 9 [11], which bind to cyclins C and T, respectively. Despite the lack of partner cyclin for CDK10, this kinase seems to play a role in the regulation of G2–M phase through the inhibition of Ets2 transactivation [12]. The CDK11 can be involved in mRNA splicing upon binding to cyclin L [13] as well as repress proliferation upon its interaction with cyclin D [14]. CDK 12 and 13 apparently bind cyclin K and subsequently maintain the genome stability, primarily by phosphorylating serine 2 in the C-terminal domain of RNA polymerase II [15]. Cyclin Y in a complex with 14-3-3 proteins activates CDK14, which is important in embryonic patterning [16]. CDK15 acts like an antiapoptotic protein by inducing phosphorylation of surviving at threonine 34 [17]. No cyclin-like, or other protein interaction is known for this kinase to date.
Most CDKs are known to play a role in different types of human cancers [4]. CDK1 is known to have a diagnostic value in esophageal and breast cancers, while the expression of CDK2 or its activity seems to have been utilized towards the prognosis of breast, ovarian and oral cancers. In other types of cancer such as the ovarian, urinary bladder, endometrial or oral, CDK4 expression is impaired. CDK5 is known to play a role in the lung cancer, while CDK6 is misexpressed in oral cancer. The polymorphisms of CDK7 are known to effect breast cancer.
Abnormalities in CDK characteristics such as its expression, activity and regulation have also been found in pathological conditions such as viral infections, neurodegenerative disorders and proliferative diseases etc. That led to a rigorous search for small-molecule CDK inhibitors for the therapeutic purposes.
The first CDK inhibitor was 2-hydroxyethylamino-6-benzylamino-9-methylpurine, which was discovered by Vesely and Meijer [18]. It was later renamed olomoucine, after Olomouc, the town of Vesely’s university in the Czech Republic. It was the first inhibitor, which had the selectivity for CDKs (IC50 = 3–7 μM) and, to a lesser extent, for MAP kinases (IC50 = 25 μM). It was found to target the ATP-binding pocket of CDKs and inhibit them by competing with ATP binding.
The next inhibitor, specific for CDKs was 2-(R)-(1-ethyl-2-hydroxyethylamino)-6-benzylamino-9-isopropylpurine, which was later renamed roscovitine (after Roscoff, where the lab which discovered the compound was located) for convenience [18]. (R)-Roscovitine is also known by the names of CYC-202 and seliciclib. It inhibits CDK1, CDK2, CDK5 and CDK7 (IC50 = 0.2–0.5 μM) but is a poor inhibitor for CDK4 and CDK6 (IC50 > 100 μM) [4]. Roscovitine was also tested in Phase I clinical trial for patients with solid tumors [19]. Results showed lack of activity, but disease stabilization was detected in eight out of 25 patients. Another Phase I trial for patients with hepatocellular carcinoma, showed partial response in one patient and tumor stabilization in six out of 25 patients involved [20].
The discovery of olomoucine and roscovitine and their co-crystal structures with CDK2 inspired the whole field of discovery of purine analogue inhibitors. That ultimately led to the identification of purvalanols which were very potent highly selective. Purvalanol A is an inhibitor selective for CDK2 (IC50 = 4–70 nM) and CDK5 (IC50 = 75 nM), but less selective toward CDK4 (IC50 = 850 nM) [4]. Purvalanol B is a selective for CDK1 (IC50 = 6 nM), CDK2 (IC50 = 6–9 nM), CDK5 (IC50 = 6 nM), but unselective towards CDK 4/6 or other kinases (IC50 > 10000).
Another type of CDK inhibitors discovered early is the flavonoids. The first discovered and most prominent of those is flavopiridol, also variously known as L-868275, HMR-1275, alvocidib or NSC-649890 (Figure 2). It is a broad-range CDK inhibitor. It inhibits CDK9 at IC50 value of 8nM and CDK1, CDK2, CDK4 and CDK7 in an IC50 value range of 0.04–0.4 μM. It less effectively inhibits epidermal growth factor receptor (IC50 = 21 μM) and protein kinase A (IC50 = 122 μM) [4]. Flavopiridol was the first CDK inhibitor used in human clinical trials. Several Phase I clinical trials showed that flavopiridol had an antitumor effect as single agent [20,21,22] or in combination with chemotherapeutic drugs [23,24,25,26,27,28,29]. Several Phase II trials were also performed, showing high response rates in chronic myeloid leukemia [30] and medium responses in pancreatic cancer [31].
Figure 2.

Flavopiridol.
Yet another compound fascaplysin, originally isolated from the sponge Fascaplysinopsis bergquist [32,33], is a kinase inhibitor selective for CDK4 (IC50 = 0.35 μM) and CDK6 (IC50 = 3.4 μM) and not selective for the other CDKs or other kinases. (IC50~500 μM) [4].
Dioxobenzothiazoles, studied for their antifungal activities, were also found to be selective and potent inhibitors of CDKs. Namely 5-arylamino-2-methyl-4,7-dioxobenzothiazoles showed incredible selectivity towards CDK4 (IC50 = 6–7μM). They were also quite cytotoxic against various cancer cells (IC50 = 0.2–3.6 μg/mL). One of these compounds was commercialized under the name of ryuvidine [34].
To date, more than 20 different CDK small molecule inhibitors have been developed [4,35], which can be subdivided into two main groups: broad-range inhibitors (such as above mentioned flavopiridol, olomoucine and roscovitine) and specific inhibitors (such as above mentioned purvalanols, fascaplysin and ryuvidine) (Table 1).
Table 1.
List of CDK inhibitors.
| Inhibitor | Alternative Names | Kinases Inhibited | In Clinical Development Yes/No | Refs. |
|---|---|---|---|---|
| 3α-Amino-5α-androstane | CDK5 | No | [36] | |
| 7x | CDK4, ARK5 | No | [37] | |
| AG-024322 | CDK1, CDK2, CDK4 | Yes | [4] | |
| AMG 925 | CDK4, FLT3 | No | [38] | |
| AT7519 | CDK1, CDK2 | Yes | [39] | |
| AZD5438 | CDK1, CDK2, CDK4, CDK5, CDK6, CDK9 | Yes | [4] | |
| BAY 1000394 | CDK1, CDK2, CDK3, CDK4, CDK7, CDK9 | No | [40] | |
| BML-259 | CDK2, CDK5 | No | [4] | |
| Compound 1 | CDK4, ABL, FGFR1, FYN, KDR, LCK, LYN, SRC | No | [41] | |
| Compound 530 | CDK4, CDK4 | No | [42] | |
| CR8 | CDK2, CDK5 | No | [43] | |
| Dinaciclib | MK-7965, SCH 727965 | CDK1, CDK2, CDK5, CDK9 | Yes | [44] |
| F07#13 | CDK2, CDK9 | No | [45] | |
| Fascaplysin | CDK4, CDK6 | No | [4] | |
| Flavopiridol | L-868275, HMR-1275, alvocidib, NSC-649890 | CDK1, CDK2, CDK4, CDK7 | Yes | [4] |
| Kenpaullone | NSC 664704, 9-bromopaullone | CDK1, CDK2, CDK5 | No | [4] |
| LY2835219 | abemaciclib | CDK4, CDK6 | Yes | [46] |
| NBI1 | CDK2 | No | [47] | |
| NU2058 | CDK1, CDK2 | No | [4] | |
| Olomoucine | CDK1, CDK2, CDK5 | No | [4] | |
| P276-00 | CDK1 | Yes | [4] | |
| PD-0332991 | CDK4, CDK6 | Yes | [4] | |
| PHA-793887 | CDK1, CDK2, CDK4 | Yes (Stopped) | [48] | |
| Purvalanol A/B | CDK1, CDK2, CDK5 | No | [4] | |
| R547 | Ro-4584820 | CDK1, CDK2, CDK4 | Yes | [4] |
| RGB-286638 | Pan-CDK | No | [49] | |
| Roscovitine | CY-202, (R)-roscovitine, seliciclib | CDK2, CDK5 | Yes | [4] |
| Ryuvidine | CDK4 | No | [4] | |
| SNS-032 | BMS-387032 | CDK2, CDK7, CDK9 | Yes | [4] |
| SU 9516 | CDK1, CDK2 | No | [4] | |
| VMY-1-101 | CDK2, CDK5, CDK7 | No | [50] | |
| VMY-1-103 | CDK2, CDK5, CDK7 | No | [50] |
2. Advances in Preclinical Studies
One of the interesting fields in CDK inhibitor research in recent years was the advance of ATP-noncompetitive inhibitors. Lo et al. identified an ATP-noncompetitive compound, using time-resolved fluorescence resonance energy transfer assay (TR-FRET) by screening more than 250,000 compounds and determining their IC50 values against CDK4 [41]. Three compounds were found to have an IC50 ratio below 2.5 μM. Compound 1 was the further tested for the specificity and it was found that is highly selective for CDK4 versus the other 34 serine/threonine kinases tested. However, it was also potent against a number of tyrosine kinases tested, such as ABL, FGFR1, FYN, KDR, LCK, LYN, and SRC. Another ATP-noncompetitive compound, an all D-amino acid hexapeptide, termed NBI1, which interferes with the formation of the CDK2/cyclin A complex, was found to induce apoptosis and inhibit proliferation of tumor cell lines [47]. Moreover, it was shown that NBI1 sensitizes erlotinib-resistant tumor cells to the treatment and erlotinib-sensitive cells to the smaller dose of erlotininb [51]. Premnath et al. used replacement with partial ligand alternatives through computational enrichment (REPLACE) approach to design ATP-noncompetitive CDK inhibitors [42]. Inhibitory peptides were used as basis, and then converted to a less peptidic inhibitor. The 3,4-diethoxy analog (compound 530) was found to be the most potent against both CDK2 (IC50 = 5.2 μM) and CDK4 (IC50 = 3 μM). Van Duyne et al. designed a new CDK9 and CDK2 inhibitor, using structure-based analysis of cyclin/CDK complexes as well as blocking peptides [45]. After screening 52 compounds, one of them, named F07#13 had an IC50 of 0.12 μM towards CDK2 and CDK9, which it achieved by disrupting interactions between CDK2/Cyclin E and CDK9/Cyclin T. Moreover, it inhibited HIV-1 viral replication in humanized mouse models. Bioluminescence resonance energy transfer (BRET)-based screening assay has also been used to identify 3α-amino-5α-androstane as an inhibitor of CDK5/p25 interaction, and thus CDK5 kinase activity (IC50 = 6 μM) [36].
Another interesting approach is a design of fluorescent CDK inhibitors. Yenugonda et al. using rational drug design, designed and fluorescent CDK inhibitors VMY-1-101 and VMY-1-103, which are based on a purvalanol B [50]. They were found to be potent inhibitors of CDK2, CDK5 and CDK7 and had an anti-proliferative activity (IC50 = 4 μM or 10–19 μM for VMY-1-103 or VMY-1-101, respectively) against breast cancer cell lines. In addition these inhibitors were easily traceable, therefore intracellular localization of the compounds could be tracked using confocal microscopy.
Recently, there were a lot of efforts studying both long-known as well as newer CDK inhibitors in animal cancer models. AMG 925, a FLT3/CDK4 dual inhibitor was found to inhibit growth of subcutaneous MOLM13 xenograft tumors and systemic MOLM13-Luc xenograft tumors in nude mice, a disease model for acute myeloid leukemia [38]. A new CDK4/ARK5 inhibitor 8-cyclopentyl-2-[4-(4-methylpiperazin-1-yl)-phenylamino]-7-oxo-7,8-dihydropyrido[2,3-d]pyrimidine-6-carbonitrile, also known as 7x, was shown to lead to a dose-dependent inhibition of tumor growth and reduction in tumor weight over a 21 day period in nude mice breast cancer model [37]. PD-0332991, a CDK4/6 inhibitor prolonged survival in the Ink4a-ARF deficient brainstem glioma model mice (42 days vs. 47 days, p = 0.033) [52]. Hayashi et al. found that flavopiridol in combination with temozolomide suppressed tumor growth in subcutaneous glioma nude mouse xenografts (p < 0.05) [53]. RGB-286638, a broad-range CDK inhibitor, was found to significantly suppress multiple myeloma tumor growth in SCID mice, with more than 80% tumor growth inhibition [49]. BAY 1000394, a pan-CDK inhibitor, was also effective in a xenograft model of mammary tumor in nude mice [40]. Martin et al. found that radiation-induced salivary gland dysfunction can be prevented by roscovitine pretreatment, which might prevent the side effects of radiation therapy in tumor surrounding normal tissues [54]. Roscovitine and its derivative CR8 were also found to inhibit renal cystogenesis and hepatic cystogenesis in Pkd1-conditional knockout mice [55]. AZD5438, a CDK1, CDK2, and CDK9 inhibitor, was found to enhance the radiosensitivity of non-small cell lung carcinoma cells in athymic nude mice [39]. The CDK4 inhibitor P276, in combination with gemcitabine, inhibited tumor growth and angiogenesis of pancreatic cancer cell xenografts in nude mice [56]. CDK inhibitors, targeting neuronal functions we also studied in animal models. Yang et al. studied roscovitine in a rat model of chronic compression of dorsal root ganglion and found that that intrathecal administration of roscovitine alleviates pain by downregulating the expression of NR2A [57]. CR8 a second-generation analog of roscovitine, was shown to provide neuroprotection in experimental traumatic brain injury by reducing cortical, hippocampal, and thalamic neuronal loss and cortical microglial and astrocyte activation [43,58].All the inhibitor information mentioned in this section is summarized in Table 2.
Table 2.
List of CDK inhibitors described in preclinical study section.
| Inhibitor | Kinases Inhibited | Tested | Disease(s) | Refs. |
|---|---|---|---|---|
| Compound 1 | CDK4, ABL, FGFR1, FYN, KDR, LCK, LYN, SRC | In vitro | n.a. | [41] |
| NBI1 | CDK2 | In vitro, cell lines HCT116, HT29, T98G, A2780, MDA-MB-468, SKBr3, MCF-7, A549, Jurkat, HL60 and Saos-2 | colon carcinoma, glioblastoma, ovarian carcinoma, breast carcinoma, acute myeloid leukemia, lung carcinoma, osteosarcoma | [47,51] |
| Compound 530 | CDK2, CDK4 | In vitro | n.a. | [42] |
| F07#13 | CDK2, CDK9 | In vitro, mouse models | AIDS | [45] |
| 3α-Amino-5α-androstane | CDK5 | In vitro, Saccharomyces cerevisiae | n.a. | [36] |
| VMY-1-101 | CDK2, CDK5, CDK7 | In vitro, cell lines MDA-MB-231, MCF-7 | breast carcinoma | [50] |
| VMY-1-103 | CDK2, CDK5, CDK7 | In vitro, cell lines MDA-MB-231, MCF-7 | breast carcinoma | [50] |
| AMG 925 | CDK4, FLT3 | xenograft mouse model | acute myeloid leukemia | [38] |
| 7x | CDK4, ARK5 | In vitro, panel of human tumor cell lines, xenograft mouse model | various human cancers (cell lines), breast carcinoma (mouse model) | [37] |
| PD 0332991 | CDK4, CDK6 | mouse model | brainstem glioma | [52] |
| Flavopiridol | CDK1, CDK2, CDK4, CDK7 | xenograft mouse model | glioma | [53] |
| RGB-286638 | pan-CDK | xenograft mouse model | multiple myeloma | [49] |
| BAY 1000394 | CDK1, CDK2, CDK3, CDK4, CDK7, CDK9 | mouse model | breast carcinoma | [40] |
| Roscovitine | CDK2, CDK5 | mouse model | radiation-induced salivary gland dysfunction, renal and hepatic cystogenesis, pain | [54,55,57] |
| CR8 | CDK2, CDK5 | mouse model | renal and hepatic cystogenesis, traumatic brain injury | [43,55,58] |
| AZD5438 | CDK1, CDK2 | xenograft mouse model | non-small cell lung carcinoma | [39] |
| P276 | CDK4 | xenograft mouse model | pancreatic carcinoma | [56] |
3. Advances in clinical studies
Flavopiridol was still popular in clinical trials of recent several years. A Phase I study of flavopirirdol as a single agent was performed on relapsed myeloma after at least two prior treatments [59]. Fifteen patients were treated and only one of them had marginal response (a decrease in his monoclonal protein >50%). Side effects included significant diarrhea, cytopenias and transaminase elevation. Another Phase I trial involved flavopiridol in combination with doxorubicin in advanced sarcomas [60]. 28 patients were involved and the combination of flavopiridol and doxorubicin achieved a considerable disease control, with 68% (19/28) achieving partial response or stable disease. Main dose-limiting toxicities were neutropenia, leukopenia, and febrile neutropenia. Yet another Phase I clinical trial was dealing with the combination of flavopiridol and imatinib mesylate in patients with Bcr-Abl+ chronic myelogenous leukemia [61]. Of 21 patients enrolled in a study, five patients responded, including four sustained responses (for 2, 4, 15 and 30 months, respectively) and four patients had stable disease. No clinically significant pharmacokinetic interactions between imatinib and flavopiridol were observed. A Phase II trial was performed on the combination of flavopiridol with cisplatin in platin-resistant ovarian and primary peritoneal carcinoma [62]. Of 40 platin-resistant patients seven patients achieved a confirmed response and ten patients maintained stable disease. Four of five platin-sensitive patients responded, but the cohort was closed. Main toxicity symptoms included neutropenia, nausea, vomiting, fatigue, thrombosis and anemia. Of nine high-risk chronic lymphocytic leukemia patients treated with flavopiridol, cyclophosphamide and rituximab combination, three achieved complete response, four-partial response, one had stable disease, and one had progressive disease [63]. The overall response rate of 46% was observed in a cohort of 112 patients with refractory or relapsed chronic lymphocytic leukemia, treated with flavopiridol as a single agent [64]. Another study of 116 patients, divided into two categories (≥70 years and <70 years) for comparison, showed feasibility of flavopiridol as single agent in elderly patients (response rates (43% vs. 47%, respectively)) [65]. Phase I trial of bortezomib and flavopiridol in patients with recurrent or refractory B-cell neoplasms, showed overall response rate of 44% [66]. Another Phase I trial of bortezomib and flavopiridol in patients with recurrent or refractory indolent B-cell neoplasms performed by the same group, showed 33% overall response rate [67].
One of the most popular CDK inhibitor in clinical trials in the recent years was dinaciclib (MK-7965, SCH 727965) (Figure 3), the inhibitor of CDK1, CDK2, CDK5, and CDK9. A Phase I trial on the effect of dinaciclib in combination with aprepitant was performed in patients with advanced malignancies [44]. Aprepitant is used for the prevention of chemotherapy-induced nausea and vomiting, is known as an inhibitor and inducer of CYP3A4, which metabolizes dinaciclib. Coadministration of dinaciclib with aprepitant resulted in no clinically significant effect on the pharmacokinetics and did not alter the safety profile of dinaciclib. The first Phase I clinical trial on dinaciclib as a single agent was performed on patients with advanced malignancies [68]. Forty-eight patients with various solid tumors were treated and 10 of them achieved prolonged stable disease for at least four treatment cycles. Adverse effects were mild, the most common being nausea, anemia, decreased appetite and fatigue. A phase II multi-center study of dinaciclib for relapsed and/or refractory AML was performed on 20 patients [69]. Temporary decrease in peripheral blood and/or bone marrow blasts was observed in 60% of patients. Four of 13 (31%) patients with circulating blasts had >50% decrease and 6 (46%) >80% decrease in the absolute blast count within 1–8 days of the first dinaciclib dose. Toxicities included diarrhea, fatigue, transaminitis, and manifestations of tumor lysis syndrome, with one patient who deceased of acute renal failure. Another Phase II study was performed of dinaciclib versus erlotinib in patients with non-small cell lung cancer [70]. Unfortunately, it was found that dinaciclib was not successful as monotherapy in non-small cell lung cancer. Most common toxicities included neutropenia, leukopenia, vomiting, and diarrhea. Yet another Phase II study was performed on dinaciclib versus capecitabine in patients with advanced breast cancer [71]. Dinaciclib treatment demonstrated antitumor activity in two of seven patients with ER-positive and ERBB 2-negative metastatic breast cancer, however efficacy was not superior to capecitabine (p = 0.991). Toxicities included neutropenia, leukopenia, increase in aspartate aminotransferase, and febrile neutropenia. Phase I nonrandomized dose-escalation trial was performed, where patients with relapsed or refractory chronic lymphocytic leukemia were treated with dinaciclib and rituximab [72]. Four out of six patients achieved stable disease, and one patient achieved complete response. Drug-related adverse events were mostly hematological, digestive and metabolic and no dose-limiting toxicities were observed. Dinaciclib was also moved into Phase III development for refractory chronic lymphocytic leukemia [73]. Phase I/II clinical trial Dinaciclib in patients with relapsed multiple myeloma showed promise as single agent [74]. The overall confirmed response rate was 3 of 27 (11%). Adverse effects included leukopenia, thrombocytopenia, gastrointestinal symptoms, alopecia, and fatigue.
Figure 3.
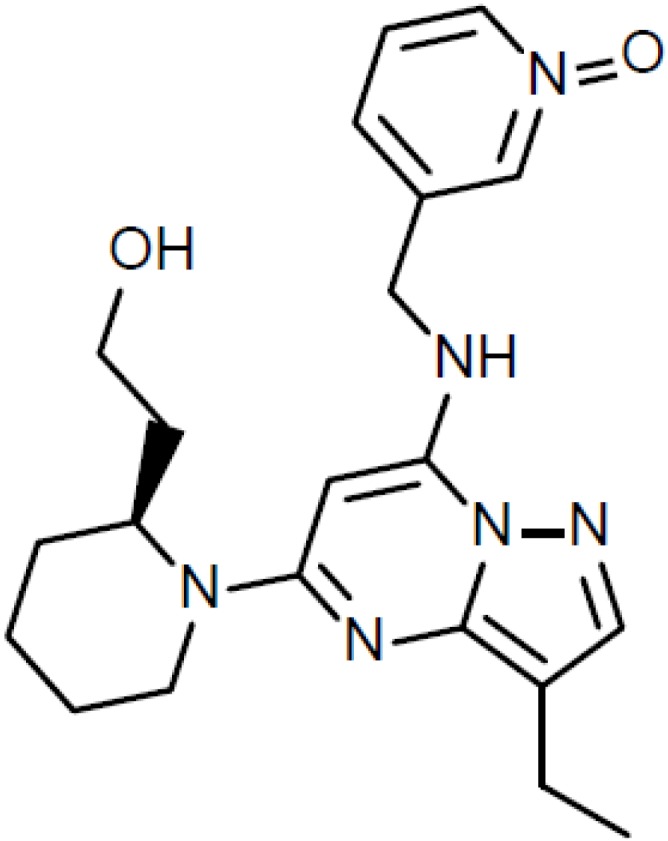
Dinaciclib.
PD 0332991, a CDK4/6 inhibitor (Figure 4), was used in a Phase I clinical trial in patients with advanced cancer [75]. Out of 37 patients 10 (27%) had stable disease for ≥4 cycles of whom six (16%) had prolonged benefit (≥10 cycles). Main toxicities included neutropenia, anemia, leukopenia, fatigue, nausea, and diarrhea. A Phase II trial in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma was also performed [76]. Of 29 patients, 19 (66%) were progression free at 12 weeks, which exceeded expected rate of 40%. Main toxicities included anemia, thrombocytopenia, neutropenia and febrile neutropenia. A Phase II trial of PD0332991 was performed in 37 patients with advanced breast cancer (mostly ER-positive ERBB2-negative), pretreated with chemotherapy [77]. Two of patients had partial response, and 18 had disease stabilization as best response. Drug was well tolerated; toxicities were uncomplicated and included neutropenia, leucopenia, lymphopenia, and thrombocytopenia. Encouraging data led to the Phase II study expansion where patients were randomized to combination therapy compared with letrozole alone as first-line treatment for breast cancer, which was designed in two parts [78]. In first part (66 patients), patients were clinically selected based on ER-positive HER2-negative breast cancer. Partial response rate and disease stabilization were higher for the letrozole and PD 0332991 combination (52% versus 32% and 35% versus 25%, respectively). This significant improvement led to the FDA breakthrough therapy designation in April 2013. In the second part (61 patients), additional eligibility criteria included cyclin D1 gene amplification and/or p16 loss. There was no correlation found between these markers and outcomes, however data is not final yet. Based on these results a Phase III trial of PD-0332991 in estrogen receptor ER-positive breast cancer is planned [73].
Figure 4.

PD 0332991.
Another CDK4/6 inhibitor, LY2835219 (abemaciclib), was shown to be beneficial as monotherapy for patients with metastatic breast cancer, mainly for those with ER/PgR—positive disease [46]. Of 47 patients, 9 (19%) had a partial response, 24 (51%) had stable disease, 11 (23%) experienced disease progression, and three could not be evaluated for a response. The most frequent toxicities included diarrhea, nausea, fatigue, neutropenia, vomiting, and decreased platelet and white-blood cell counts.
Unfortunate fate awaited the CDK1, CDK2 and CDK4 inhibitor PHA-793887 in a Phase I clinical study [48]. It was shown that PHA-793887 induces severe, dose-related hepatic toxicity, and any further clinical development was stopped. All the inhibitor information, mentioned in this section is summarized in Table 3.
Table 3.
List of CDK inhibitors described in clinical study section.
| Treatment | Clinical Trial Phase | Disease(s) | Response Rate | Adverse Effects | Refs. |
|---|---|---|---|---|---|
| Flavopiridol | Phase I | relapsed myeloma | 1/50 (2%) | diarrhea, cytopenias, transaminase elevation | [59] |
| Flavopiridol in combination with doxorubicin | Phase I | advanced sarcomas | 19/28 (68%) | neutropenia, leukopenia, febrile neutropenia | [60] |
| Flavopiridol in combination with imatinib mesylate | Phase I | Bcr-Abl + chronic myelogenous leukemia | 5/21 (24%) | anemia, leukopenia, lymphopenia, thrombocytopenia | [61] |
| Flavopiridol in combination with cisplatin | Phase II | platin-resistant ovarian and primary peritoneal carcinoma | 17/40 (43%) platin-resistant patients; 4/5 (80%) platin-sensitive patients | neutropenia, nausea, vomiting, fatigue, thrombosis, anemia | [62] |
| Flavopiridol in combination with cyclophosphamide and rituximab | Phase I | chronic lymphocytic leukemia | 7/9 (78%) | fatigue, electrolyte disturbances, diarrhea, abdominal discomfort, nausea/vomiting, liver dysfunction, anemia, leukopenia, neutropenia, thrombocytopenia | [63] |
| Flavopiridol | Phase I/II | chronic lymphocytic leukemia | 112/52 (46%) | n.a. | [64] |
| Flavopiridol | ? | chronic lymphocytic leukemia | 41/95 (43%) ≥70 years old; 10/21 (47%) <70 years old | tumor lysis syndrome, cytokine release syndrome, neutropenia, diarrhea, fatigue | [65] |
| Flavopiridol in combination with bortezomib | Phase I | refractory B-cell neoplasms | 7/16 (44%) | neutropenia, lymphopenia, and thrombocytopenia | [66] |
| Flavopiridol in combination with bortezomib | Phase I | Refractory indolent B-cell neoplasms | 13/39 (33%) | leukopenia, lymphopenia, neutropenia, thrombocytopenia, diarrhea, fatigue, sensory neuropathy | [67] |
| Dinaciclib in combination with aprepitant | Phase I | advanced malignancies | n.a. | no change in safety profile of dinaciclib | [44] |
| Dinaciclib | Phase I | advanced malignancies | 10/48 (21%) | nausea, anemia, decreased appetite and fatigue | [68] |
| Dinaciclib | Phase I | relapsed and/or refractory acute myeloid leukemia | 12/20 (60%) | diarrhea, fatigue, transaminitis, manifestations of tumor lysis syndrome; one patient deceased of acute renal failure | [69] |
| Dinaciclib vs. erlotinib | Phase II | non-small cell lung cancer | Not successful | neutropenia, leukopenia, vomiting, diarrhea | [70] |
| Dinaciclib vs. capecitabine | Phase II | advanced breast cancer | 2/7 (29%) (not superior to capecitabine) | neutropenia, leukopenia, increase in aspartate aminotransferase, febrile neutropenia | [71] |
| Dinaciclib vs. capecitabine | Phase I | chronic lymphocytic leukemia | 5/6 (83%) | hematological, digestive and metabolic; no dose-limiting toxicities | [72] |
| Dinaciclib | Phase I/II | relapsed multiple myeloma | 3/27 (11%) | leukopenia, thrombocytopenia, gastrointestinal symptoms, alopecia, fatigue | [74] |
| PD 0332991 | Phase I | advanced cancer | 10/37 (27%) | neutropenia, anemia, leukopenia, fatigue, nausea, diarrhea | [75] |
| PD 0332991 | Phase I | advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma | 19/29 (66%) | anemia, thrombocytopenia, neutropenia, febrile neutropenia | [76] |
| PD 0332991 | Phase II | advanced breast cancer | 20/37 (%) | neutropenia, leucopenia, lymphopenia, thrombocytopenia | [77] |
| PD 0332991 in combination with letrozole vs. letrozole alone | Phase II | advanced breast cancer | 87% vs. 57% (66 patients) | neutropenia, leukopenia, and fatigue | [78] |
| LY2835219 | Phase I | metastatic breast cancer | 33/47 (70%) | diarrhea, nausea, fatigue, neutropenia, vomiting, decreased platelet and white-blood cell counts | [46] |
| PHA-793887 | Phase I | solid tumors | n.a. | severe, dose-related hepatic toxicity | [48] |
4. Perspectives
Recent developments in the small-molecule CDK inhibitor field led to quite a number of compounds with a different range of the inhibited targets, anticancer potencies both in vitro and in vivo and modes of molecular action. However, there is still much room for improvement. First of all, the specificity of the inhibitors, which inhibit close isoforms, is not fully achieved. Especially that is relevant for CDK5, which is involved in neurodegenerative diseases and CDK9, involved in viral replication. Structure and bioinformatics—based approaches should help tackle this problem. On the other hand it is still unclear which CDK or range of CDKs should be targeted in a clinical setup. Clinical (and maybe also preclinical) studies should help to determine whether it is better to use highly selective or rather broad range CDK inhibitors as anticancer therapy.
It also seems that combination therapies with CDK inhibitors are more promising than monotherapies, therefore more chemotherapeutic agents should be evaluated in combination with CDK inhibitors. Other targeted drugs seem to work nicely in combination; therefore this field of clinical research should also be further pursued.
Acknowledgements
Jonas Cicenas is grateful to Swiss Institute of Bioinformatics for the continuous support, as well as to “Enterprise Lithuania” for fruitful collaboration. Mindaugas Valius and Algirdas Kaupinis are grateful for financial support of the project “High throughput proteomics for cancer cell surface protein recognition by quantum dots” (MIP-033/2014) funded by Science Council of Lithuania. We apologize if we inadvertently left out any major contribution in this field.
Author Contributions
Jonas Cicenas has written most of the article. Karthik Kalyan has written first draft of the overview of CDKs. All other authors contributed to the preclinical and clinical advance sections, and/or tables. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Diallo A., Prigent C. The serine/threonine kinases that control cell cycle progression as therapeutic targets. Bull. Cancer. 2011;98:1335–1345. doi: 10.1684/bdc.2011.1467. [DOI] [PubMed] [Google Scholar]
- 2.Doonan J.H., Kitsios G. Functional evolution of cyclin-dependent kinases. Mol. Biotechnol. 2009;42:14–29. doi: 10.1007/s12033-008-9126-8. [DOI] [PubMed] [Google Scholar]
- 3.Pines J. Cyclins and cyclin-dependent kinases: Take your partners. Trends Biochem. Sci. 1993;18:195–197. doi: 10.1016/0968-0004(93)90185-P. [DOI] [PubMed] [Google Scholar]
- 4.Cicenas J., Valius M. The CDK inhibitors in cancer research and therapy. J. Cancer Res. Clin. Oncol. 2011;137:1409–1418. doi: 10.1007/s00432-011-1039-4. [DOI] [PubMed] [Google Scholar]
- 5.Malumbres M., Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005;30:630–641. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 6.Malumbres M., Harlow E., Hunt T., Hunter T., Lahti J.M., Manning G., Morgan D.O., Tsai L.H., Wolgemuth D.J. Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009;11:1275–1276. doi: 10.1038/ncb1109-1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hengstschläger M., Braun K., Soucek T., Miloloza A., Hengstschläger-Ottnad E. Cyclin-dependent kinases at the G1-S transition of the mammalian cell cycle. Mutat. Res. 1999;436:1–9. doi: 10.1016/S1383-5742(98)00022-2. [DOI] [PubMed] [Google Scholar]
- 8.Dhariwala F.A., Rajadhyaksha M.S. An unusual member of the CDK family: CDK5. Cell. Mol. Neurobiol. 2008;28:351–369. doi: 10.1007/s10571-007-9242-1. [DOI] [PubMed] [Google Scholar]
- 9.Fisher R.P. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J. Cell Sci. 2005;118:5171–5180. doi: 10.1242/jcs.02718. [DOI] [PubMed] [Google Scholar]
- 10.Leclerc V., Léopold P. The cyclin C/CDK8 kinase. Prog. Cell Cycle Res. 1996;2:197–204. doi: 10.1007/978-1-4615-5873-6_19. [DOI] [PubMed] [Google Scholar]
- 11.Napolitano G., Majello B., Lania L. Role of cyclinT/CDK9 complex in basal and regulated transcription (review) Int. J. Oncol. 2002;21:171–177. [PubMed] [Google Scholar]
- 12.Kasten M., Giordano A. Cdk10, a Cdc2-related kinase, associates with the Ets2 transcription factor and modulates its transactivation activity. Oncogene. 2001;20:1832–1838. doi: 10.1038/sj.onc.1204295. [DOI] [PubMed] [Google Scholar]
- 13.Loyer P., Trembley J.H., Grenet J.A., Busson A., Corlu A., Zhao W., Kocak M., Kidd V.J., Lahti J.M. Characterization of cyclin L1 and L2 interactions with CDK11 and splicing factors: Influence of cyclin L isoforms on splice site selection. J. Biol. Chem. 2008;283:7721–7732. doi: 10.1074/jbc.M708188200. [DOI] [PubMed] [Google Scholar]
- 14.Duan Y., He X., Yang H., Ji Y., Tao T., Chen J., Hu L., Zhang F., Li X., Wang H., et al. Cyclin D3/CDK11(p58) complex involved in Schwann cells proliferation repression caused by lipopolysaccharide. Inflammation. 2010;33:189–199. doi: 10.1007/s10753-009-9173-8. [DOI] [PubMed] [Google Scholar]
- 15.Kohoutek J., Blazek D. Cyclin K goes with CDK12 and CDK13. Cell Div. 2012;7:12. doi: 10.1186/1747-1028-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li S., Jiang M., Wang W., Chen J. 14-3-3 Binding to Cyclin Y contributes to cyclin Y/CDK14 association. Acta Biochim. Biophys. Sin. Shanghai. 2014;46:299–304. doi: 10.1093/abbs/gmu005. [DOI] [PubMed] [Google Scholar]
- 17.Park M.H., Kim S.Y., Kim Y.J., Chung Y.H. ALS2CR7 (CDK15) attenuates TRAIL induced apoptosis by inducing phosphorylation of survivin Thr34. Biochem. Biophys. Res. Commun. 2014;450:129–134. doi: 10.1016/j.bbrc.2014.05.070. [DOI] [PubMed] [Google Scholar]
- 18.Meijer L., Raymond E. Roscovitine and other purines as kinase inhibitors. From starfish oocytes to clinical trials. Acc. Chem. Res. 2003;36:417–425. doi: 10.1021/ar0201198. [DOI] [PubMed] [Google Scholar]
- 19.Benson C., White J., de Bono J., O’Donnell A., Raynaud F., Cruickshank C., McGrath H., Walton M., Workman P., Kaye S., et al. A phase I trial of the selective oral cyclin-dependent kinase inhibitor seliciclib (CYC202; R-Roscovitine), administered twice daily for 7 daysevery 21 days. Br. J. Cancer. 2007;96:29–37. doi: 10.1038/sj.bjc.6603509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whitlock J.A., Krailo M., Reid J.M., Ruben S.L., Ames M.M., Owen W., Reaman G. Phase I clinical and pharmacokinetic study of Flavopiridol in children with refractory solid tumors: A children’s oncology group study. J. Clin. Oncol. 2005;23:9179–9186. doi: 10.1200/JCO.2004.01.0660. [DOI] [PubMed] [Google Scholar]
- 21.Phelps M.A., Lin T.S., Johnson A.J., Hurh E., Rozewski D.M., Farley K.L., Wu D., Blum K.A., Fischer B., Mitchell S.M., Moran M.E., et al. Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia. Blood. 2009;113:2637–2645. doi: 10.1182/blood-2008-07-168583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blum W., Phelps M.A., Klisovic R.B., Rozewski D.M., Ni W., Albanese K.A., Rovin B., Kefauver C., Devine S.M., Lucas D.M., et al. Phase I clinical and pharmacokinetic study of a novel schedule of flavopiridol in relapsed or refractory acute leukemias. Haematologica. 2010;95:1098–1105. doi: 10.3324/haematol.2009.017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El-Rayes B.F., Gadgeel S., Parchment R., Lorusso P., Philip P.A. A phase I study of Flavopiridol and docetaxel. Investig. New Drugs. 2006;24:305–310. doi: 10.1007/s10637-005-4343-5. [DOI] [PubMed] [Google Scholar]
- 24.Fornier M.N., Rathkopf D., Shah M., Patil S., O’Reilly E., Tse A.N., Hudis C., Lefkowitz R., Kelsen D.P., Schwartz G.K. Phase I dose-Wnding study of weekly docetaxel followed by Flavopiridol for patients with advanced solid tumors. Clin. Cancer Res. 2007;13:5841–5846. doi: 10.1158/1078-0432.CCR-07-1218. [DOI] [PubMed] [Google Scholar]
- 25.George S., Kasimis B.S., Cogswell J., Schwarzenberger P., Shapiro G.I., Fidias P., Bukowski R.M. Phase I study of flavopiridol in combination with paclitaxel and carboplatin in patients with nonsmall-cell lung cancer. Clin. Lung Cancer. 2008;9:160–165. doi: 10.3816/CLC.2008.n.024. [DOI] [PubMed] [Google Scholar]
- 26.Rathkopf D., Dickson M.A., Feldman D.R., Carvajal R.D., Shah M.A., Wu N., Lefkowitz R., Gonen M., Cane L.M., Dials H.J., et al. Phase I study of Flavopiridol with oxaliplatin and Fluorouracil/leucovorin in advanced solid tumors. Clin. Cancer Res. 2009;15:7405–7411. doi: 10.1158/1078-0432.CCR-09-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fekrazad H.M., Verschraegen C.F., Royce M., Smith H.O., Chyi Lee F., Rabinowitz I. A phase I study of flavopiridol in combination with gemcitabine and irinotecan in patients with metastatic cancer. Am. J. Clin. Oncol. 2010;33:393–397. doi: 10.1097/COC.0b013e3181b2043f. [DOI] [PubMed] [Google Scholar]
- 28.Dickson M.A., Rathkopf D.E., Carvajal R.D., Grant S., Roberts J.D., Reid J.M., Ames M.M., McGovern R.M., Lefkowitz R.A., Gonen M., et al. A phase I pharmacokinetic study of pulse-dose vorinostat with flavopiridol in solid tumors. Investig. New Drugs. 2011;29:1004–1012. doi: 10.1007/s10637-010-9447-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karp J.E., Smith B.D., Resar L.S., Greer J.M., Blackford A., Zhao M., Moton-Nelson D., Alino K., Levis M.J., Gore S.D., et al. Phase 1 and pharmacokinetic study of bolus-infusion flavopiridol followed by cytosine arabinoside and mitoxantrone for acute leukemias. Blood. 2011;117:3302–3310. doi: 10.1182/blood-2010-09-310862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin T.S., Ruppert A.S., Johnson A.J., Fischer B., Heerema N.A., Andritsos L.A., Blum K.A., Flynn J.M., Jones J.A., Hu W., et al. Phase II study of flavopiridol in relapsed chronic lymphocytic leukemia demonstrating high response rates in genetically high-risk disease. J. Clin. Oncol. 2009;27:6012–6018. doi: 10.1200/JCO.2009.22.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carvajal R.D., Tse A., Shah M.A., Lefkowitz R.A., Gonen M., Gilman-Rosen L., Kortmansky J., Kelsen D.P., Schwartz G.K., O’Reilly E.M. A phase II study of flavopiridol (Alvocidib) in combination with docetaxel in refractory, metastatic pancreatic cancer. Pancreatology. 2009;9:404–409. doi: 10.1159/000187135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roll D.M., Ireland C.M., Lu H.S.M., Clardy J. Fascaplysin, an unusual antimicrobial pigment from the marine sponge Fascaplysinopsis sp. J. Org. Chem. 1988;53:3276–3278. doi: 10.1021/jo00249a025. [DOI] [Google Scholar]
- 33.Bharate S.B., Manda S., Mupparapu N., Battini N., Vishwakarma R.A. Chemistry and biology of fascaplysin, a potent marine-derived CDK-4 inhibitor. Mini Rev. Med. Chem. 2012;12:650–664. doi: 10.2174/138955712800626719. [DOI] [PubMed] [Google Scholar]
- 34.Ryu C.K., Kang H.Y., Lee S.K., Nam K.A., Hong C.Y., Ko W.G., Lee B.H. 5-Arylamino-2-methyl-4,7-dioxobenzothiazoles as inhibitors of cyclin-dependent kinase 4 and cytotoxic agents. Bioorg. Med. Chem. Lett. 2000;10:461–464. doi: 10.1016/S0960-894X(00)00014-7. [DOI] [PubMed] [Google Scholar]
- 35.Abate A.A., Pentimalli F., Esposito L., Giordano A. ATP-noncompetitive CDK inhibitors for cancer therapy: An overview. Expert Opin. Investig. Drugs. 2013;22:895–906. doi: 10.1517/13543784.2013.798641. [DOI] [PubMed] [Google Scholar]
- 36.Corbel C., Wang Q., Bousserouel H., Hamdi A., Zhang B., Lozach O., Ferandin Y., Tan V.B., Guéritte F., Colas P., et al. First BRET-based screening assay performed in budding yeast leads to the discovery of CDK5/p25 interaction inhibitors. Biotechnol. J. 2011;6:860–870. doi: 10.1002/biot.201100138. [DOI] [PubMed] [Google Scholar]
- 37.Reddy M.V., Akula B., Cosenza S.C., Athuluridivakar S., Mallireddigari M.R., Pallela V.R., Billa V.K., Subbaiah D.R., Bharathi E.V., Vasquez-Del Carpio R., et al. Discovery of 8-cyclopentyl-2-[4-(4-methyl-piperazin-1-yl)-phenylamino]-7-oxo-7,8-dihydro-pyrido[2,3-d]pyrimidine-6-carbonitrile (7x) as a potent inhibitor of cyclin-dependent kinase 4 (CDK4) and AMPK-related kinase 5 (ARK5) J. Med. Chem. 2014;57:578–599. doi: 10.1021/jm401073p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keegan K., Li C., Li Z., Ma J., Ragains M., Coberly S., Hollenback D., Eksterowicz J., Liang L., Weidner M., et al. Preclinical evaluation of AMG 925, a FLT3/CDK4 dual kinase inhibitor for treating acute myeloid leukemia. Mol. Cancer Ther. 2014;13:880–889. doi: 10.1158/1535-7163.MCT-13-0858. [DOI] [PubMed] [Google Scholar]
- 39.Raghavan P., Tumati V., Yu L., Chan N., Tomimatsu N., Burma S., Bristow R.G., Saha D. AZD5438, an inhibitor of CDK1, 2, and 9, enhances the radiosensitivity of non-small cell lung carcinoma cells. Int. J. Radiat. Oncol. Biol. Phys. 2012;84:e507–e514. doi: 10.1016/j.ijrobp.2012.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lücking U., Jautelat R., Krüger M., Brumby T., Lienau P., Schäfer M., Briem H., Schulze J., Hillisch A., Reichel A., et al. The lab oddity prevails: Discovery of pan-CDK inhibitor (R)-S-cyclopropyl-S-(4-{[4-{[(1R,2R)-2-hydroxy-1-methylpropyl]oxy}-5-(trifluorome thyl)pyrimidin-2-yl]amino}phenyl)sulfoximide (BAY 1000394) for the treatment of cancer. Chem. Med. Chem. 2013;8:1067–1085. doi: 10.1002/cmdc.201300096. [DOI] [PubMed] [Google Scholar]
- 41.Lo M.C., Ngo R., Dai K., Li C., Liang L., Lee J., Emkey R., Eksterowicz J., Ventura M., Young S.W., et al. Development of a time-resolved fluorescence resonance energy transfer assay for cyclin-dependent kinase 4 and identification of its ATP-noncompetitive inhibitors. Anal. Biochem. 2012;421:368–377. doi: 10.1016/j.ab.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 42.Premnath P.N., Liu S., Perkins T., Abbott J., Anderson E., McInnes C. Fragment based discovery of arginine isosteres through REPLACE: Towards non-ATP competitive CDK inhibitors. Bioorg. Med. Chem. 2014;22:616–622. doi: 10.1016/j.bmc.2013.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kabadi S.V., Stoica B.A., Hanscom M., Loane D.J., Kharebava G., Murray Ii M.G., Cabatbat R.M., Faden A.I. CR8, a selective and potent CDK inhibitor, provides neuroprotection in experimental traumatic brain injury. Neurotherapeutics. 2012;9:405–421. doi: 10.1007/s13311-011-0095-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang D., Mita M., Shapiro G.I., Poon J., Small K., Tzontcheva A., Kantesaria B., Zhu Y., Bannerji R., Statkevich P. Effect of aprepitant on the pharmacokinetics of the cyclin-dependent kinase inhibitor dinaciclib in patients with advanced malignancies. Cancer Chemother. Pharmacol. 2012;70:891–898. doi: 10.1007/s00280-012-1967-y. [DOI] [PubMed] [Google Scholar]
- 45.Van Duyne R., Guendel I., Jaworski E., Sampey G., Klase Z., Chen H., Zeng C., Kovalskyy D., El Kouni M.H., Lepene B., et al. Effect of mimetic CDK9 inhibitors on HIV-1-activated transcription. J. Mol. Biol. 2013;425:812–829. doi: 10.1016/j.jmb.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Metastatic breast cancer responds to CDK4/6 inhibitor. [(accessed on 2 May 2014)]. Available online: http://cancerdiscovery.aacrjournals.org/content/early/2014/04/22/2159-8290.CD-NB2014-062.full.
- 47.Canela N., Orzáez M., Fucho R., Mateo F., Gutierrez R., Pineda-Lucena A., Bachs O., Pérez-Payá E. Identification of an hexapeptide that binds to a surface pocket in cyclin A and inhibits the catalytic activity of the complex cyclin-dependent kinase 2-cyclin A. J. Biol. Chem. 2006;281:35942–35953. doi: 10.1074/jbc.M603511200. [DOI] [PubMed] [Google Scholar]
- 48.Massard C., Soria J.C., Anthoney D.A., Proctor A., Scaburri A., Pacciarini M.A., Laffranchi B., Pellizzoni C., Kroemer G., Armand J.P., et al. A first in man, phase I dose-escalation study of PHA-793887, an inhibitor of multiple cyclin-dependent kinases (CDK2, 1 and 4) reveals unexpected hepatotoxicity in patients with solid tumors. Cell Cycle. 2011;10:963–970. doi: 10.4161/cc.10.6.15075. [DOI] [PubMed] [Google Scholar]
- 49.Cirstea D., Hideshima T., Santo L., Eda H., Mishima Y., Nemani N., Hu Y., Mimura N., Cottini F., Gorgun G., et al. Small-molecule multi-targeted kinase inhibitor RGB-286638 triggers P53-dependent and -independent anti-multiple myeloma activity through inhibition of transcriptional CDKs. Leukemia. 2013;27:2366–2375. doi: 10.1038/leu.2013.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yenugonda V.M., Deb T.B., Grindrod S.C., Dakshanamurthy S., Yang Y., Paige M., Brown M.L. Fluorescent cyclin-dependent kinase inhibitors block the proliferation of human breast cancer cells. Bioorg. Med. Chem. 2011;19:2714–2725. doi: 10.1016/j.bmc.2011.02.052. [DOI] [PubMed] [Google Scholar]
- 51.Orzáez M., Guevara T., Sancho M., Pérez-Payá E. Intrinsic caspase-8 activation mediates sensitization of erlotinib-resistant tumor cells to erlotinib/cell-cycle inhibitors combination treatment. Cell Death Dis. 2012;3:e415. doi: 10.1038/cddis.2012.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barton K.L., Misuraca K., Cordero F., Dobrikova E., Min H.D., Gromeier M., Kirsch D.G., Becher O.J. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. PLoS One. 2013;8:e77639. doi: 10.1371/journal.pone.0077639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hayashi T., Adachi K., Ohba S., Hirose Y. The CDK inhibitor flavopiridol enhances temozolomide-induced cytotoxicity in human glioma cells. J. Neurooncol. 2013;115:169–178. doi: 10.1007/s11060-013-1220-5. [DOI] [PubMed] [Google Scholar]
- 54.Martin K.L., Hill G.A., Klein R.R., Arnett D.G., Burd R., Limesand K.H. Prevention of radiation-induced salivary gland dysfunction utilizing a CDK inhibitor in a mouse model. PLoS One. 2012;7:e51363. doi: 10.1371/journal.pone.0051363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bukanov N.O., Moreno S.E., Natoli T.A., Rogers K.A., Smith L.A., Ledbetter S.R., Oumata N., Galons H., Meijer L., Ibraghimov-Beskrovnaya O. CDK inhibitors R-roscovitine and S-CR8 effectively block renal and hepatic cystogenesis in an orthologous model of ADPKD. Cell Cycle. 2012;11:4040–4046. doi: 10.4161/cc.22375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Subramaniam D., Periyasamy G., Ponnurangam S., Chakrabarti D., Sugumar A., Padigaru M., Weir S.J., Balakrishnan A., Sharma S., Anant S. CDK-4 inhibitor P276 sensitizes pancreatic cancer cells to gemcitabine-induced apoptosis. Mol. Cancer Ther. 2012;11:1598–1608. doi: 10.1158/1535-7163.MCT-12-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang L., Gu X., Zhang W., Zhang J., Ma Z. CDK5 inhibitor roscovitine alleviates neuropathic pain in the dorsal root ganglia by downregulating N-methyl-d-aspartate receptor subunit 2A. Neurol. Sci. 2014;35:1365–1371. doi: 10.1007/s10072-014-1713-9. [DOI] [PubMed] [Google Scholar]
- 58.Kabadi S.V., Stoica B.A., Loane D.J., Luo T., Faden A.I. CR8, a novel inhibitor of CDK, limits microglial activation, astrocytosis, neuronal loss, and neurologic dysfunction after experimental traumatic brain injury. J. Cereb. Blood Flow Metab. 2014;34:502–513. doi: 10.1038/jcbfm.2013.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hofmeister C.C., Poi M., Bowers M.A., Zhao W., Phelps M.A., Benson D.M., Kraut E.H., Farag S., Efebera Y.A., Sexton J., et al. A phase I trial of flavopiridol in relapsed multiple myeloma. Cancer Chemother. Pharmacol. 2014;73:249–257. doi: 10.1007/s00280-013-2347-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luke J.J., D’Adamo D.R., Dickson M.A., Keohan M.L., Carvajal R.D., Maki R.G., de Stanchina E., Musi E., Singer S., Schwartz G.K. The cyclin-dependent kinase inhibitor flavopiridol potentiates doxorubicin efficacy in advanced sarcomas: Preclinical investigations and results of a phase I dose-escalation clinical trial. Clin. Cancer Res. 2012;18:2638–2647. doi: 10.1158/1078-0432.CCR-11-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bose P., Perkins E.B., Honeycut C., Wellons M.D., Stefan T., Jacobberger J.W., Kontopodis E., Beumer J.H., Egorin M.J., Imamura C.K., et al. Phase I trial of the combination of flavopiridol and imatinib mesylate in patients with Bcr-Abl+ hematological malignancies. Cancer Chemother. Pharmacol. 2012;69:1657–1667. doi: 10.1007/s00280-012-1839-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bible K.C., Peethambaram P.P., Oberg A.L., Maples W., Groteluschen D.L., Boente M., Burton J.K., Gomez Dahl L.C., Tibodeau J.D., Isham C.R., et al. A phase 2 trial of flavopiridol (Alvocidib) and cisplatin in platin-resistant ovarian and primary peritoneal carcinoma: MC0261. Gynecol. Oncol. 2012;127:55–62. doi: 10.1016/j.ygyno.2012.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stephens D.M., Ruppert A.S., Maddocks K., Andritsos L., Baiocchi R., Jones J., Johnson A.J., Smith L.L., Zhao Y., Ling Y., et al. Cyclophosphamide, alvocidib (flavopiridol), and rituximab, a novel feasible chemoimmunotherapy regimen for patients with high-risk chronic lymphocytic leukemia. Leuk. Res. 2013;37:1195–1199. doi: 10.1016/j.leukres.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Woyach J.A., Lozanski G., Ruppert A.S., Lozanski A., Blum K.A., Jones J.A., Flynn J.M., Johnson A.J., Grever M.R., Heerema N.A., et al. Outcome of patients with relapsed or refractory chronic lymphocytic leukemia treated with flavopiridol: Impact of genetic features. Leukemia. 2012;26:1442–1444. doi: 10.1038/leu.2011.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stephens D.M., Ruppert A.S., Blum K., Jones J., Flynn J.M., Johnson A.J., Ji J., Phelps M.A., Grever M.R., Byrd J.C. Flavopiridol treatment of patients aged 70 or older with refractory or relapsed chronic lymphocytic leukemia is a feasible and active therapeutic approach. Haematologica. 2012;97:423–427. doi: 10.3324/haematol.2011.047324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Holkova B., Perkins E.B., Ramakrishnan V., Tombes M.B., Shrader E., Talreja N., Wellons M.D., Hogan K.T., Roodman G.D., Coppola D., et al. Phase I trial of bortezomib (PS-341; NSC 681239) and alvocidib (flavopiridol; NSC 649890) in patients with recurrent or refractory B-cell neoplasms. Clin. Cancer Res. 2011;17:3388–3397. doi: 10.1158/1078-0432.CCR-10-2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Holkova B., Kmieciak M., Perkins E.B., Bose P., Baz R., Roodman G.D., Stuart R.K., Ramakrishnan V., Wan W., Peer C.J., et al. Phase I trial of bortezomib and “non-hybrid”(bolus) infusion schedule of alvocidib (flavopiridol) in patients with recurrent or refractory indolent B-cell neoplasms. Clin. Cancer Res. 2014 doi: 10.1158/1078-0432.CCR-14-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nemunaitis J.J., Small K.A., Kirschmeier P., Zhang D., Zhu Y., Jou Y.M., Statkevich P., Yao S.L., Bannerji R. A first-in-human, phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J. Transl. Med. 2013;11 doi: 10.1186/1479-5876-11-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gojo I., Sadowska M., Walker A., Feldman E.J., Iyer S.P., Baer M.R., Sausville E.A., Lapidus R.G., Zhang D., Zhu Y., et al. Clinical and laboratory studies of the novel cyclin-dependent kinase inhibitor dinaciclib (SCH 727965) in acute leukemias. Cancer Chemother. Pharmacol. 2013;72:897–908. doi: 10.1007/s00280-013-2249-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stephenson J.J., Nemunaitis J., Joy A.A., Martin J.C., Jou Y.M., Zhang D., Statkevich P., Yao S.L., Zhu Y., Zhou H., et al. Randomized phase 2 study of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus erlotinib in patients with non-small cell lung cancer. Lung Cancer. 2014;83:219–223. doi: 10.1016/j.lungcan.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 71.Mita M.M., Joy A.A., Mita A., Sankhala K., Jou Y.M., Zhang D., Statkevich P., Zhu Y., Yao S.L., Small K., et al. Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin. Breast Cancer. 2014;14:169–176. doi: 10.1016/j.clbc.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 72.Fabre C., Gobbi M., Ezzili C., Zoubir M., Sablin M.P., Small K., Im E., Shinwari N., Zhang D., Zhou H., et al. Clinical study of the novel cyclin-dependent kinase inhibitor dinaciclib in combination with rituximab in relapsed/refractory chronic lymphocytic leukemia patients. Cancer Chemother. Pharmacol. 2014 doi: 10.1007/s00280-014-2583-9. [DOI] [PubMed] [Google Scholar]
- 73.Guha M. Cyclin-dependent kinase inhibitors move into Phase III. Nat. Rev. Drug Discov. 2012;11:892–894. doi: 10.1038/nrd3908. [DOI] [PubMed] [Google Scholar]
- 74.Kumar S.K., LaPlant B.R., Chng W.J., Zonder J.A., Callander N., Roy V., Furth B., Erlichman C., Stewart K. Phase 1/2 Trial of a novel CDK inhibitor dinaciclib (SCH727965) in patients with relapsed multiple myeloma demonstrates encouraging single agent activity; Proceedings of the 54th ASH Meeting; Atlanta, GA, USA. 9 December 2012. [Google Scholar]
- 75.Flaherty K.T., Lorusso P.M., Demichele A., Abramson V.G., Courtney R., Randolph S.S., Shaik M.N., Wilner K.D., O’Dwyer P.J., Schwartz G.K. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin. Cancer Res. 2012;18:568–576. doi: 10.1158/1078-0432.CCR-11-0509. [DOI] [PubMed] [Google Scholar]
- 76.Dickson M.A., Tap W.D., Keohan M.L., D’Angelo S.P., Gounder M.M., Antonescu C.R., Landa J., Qin L.X., Rathbone D.D., Condy M.M., et al. Phase II trial of the CDK4 inhibitor PD0332991 in patients with advanced CDK4-amplified well-differentiated or dedifferentiated liposarcoma. J. Clin. Oncol. 2013;31:2024–2028. doi: 10.1200/JCO.2012.46.5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.DeMichele A., Sanders Clark A., Heitjan D., Randolph S., Gallagher M., Lal P., Feldman M.D., Zhang P.J., Schnader A., Zafman K., et al. A phase II trial of an oral CDK 4/6 inhibitor, PD0332991, in advanced breast cancer; Proceedings of the 2013 ASCO Annual Meeting; Chicago, IL, USA. 31 May–4 June 2013. [Google Scholar]
- 78.Finn R.S., Crown J.P., Boer K., Lang I., Parikh R.J., Breazna A., Ho S.N., Kim S.T., Randolph S., Slamon D.J. Results of a randomized phase 2 study of PD 0332991, a Cyclin-Dependent Kinase (CDK) 4/6 inhibitor, in combination with letrozole vs. letrozole alone for first-line treatment of ER+, HER2– advanced breast cancer (TRIO-18); Proceedings of the AACR Annual Meeting 2014; San Diego, CA, USA. 6 April 2014. [Google Scholar]