

Abstract
The amyloid cascade model for the origin of sporadic forms of Alzheimer's disease (AD) posits that the imbalance in the production and clearance of beta-amyloid is a necessary condition for the disease. A competing theory called the entropic selection hypothesis asserts that the primary cause of sporadic AD is age-induced mitochondrial dysregulation and the following cascade of events: (i) metabolic reprogramming—the upregulation of oxidative phosphorylation in compensation for insufficient energy production in neurons, (ii) natural selection—competition between intact and reprogrammed neurons for energy substrates and (iii) propagation—the spread of the disease due to the selective advantage of neurons with upregulated metabolism. Experimental studies to evaluate the predictions of the amyloid cascade model are being continually retuned to accommodate conflicts of the predictions with empirical data. Clinical trials of treatments for AD based on anti-amyloid therapy have been unsuccessful. We contend that these anomalies and failures stem from a fundamental deficit of the amyloid hypothesis: the model derives from a nuclear-genomic perspective of sporadic AD and discounts the bioenergetic processes that characterize the progression of most age-related disorders. In this article, we review the anomalies of the amyloid model and the theoretical and empirical support for the entropic selection theory. We also discuss the new therapeutic strategies based on natural selection which the model proposes.
Keywords: Alzheimer's disease, natural selection, neuroenergetics
1. Introduction
The challenge of elucidating the origin of Alzheimer's disease (AD) and developing neuroprotective strategies is one of the most daunting in modern medical history. Intense research over the past 25 years has produced a detailed picture of early-onset AD, a genetic disease caused by mutations in the nuclear genes that regulate the processing of the amyloid precursor protein (APP) [1,2]. This led to the hypothesis that most cases of AD are caused by abnormalities in the production or clearance of beta-amyloid (Aβ), and that this is a necessary and sufficient condition for its development [3]. The hypothesis has undergone some revision over time due to the poor association between Aβ plaque deposition and cognitive decline [4]. There is strong support for this hypothesis in the case of familial AD. As the histopathology of early- and late-onset forms is indistinguishable, this neuron-centric, genetic paradigm has also dominated thinking about sporadic AD, which accounts for 95% of cases.
But shared pathology does not imply the same cause, and studies of late-onset AD suggest the existence of multiple aetiologies [5]. The function of Aβ is unknown, but it is normally released at the synapse and accumulates quickly with increased neuronal activity [6]. Increased Aβ production is detected to some extent in normal ageing [7], and in response to age-related pathologies, including stroke, trauma, oxidative stress and inflammation [8–11]. Elderly patients with normal cognition often have substantial Aβ deposition, as detected by in vivo amyloid imaging or at autopsy [12,13]. Clinical studies to evaluate the predictions of the amyloid hypothesis have consistently failed [14–18]. The failure has led some to conclude that Aβ pathology may not be the primary cause of sporadic AD [19,20], but proponents of the model have altered some of its basic premises in an effort to align prediction with empirical observation. For example, it is now postulated that neuronal loss derives from the toxicity of tau, the accumulation of which is assumed to be triggered by overproduction of amyloid [4]. Recent experiments in neural culture systems which show that Aβ drives tauopathy have been promoted as providing experimental validation of the amyloid hypothesis [21]. However, these reports have failed to remark that in elderly patients there is often no strong relationship between the severity of dementia and the prevalence of either Aβ or tau [22,23]. The ineffectiveness of anti-amyloid therapy has also been ascribed to late implementation, and ongoing trials now focus on disease prevention in individuals with normal cognition who have evidence of Aβ accumulation.
We contend that the experimental anomalies and clinical failures that currently characterize the amyloid hypothesis derive from basic conceptual deficits in the model. The amyloid hypothesis is based on the assumption that the disease is due to nuclear-genomic defects which result in the accumulation of abnormal forms of Aβ and tau, which in large concentrations become toxic to neurons. This nuclear-genomic perspective implicitly ignores the age-related bioenergetic changes prescribed by the thermodynamic laws of energy transduction in living organisms. While attractive, the idea that late-onset AD could be prevented or cured by targeting one or two abnormal proteins ignores the most striking fact about the disease—its exponential increase with age.
In the conceptual framework of the amyloid hypothesis, age is considered only in the chronological sense; namely, the time it takes for Aβ to attain sufficient concentration to induce synaptic damage and ultimately neuronal loss. But as we shall illustrate, ageing is a physiological phenomenon, consistent with the random emergence of molecular disorder—an intrinsic property of the thermodynamic instability of large biomolecules [24]. Normal ageing is characterized by the random loss of molecular fidelity which preferentially affects the energy-producing organelles. The molecular dysregulation which thermodynamic instability generates is a stochastic process: certain neurons will remain largely intact and other neurons will become impaired and reprogramme their metabolic activity to compensate for the decline in energy production [25]. This sequential pattern defined by mitochondrial defect and metabolic alteration ultimately leads to the development of two classes of neurons—relatively intact cells with a normal mode of energy processing, and impaired cells with reprogrammed metabolic machinery.
Our model posits that competition between relatively intact cells and impaired cells with upregulated oxidative phosphorylation (OxPhos) will ultimately lead to a quasi-stable state characterized by a high frequency of intact neurons and a comparatively low frequency of impaired neurons. This equilibrium is maintained with the help of energy and anti-oxidant precursors produced by astrocytes, although this resource is also limited due to age-related changes in astrocyte function [26,27]. Pathological ageing is caused by a disruption of this quasi-stable state and the subsequent decline in frequency of intact neurons on account of their selective disadvantage in the neuronal microenvironment. The transition from normal to pathological ageing is presumed to be the action of various internal and external factors which will induce a large change in the selective advantage of impaired neurons. These factors could include age-related changes in cerebrovascular health, lifestyle factors, trauma and various forms of stress. Thus, the model asserts that the imbalance between the generation of intact and impaired mitochondria is a necessary but not sufficient condition for the development of AD.
This neuroenergetic perspective of the origin of AD resolves several features of the sporadic forms of AD which seemed intractable in the context of the amyloid cascade model. These features include the absence of dementia in individuals with large amounts of Aβ in their brain [12,13], the selective vulnerability of certain neurons to AD [28] and the inverse comorbidity between AD and cancer [29,30]. This model is also consistent with the epidemiologic fact that the incidence rate of the sporadic form of AD increases exponentially with age.
This article will describe the theoretical and empirical support for a neuroenergetic model of AD—the entropic selection hypothesis. The entropy, in this context, refers to the statistical measure evolutionary entropy [31–33]. This measure describes the dynamic organization that defines an aggregate of interacting metabolic entities. Typical examples in the case of biochemical networks are the glycolytic cycle, or the citric acid cycle in OxPhos. Evolutionary entropy, which pertains to networks at the molecular, cellular and organismic levels, is a generic concept which describes the number of pathways of energy flow within the network. The quantity is a non-equilibrium analogue of the statistical measure thermodynamic entropy, which pertains to aggregates of non-metabolic entities (solids, liquids and gases). Thermodynamic entropy describes the number of energy states in the aggregate consistent with a particular macroscopic configuration. The term entropic selection refers to the analytical and empirical fact that the statistical measure evolutionary entropy predicts the outcome of competition between intact neurons; neurons with normal energy processing, and impaired neurons; cells defined by reprogrammed metabolic machinery [31,34].
We will also invoke the entropic selection hypothesis to propose new strategies to prevent and ameliorate the disease. These strategies are based on metabolic interventions to maintain a homeostatic balance between intact and impaired neurons which characterize the quasi-stable state of normal ageing. Our work builds on the observations of others who have argued that late-onset AD is driven by age-related changes in the energy-producing organelles of the brain [24,35–39]. The properties of age and energy thus play a critical role in our analysis. In order to introduce these properties in our model, we will draw from certain notions and methodology from the physical sciences and evolutionary theory which may not be part of a neuroscientist's usual lexicon [26,34]. These concepts, as we will observe, are critical in providing a quantitative analysis of the origin of a disease whose understanding has resisted an approach based on traditional biochemical methods.
2. Darwin and disease
It is no surprise to the evolutionary biologist that early- and late-onset diseases can have distinct causes while sharing a similar phenotype. Before the age of reproductive maturity, there is strong selection to maintain metabolic integrity and to enhance viability and ensure the reproductive success of the organism. Perturbations in the energy-producing networks of cells will be efficiently repaired and diseases will be strongly influenced by mutations in the nuclear genome. Thus, early-onset diseases are primarily genetic; they can be attributed to defects in one gene or a few genes and are quite rare due to strong selective pressure to ensure survival at least until reproductive maturity. While mutations in mitochondrial genes are also inherited, although the inheritance rules are non-Mendelian. The age at which inherited mitochondrial disease presents depends on the severity and prevalence of the defect across the inherited mitochondria.
By contrast, selection to maintain metabolic integrity will be weak in post-reproductive years, as there is no longer a great need to invest in individual viability. Consequently, damage in mitochondria and other metabolic networks will not be repaired, will increase with age and their effect will be cumulative. There is abundant evidence that mitochondrial DNA is uniquely vulnerable to sporadic mutation, and that mitochondrial function declines with age [35,40]. Since familial disease-linked mutations remain rare, late-onset sporadic diseases will primarily be metabolic and, due to the high incidence of mitochondrial mutations with age, will be common. This perspective correctly predicts that the incidence curve of familial AD is bell-shaped, while that of sporadic AD increases exponentially after age 65 [38].
Each individual has a different physiological capacity, or reserve, at the time of reproductive maturity that is influenced by genetic, lifestyle and environmental factors, and which collectively determines their potential longevity. However, the incidence of sporadic AD and other age-related diseases is ultimately determined by the cumulative effect of the ageing process, which unfolds largely independent of genetic make-up. Thus, the theory of natural selection predicts that abnormal accumulation of Aβ before and during the reproductive years is primarily due to genetic defects, but afterwards is primarily driven by the ageing process itself. If amyloid deposition is a secondary process in sporadic AD, what is the primary defect?
3. Energetics, neural networks and alzheimer's disease
There has been a growing appreciation of the complexity of sporadic AD and its tremendous overlap with ageing-related changes, including the decline in mitochondrial function and cellular energy metabolism [36]. Functional neuroimaging studies reveal that AD is characterized by both hypo-metabolism and atrophy (a marker of neuronal loss) beginning in the medial temporal lobe (MTL) and extending to other anatomically connected areas [41,42]. The pyramidal cells of the hippocampus are known to require the most energy of any cell in the brain, and this is the first area affected in AD [43]. This has led some to propose that bioenergetic failure in vulnerable neurons may be the primary driver of sporadic AD [44,45]. AD targets specific neural networks that have high metabolic demand, and areas within those networks that function as ‘connectivity hubs' with other brain regions are particularly at risk, including the posterior cingulate and precuneous, the medial frontal cortex, and the lateral frontal and parietal regions [46,47].
It is intriguing that in functional imaging studies of older adults with normal cognition, these same regions show evidence of initial hyper-metabolism and increased neuronal activity as well as increased Aβ deposition [48–51]. This increased activity is associated with preservation of brain function, suggesting it serves as a compensatory response. However, there is evidence that greater hippocampal activation at baseline correlates with cognitive decline on follow-up [48]. In another study of healthy adults, individuals with increased risk of AD due to APOE4 genotype had a higher MTL activation than those with other genotypes, also suggesting compensation [50]. Interestingly, this hyper-metabolic phase has also been documented in subjects with familial AD about 25 years before symptom onset, prior to both amyloid deposition and atrophy [52]. Together, these results suggest that the hypo-metabolism seen in AD is preceded by a period of compensatory hyper-metabolism, or upregulation of energy production [51]. Since there is direct evidence that increased neural activity leads to increased Aβ release [6,53,54], Aβ deposition may serve as a marker of this increased synaptic activity. These observations support the hypothesis that energy failure in some hippocampal neurons leads to recruitment of others, which must in turn invoke compensatory mechanisms to meet their increased energy demands, and that this metabolic upregulation eventually leads to neuronal dysfunction and loss [44]. This bioenergetic perspective explains key features of the disease including the selective vulnerability of affected neurons and the pattern of spread of neuronal loss. As age-related energy decline is a random process, diseased and healthy neurons will be in close proximity, a core feature of AD pathology.
4. Ageing: thermodynamic and evolutionary entropy
Full appreciation of the effect of ageing on living organisms requires an understanding of the concept of entropy (figure 1) [55]. Complex biomolecules, such as proteins, achieve their function by means of the stability of their three-dimensional folded structures. Biological ageing has its origin in the vulnerability of these structures to random perturbations in the interaction and activity of the molecular components [56]. These perturbations are an intrinsic property of all material objects and have effects at the level of both molecules and cells.
Figure 1.

Understanding entropy. Thermodynamic entropy refers to the spreading of energy within an inanimate matter—solid, liquid or gas. The quantity describes the number of accessible energy states within the aggregate. The molecules in a solid occupy a fixed position and the number of accessible energy states is small. A solid has low entropy. The molecules of a gas enclosed within a container are free to move about and have access to a large number of energy states. A gas has high entropy. Evolutionary entropy refers to the diversity of pathways within a metabolic network. The quantity describes the number of accessible pathways of energy flow. Glycolysis has a simple pathway of energy flow and the rate at which energy is taken from the environment and reinvested into metabolic activities is low, giving it low evolutionary entropy. By contrast, oxidative phosphorylation has several distinct pathways of energy flow and includes a large number of interacting enzymes. The rate at which energy precursors are reinvested in metabolic activity is high. Thus, OxPhos has high evolutionary entropy. G6P, glucose-6-phosphate; F6P, fructose-6-phosphate; AcetylCo-A, acetyl coenzyme A; TCA, tricarboxylic acid; NADH, nicotinamide adenine dinucleotide; ATP, adenosine triphosphate. (Online version in colour.)
At the molecular level, the increase in disorder can be analytically described in terms of an increase in thermodynamic entropy. This concept refers to any molecular aggregate (solid, liquid or gas) and describes the number of accessible energy states within the aggregate, or equivalently, the extent to which energy is spread throughout the aggregate and its microscopic storage modes. This type of molecular disorder can induce a critical effect on protein folding and the formation of amyloid. The driving force of protein folding is the search for a conformation of minimal free energy. This state is a function of the conformation defined by the interaction between the amino acid residues. Small random fluctuations in the force of interaction between these residues, an intrinsic property of large molecules, may alter the surface of the energy landscape, the configuration space of energy available to a protein from its denatured state to its folded state.
Single-domain proteins follow two-state kinetics. The folding pathways have no detectable intermediates and the proteins adopt the correct conformation on thermodynamic grounds. The folding pathway in multiple-domain proteins involves several intermediates. The fluctuations induced by thermodynamic instability will induce deviations in the topology of the free-energy landscape. Molecular chaperones play a critical role in re-arranging these alterations in order that the free energy minimum is attained. Chaperones iteratively bind and release their substrates, thus enabling them to escape from wrong folding pathways. However, the efficiency of the chaperones declines with age, and the capacity to ensure that the system attains its global energy minimum is impaired [57]. Consequently, a new local or global free energy minimum may be attained. Accordingly, misfolding or the formation of aggregated structures will result. The formation of amyloid, a dysfunctional aggregate structure, can be considered an inherent property of the thermodynamic instability of molecular structures. This instability affects the quality control systems designed to rearrange misfolded structures and increases with age. The amyloids that arise from this age-dependent misfolding may be differentiated in both sequence and toxicity from the molecules that arise from genetic mutation [58].
At the metabolic level, the random loss of molecular fidelity entails a decline in enzymatic kinetic activity and a decrease in evolutionary entropy [31]. This concept describes the number of accessible pathways of energy flow within a metabolic network. It also characterizes the rate at which energy is appropriated from the external environment and transformed into metabolic activities. OxPhos, an extremely complex metabolic network with several distinct pathways of energy flow, has high evolutionary entropy. It is a slow but efficient metabolic process, converting one molecule of glucose into 36 ATP. Glycolysis, which involves a much smaller number of enzymes and a more linear pathway, has lower evolutionary entropy. It yields only two molecules of ATP but is 100 times faster than OxPhos. The inherent trade-off between the yield and rate of ATP production has played an important role in the evolution of bioenergetic systems and in characterizing the outcome of selection between cells whose activity is regulated by different biochemical networks.
It should be emphasized here that the process of natural selection underlies almost all the diversity observed in the world of molecules, cells and higher organisms. Selection does not only happen at the level of reproduction—it is also at work in the survival of individual cells, and accordingly as the arbiter of competition for energy between post-mitotic cells such as neurons. Neurons with different levels of OxPhos activity will compete for the glucose or lactate that is available. The selective outcome can be described in terms of a general rule, called the entropic selection principle. This rule states that the outcome of competition between cells is predicted by evolutionary entropy and depends on the availability of resources [31]. When energy precursors are constant but limited in abundance, a high entropy strategy that maximizes ATP yield will promote survival. High entropy systems are robust and stable networks, insensitive to endogenous and exogenous perturbations. These systems cannot readily adapt to changes in the availability of resources. When there are large variations in resource abundance, a lower entropy strategy is more advantageous as it can fluctuate more easily to match resources.
In the case of a cancer cell, if the abnormal tumour microenvironment is characterized by a large variation in resource availability, glycolysis will confer a selective advantage. On the other hand, if nutrient levels are low but constant in the tumour, OxPhos is advantageous as it produces more energy per molecule of glucose. Cancer cells that are unable to upregulate OxPhos in this setting have a survival disadvantage and are more vulnerable to cell death from low glucose levels if treated with metformin, an anti-diabetic medication [59].
In the ageing brain, energy resources are constant but limited, owing to the age-related decline in energy uptake and production. Neurons with an ‘energy crisis’ will increase their metabolic rate, and concomitantly the evolutionary entropy, by upregulating OxPhos. In this microenvironment, defined by limited energy precusors, they will out-compete neurons with normal OxPhos activity and metabolic rate for limited energy precursors.
5. Metabolic reprogramming and cellular survival
The entropic selection principle is a mathematical principle which was originally derived by applying the ergodic theory of dynamical systems to the study of competition between related biological networks [34]. The principle has empirical support based on laboratory populations [31] and has also been validated by numerical studies of metabolic networks [60]. Recent studies of metabolic reprogramming in cancer cells are also consistent with the principle [59]. OxPhos and glycolysis are the two major pathways of cellular energy production [61]. Under normal conditions, most mammalian cells primarily metabolize glucose by the more efficient mechanism of OxPhos, as explained in figure 2a. Cells adapt to conditions of low oxygen tension by upregulating glycolysis and also tend to use glycolysis when proliferating. Even if surrounded by abundant energy precursors, normal cells produce only what they need to maintain their basic functions. In 1925, Otto Warburg observed that cancer cells metabolize glucose to lactate even under aerobic conditions, a phenomenon known as aerobic glycolysis or the ‘Warburg effect’ (figure 2b) [62,63]. This metabolic shift allows pre-cancerous and malignant cells to switch into ‘growth mode’ in the absence of normal growth signals. As explained above, the shift to glycolysis, in view of its lower evolutionary entropy, entails a survival advantage in an environment where resources are subject to large fluctuations in abundance. The super-active glucose metabolism which this selective advantage confers is detected on PET scans, making enhanced glycolytic activity a useful tool for identifying malignancy. In addition to producing energy quickly, glycolysis provides key materials for biosynthesis such as nucleic acids, proteins and lipids [64].
Figure 2.

Normal and pathological energy metabolism. (a) Normal cell. Glucose, the primary substrate of ATP production, is converted to pyruvate in the cytosol through glycolytic enzymes (figure 1). Pyruvate can be converted anaerobically to lactate to complete glycolysis or transported to mitochondria, where it is becomes the substrate for oxidative phosphorylation (OxPhos). OxPhos has a higher yield of ATP but is significantly slower than glycolysis. In a typical cell, OxPhos provides 88% of cellular energy when compared with only 12% from glycolysis. (b) The Warburg effect. Cancer cells switch from OxPhos to aerobic glycolysis to meet their demands for energy and biomass and upregulate glycolytic enzymes such as 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase, isoform 3 (PFKβ3). This is an early and necessary step in carcinogenesis. Cancer cells that have functional mitochondria can also upregulate OxPhos. (c) The inverse Warburg effect. Neurons have weak glycolytic capacity and thus primarily upregulate OxPhos in response to age-related energy decline by mechanisms such as increased transcription of cytochrome oxidase complexes III and IV and mitochondrial fusion and fission. (Online version in colour.)
This switch to aerobic glycolysis, an early and necessary step in carcinogenesis, led Warburg to conclude that cancer is essentially a ‘metabolic disease’. The increased proliferation of cancer cells due to the Warburg effect has been identified as a major mechanism of resistance to current cancer therapies and has led to the pursuit of a new class of agents based on reducing the selective growth and survival advantage of these cells [65].
In the context of human brain metabolism, aerobic glycolysis is predominant during in utero development of the brain to support the demands of neuronal proliferation. Later, during postnatal development, aerobic glycolysis may persist to support the maturational changes of neurons, including axonal elongation, synaptogenesis and myelination. It has been speculated that aerobic glycolysis may persist in restricted regions of the brain throughout adult life for the purposes of activity-related changes at the synapse that accompany learning and memory [66]. Interestingly, the areas of the brain with the highest levels of glycolysis in adulthood are those that have the highest susceptibility to AD, suggesting they are more vulnerable to energy failure [67].
6. Neuronal energy metabolism and the inverse warburg effect
Neurons are post-mitotic cells whose mission requires that they survive for the lifespan of the organism and maintain their connections with other neurons indefinitely. The cortical neuron is highly specialized for information processing, a task that requires enormous energy. Despite its small size relative to the rest of the body, the brain consumes 20% of total blood glucose and oxygen [68]. When at rest, neurons preferentially metabolize glucose via OxPhos and use only about 25% of their respiratory capacity [69]. During heavy synaptic activity, they use up to 80% of their maximal respiratory capacity, and rely on lactate as an additional substrate for OxPhos [70]. Whether neurons produce this lactate themselves or it is ‘shuttled’ to them by astrocytes is a topic of debate [71]. Despite these controversies, it seems clear that neurons primarily use OxPhos to meet energy demands, are supported metabolically to some degree by astrocytes and are uniquely vulnerable to metabolic exhaustion.
It stands to reason that neurons with the highest energy demands will be the most affected by age-related mitochondrial dysfunction, and there is evidence that affected neurons attempt to increase their respiratory capacity. Immunofluorescence studies suggest that the sub-population of neurons affected by mitochondrial abnormalities and oxidative damage overexpress cytochrome oxidase to compensate for energetic insufficiencies [72–74]. Another mechanism for adapting to bioenergetic stress is the cycle of mitochondrial fusion, fission and mitophagy that helps to increase both the number and quality of mitochondria [25,75]. We call this compensatory metabolic shift the ‘inverse Warburg effect’, as neurons upregulate OxPhos while cancer cells switch to aerobic glycolysis (figure 2c). The reprogramming leads to the development of two populations: relatively intact neurons with normal OxPhos activity (Type 1) and highly impaired neurons with upregulated OxPhos activity (Type 2). In the neuronal microenvironment, characterized by constant but limited energy resources, the Type 2 neurons have a selective advantage due to their increased evolutionary entropy. They will take up energy precursors at a faster rate due to their increased metabolism and use them as substrate for OxPhos. Some Type 1 cells will die from bioenergetic failure as the result of this competition, but eventually a steady state will be reached and the two populations will coexist, but with a certain level of neuronal dysfunction and vulnerability.
7. From physiological to pathological ageing: neuronal energy crisis?
Our model contends that the transition to disease arises when the steady-state condition involving Types 1 and 2 neurons is disrupted, and Type 1 neurons are no longer able to appropriate sufficient energy to maintain their viability (figure 3). The forces that trigger this shift from normal to pathological ageing may be environmental, a stroke or similar vascular insult, a metabolic disease such as diabetes or genetic inheritance, which demarcates a baseline mitochondrial function and metabolic stability which prescribes the rate at which an individual ages. Vascular pathology and cerebral hypoperfusion seem to play a central role in pathologic brain ageing, and in the pathophysiology of sporadic AD [76]. Indeed, the most prominent modifiable risk factors for sporadic AD are those for vascular disease [77–80]. Reduced glucose and oxygen delivery to impaired neurons further depletes energy reserve and promotes cellular dysfunction. In this setting, more and more neurons will need to upregulate OxPhos in order to survive and will out-compete those that do not. Thus, the propagation of disease is by way of natural selection and the disorder spreads from neurons with the highest energy requirements to those with lower energy demands.
Figure 3.

Metabolic alteration and natural selection lead to pathological brain ageing. (a) Normal (Type 1) neurons primarily use OxPhos for ATP production. Neurons use glucose preferentially when at rest but rely on lactate to meet energy needs when synaptically active. Age-related damage leads to decreased energy production from mitochondrial dysfunction and decreased energy precursors. Neurons with impaired energetics compensate by upregulating OxPhos (the inverse Warburg effect). (b) These impaired (Type 2) neurons now compete with (Type 1) neurons for limited lactate. In normal ageing, these two populations are able to coexist in a quasi-stable state. (b) Under additional metabolic stress from diverse processes such as vascular disease, cerebral trauma and diabetes, more and more neurons will convert to Type 2, as they have a selective advantage and their higher evolutionary entropy gives them higher viability under conditions of a scarce energy resource. (c) Eventually, Type 1 neurons will lose this energy competition and begin to die. Owing to their OxPhos upregulation, Type 2 neurons will incur progressive oxidative stress. While this benefits survival in the short term, eventually it causes progressive oxidative damage and neuronal loss. (Online version in colour.)
The pathway to neuronal loss that begins with disrupted energy metabolism proceeds by way of increased oxidative stress, an imbalance between the generation and detoxification of ROS. The increase in mitochondria and electron carriers to compensate for energy crisis in impaired neurons leads to an increase in ROS production. The brain is very rich in easily peroxidizable fatty acids and is relatively poor in anti-oxidant systems [81]. Oxidative stress manifests itself as protein, RNA and DNA oxidation and lipid peroxidation. These metabolic changes can have critical effects on neuronal dynamics and the capacity of neurons to maintain synaptic connections and signal effectively. Downstream events of oxidative insults include progressive DNA damage, disruption of neurotrophic factors, Aβ oligomer formation and hyperphosphorylation and aggregation of tau [26]. Oxidative stress can also prompt damaged neurons to enter a dysfunctional cell cycle, leading to additional oxidative damage, tau hyperphosphorylation and ultimately apoptosis [82].
8. Therapeutic implications: suppressing the inverse warburg effect
The bioenergetic model we have discussed suggests that metabolic interventions might prevent AD or inhibit its spread to other areas of the cortex. The first approach to AD prevention is to slow the age-related damage that creates the neuronal energy crisis in the first place. Interventions that preserve metabolic health and promote healthy ageing, such as a healthy diet and sufficient exercise, are perhaps the most important preventive interventions for AD and a host of other age-related diseases [83–85].
Another important approach, an immediate derivative of the entropic selection theory, is by means of natural selection: the reduction of the selective advantage of impaired neurons in competition with intact neurons for energy precursors. One strategy is to prevent the microenvironment that fosters metabolic reprogramming by increasing the availability of lactate for neuronal energy needs. This could be done by enhancing the function of key glycolytic enzymes [86]. Interestingly, exercise-induced increases in systemic lactate levels have been shown to elicit positive effects on memory [87]. Lactate also appears to have a number of additional neuroprotective properties. Lactate infusion can attenuate ischaemic damage and increase the expression of neuroprotective brain-derived neurotrophic factor in mouse models [88,89]. It also seems to be an important signalling mechanism for cerebral blood flow and can stabilize hypoxia-inducible factor-1, which in turn increases glycolysis [90,91].
Another approach to increasing neuronal energy precursors is to bypass abnormal glucose metabolism and supplement the neuron with an alternative energy substrate such as fat. During fasting, the body metabolizes fatty acids to ketone bodies, which can enter the TCA cycle and generate ATP. Ketone metabolism results in production of fewer ROS and enhanced mitochondrial efficiency as it leads to a lower mitochondrial membrane potential [92,93]. The ketogenic diet is an established therapy for epilepsy and some disorders of glucose metabolism. Clinical trials of supplementation with a medium-chain triglyceride have demonstrated improvements in cognition in patients with AD and age-associated memory impairment [94].
Agents that improve astrocyte function might also be neuroprotective [95]. Activated astrocytes quickly deplete glutathione precursors such as cysteine, and can no longer supply neurons with sufficient anti-oxidants. A clinical trial of n-acetyl cysteine supplementation resulted in improvement across a wide range of outcomes in patients with probable AD [96]. Anti-oxidant treatment may also improve the uptake of glutamate, an excitotoxic neurotransmitter [97]. Finally, drugs that prevent or suppress astroglial activation would keep these cells in their supportive role and decrease the level of inflammation in the neuronal milieu [95].
Other metabolic therapies, such as anti-diabetic drugs that enhance glucose uptake and metabolic processing may improve the metabolism of both astrocytes and neurons and show promise as chemoprevention and treatment for AD. Intranasal insulin improves memory and attention in normal subjects and those with mild cognitive impairment [98–100]. Mimetics of glucagon-like peptide 1 (GLP-1) have impressive neuroprotective effects in animal models through multiple mechanisms, including increased insulin sensitivity cellular metabolism and inhibition of inflammation and apoptosis [101]. These agents can be delivered intra-nasally and cross the blood–brain barrier.
9. Conclusion
For over 25 years, AD research has been driven by a neuron-centric model in which disease is considered to be the result of mutations in the nuclear genome leading to toxic species of Aβ and tau. This model has singularly failed to explain the origin and progression of the late-onset form. James Watson lamented that the exciting work of biochemists such as Warburg, who discovered the metabolic basis of sporadic forms of cancer, was de-emphasized after the discovery of the double helix. Watson called for a new generation of biochemists who could reinvigorate cancer research by investigating the metabolic basis of the disease [102]. The processes that drive the dynamics of living organisms can be considered in terms of two complementary viewpoints—genetic and bioenergetic. Together, these viewpoints capture the essence of life: the organization and regulation of genetic information and the conversion of energy from the environment into a form that can do cellular work [103]. According to the genetic mode (figure 4a), disease states are characterized in terms of defects in specific proteins that result from mutations in the nuclear genome. Disease states in the context of the bioenergetic model are described by defects in the normal energy transduction process (figure 4b)—a condition induced by mitochondrial dysregulation. The argument reviewed in this paper revolves around a distinction between early- and late-onset diseases. Early-onset diseases are rare, as selection to eliminate these disorders is strong. Late-onset diseases are common, as selection to eliminate these disorders is weak. Warburg evidently recognized the distinction between the genetic and the bioenergetic perspectives. His claim that the sporadic forms of cancer are metabolic diseases—a claim that was largely ignored—has been recently acknowledged, as indicated by the significant role the Warburg effect now plays in current cancer research.
Figure 4.
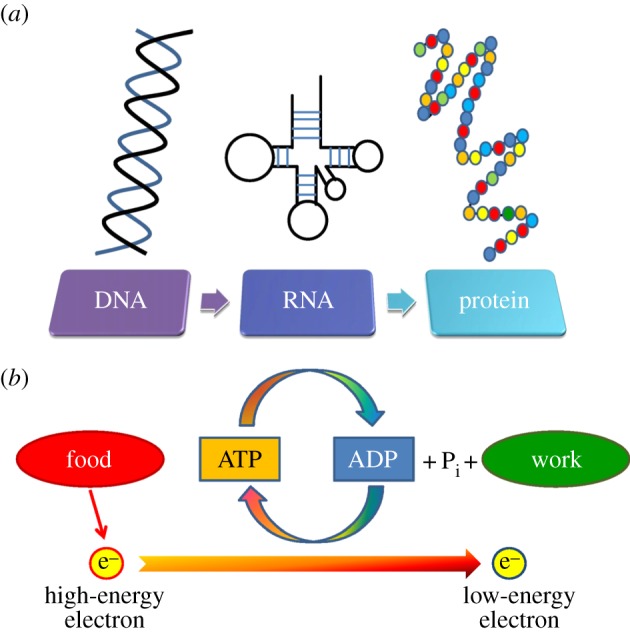
Genetic and bioenergetic models of health and disease. Life requires the flow of both information and energy. Disease and death result from dysfunction of either of these processes. (a) The organization, maintenance and expression of the information encoded in DNA is critical to surival. Information flows from DNA to RNA to proteins. DNA is moulded by mutation, recombination and natural selection. Genetic diseases are caused by specific defects in the nuclear genome that result in impaired or absent protein function. Disease can be directly attributed to the abnormal gene/protein. These defects tend to present early in life, be severe and are strongly selected against. They are therefore rare. (b) Life requires energy. The figure shows the flow of energy as the fall of electrons from a higher to a lower energy state. High-energy electrons are derived from organic food and the final electron acceptor is oxygen. The coupled assembly of ATP from ADP and organic phosphate is supported by the shift from a high-energy electron to a low-level state. The cell uses ATP to do work. The bioenergetic model of diseases is based on the concept that the bioenergetic machinery is vulnerable to thermodynamic entropy and somatic mutation. Selection to maintain bioenergetic systems in the post-reproductive years is weak. The age-related decline in bioenergetics makes cells vulnerable to various forms of stress, but additonal forces are needed for pathological ageing to occur. (Online version in colour.)
The nuclear-genomic perspective denies current facts about the sporadic forms of AD. Age in the context of this model is considered uniquely in terms of its influence on the toxicity of the defective protein. According to this viewpoint, sporadic forms of AD are also governed by common DNA variants (such as single-nucleotide polymorphisms). These variants are assumed to be modulated by the action of various environmental factors which significantly increase disease risk. The progress of the disease is assumed to be determined by the incidence of biochemical markers such as Aβ and tau, and progression of the disease is conceived as a linear process that tracks with the accumulation of these markers. The validity of this prediction has been compromised by the prevalence of individuals who show no sign of dementia yet have substantial Aβ deposition. There is now strong support that, at least among the oldest old, dementia severity is completely dissociated from Aβ and tau neuropathology. Furthermore, over a decade of therapeutic strategies that target Aβ have all failed to show any clinical improvement.
The bioenergetic model reviewed in this article considers age in terms of the increase in molecular disorder it generates and the effect of this disorder on the efficiency of biochemical processes. According to this bioenergetic model, the sporadic form of AD is governed by the dynamics of the mitochondria in neurons. The model contends that the progression of AD is a nonlinear, age-dependent process which is triggered by mitochondrial dysregulation and can be described in terms of three phases: (1) an upregulation of OxPhos to compensate for the crisis in neurons with impaired energy production; (2) a quasi-stable state of energy production that results from competition between neurons with different energy production capacities, and (3) a global decline in energy production triggered by forces that disrupt this quasi-equilibrium. Our model contends that the second stage defines normal or physiological ageing, and that the transition to pathological ageing and progression towards AD is a multifactorial process in which vascular disease and cerebral hypoperfusion play a prominent role. The empirical studies we have cited are consistent with these stages and our model is also consistent with the epidemiological observation that sporadic AD is an age-related disorder whose incidence increases, not linearly, but exponentially with age. The model also explains why AD is sporadic, and why individual genes seem to contribute little to its pathophysiology. It is consistent with the patterns of neuronal vulnerability and propagation of disease. The stochastic nature of bioenergetic damage helps explain the observation that diseased neurons are found in proximity to healthy neurons in the brains of people with AD. While not every element of our hypothesis is based on experimental data, all of its aspects are testable.
While the formal analysis of the model involves notions that derive from non-medical disciplines, the concepts of age as physiological time, energy as a metabolic entity and competition as a dynamic process are less abstract and should be extremely helpful in elucidating the pathogenesis of AD and other complex, late-onset diseases. Understanding sporadic AD as a metabolic disease will help clinicians promote effective ‘metabolic’ interventions such as exercise and healthy dietary habits, the value of which cannot be over-emphasized. We sincerely hope that the mechanism of disease we have outlined will inspire those who fund and conduct AD research to seek metabolic therapies that can enhance the survival of intact neurons during the ageing process, thus turning natural selection to our patients' advantage.
Acknowledgements
L.A.D. generated the idea for the manuscript, performed literature searches and helped to draft and edit it. J.A.D. also helped to generate ideas, performed literature searches, helped to draft and edit the manuscript and prepared the figures.
Funding statement
Support from the Max Planck Institute for Molecular Genetics, Berlin, Germany, is gratefully acknowledged. J.A.D. is supported by a Veteran's Administration Merit Award CSR&D I01CX000934–01A1.
Conflict of interests
The authors have no financial or personal relationships with other people or organizations that might inappropriately influence or bias the work.
References
- 1.Goate A, et al. 1991. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 349, 704–706. ( 10.1038/349704a0) [DOI] [PubMed] [Google Scholar]
- 2.Gooch MD, Stennett DJ. 1996. Molecular basis of Alzheimer's disease. Am. J. Health Syst. Pharm. 53, 1545–1557; quiz 603–604. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J, Selkoe DJ. 2002. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. ( 10.1126/science.1072994) [DOI] [PubMed] [Google Scholar]
- 4.Hardy J. 2006. Alzheimer's disease: the amyloid cascade hypothesis: an update and reappraisal. J. Alzheimer's Dis. 9(Suppl. 3), 151–153. [DOI] [PubMed] [Google Scholar]
- 5.Rao VS, van Duijn CM, Connor-Lacke L, Cupples LA, Growdon JH, Farrer LA. 1994. Multiple etiologies for Alzheimer disease are revealed by segregation analysis. Am. J. Hum. Genet. 55, 991–1000. [PMC free article] [PubMed] [Google Scholar]
- 6.Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee J-M, Holtzman DM. 2011. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat. Neurosci. 14, 750–756. ( 10.1038/nn.2801) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies L, Wolska B, Hilbich C, Multhaup G, Martins R, Simms G, Beyreuther K, Masters CL. 1988. A4 amyloid protein deposition and the diagnosis of Alzheimer's disease: prevalence in aged brains determined by immunocytochemistry compared with conventional neuropathologic techniques. Neurology 38, 1688–1693. ( 10.1212/WNL.38.11.1688) [DOI] [PubMed] [Google Scholar]
- 8.Bulbarelli A, Lonati E, Brambilla A, Orlando A, Cazzaniga E, Piazza F, Ferrarese C, Masserini M, Sancini G. 2012. Aβ42 production in brain capillary endothelial cells after oxygen and glucose deprivation. Mol. Cell. Neurosci. 49, 415–422. ( 10.1016/j.mcn.2012.01.007) [DOI] [PubMed] [Google Scholar]
- 9.Daulatzai MA. 2013. Death by a thousand cuts in Alzheimer's disease: hypoxia—the prodrome. Neurotox. Res. 24, 216–243. ( 10.1007/s12640-013-9379-2) [DOI] [PubMed] [Google Scholar]
- 10.Pimentel-Coelho PM, Rivest S. 2012. The early contribution of cerebrovascular factors to the pathogenesis of Alzheimer's disease. Eur. J. Neurosci. 35, 1917–1937. ( 10.1111/j.1460-9568.2012.08126.x) [DOI] [PubMed] [Google Scholar]
- 11.Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. 1994. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry 57, 419–425. ( 10.1136/jnnp.57.4.419) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klunk WE, et al. 2004. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann. Neurol. 55, 306–319. ( 10.1002/ana.20009) [DOI] [PubMed] [Google Scholar]
- 13.Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, Markesbery WR. 2000. ‘Preclinical’ AD revisited: neuropathology of cognitively normal older adults. Neurology 55, 370–376. ( 10.1212/WNL.55.3.370) [DOI] [PubMed] [Google Scholar]
- 14.Doody RS, et al. 2014. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer's disease. N Engl. J. Med. 370, 311–321. ( 10.1056/NEJMoa1312889) [DOI] [PubMed] [Google Scholar]
- 15.Fleisher AS, et al. 2008. Phase 2 safety trial targeting amyloid beta production with a gamma-secretase inhibitor in Alzheimer disease. Arch. Neurol. 65, 1031–1038. ( 10.1001/archneur.65.8.1031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green RC, et al. 2009. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. J. Am. Med. Assoc. 302, 2557–2564. ( 10.1001/jama.2009.1866) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holmes C, et al. 2008. Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 372, 216–223. ( 10.1016/S0140-6736(08)61075-2) [DOI] [PubMed] [Google Scholar]
- 18.Salloway S, et al. 2014. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. N Engl. J. Med. 370, 322–333. ( 10.1056/NEJMoa1304839) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pimplikar SW. 2009. Reassessing the amyloid cascade hypothesis of Alzheimer's disease. Int. J. Biochem. Cell Biol. 41, 1261–1268. ( 10.1016/j.biocel.2008.12.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Struble RG, Ala T, Patrylo PR, Brewer GJ, Yan XX. 2010. Is brain amyloid production a cause or a result of dementia of the Alzheimer's type? J. Alzheimer's Dis. 22, 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi SH, et al. 2014. A three-dimensional human neural cell culture model of Alzheimer's disease. Nature 515, 274–278. ( 10.1038/nature13800) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haroutunian V, et al. 2008. Role of the neuropathology of Alzheimer disease in dementia in the oldest-old. Arch. Neurol. 65, 1211–1217. ( 10.1001/archneur.65.9.1211) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prohovnik I, Perl DP, Davis KL, Libow L, Lesser G, Haroutunian V. 2006. Dissociation of neuropathology from severity of dementia in late-onset Alzheimer disease. Neurology 66, 49–55. ( 10.1212/01.wnl.0000191298.68045.50) [DOI] [PubMed] [Google Scholar]
- 24.Hayflick L. 2007. Entropy explains aging, genetic determinism explains longevity, and undefined terminology explains misunderstanding both. PLOS Genet. 3, e220 ( 10.1371/journal.pgen.0030220) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Demetrius LA, Simon DK. 2012. An inverse-Warburg effect and the origin of Alzheimer's disease. Biogerontology 13, 583–594. ( 10.1007/s10522-012-9403-6) [DOI] [PubMed] [Google Scholar]
- 26.Demetrius LA, Driver J. 2013. Alzheimer's as a metabolic disease. Biogerontology 14, 641–649. ( 10.1007/s10522-013-9479-7) [DOI] [PubMed] [Google Scholar]
- 27.Demetrius LA, Simon DK. 2013. The inverse association of cancer and Alzheimer's: a bioenergetic mechanism. J. R. Soc. Interface 10, 20130006 ( 10.1098/rsif.2013.0006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mattson MP, Magnus T. 2006. Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 7, 278–294. ( 10.1038/nrn1886) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Driver JA, Beiser A, Au R, Kreger BE, Splansky GL, Kurth T, Kiel DP, Lu KP, Seshadri S. 2012. Inverse association between cancer and Alzheimer's disease: results from the Framingham Heart Study. Br. Med. J. 344, e1442 ( 10.1136/bmj.e1442) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tabares-Seisdedos R, Rubenstein JL. 2013. Inverse cancer comorbidity: a serendipitous opportunity to gain insight into CNS disorders. Nat. Rev. Neurosci. 14, 293–304. ( 10.1038/nrn3464) [DOI] [PubMed] [Google Scholar]
- 31.Demetrius L, Legendre S. 2013. Evolutionary entropy predicts the outcome of selection: competition for resources that vary in abundance and diversity. Theor. Popul. Biol. 83, 39–54. ( 10.1016/j.tpb.2012.10.004) [DOI] [PubMed] [Google Scholar]
- 32.Demetrius L. 1974. Demographic parameters and natural selection. Proc. Natl Acad. Sci. USA 71, 4645–4647. ( 10.1073/pnas.71.12.4645) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demetrius L. 2013. Boltzmann, Darwin and directionality theory. Phys. Rep. 530, 1–85. ( 10.1016/j.physrep.2013.04.001) [DOI] [Google Scholar]
- 34.Demetrius L. 1997. Directionality principles in thermodynamics and evolution. Proc. Natl Acad. Sci. USA 94, 3491–3498. ( 10.1073/pnas.94.8.3491) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beal MF. 1995. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann. Neurol. 38, 357–366. ( 10.1002/ana.410380304) [DOI] [PubMed] [Google Scholar]
- 36.Drachman DA. 2006. Aging of the brain, entropy, and Alzheimer disease. Neurology 67, 1340–1352. ( 10.1212/01.wnl.0000240127.89601.83) [DOI] [PubMed] [Google Scholar]
- 37.Schmitt K, Grimm A, Kazmierczak A, Strosznajder JB, Gotz J, Eckert A. 2012. Insights into mitochondrial dysfunction: aging, amyloid-beta, and tau-A deleterious trio. Antioxid. Redox Signal. 16, 1456–1466. ( 10.1089/ars.2011.4400) [DOI] [PubMed] [Google Scholar]
- 38.Swerdlow RH. 2012. Mitochondria and cell bioenergetics: increasingly recognized components and a possible etiologic cause of Alzheimer's disease. Antioxid. Redox Signal. 16, 1434–1455. ( 10.1089/ars.2011.4149) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wallace DC, Fan W, Procaccio V. 2010. Mitochondrial energetics and therapeutics. Annu. Rev. Pathol. 5, 297–348. ( 10.1146/annurev.pathol.4.110807.092314) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wallace DC. 1992. Mitochondrial genetics: a paradigm for aging and degenerative diseases? Science 256, 628–632. ( 10.1126/science.1533953) [DOI] [PubMed] [Google Scholar]
- 41.Bakkour A, Morris JC, Dickerson BC. 2009. The cortical signature of prodromal AD: regional thinning predicts mild AD dementia. Neurology 72, 1048–1055. ( 10.1212/01.wnl.0000340981.97664.2f) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dickerson BC, et al. 2009. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb. Cortex. 19, 497–510. ( 10.1093/cercor/bhn113) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.LaManna JC, Harik SI. 1985. Regional comparisons of brain glucose influx. Brain Res. 326, 299–305. ( 10.1016/0006-8993(85)90039-3) [DOI] [PubMed] [Google Scholar]
- 44.Kapogiannis D, Mattson MP. 2011. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer's disease. Lancet Neurol. 10, 187–198. ( 10.1016/S1474-4422(10)70277-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swerdlow RH, Burns JM, Khan SM. 2010. The Alzheimer's disease mitochondrial cascade hypothesis. J. Alzheimer's Dis. 20(Suppl. 2), S265–S279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arnold SE, Hyman BT, Flory J, Damasio AR, Van Hoesen GW. 1991. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer's disease. Cereb. Cortex 1, 103–116. ( 10.1093/cercor/1.1.103) [DOI] [PubMed] [Google Scholar]
- 47.Buckner RL, et al. 2009. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer's disease. J. Neurosci. 29, 1860–1873. ( 10.1523/JNEUROSCI.5062-08.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dickerson BC, et al. 2004. Medial temporal lobe function and structure in mild cognitive impairment. Ann. Neurol. 56, 27–35. ( 10.1002/ana.20163) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jacobs HI, Van Boxtel MP, Heinecke A, Gronenschild EH, Backes WH, Ramakers IH, Jolles J, Verhey FRJ. 2012. Functional integration of parietal lobe activity in early Alzheimer disease. Neurology 78, 352–360. ( 10.1212/WNL.0b013e318245287d) [DOI] [PubMed] [Google Scholar]
- 50.Nichols LM, et al. 2012. Interactive effect of apolipoprotein E genotype and age on hippocampal activation during memory processing in healthy adults. Arch. Gen. Psychiatry 69, 804–813. ( 10.1001/archgenpsychiatry.2011.1893) [DOI] [PubMed] [Google Scholar]
- 51.Oh H, Habeck C, Madison C, Jagust W. 2014. Covarying alterations in Aβ deposition, glucose metabolism, and gray matter volume in cognitively normal elderly. Hum. Brain Mapp. 35, 297–308. ( 10.1002/hbm.22173) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benzinger TL, et al. 2013. Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc. Natl Acad. Sci. USA 110, E4502–E4509. ( 10.1073/pnas.1317918110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cirrito JR, et al. 2005. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922. ( 10.1016/j.neuron.2005.10.028) [DOI] [PubMed] [Google Scholar]
- 54.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K. 1987. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736. ( 10.1038/325733a0) [DOI] [PubMed] [Google Scholar]
- 55.Hayflick L. 2007. Biological aging is no longer an unsolved problem. Ann. NY Acad. Sci. 1100, 1–13. ( 10.1196/annals.1395.001) [DOI] [PubMed] [Google Scholar]
- 56.Demetrius L. 2002. Thermodynamics and kinetics of protein folding: an evolutionary perspective. J. Theor. Biol. 217, 397–411. ( 10.1006/jtbi.2002.3006) [DOI] [PubMed] [Google Scholar]
- 57.Csermely P. 2001. Chaperone overload is a possible contributor to ‘civilization diseases’. Trends Genet. 17, 701–704. ( 10.1016/S0168-9525(01)02495-7) [DOI] [PubMed] [Google Scholar]
- 58.Chiti F, Dobson CM. 2006. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366. ( 10.1146/annurev.biochem.75.101304.123901) [DOI] [PubMed] [Google Scholar]
- 59.Birsoy K, et al. 2014. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508, 108–112. ( 10.1038/nature13110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pfeiffer T, Bonhoeffer S. 2002. Evolutionary consequences of tradeoffs between yield and rate of ATP production. Z. Physikalische Chemie 216, 51–63. ( 10.1524/zpch.2002.216.1.051) [DOI] [Google Scholar]
- 61.Koopman WJ, Distelmaier F, Smeitink JA, Willems PH. 2013. OXPHOS mutations and neurodegeneration. EMBO J. 32, 9–29. ( 10.1038/emboj.2012.300) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vander Heiden MG, Cantley LC, Thompson CB. 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. ( 10.1126/science.1160809) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Warburg O. 1956. On the origin of cancer cells. Science 123, 309–314. ( 10.1126/science.123.3191.309) [DOI] [PubMed] [Google Scholar]
- 64.Lunt SY, Vander Heiden MG. 2011. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27, 441–464. ( 10.1146/annurev-cellbio-092910-154237) [DOI] [PubMed] [Google Scholar]
- 65.Zhao Y, Butler EB, Tan M. 2013. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 4, e532 ( 10.1038/cddis.2013.60) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bauernfeind AL, Barks SK, Duka T, Grossman LI, Hof PR, Sherwood CC. 2013. Aerobic glycolysis in the primate brain: reconsidering the implications for growth and maintenance. Brain Struct. Funct. 219, 1149–1167. ( 10.1007/s00429-013-0662-z) [DOI] [PubMed] [Google Scholar]
- 67.Goyal MS, Hawrylycz M, Miller JA, Snyder AZ, Raichle ME. 2014. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 19, 49–57. ( 10.1016/j.cmet.2013.11.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Magistretti PJ. 2006. Neuron–glia metabolic coupling and plasticity. J. Exp. Biol. 209, 2304–2311. ( 10.1242/jeb.02208) [DOI] [PubMed] [Google Scholar]
- 69.Nicholls DG. 2002. Mitochondrial function and dysfunction in the cell: its relevance to aging and aging-related disease. Int. J. Biochem. Cell Biol. 34, 1372–1381. ( 10.1016/S1357-2725(02)00077-8) [DOI] [PubMed] [Google Scholar]
- 70.Hu Y, Wilson GS. 1997. A temporary local energy pool coupled to neuronal activity: fluctuations of extracellular lactate levels in rat brain monitored with rapid-response enzyme-based sensor. J. Neurochem. 69, 1484–1490. ( 10.1046/j.1471-4159.1997.69041484.x) [DOI] [PubMed] [Google Scholar]
- 71.Stobart JL, Anderson CM. 2013. Multifunctional role of astrocytes as gatekeepers of neuronal energy supply. Front. Cell. Neurosci. 7, 38 ( 10.3389/fncel.2013.00038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.de la Monte SM, Luong T, Neely TR, Robinson D, Wands JR. 2000. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer's disease. Lab. Invest. J. Tech. Methods Pathol. 80, 1323–1335. ( 10.1038/labinvest.3780140) [DOI] [PubMed] [Google Scholar]
- 73.Manczak M, Park BS, Jung Y, Reddy PH. 2004. Differential expression of oxidative phosphorylation genes in patients with Alzheimer's disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromol. Med. 5, 147–162. ( 10.1385/NMM:5:2:147) [DOI] [PubMed] [Google Scholar]
- 74.Nagy Z, Esiri MM, LeGris M, Matthews PM. 1999. Mitochondrial enzyme expression in the hippocampus in relation to Alzheimer-type pathology. Acta Neuropathol. 97, 346–354. ( 10.1007/s004010050997) [DOI] [PubMed] [Google Scholar]
- 75.Youle RJ, van der Bliek AM. 2012. Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. ( 10.1126/science.1219855) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.de la Torre JC. 2008. Pathophysiology of neuronal energy crisis in Alzheimer's disease. Neuro-Degener. Dis. 5, 126–132. ( 10.1159/000113681) [DOI] [PubMed] [Google Scholar]
- 77.Breteler MM. 2000. Vascular risk factors for Alzheimer's disease: an epidemiologic perspective. Neurobiol. Aging 21, 153–160. ( 10.1016/S0197-4580(99)00110-4) [DOI] [PubMed] [Google Scholar]
- 78.Elias MF, Wolf PA, D'Agostino RB, Cobb J, White LR. 1993. Untreated blood pressure level is inversely related to cognitive functioning: the Framingham Study. Am. J. Epidemiol. 138, 353–364. [DOI] [PubMed] [Google Scholar]
- 79.Ivan CS, Seshadri S, Beiser A, Au R, Kase CS, Kelly-Hayes M, Kelly-Hayes M, Wolf PA. 2004. Dementia after stroke: the Framingham Study. Stroke J. Cereb. Circulation 35, 1264–1268. ( 10.1161/01.STR.0000127810.92616.78) [DOI] [PubMed] [Google Scholar]
- 80.Ruitenberg A, den Heijer T, Bakker SL, van Swieten JC, Koudstaal PJ, Hofman A, Breteler MMB. 2005. Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann. Neurol. 57, 789–794. ( 10.1002/ana.20493) [DOI] [PubMed] [Google Scholar]
- 81.Coyle JT, Puttfarcken P. 1993. Oxidative stress, glutamate, and neurodegenerative disorders. Science 262, 689–695. ( 10.1126/science.7901908) [DOI] [PubMed] [Google Scholar]
- 82.Folch J, Junyent F, Verdaguer E, Auladell C, Pizarro JG, Beas-Zarate C, Camins A. 2012. Role of cell cycle re-entry in neurons: a common apoptotic mechanism of neuronal cell death. Neurotox. Res. 22, 195–207. ( 10.1007/s12640-011-9277-4) [DOI] [PubMed] [Google Scholar]
- 83.Baker LD, et al. 2010. Effects of aerobic exercise on mild cognitive impairment: a controlled trial. Arch. Neurol. 67, 71–79. ( 10.1001/archneurol.2009.307) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Etgen T, Sander D, Huntgeburth U, Poppert H, Forstl H, Bickel H. 2010. Physical activity and incident cognitive impairment in elderly persons: the INVADE study. Arch. Intern. Med. 170, 186–193. ( 10.1001/archinternmed.2009.498) [DOI] [PubMed] [Google Scholar]
- 85.Geda YE, et al. 2010. Physical exercise, aging, and mild cognitive impairment: a population-based study. Arch. Neurol. 67, 80–86. ( 10.1001/archneurol.2009.297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brix B, Mesters JR, Pellerin L, Johren O. 2012. Endothelial cell-derived nitric oxide enhances aerobic glycolysis in astrocytes via HIF-1alpha-mediated target gene activation. J. Neurosci. 32, 9727–9735. ( 10.1523/JNEUROSCI.0879-12.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Roig M, Skriver K, Lundbye-Jensen J, Kiens B, Nielsen JB. 2012. A single bout of exercise improves motor memory. PLoS ONE 7, e44594 ( 10.1371/journal.pone.0044594) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Berthet C, Castillo X, Magistretti PJ, Hirt L. 2012. New evidence of neuroprotection by lactate after transient focal cerebral ischaemia: extended benefit after intracerebroventricular injection and efficacy of intravenous administration. Cerebrovasc. Dis. 34, 329–335. ( 10.1159/000343657) [DOI] [PubMed] [Google Scholar]
- 89.Coco M, Caggia S, Musumeci G, Perciavalle V, Graziano AC, Pannuzzo G, Cardile V. 2013. Sodium l-lactate differently affects brain-derived neurothrophic factor, inducible nitric oxide synthase, and heat shock protein 70 kDa production in human astrocytes and SH-SY5Y cultures. J. Neurosci. Res. 91, 313–320. ( 10.1002/jnr.23154) [DOI] [PubMed] [Google Scholar]
- 90.Newington JT, Pitts A, Chien A, Arseneault R, Schubert D, Cumming RC. 2011. Amyloid beta resistance in nerve cell lines is mediated by the Warburg effect. PLoS ONE 6, e19191 ( 10.1371/journal.pone.0019191) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Soucek T, Cumming R, Dargusch R, Maher P, Schubert D. 2003. The regulation of glucose metabolism by HIF-1 mediates a neuroprotective response to amyloid beta peptide. Neuron 39, 43–56. ( 10.1016/S0896-6273(03)00367-2) [DOI] [PubMed] [Google Scholar]
- 92.Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM. 2007. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 145, 256–264. ( 10.1016/j.neuroscience.2006.11.065) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sato K, et al. 1995. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. 9, 651–658. [DOI] [PubMed] [Google Scholar]
- 94.Costantini LC, Barr LJ, Vogel JL, Henderson ST. 2008. Hypometabolism as a therapeutic target in Alzheimer's disease. BMC Neurosci. 9(Suppl. 2), S16 ( 10.1186/1471-2202-9-S2-S16) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fuller S, Munch G, Steele M. 2009. Activated astrocytes: a therapeutic target in Alzheimer's disease? Expert Rev. Neurother. 9, 1585–1594. ( 10.1586/ern.09.111) [DOI] [PubMed] [Google Scholar]
- 96.Adair JC, Knoefel JE, Morgan N. 2001. Controlled trial of N-acetylcysteine for patients with probable Alzheimer's disease. Neurology 57, 1515–1517. ( 10.1212/WNL.57.8.1515) [DOI] [PubMed] [Google Scholar]
- 97.Begni B, et al. 2004. Oxidative stress impairs glutamate uptake in fibroblasts from patients with Alzheimer's disease. Free Rad. Biol. Med. 37, 892–901. ( 10.1016/j.freeradbiomed.2004.05.028) [DOI] [PubMed] [Google Scholar]
- 98.Benedict C, Kern W, Schultes B, Born J, Hallschmid M. 2008. Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J. Clin. Endocrinol. Metabol. 93, 1339–1344. ( 10.1210/jc.2007-2606) [DOI] [PubMed] [Google Scholar]
- 99.Craft S, et al. 2012. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch. Neurol. 69, 29–38. ( 10.1001/archneurol.2011.233) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shemesh E, Rudich A, Harman-Boehm I, Cukierman-Yaffe T. 2012. Effect of intranasal insulin on cognitive function: a systematic review. J. Clin. Endocrinol. Metab. 97, 366–376. ( 10.1210/jc.2011-1802) [DOI] [PubMed] [Google Scholar]
- 101.Hölscher C. 2013. Central effects of GLP-1: new opportunities for treatments of neurodegenerative diseases. J. Endocrinol. 221, T31–T41. ( 10.1530/JOE-13-0221) [DOI] [PubMed] [Google Scholar]
- 102.Watson JD. 2009. To fight cancer, know the enemy. New York Times. 6 August 2009, p. A29. See http://www.nytimes.com/2009/08/06/opinion/06watson.html?pagewanted=all&_r=0.
- 103.Kennedy SR, Loeb LA, Herr AJ. 2012. Somatic mutations in aging, cancer and neurodegeneration. Mech. Ageing Dev. 133, 118–126. ( 10.1016/j.mad.2011.10.009) [DOI] [PMC free article] [PubMed] [Google Scholar]