

Abstract
Identifying dysregulated pathways from high-throughput experimental data in order to infer underlying biological insights is an important task. Current pathway-identification methods focus on single pathways in isolation; however, consideration of crosstalk between pathways could improve our understanding of alterations in biological states. We propose a novel method of pathway analysis based on global influence (PAGI) to identify dysregulated pathways, by considering both within-pathway effects and crosstalk between pathways. We constructed a global gene–gene network based on the relationships among genes extracted from a pathway database. We then evaluated the extent of differential expression for each gene, and mapped them to the global network. The random walk with restart algorithm was used to calculate the extent of genes affected by global influence. Finally, we used cumulative distribution functions to determine the significance values of the dysregulated pathways. We applied the PAGI method to five cancer microarray datasets, and compared our results with gene set enrichment analysis and five other methods. Based on these analyses, we demonstrated that PAGI can effectively identify dysregulated pathways associated with cancer, with strong reproducibility and robustness. We implemented PAGI using the freely available R-based and Web-based tools (http://bioinfo.hrbmu.edu.cn/PAGI).
Keywords: dysregulated pathway identification, pathway crosstalk, global influence, random walk
1. Introduction
The development of high-throughput experimental techniques such as gene expression microarrays, mass spectrometry and large-scale mutagenesis has led to the identification of many interesting genes and gene products. In order to interpret these high-throughput experimental data more thoroughly, researchers often study the functional relationships among these genes or gene products systematically. Such studies have shown that canonical biological pathways can help us to understand high-level biological functions at the system level [1], and understanding the inherent interdependency among canonical biological pathways and altered cellular states has become a significant research task. Several computational methods have been developed to identify dysregulated pathways associated with disease states. The classical approaches use statistical models (e.g. the hypergeometric test) to calculate the probability of observing the actual number of differentially expressed genes in a given pathway by chance. These methods consider all differentially expressed genes equally; however, prioritizing genes according to the extent of differential expression may help to identify dysregulated pathways more effectively.
An improved method, gene set enrichment analysis (GSEA), ranks all genes according to the correlations between gene expression profiles and phenotype, and then calculates an enrichment score to determine whether the genes from a predefined pathway cluster at the top or the bottom of the list [2]. However, despite its wide application in pathway analysis, GSEA considers only sets of genes belonging to the pathways and does not take advantage of the important topological relationships between genes embedded in different pathways. Pathway databases, such as the Kyoto Encyclopaedia of Genes and Genomes (KEGG), provide useful pathway topology information. Exploiting this topology information in pathway-identification analysis would improve our understanding of delicate pathway functions and would be meaningful from a systems-biology perspective [3].
Several recent techniques have adopted pathway topology for the identification of dysregulated pathways. ScorePage calculates the significance of changes in activity of metabolic pathways by integrating the shortest distances between genes in pathways [4]. Pathway enrichment analysis (PWEA) incorporates the shortest distance between each pair of genes and uses gene–gene correlations to identify dysregulated pathways [5]. Signalling pathway impact analysis (SPIA) prioritizes pathways by combining classical over-representation evidence with the positions and interactions of genes in the given signalling pathways [6]. These approaches improve the identification of dysregulated pathways from gene expression data; however, they invariably focus on the internal effects of single pathways and fail to consider crosstalk between pathways. Pathway crosstalk refers to the phenomenon of interaction or cooperation between pathways. Understanding crosstalk between pathways will help us to understand the comprehensive biological functions of complex systems [7].
Although latent pathway-identification analysis (LPIA) constructs pathway networks based on shared gene ontology (GO) functions to look for evidence of dysregulated pathways [8], these links between pathways do not consider the overlap between pathways, and the internal effects of pathways are ignored. Pathways based on network information (PathNet) is another interesting method for identifying pathways, which uses inter- and intrapathway relationships to calculate the enrichment of non-metabolic pathways [9]. In PathNet, the association of each gene with a disease phenotype depends on the differential expression level of the gene and its direct pathway neighbours, and thus the effect of other non-neighbour genes may be neglected. From a systems-biology perspective, dysregulated genes may iteratively alter the properties of many other genes, both within and outside a given pathway, via both internal pathway effects and pathway crosstalk. Complex diseases such as cancer are caused by the joint effects of multiple dysregulated genes in pathways and the crosstalk between pathways [10]. The identification of dysregulated pathways, taking into account both internal pathway effects and crosstalk between pathways, thus presents a challenge.
In this study, we propose a novel computational approach to pathway analysis based on global influence (PAGI) to identify dysregulated pathways associated with the initiation or progression of complex diseases. PAGI uses a network-based approach to detect latent dysregulated pathways by considering the global influence of both the internal effect of pathways and crosstalk between pathways. We initially constructed a global gene–gene network based on the relationships of genes extracted from all pathways in the KEGG database and the genes that overlap between pathways. We then evaluated the extent of differential expression for each gene, and all genes represented in the expression data were mapped to the global network. A global dysregulated score (GDS) was defined to represent the extent to which genes were affected by global influence from both internal pathway effects and crosstalk between pathways. The random walk with restart (RWR) algorithm was used to calculate the GDS by integrating the extent of differential gene expression and the global network topology. Finally, we used cumulative distribution functions (CDFs) to evaluate each pathway in the pathway database. We applied PAGI to datasets on breast, prostate and lung cancer and demonstrated its ability to produce biologically meaningful outcomes.
2. Material and methods
We developed a novel computational approach, PAGI, to identify dysregulated pathways by considering the global influence of both the internal effects of pathways and crosstalk between pathways. A flow diagram of the PAGI methodology is shown in figure 1. PAGI was implemented as a freely available R-based tool (http://cran.r-project.org/web/packages/PAGI) and a Web-based tool (http://bioinfo.hrbmu.edu.cn/PAGI or http://202.97.205.78:8080/PAGI).
Figure 1.

Flow diagram of the methodology. A global gene–gene network was initially constructed on the basis of the relationships among genes extracted from all pathways. The extent of differential expression for each gene was evaluated by t-test, and all genes represented in the expression data were mapped to the global network. The RWR algorithm was then used to evaluate the number of genes affected by global influence from both internal effects of the pathways and crosstalk between pathways. Finally, cumulative distribution functions were used to identify dysregulated pathways. (Online version in colour.)
2.1. Datasets
Expression datasets were obtained from the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/). We analysed five cancer datasets: one breast cancer, one prostate cancer and three lung cancer datasets. The breast cancer expression dataset was published by Pau Ni et al. [11] (GSE15852) and consisted of data on 43 human breast tumours and 43 normal tissues. The prostate cancer dataset was obtained from Nanni et al. [12] (GSE3868) and included information on 22 prostate cancer samples and eight control samples derived from normal/hyperplastic tissues and prostate epithelial cells. The three lung cancer datasets [13–15] (GSE7670; GSE10072; GSE2514) each included data on cancer samples and normal controls. Details of these datasets are listed in the electronic supplementary material, table S1. The raw data were log transformed for downstream analysis.
2.2. Constructing the global gene–gene network
The KEGG database provides copious and complex pathway structure information and is widely used in pathway analysis [4–6]. We analysed all 219 pathways, including those for metabolism, genetic information processing, environmental information processing, cellular processes, organism systems and human diseases, from the KEGG database (release 56.1). Each pathway can be converted to a gene–gene network on the basis of the relationships such as reactions, modifications and binding involved in the pathway. Information on pathway crosstalk can be derived from the genes that overlap between pathways [16,17]. We constructed a global gene–gene network as follows. The relationships of genes from the XML files for each pathway in KEGG were extracted (ftp://ftp.genome.jp/pub/kegg/xml). Two genes were connected by an edge if there was a common compound in their corresponding reaction in a metabolic pathway, or if they had a relationship such as interaction, binding or modification indicated in the relation element of the XML file for a non-metabolic pathway. We then merged the genes that overlapped between each pair of pathways and retained the topological relationships of each pathway. A global gene–gene network reflecting the relationships both within and between pathways was therefore constructed using our developed ‘iSubpathwayMiner’ system [18,19]. This global network consisted of 4001 nodes and 32 940 edges, and could be represented by an undirected graph G.
2.3. Calculating the global dysregulated score
A GDS was defined to reflect the extent to which genes were affected by global influence from both internal pathway effects and crosstalk between pathways. We used the RWR algorithm [20] to calculate the GDS in the context of the gene expression data. The RWR algorithm simulates an iterative random walker that starts from a set of source nodes and travels to its immediate neighbours, or goes back to the source nodes at each time step in the graph. This algorithm, which captures global relationships within a network, can be used to compute the proximity of a node to a set of source nodes. From a systems-biology perspective, the genes that are located close to the dysregulated genes in the gene interaction network may be liable to perturbation. The properties of the RWR algorithm allow the identification of candidate perturbed genes in the network when a set of dysregulated genes is known [21].
In our application, the two-sample t-test was performed to evaluate the extent of differential expression (t-score) by comparing the expression values between normal and diseased samples. To reflect the information on specific disease processes, all genes represented in the expression data were mapped to the global network as source nodes. We modified the RWR algorithm by combining the extent of differential expression and the global network topology; the combination was used to calculate the GDS, reflecting the global influence of the gene on the source nodes. The formula for this modified algorithm is given as
| 2.1 |
where M is the column-normalized adjacency matrix of the global network graph G; 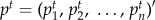 is the vector of nodes at time step t, and its ith element
is the vector of nodes at time step t, and its ith element 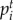 holds the probability of being at node i at time step t; n is the number of nodes (genes) in graph G (the global network).
holds the probability of being at node i at time step t; n is the number of nodes (genes) in graph G (the global network).
To start this algorithm, the initial probability vector 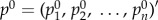 was constructed by assigning to each node its |t-score| (nodes with no t-score were assigned a value of zero), and normalized to a unit vector with
was constructed by assigning to each node its |t-score| (nodes with no t-score were assigned a value of zero), and normalized to a unit vector with 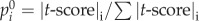 . This is equivalent to letting the random walker begin from a certain gene i with probability
. This is equivalent to letting the random walker begin from a certain gene i with probability 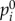 , which is proportional to its t-score. A gene i with higher
, which is proportional to its t-score. A gene i with higher 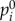 indicates that this gene possesses a greater likelihood of perturbing other genes. The parameter r is the restart probability, which controls the degree to which the random walker returns to the source nodes at every iteration. Within the interval (0.1–0.9), r has been demonstrated to have only a slight effect on the results of the RWR algorithm [21]. The restart probability r was set at 0.7 in this study. After certain steps, the probability pt will converge to a unique steady-state p∞, which was obtained by performing iterations until the difference between pt and p t+1 fell below 10−10. The global influence of genes (GDS) can be measured by the steady-state p∞. When
indicates that this gene possesses a greater likelihood of perturbing other genes. The parameter r is the restart probability, which controls the degree to which the random walker returns to the source nodes at every iteration. Within the interval (0.1–0.9), r has been demonstrated to have only a slight effect on the results of the RWR algorithm [21]. The restart probability r was set at 0.7 in this study. After certain steps, the probability pt will converge to a unique steady-state p∞, which was obtained by performing iterations until the difference between pt and p t+1 fell below 10−10. The global influence of genes (GDS) can be measured by the steady-state p∞. When 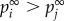 , the strength of the node (gene) i affected by global influence of internal pathway effects and pathway crosstalk is larger than node (gene) j. The GDS for gene i was assigned by the normalized
, the strength of the node (gene) i affected by global influence of internal pathway effects and pathway crosstalk is larger than node (gene) j. The GDS for gene i was assigned by the normalized 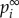 with:
with: 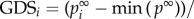
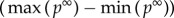 . In this way, each gene in the global network obtained a GDS. The GDSs for all other genes in the expression profiles, but not in the global network, were set at zero. The final GDS may reflect the extent to which genes are affected by both internal pathway effects and crosstalk between pathways.
. In this way, each gene in the global network obtained a GDS. The GDSs for all other genes in the expression profiles, but not in the global network, were set at zero. The final GDS may reflect the extent to which genes are affected by both internal pathway effects and crosstalk between pathways.
2.4. Identifying statistically significant dysregulated pathways
A gene list L = {g1, g2, … ,gn} was constructed by ranking all genes in the expression profiles by 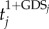 , where tj is the |t-score| of gene j and GDSj is the GDS of gene j. This ranked gene list reflects both differential expression between two classes and the influence of internal pathway effects and pathway crosstalk. Genes located higher in the list may be more correlated with a given phenotype. We used CDFs to calculate a dysregulated score for each pathway, which reflects the degree to which genes in a pathway are overrepresented at the top of the ranked list. Specifically, the genes in a given pathway P were mapped to the ranked list L. The CDFs of InP and NotP are used to evaluate the fraction of genes in P weighted by their correlation (
, where tj is the |t-score| of gene j and GDSj is the GDS of gene j. This ranked gene list reflects both differential expression between two classes and the influence of internal pathway effects and pathway crosstalk. Genes located higher in the list may be more correlated with a given phenotype. We used CDFs to calculate a dysregulated score for each pathway, which reflects the degree to which genes in a pathway are overrepresented at the top of the ranked list. Specifically, the genes in a given pathway P were mapped to the ranked list L. The CDFs of InP and NotP are used to evaluate the fraction of genes in P weighted by their correlation (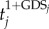 ), and the fraction of genes not in P present up to a given position i in L. The formulae are given as follows:
), and the fraction of genes not in P present up to a given position i in L. The formulae are given as follows:
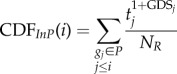 |
2.2 |
and
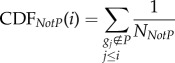 |
2.3 |
where 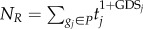 ; NNotP represents the number of genes in L not in P. With the position i walking down the list L, the dysregulated score of pathway P was calculated by
; NNotP represents the number of genes in L not in P. With the position i walking down the list L, the dysregulated score of pathway P was calculated by
| 2.4 |
It can be seen that when the GDSs for all genes are set at zero, the PAGI will be reduced to the GSEA [2].
To estimate the statistical significance (p-value) of the observed score, we performed a gene-based permutation test procedure that preserved the gene expression profiles in the data structure and permutated gene labels. Specifically, we redistributed the t-scores of the genes and recomputed the score of each pathway for the permutated data. The background distribution was generated after performing n permutations. The p-value was computed as p-value = m/n, where m is the number of scores greater than the observed score in the background distribution. We set n at 5000 times in this study. Because several pathways were involved in this analysis, it was necessary to perform multiple hypothesis-testing methods to control the proportion of false positives. We applied the false discovery rate (FDR) method proposed by Benjamini & Hochberg [22] to account for false positives.
3. Results
3.1. Evaluating the effect of the global dysregulated score
The GDS was defined to reflect the extent to which genes were affected by global influence from both internal pathway effects and crosstalk between pathways. We used the GDS to calculate the score of dysregulated pathways. In this study, we constructed a common global gene–gene network including 4001 nodes and 32 940 edges (figure 2a). The RWR algorithm, which exploits the complete network topology, was used to calculate the GDS. Each gene in the global network was assigned a GDS. For the breast cancer dataset (GSE15852), the GDSs across all genes in the global network ranged from 0 to 1, and the average GDS was 0.08 ± 0.07.
Figure 2.

Global gene–gene network. (a) Each gene was assigned a global dysregulation score calculated from the breast cancer dataset. The scale of the global scores ranged from 0 to 1, and genes with larger scores were marked with darker colour. Two blue ellipses correspond to the regions of the insulin signalling pathway and PPAR signalling pathway, respectively. (b–d) The zoom-in plots correspond to the regions around GNAL, RXRA and CTNNB1, respectively. (Online version in colour.)
Our results demonstrated that the GDS was able to reveal the GDS from both internal pathway effects and crosstalk between pathways. Of the 10 genes with the highest GDSs, four genes did not have high rank according to the |t-score|; however, their functions were associated with roles in cancer such as signal transduction, apoptosis and the inflammatory response, according to GO and KEGG annotation (electronic supplementary material, table S2). These results indicate that the GDS was not determined exclusively by the extent of association between genes and phenotype, and the GDS may thus provide further meaningful biological results.
We further tested the topological properties of the genes with high GDS values, but low t-score in the global network. Specifically, we noted that these genes, including the guanine nucleotide-binding protein G subunit α (GNAL), cadherin-associated protein (CTNNB1) and retinoid X receptor α (RXRA), tended to be of high degree in the global network (figure 2b–d). Moreover, these genes were located close to the dysregulated genes. For example, the gene with the third highest GDS, CTNNB1, interacted with up to 50 genes (figure 2d). Its neighbour genes, insulin-like growth factor receptor (IGF1R) and epidermal growth factor receptor (EGFR), obtained high GDSs of 0.24 and 0.33, respectively. Interestingly, IGF1R and EGFR are implicated in tumour development through their effects on cell proliferation, angiogenesis and inflammation, and their crosstalk increases the metastatic potential of breast tumours [23,24]. These results indicate that genes with high GDS but low t-score may be iteratively perturbed by multiple dysregulated genes, both within and outside the pathway. Although some of these genes had low t-scores, their functions may be altered by interaction and crosstalk during the process of cancer development.
3.2. Identifying dysregulated pathways associated with breast cancer
We first applied PAGI to a breast cancer dataset (GSE15852) to demonstrate the effectiveness of the method in identifying dysregulated pathways. We also analysed this dataset using GSEA for comparison. PAGI identified 21 pathways with FDR values less than 0.01 after 5000 permutations, of which 85% (18/21) are supported by the existing literature (table 1). The full list of ranked pathways and the GDSs of the genes in each significant pathway are presented in the electronic supplementary material, table S3. We also applied GSEA to the same dataset and identified 10 significant pathways (FDR < 0.01). Comparison of the PAGI and GSEA results showed that these two methods shared five pathways, whereas PAGI exclusively identified 16 pathways (electronic supplementary material, figure S1). Interestingly, 13 of the 16 pathways identified exclusively by PAGI have previously been associated with the initiation and progression of breast cancer in the literature (table 1). For example, alterations in the pathway of retinol metabolism in cells play an important role in the differential responsiveness to retinoid of normal human mammary epithelial cells and breast cancer cells [25], whereas the tryptophan metabolism pathway is linked to tumoral immunoresistance and malignant progression in breast cancer [26]. Moreover, most of the pathways identified simultaneously by both methods were more significant in PAGI than in GSEA (electronic supplementary material, figure S1). These results suggest that PAGI was able to identify more pathways associated with breast cancer than was GSEA. There were also five pathways, namely spliceosome, ribosome, proteasome, base excision repair and aminoacyl–tRNA biosynthesis pathways, that were identified by GSEA, but not by PAGI. This is because PAGI uses topological information to identify pathways, and there were very few relationships such as reactions, modifications or binding between the genes in these pathways. Moreover, most genes in these pathways were not included in the global gene–gene network. These results suggest that the genes in these pathways may not be affected by both internal pathway effects and pathway crosstalk. Thus, PAGI may fail to identify pathways if there is incomplete information on genes or the topological relationships among genes in the pathways.
Table 1.
Pathways identified by PAGI with FDR < 0.01 in the breast cancer dataset.
| pathway | sizea | score | FDR | reference (PMID) |
|---|---|---|---|---|
| PPAR signalling pathwayb | 61 | 0.79 | <0.001 | 18645617 |
| insulin signalling pathway | 128 | 0.59 | <0.001 | — |
| adipocytokine signalling pathwayb | 61 | 0.61 | <0.001 | 16436010; 15245384 |
| pathways in cancer | 307 | 0.48 | <0.001 | — |
| thyroid cancer | 29 | 0.75 | <0.001 | — |
| fatty acid metabolismb | 39 | 0.70 | <0.001 | 17902053 |
| pyruvate metabolism | 34 | 0.69 | <0.001 | 19826085; 22236875 |
| propanoate metabolism | 28 | 0.73 | <0.001 | 20831783 |
| focal adhesion | 191 | 0.50 | 0.0016 | 21832234 |
| tyrosine metabolism | 38 | 0.68 | 0.0016 | 21376233; 22388088 |
| glutathione metabolism | 41 | 0.63 | 0.0026 | 22545423; 11414197 |
| retinol metabolism | 43 | 0.63 | 0.0026 | 12038710; 9377581 |
| metabolism of xenobiotics by cytochrome P450b | 50 | 0.64 | 0.0036 | 9472688 |
| ECM–receptor interaction | 80 | 0.55 | 0.0045 | 18177501; 21718500 |
| Fc gamma R-mediated phagocytosis | 83 | 0.55 | 0.0052 | 7909275 |
| MAPK signalling pathway | 247 | 0.45 | 0.0068 | 14623520; 21258408 |
| cell cycle | 115 | 0.51 | 0.0068 | 9652762; 16267837 |
| complement and coagulation cascadesb | 66 | 0.57 | 0.0068 | 21718500 |
| progesterone-mediated oocyte maturation | 77 | 0.55 | 0.0068 | 20540763 |
| tryptophan metabolism | 35 | 0.63 | 0.0068 | 21615916 |
| glycerolipid metabolism | 39 | 0.64 | 0.0068 | 18606873 |
aThe number of genes which were mapped to the pathway from gene expression profiles.
bThe pathways identified by GSEA.
The most significant pathway was the peroxisome proliferator-activated receptor (PPAR) signalling pathway. The PPAR signalling pathway has recently been found to be related to cell growth and to induce apoptosis in breast cancer [27]. We found that most of the genes in this pathway had high GDSs (right blue ellipse in figure 2a). Moreover, most of these high GDS genes were associated with breast cancer development. For example, the PPAR γ and PPAR α (indicated by blue stars in figure 3) had high GDSs, 0.63 and 0.25, respectively, compared with an average GDS across all genes in the global network of 0.08. PPAR γ and PPAR α are ligand-inducible transcription factors that are implicated in a diverse range of biological processes such as cancer development [27]. Moreover, the retinoid X receptor RXRA is bound by PPAR γ as a heterodimeric partner to specific DNA sequence elements in this pathway (figure 3). RXRA, which had a high GDS of 0.48, has been reported to be a therapeutic target in breast cancer cell lines [28]. In addition, genes such as ANGPTL4, LPL and PLIN1 that are affected by PPAR γ and RXRA also showed high GDSs (0.47, 0.46 and 0.41, respectively). The large number of genes with high GDSs indicates significant dysregulation associated with cancer, and suggests that PAGI, which weights the genes according to GDS, is a suitable method for identifying the PPAR signalling pathway.
Figure 3.

PPAR signalling pathway. Genes with global dysregulation scores (GDSs) are annotated. The GDSs ranged from 0 to 1, and nodes with higher GDS are indicated by darker colour. (Online version in colour.)
The insulin signalling pathway was also significant, and many genes in this pathway also had high GDSs (left blue ellipse in figure 2a); most of them have been associated with breast cancer in the literature. Specifically, acetyl-CoA carboxylase B (ACACB) and phosphodiesterase 3B (PDE3B; indicated by blue stars in the electronic supplementary material, figure S2) both had high GDSs of 0.44, and both have been implicated in breast cancer progression [29,30]. Other genes such as PCK1, SORBS1and FOXO1 also had high GDSs, 0.42, 0.41 and 0.31, respectively. The presence of multiple genes with high GDSs indicates a significant perturbation in the pathway associated with breast tumours. Moreover, it was notable that genes in this pathway (left blue ellipse) had extensive connections with genes outside the pathway (figure 2a), suggesting that the genes within this pathway are susceptible to perturbation by genes in other pathways. Pathway crosstalk may play a major role in altering the activity of this pathway involved in breast cancer progression. However, this pathway was not identified as significant by GSEA, indicating that the consideration of pathway crosstalk incorporated in PAGI allowed the effective identification of the insulin signalling pathway.
3.3. Identifying dysregulated pathways associated with prostate cancer
We also illustrated the effectiveness of PAGI in identifying dysregulated pathways in a prostate cancer dataset (GSE3868). In this dataset, the average GDS across all genes in the network was 0.09 ± 0.08. PAGI identified nine pathways with FDR values less than 0.01 (see table 2 and electronic supplementary material, table S4), whereas GSEA failed to identify any pathways. Of the nine significant pathways identified by PAGI, five are supported by evidence from the existing literature (table 2). To compare the ability of PAGI and GSEA to identify dysregulated pathways, we also examined the ranks of the five pathways associated with prostate cancer in GSEA, and found that most were ranked more than 25 (table 2). These dysregulated pathways might therefore be ignored by GSEA from a rank-based perspective.
Table 2.
Pathways identified by PAGI with FDR < 0.01 in the prostate cancer dataset.
| pathway | PAGI FDR | GSEA FDR | PAGI rank | GSEA rank | reference (PMID) |
|---|---|---|---|---|---|
| MAPK signalling pathway | <0.001 | 0.88 | 1 | 96 | 9927031;12466969 |
| focal adhesion | <0.001 | 0.47 | 2 | 26 | 20160039;18922979 |
| ECM–receptor interaction | <0.001 | 0.15 | 3 | 6 | 18792917;14711377 |
| protein digestion and absorption | <0.001 | 0.14 | 4 | 5 | — |
| amoebiasis | <0.001 | 0.79 | 5 | 76 | — |
| pathways in cancer | <0.001 | 0.79 | 6 | 77 | — |
| Wnt signalling pathway | 0.005 | 0.70 | 7 | 42 | 15809669;18673243 |
| Jak–STAT signalling pathway | 0.007 | 0.73 | 8 | 59 | 11948098;10728680 |
| aldosterone-regulated sodium reabsorption | 0.007 | 0.63 | 9 | 31 | — |
The most significant pathway identified by PAGI was the mitogen-activated protein kinase (MAPK) signalling pathway. In this pathway, a high proportion of genes have relatively large differential expression level (|t-score|) and GDS (electronic supplementary material, table S5). In detail, up to 62% of genes had higher GDSs than the average value across all genes in the network (0.09). The fact that the pathway includes multiple genes with high GDSs indicates that the pathway may tend to be influenced by both pathway crosstalk and internal pathway effects. Moreover, these genes with high GDSs had a relatively large amount of differential expression (|t-scores|). Interestingly, we found that the genes in this pathway significantly clustered at the top of the ranked gene list L (electronic supplementary material, figure S3). These observations indicate a strong connection between prostate cancer and the MAPK signalling pathway. Indeed, it has been demonstrated that activation of this pathway and increased MAPK levels induce androgen-independent prostate cancer progression [31].
We further tested the effect of crosstalk between significant pathways (electronic supplementary material, figure S4) and found that the MAPK signalling pathway and focal adhesion pathway shared a relatively high number of genes, particularly genes encoding members of the MAPK family, such as MAPK1, MAPK2 and MAPK10. These genes had significantly high GDSs (0.41, 0.40 and 0.27, respectively). Interestingly, MAPKs play a prominent role in regulating focal adhesion signalling proteins in prostate cancer cells, and crosstalk between the MAPK signalling pathway and the focal adhesion pathway might control invasive and clonogenic phenotypes in androgen-independent prostate cancer [32]. Moreover, the focal adhesion pathway and extracellular matrix (ECM)–receptor interaction pathway also shared a significant number of genes (electronic supplementary material, figure S4), most of which, such as integrin α6 (ITGA6), collagen 1A1 (COL1A1) and collagen 1A2 (COL1A2), had significantly high GDSs (0.32, 0.22 and 0.32, respectively) and have been implicated in the metastasis of prostate cancer [33,34]. Interestingly, the crosstalk between these two pathways may play an important role in the regulation of prostate tumour cell migration [35].
3.4. Comparison of pathway analysis based on global influence with other methods
To confirm the power of PAGI in identifying dysregulated pathways, we also applied hypergeometric tests, GSEA, SPIA, PWEA, LPIA and PathNet to the prostate cancer dataset (GSE3868) and breast cancer dataset (GSE15852). With FDR < 0.01 as the pathway significance threshold, 16 statistically significant pathways were identified by all the above methods in the prostate cancer dataset (table 3). In detail, the hypergeometric test identified four significant pathways, all of which were also identified by PAGI. GSEA and PWEA did not identify any statistically significant pathways. However, PAGI identified nine significant pathways. PAGI, which integrates both internal pathway effects and pathway crosstalk, may improve the power for the identification of dysregulated pathways. Meanwhile, SPIA identified six significant pathways, three of which were identified by PAGI; however, PAGI identified an additional six pathways not identified by SPIA. The power of LPIA and PathNet also seemed to be limited. LPIA and PathNet found only one and four significant pathways, respectively. By comparing the results of PAGI with other methods in the prostate cancer dataset, we found that PAGI identified four statistically significant pathways with FDR < 0.01, which were simultaneously missed by other methods (table 3). Surprisingly, most of these pathways, such as the MAPK signalling pathway, Wnt signalling pathway and Jak–STAT signalling pathway, have been well reported to be associated with prostate cancer [31,36,37]. Similarly, in the breast cancer dataset, PAGI identified six statistically significant pathways which were simultaneously missed by the other methods (electronic supplementary material, table S6).
Table 3.
Pathways identified by seven methods (hypergeometric test, GSEA, SPIA, PWEA, LPIA, PathNet, PAGI) with FDR < 0.01 in the prostate cancer dataset.
| pathway | hypergeometric testa | GSEA | SPIA | PWEA | LPIA | PathNet | PAGI |
|---|---|---|---|---|---|---|---|
| MAPK signalling pathway | √ | ||||||
| focal adhesion | √ | √ | √ | ||||
| ECM–receptor interaction | √ | √ | √ | ||||
| protein digestion and absorption | √ | √ | |||||
| amoebiasis | √ | √ | √ | ||||
| pathways in cancer | √ | √ | |||||
| Wnt signalling pathway | √ | ||||||
| Jak–STAT signalling pathway | √ | ||||||
| aldosterone-regulated sodium reabsorption | √ | ||||||
| melanoma | √ | ||||||
| NF-kappa B signalling pathway | √ | ||||||
| osteoclast differentiation | √ | ||||||
| cell adhesion molecules (CAMs) | √ | ||||||
| leucocyte transendothelial migration | √ | ||||||
| ribosome | √ | ||||||
| tight junction | √ |
aThe t-test was used to perform differential expression analysis and genes with FDR < 0.05 were used in the hypergeometric test.
3.5. Reproducibility of the pathway analysis based on global influence method
To test the reproducibility of the results across different datasets, we applied the PAGI method to three independent lung cancer datasets (GSE7670, GSE10072 and GSE2514). With FDR < 0.01 as the pathway significance threshold, PAGI identified 29, 33 and 26 significant pathways, respectively, 15 of which were reproducible across these results (electronic supplementary material, table S7). Most of these reproducible pathways have been reported to be associated with the occurrence and development of lung cancer. For example, Soini et al. [38] proposed that tight junctions and their proteins may influence lung tumour spread, and Hembruff & Cheng [39] explained that the chemokine signalling pathway plays an important role in regulating the cancer microenvironment and cancer progression. We further calculated the ratio of reproducible pathways to statistically significant pathways for each dataset, and the results showed that the average ratio for PAGI was up to 52%. Moreover, we compared the reproducibility of PAGI with that of the other methods (GSEA, SPIA, PWEA, LPIA and PathNet). We applied each of these methods to the three lung cancer datasets, and the top 30 pathways from each lung dataset were used to test how many pathways were reproducible. PAGI identified 16 reproducible pathways across the three lung cancer datasets, more than the other methods (electronic supplementary material, figure S5). We also compared the reproducibility of PAGI with that of the other methods in three breast cancer datasets (GSE15852, GSE29431 and GSE42568) and three prostate cancer datasets (GSE3868, GSE3325 and GSE26910). The results showed that PAGI had greater reproducibility than the other methods (electronic supplementary material, figure S5).
3.6. Robustness of the pathway analysis based on global influence method
We tested the robustness of the PAGI method by performing data removal tests using the prostate cancer dataset (GSE3868), breast cancer dataset (GSE15852) and lung cancer dataset (GSE7670). For each set of gene expression data, we removed the gene expression values from 5% to 30%, at 5% intervals, and repeated the PAGI method 20 times for each removal (electronic supplementary material, figure S6). In the prostate cancer dataset, the number of overlapped significant pathways (FDR < 0.01) fell slowly compared with the original data, and the ratio of overlapped pathways to original significant pathways remained above 75%, even after removal of up to 30% of the expression data (electronic supplementary material, figure S6a). These results indicate that the PAGI method is robust to data removal. We also performed the same operation using the other methods (GSEA, SPIA, PWEA, LPIA and PathNet) in the prostate cancer dataset. Although some of these methods also seemed to be robust, the number of overlapped significant pathways tended to zero when the percentage of removed gene expression values tended to 30% (electronic supplementary material, figure S6a). Moreover, we tested the robustness of the PAGI and other methods in the breast cancer and lung cancer datasets, and obtained similar results (electronic supplementary material, figure S6b,c).
4. Discussion
Complex diseases are currently thought to arise from multiple dysregulated genes rather from than individual genes, and these dysregulated genes may jointly alter some biological functions of pathways. Network-based methods have been demonstrated to be effective in detecting the correlations between pathways (or genes) and disease phenotypes from high-throughput experimental data [40,41]. We propose a novel network-based approach, PAGI, to detect latent dysregulated pathways by considering the global influence of both the internal effects of pathways and crosstalk between pathways. In this study, considering the dysregulated genes result in the occurrence and development of complex diseases, we performed the two-sample t-test to evaluate the extent of differential expression (t-score). We then produced a global gene–gene network by collecting the inherent relationships among genes embedded in all the pathways and the genes that overlap between pathways. This global network can reflect the inter- and intrapathway relationships. To reflect the information on specific disease processes, we mapped all genes represented in the expression data to the global network. In biology, the dysregulated genes will iteratively influence the status of other genes in and out of the pathways through both the internal effects of pathways and crosstalk between pathways. And the genes with a larger differential expression level will more strongly influence other genes. The modified RWR algorithm, which combines the global network topology and the extent of differential gene expression, was used to calculate the extent to which genes were affected by the global influence (internal effects of pathways and pathway crosstalk). The fact that a pathway includes multiple genes with a large differential expression level and global influence extent indicates that the pathway may tend to be correlated with a given phenotype. The CDFs, deemed to be a modification of GSEA, were used to determine the significance of the pathways (see Material and methods).
From the biological-systems perspective, pathways are not isolated and usually interact with each other [3]. Pathway crosstalk, which refers to the phenomenon of interaction or cooperation between pathways, provides the necessary insights into linked cellular processes and can further our understanding of alterations of biological states [7,8]. To study pathway crosstalk, we used a more direct strategy, which was based on the genes that overlap between pathways, to connect each pair of pathways. The overlapped genes were used as media for propagating the influence of the dysregulated genes from one pathway to another pathway. Although the overlapped genes were shared by different pathways, they would perform a different biological function with other genes in different pathways. The RWR algorithm simulates a random walk that starts from a set of source nodes, and iteratively propagates to other nodes in the global network. We used this algorithm to evaluate the GDS, which reflects the extent to which genes were affected by both pathway crosstalk and internal pathway effects. Although the pathway crosstalk can also be evaluated by the interactions among genes/proteins from different pathways, the RWR algorithm is able to evaluate this kind of pathway crosstalk effect on the basis of the propagation of dysregulated genes in the global network.
Recent pathway-identification methods, such as PWEA [5] and SPIA [6], have proven useful and efficient in identifying dysregulated pathways; however, most of these focus on the internal effects of a single pathway and fail to take account of crosstalk (interaction or cooperation) between pathways. Although LPIA [8] and PathNet [9] consider the relationships between pathways in identifying dysregulated pathways, LPIA does not consider pathway topology within the pathways and PathNet considers only the association between genes based on their direct pathway neighbours and neglects the effect of other genes. PAGI, which combines internal pathway effects and pathway crosstalk, is able to recall dysregulated pathways associated with complex diseases effectively, with strong reproducibility and robustness. By comparing the results of PAGI and other methods (the hypergeometric test, GSEA, PWEA, SPIA, LPIA and PathNet) in prostate cancer and breast cancer datasets (table 3 and electronic supplementary material, table S6), we showed that PAGI possessed greater power to identify dysregulated pathways. PAGI may thus complement existing pathway-identification methods, and may help to characterize the comprehensive alterations occurring during disease progression.
PAGI identifies dysregulated pathways using pathway topological information. Pathway topologies can help to provide more detailed and comprehensive biological insights. However, we also found that, although most pathways in the pathway database have good topological information, a few pathways, such as the spliceosome, ribosome and proteasome pathways, still lack information on the relationships among genes. PAGI may therefore fail to identify these pathways effectively because of incomplete information on pathway topologies. This is similar to the challenge faced by other pathway-identification methods that apply topological pathway information [5]. Improved ways to use pathway topologies to identify dysregulated pathways have become a major focus of research, and the sensitivity of PAGI will be further improved by the addition of new pathway topological information. We have implemented our method as an R-based package, which is publicly available on CRAN (http://cran.r-project.org/web/packages/PAGI), and as a Web-based tool (http://bioinfo.hrbmu.edu.cn/PAGI). Input of expression profiles with two biological states can produce information on dysregulated pathways within a few minutes. PAGI based on the internal effect of pathways and crosstalk between pathways will be a valuable tool to help biologists identify dysregulated pathways associated with complex diseases.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgements
We thank all the research staff for their contributions to this project.
Funding statement
This work was supported in part by the National Natural Science Foundation of China (grant nos. 61170154, 31200996 and 31401127) and the Heilongjiang Province Health Department of Scientific Research Projects (grant no. 2012-804).
References
- 1.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. 2012. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114. ( 10.1093/nar/gkr988) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Subramanian A, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15 545–15 550. ( 10.1073/pnas.0506580102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khatri P, Sirota M, Butte AJ. 2012. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput. Biol. 8, e1002375 ( 10.1371/journal.pcbi.1002375) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rahnenfuhrer J, Domingues FS, Maydt J, Lengauer T. 2004. Calculating the statistical significance of changes in pathway activity from gene expression data. Stat. Appl. Genet. Mol. Biol. 3, 16 ( 10.2202/1544-6115.1055) [DOI] [PubMed] [Google Scholar]
- 5.Hung JH, Whitfield TW, Yang TH, Hu Z, Weng Z, DeLisi C. 2010. Identification of functional modules that correlate with phenotypic difference: the influence of network topology. Genome Biol. 11, R23 ( 10.1186/gb-2010-11-2-r23) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tarca AL, Draghici S, Khatri P, Hassan SS, Mittal P, Kim JS, Kim CJ, Kusanovic JP, Romero R. 2009. A novel signaling pathway impact analysis. Bioinformatics 25, 75–82. ( 10.1093/bioinformatics/btn577) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, Agarwal P, Rajagopalan D. 2008. A global pathway crosstalk network. Bioinformatics 24, 1442–1447. ( 10.1093/bioinformatics/btn200) [DOI] [PubMed] [Google Scholar]
- 8.Pham L, Christadore L, Schaus S, Kolaczyk ED. 2011. Network-based prediction for sources of transcriptional dysregulation using latent pathway identification analysis. Proc. Natl Acad. Sci. USA 108, 13 347–13 352. ( 10.1073/pnas.1100891108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dutta B, Wallqvist A, Reifman J. 2012. PathNet: a tool for pathway analysis using topological information. Source Code Biol. Med. 7, 10 ( 10.1186/1751-0473-7-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang JM, Wu JT, Sun DK, Zhang P, Wang L. 2012. Pathway crosstalk analysis based on protein-protein network analysis in prostate cancer. Eur. Rev. Med. Pharmacol. Sci. 16, 1235–1242. [PubMed] [Google Scholar]
- 11.Pau Ni IB, Zakaria Z, Muhammad R, Abdullah N, Ibrahim N, Aina Emran N, Hisham Abdullah N, Syed Hussain SN. 2010. Gene expression patterns distinguish breast carcinomas from normal breast tissues: the Malaysian context. Pathol. Res. Pract. 206, 223–228. ( 10.1016/j.prp.2009.11.006) [DOI] [PubMed] [Google Scholar]
- 12.Nanni S, et al. 2006. Epithelial-restricted gene profile of primary cultures from human prostate tumors: a molecular approach to predict clinical behavior of prostate cancer. Mol. Cancer Res. 4, 79–92. ( 10.1158/1541-7786.MCR-05-0098) [DOI] [PubMed] [Google Scholar]
- 13.Su LJ, et al. 2007. Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics 8, 140 ( 10.1186/1471-2164-8-140) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Landi MT, et al. 2008. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS ONE 3, e1651 ( 10.1371/journal.pone.0001651) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stearman RS, et al. 2005. Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen-induced murine model. Am. J. Pathol. 167, 1763–1775. ( 10.1016/S0002-9440(10)61257-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parikh JR, Xia Y, Marto JA. 2012. Multi-edge gene set networks reveal novel insights into global relationships between biological themes. PLoS ONE 7, e45211 ( 10.1371/journal.pone.0045211) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu ZP, Wang Y, Zhang XS, Chen L. 2010. Identifying dysfunctional crosstalk of pathways in various regions of Alzheimer's disease brains. BMC Syst. Biol. 4 (Suppl. 2), S11 ( 10.1186/1752-0509-4-S2-S11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li C, et al. 2009. SubpathwayMiner: a software package for flexible identification of pathways. Nucleic Acids Res. 37, e131 ( 10.1093/nar/gkp667) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li C, et al. 2013. Subpathway-GM: identification of metabolic subpathways via joint power of interesting genes and metabolites and their topologies within pathways. Nucleic Acids Res. 41, e101 ( 10.1093/nar/gkt161) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Can T, Çamoğlu O, Singh AK. 2005. Analysis of protein–protein interaction networks using random walks. In BIOKDD '05: Proc. 5th Int. Workshop on Bioinformatics, Chicago, IL, 21 August 2005, pp. 61–68. New York, NY: Association for Computing Machinery. [Google Scholar]
- 21.Kohler S, Bauer S, Horn D, Robinson PN. 2008. Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet. 82, 949–958. ( 10.1016/j.ajhg.2008.02.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B (Methodol.) 57, 289–300. [Google Scholar]
- 23.van der Veeken J, Oliveira S, Schiffelers RM, Storm G, van Bergen En Henegouwen PM, Roovers RC. 2009. Crosstalk between epidermal growth factor receptor- and insulin-like growth factor-1 receptor signaling: implications for cancer therapy. Curr. Cancer Drug Targets 9, 748–760. ( 10.2174/156800909789271495) [DOI] [PubMed] [Google Scholar]
- 24.Riedemann J, Takiguchi M, Sohail M, Macaulay VM. 2007. The EGF receptor interacts with the type 1 IGF receptor and regulates its stability. Biochem. Biophys. Res. Commun. 355, 707–714. ( 10.1016/j.bbrc.2007.02.012) [DOI] [PubMed] [Google Scholar]
- 25.Hayden LJ, Satre MA. 2002. Alterations in cellular retinol metabolism contribute to differential retinoid responsiveness in normal human mammary epithelial cells versus breast cancer cells. Breast Cancer Res. Treat. 72, 95–105. ( 10.1023/A:1014815112078) [DOI] [PubMed] [Google Scholar]
- 26.Juhasz C, et al. 2012. Tryptophan metabolism in breast cancers: molecular imaging and immunohistochemistry studies. Nucl. Med. Biol. 39, 926–932. ( 10.1016/j.nucmedbio.2012.01.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baranova A. 2008. PPAR ligands as potential modifiers of breast carcinoma outcomes. PPAR Res. 2008, 230893 ( 10.1155/2008/230893) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crowe DL, Chandraratna RA. 2004. A retinoid X receptor (RXR)-selective retinoid reveals that RXR-alpha is potentially a therapeutic target in breast cancer cell lines, and that it potentiates antiproliferative and apoptotic responses to peroxisome proliferator-activated receptor ligands. Breast Cancer Res. 6, R546–R555. ( 10.1186/bcr913) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hadad SM, et al. 2009. Histological evaluation of AMPK signalling in primary breast cancer. BMC Cancer 9, 307 ( 10.1186/1471-2407-9-307) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hadad S, et al. 2011. Evidence for biological effects of metformin in operable breast cancer: a pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res. Treat. 128, 783–794. ( 10.1007/s10549-011-1612-1) [DOI] [PubMed] [Google Scholar]
- 31.Gioeli D, Mandell JW, Petroni GR, Frierson HF, Jr, Weber MJ. 1999. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res. 59, 279–284. [PubMed] [Google Scholar]
- 32.Johnson TR. et al 2008. Focal adhesion kinase controls aggressive phenotype of androgen-independent prostate cancer. Mol. Cancer Res. 6, 1639–1648. ( 10.1158/1541-7786.MCR-08-0052) [DOI] [PubMed] [Google Scholar]
- 33.Ports MO, Nagle RB, Pond GD, Cress AE. 2009. Extracellular engagement of alpha6 integrin inhibited urokinase-type plasminogen activator-mediated cleavage and delayed human prostate bone metastasis. Cancer Res. 69, 5007–5014. ( 10.1158/0008-5472.CAN-09-0354) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kiefer JA, Farach-Carson MC. 2001. Type I collagen-mediated proliferation of PC3 prostate carcinoma cell line: implications for enhanced growth in the bone microenvironment. Matrix Biol. 20, 429–437. ( 10.1016/S0945-053X(01)00159-7) [DOI] [PubMed] [Google Scholar]
- 35.Slack JK, Adams RB, Rovin JD, Bissonette EA, Stoker CE, Parsons JT. 2001. Alterations in the focal adhesion kinase/Src signal transduction pathway correlate with increased migratory capacity of prostate carcinoma cells. Oncogene 20, 1152–1163. ( 10.1038/sj.onc.1204208) [DOI] [PubMed] [Google Scholar]
- 36.Robinson DR, Zylstra CR, Williams BO. 2008. Wnt signaling and prostate cancer. Curr. Drug Targets 9, 571–580. ( 10.2174/138945008784911831) [DOI] [PubMed] [Google Scholar]
- 37.Buettner R, Mora LB, Jove R. 2002. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. 8, 945–954. [PubMed] [Google Scholar]
- 38.Soini Y. 2012. Tight junctions in lung cancer and lung metastasis: a review. Int. J. Clin. Exp. Pathol. 5, 126–136. [PMC free article] [PubMed] [Google Scholar]
- 39.Hembruff SL, Cheng N. 2009. Chemokine signaling in cancer: implications on the tumor microenvironment and therapeutic targeting. Cancer Ther. 7, 254–267. [PMC free article] [PubMed] [Google Scholar]
- 40.Liu ZP, Zhang W, Horimoto K, Chen L. 2013. Gaussian graphical model for identifying significantly responsive regulatory networks from time course high-throughput data. IET Syst. Biol. 7, 143–152. ( 10.1049/iet-syb.2012.0062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu ZP, Wang Y, Zhang XS, Chen L. 2012. Network-based analysis of complex diseases. IET Syst. Biol. 6, 22–33. ( 10.1049/iet-syb.2010.0052) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.